

ABSTRACT
As new pathogenic viruses continue to emerge, it is paramount to have intervention strategies that target a common denominator in these pathogens. The fusion of viral and cellular membranes during viral entry is one such process that is used by many pathogenic viruses, including chikungunya virus, West Nile virus, and influenza virus. Obatoclax, a small-molecule antagonist of the Bcl-2 family of proteins, was previously determined to have activity against influenza A virus and also Sindbis virus. Here, we report it to be active against alphaviruses, like chikungunya virus (50% effective concentration [EC50] = 0.03 μM) and Semliki Forest virus (SFV; EC50 = 0.11 μM). Obatoclax inhibited viral entry processes in an SFV temperature-sensitive mutant entry assay. A neutral red retention assay revealed that obatoclax induces the rapid neutralization of the acidic environment of endolysosomal vesicles and thereby most likely inhibits viral fusion. Characterization of escape mutants revealed that the L369I mutation in the SFV E1 fusion protein was sufficient to confer partial resistance against obatoclax. Other inhibitors that target the Bcl-2 family of antiapoptotic proteins inhibited neither viral entry nor endolysosomal acidification, suggesting that the antiviral mechanism of obatoclax does not depend on its anticancer targets. Obatoclax inhibited the growth of flaviviruses, like Zika virus, West Nile virus, and yellow fever virus, which require low pH for fusion, but not that of pH-independent picornaviruses, like coxsackievirus A9, echovirus 6, and echovirus 7. In conclusion, obatoclax is a novel inhibitor of endosomal acidification that prevents viral fusion and that could be pursued as a potential broad-spectrum antiviral candidate.
KEYWORDS: Zika virus, chikungunya virus, envelope protein, resistant mutant, virus entry, antiviral
INTRODUCTION
The Alphavirus genus of plus-strand RNA viruses belongs to the Togaviridae family and includes a number of human pathogens, like Sindbis virus (SINV) and chikungunya virus (CHIKV), and other zoonotic threats, like Venezuelan equine encephalitis virus. These mosquito-borne viruses cause either polyarthritis or encephalitis and can result in large-scale outbreaks in immunologically naive populations (1). In recent years, CHIKV has been in the limelight because of its spread to the Americas and has infected more than 1.5 million people since 2013 (2). There are no licensed vaccines or therapeutic drugs currently available to counter these viruses (3).
Semliki Forest virus (SFV) is a relatively less pathogenic well-studied alphavirus. Most of our knowledge about the composition and structure of the alphavirus particle and the functions of its different proteins stems from work done with SFV. The alphavirus particle is 70 nm in diameter and contains a nucleocapsid core surrounded by a lipid membrane envelope embedded with 80 trimeric spikes, with each spike being made up of three E1-E2 envelope protein heterodimers. The E2 envelope protein mediates viral entry by attachment to cellular receptors, followed by clathrin-mediated endocytic uptake, which delivers the viral particle to early endosomes. The low-pH environment of the endosome triggers a sequence of events starting with dissociation of the E1-E2 dimer and conformational changes in the E1 membrane fusion protein. This leads to insertion of the E1 fusion protein in the target membrane and homotrimer formation, steps that ultimately result in the formation of a fusion pore and release of the viral nucleocapsid into the cytosol (reviewed in references 4 and 5). This is followed by the intracellular steps of the viral infectious cycle, which culminate in progeny virions budding out from the infected cell.
The low-pH-mediated fusion of viral and cellular membranes is a common theme in many enveloped viruses from different families and has been explored as a target for antiviral therapy (6). The influenza A virus (IAV) hemagglutinin requires the low pH of the endosome for rearrangement and exposure of its fusion peptide (7). The acidic environment of the endosome also induces conformational changes in the flavivirus E glycoprotein (8), similar to the findings for alphavirus E1. Some nonenveloped viruses from the Picornaviridae family, like different strains of the human rhinoviruses and foot-and-mouth disease virus, also use low-pH cues for the uncoating of their capsid proteins and genome release, while some others, like poliovirus and coxsackievirus A9 (CV-A9), are pH independent (9, 10). Different classes of acidification inhibitors, like weak bases, ionophores, and vacuolar proton pump inhibitors, interfere with alphavirus infection (4) and have been important tools that have helped to decipher the low-pH requirement in the life cycles of different viruses. Previously, the antimalarial drug chloroquine, which is capable of elevating the endosomal pH, was shown to possess in vitro antiviral activity against CHIKV (11) and a number of other viruses, like the severe acute respiratory syndrome coronavirus (12), HIV, Ebola virus (EBOV) (13), and dengue virus (DENV). While chloroquine did not offer any protection or added benefit against CHIKV in rhesus macaques or human clinical trials (14), it has worked better against HIV in clinical trials (15) and showed in vivo activity against DENV (16) and EBOV (13). Niclosamide, an anthelminthic drug in clinical use, was described to inhibit the infection of pH-dependent human rhinoviruses and IAV by a similar mechanism (17) and was recently also shown to inhibit CHIKV (18). This suggests that the inhibition of endosomal acidification represents a target for the development of broad-spectrum antiviral compounds.
Obatoclax (OLX) is an anticancer compound that antagonizes the prosurvival Mcl-1 protein belonging to the Bcl-2 family of proteins. It triggers apoptosis in cancer cells by occupying a hydrophobic pocket in the BH3 binding groove of Bcl-2 proteins and interferes in their interaction with the proapoptotic Bak protein (19). Earlier, Denisova et al. identified OLX to be an antiviral compound in a targeted screen of host-directed compounds with activity against reporter IAV infection (20). They proposed that OLX inhibits IAV entry and uptake by inhibiting Mcl-1. In the same study, proof-of-principle results showing the antiviral action of OLX on SINV and, to a lesser extent, SFV were presented (20). Here, we endeavored to thoroughly evaluate the antiviral mechanism of OLX and show that OLX is equally effective against other alphaviruses, SFV and CHIKV, at submicromolar concentrations with high selectivity indices. OLX was found to affect early events in virus infection in an entry assay utilizing the SFV-ts9 temperature-sensitive mutant. Contrary to the previously proposed mechanism of OLX, we show here that OLX neutralizes the acidic environment of endocytic organelles and most likely inhibits the fusion of viral and endosomal membranes. In support of this mechanism, an escape mutation in the SFV E1 membrane fusion protein could confer partial resistance against OLX.
RESULTS
Obatoclax is an antiviral with activity against different alphaviruses.
OLX has previously been shown to act as a novel antiviral drug with activity against IAV. In the same study, OLX at a low concentration of 0.3 μM was suggested to be effective against SINV but not against SFV (20). In this study, we tested the activity of OLX against the reemerging CHIKV and used SINV and SFV as controls. BHK-21 cells were infected with wild-type SFV, SINV, and CHIKV at a multiplicity of infection (MOI) of 0.01 PFU/cell for 16 h in the presence or absence of 0.5 μM OLX. Viral titers from the supernatants of infected cells indicated that both SINV and CHIKV were inhibited by 5 logs and, contrary to the findings of the earlier study, SFV was inhibited by 3 to 4 logs (Fig. 1A). Next, we tested the activity of OLX against CHIKV at a higher MOI of 10 PFU/cell in BHK-21 cells for 6 h postinfection (p.i.) to assess the effectiveness of the compound in a single replication cycle. We employed OLX in a 2-fold dilution series at concentrations ranging from 2 μM to 0.125 μM in an assay in which OLX was present throughout the infection and compared the viral titers and the levels of viral RNA and protein synthesis to those for untreated samples and samples treated with 0.1% dimethyl sulfoxide (DMSO). Viral progeny production was reduced by 3 logs for the highest concentration tested of 2 μM, and a significant reduction was observed even at the lowest concentration tested of 0.125 μM (Fig. 1B). Expression of viral nonstructural and structural proteins was also severely diminished in a dose-dependent manner at concentrations down to 0.5 μM (Fig. 1C). Viral genomic and subgenomic RNA synthesis was also reduced to a large extent, with a similar trend of concentration-dependent inhibition being seen (Fig. 1D). Overall, these results indicate that OLX is a potent inhibitor of alphaviruses and dose dependently reduces viral titers and the levels of RNA and protein synthesis even in a high-MOI setting.
FIG 1.

Obatoclax is an effective antiviral against alphaviruses. (A) BHK-21 cells were infected with wild-type SFV, SINV, and CHIKV at an MOI of 0.01 PFU/cell with or without 0.5 μM OLX, which was present throughout the experiment when it was added. Plaque assay titers in cell culture supernatants collected at 16 h p.i. were determined in duplicate (data are means from three independent experiments ± SEMs). (B) BHK-21 cells were infected with wild-type CHIKV at an MOI of 10 PFU/cell for 6 h. Throughout the experiment, OLX was present in a 2-fold dilution series at concentrations ranging from 2 μM to 0.125 μM. Plaque assay titers in cell culture supernatants collected at 6 h p.i. were determined in duplicate (data are means from three independent experiments ± SEMs). Statistical significance was determined using a one-way ANOVA test (****, P < 0.0001). (C) Cell lysates were analyzed by Western blotting for the presence of the indicated CHIKV proteins. Actin was used as a loading control. (D) Total RNA was isolated from parallel wells, Northern blot analysis was done using probes specific to the 3′ UTR of the CHIKV genome, and the viral genomic and subgenomic RNAs were detected. The results are representative of those from two independent experiments.
Obatoclax is effective in multiple cell types at submicromolar concentrations.
Next, we assessed if OLX inhibits alphaviruses in multiple cell types. We used reporter SFV and CHIKV expressing Renilla luciferase (Rluc), and inhibition of the luciferase signal served as a readout of antiviral activity. Dose-response antiviral activity assays were performed in BHK-21 cells (MOI, 0.01 PFU/cell), Huh 7.5 cells (MOI, 0.1 PFU/cell), and HOS cells (MOI, 1 PFU/cell) using OLX at concentrations ranging from 100 μM to 0.006 μM. The time points used for the assays were chosen on the basis of the luciferase signals obtained for the different viruses: for BHK-21 cells, SFV and CHIKV were tested at 16 h; for Huh 7.5 cells, SFV was tested at 12 h and CHIKV was tested at 18 h; and for HOS cells, SFV was tested at 16 h and CHIKV was tested at 20 h. The concentrations causing 50% inhibition of viral replication (or the 50% effective concentration [EC50s]) were observed to be at submicromolar levels for OLX against both SFV and CHIKV in the different cell lines tested (Table 1), with the lowest EC50 of 0.03 μM (selectivity index = 670) being obtained against CHIKV in BHK-21 cells (Table 1) (inhibition curves are shown in Fig. S1 in the supplemental material). Cytotoxicity assays were done in parallel at the same concentrations to determine the concentration causing a 50% reduction in cell survival (or the 50% cytotoxic concentration [CC50]) for OLX in the different cell lines used. OLX showed time-dependent and cell type-specific toxicity, with Huh 7.5 cells treated for 18 h being the most sensitive (CC50 = 13.3 μM; Table 1). HOS cells were the least affected by OLX treatment and had a high CC50 value after 16 h of treatment (102.8 μM; Table 1) (cell survival curves are shown in Fig. S2). In summary, OLX is a novel antiviral with activity against alphaviruses and shows activity at submicromolar concentrations.
TABLE 1.
EC50s of obatoclax against SFV and CHIKV in different cell lines
| Cell line | SFV |
CHIKV |
||||
|---|---|---|---|---|---|---|
| EC50a (μM) | CC50b (μM) | SIc | EC50 (μM) | CC50 (μM) | SI | |
| BHK-21 | 0.23 ± 0.02 | 20.1 ± 4.8 | 87.4 | 0.03 ± 0.01 | 20.1 ± 4.8 | 670 |
| Huh 7.5 | 0.11 ± 0.01 | 42.7 ± 4.7 | 388.2 | 0.13 ± 0.01 | 13.1 ± 3.3 | 100.8 |
| HOS | 0.2 ± 0.06 | 102.8 ± 15.2 | 514 | 0.09 ± 0.01 | 45.4 ± 9.3 | 504.4 |
EC50, concentration causing 50% inhibition of viral replication.
CC50, concentration causing a 50% reduction in cell survival.
SI, selectivity index, which is the ratio of the CC50 to the EC50.
Obatoclax inhibits the early stages of alphavirus replication.
Next, we performed a time-of-addition assay with wild-type SFV and CHIKV to determine the stage of the viral life cycle being inhibited. In order to ensure that most of the cells were simultaneously infected, infections were done at an MOI of 1 PFU/cell. A total of 6 different times of addition were used: BHK-21 cells were treated with 0.5 μM OLX 2 h prior to infection, with treatment being continued throughout the course of the infection; only 2 h before infection; at the time of infection, with treatment being continued throughout the course of the infection; or at the time of infection but with OLX being present only during virus adsorption for 1 h p.i. The final two times of addition were chosen on the basis of the respective virus production curves. For SFV, OLX was added at 2 h and 4 h p.i. and the cell culture supernatants were harvested 8 h p.i. For CHIKV, the compound was added at 4 h and 8 h p.i. and the cell culture supernatants were collected 14 h p.i. (a schematic representation of the experimental layout is shown in Fig. 2A and B). The viral titers obtained in the supernatants of the respective samples were compared to those in the supernatants of untreated samples collected at the same time points. For SFV, the reduction in viral titers was most efficient when the cells were pretreated with OLX or when OLX was added simultaneously with the infectious inoculum and treatment with OLX was continued throughout the course of the infection (Fig. 2C). The inhibition was gradually reduced when the drug was added at later time points. Strong inhibition was seen when OLX was present only at early time points and not later, suggesting an effect on the early phase of the viral life cycle. A similar trend was also seen for CHIKV (Fig. 2D), suggesting that OLX has a similar mode of action against both SFV and CHIKV. Due to the predominant effect of OLX on viral titers when it was it was added prior to infection and also when it was present for only 1 h p.i. during adsorption of the infectious inoculum, we wondered if OLX could be virucidal in nature, inactivating the virus particle and thereby preventing the initiation of a productive infection. To this end, an undiluted CHIKV stock was treated with 0.5 μM OLX, 0.1% DMSO (negative control), or 0.1% disinfectant containing potassium peroxymonosulfate, sodium dodecylbenzenesulfonate, sulfamic acid, and inorganic buffers (Virkon; positive control) for 30 min at 37°C, followed by plaque titration on BHK-21 cells. Compared to the significant reduction in titers observed with 0.1% disinfectant treatment, no such difference was seen with OLX treatment (Fig. S3).
FIG 2.

Obatoclax inhibits early steps in alphavirus replication. (A and B) Layout of time-of-addition experiment with SFV and CHIKV. BHK-21 cells were infected with SFV (A) and CHIKV (B) at an MOI of 1 PFU/cell for 8 h and 14 h, respectively. OLX was present at 0.5 μM for the different times indicated for SFV (A) and CHIKV (B) infections. (C and D) Samples were collected from cultures receiving the different treatments at 8 h p.i. for SFV (C) and at 14 h p.i. for CHIKV (D) and subjected to titration by the plaque assay. The titers were compared to those of the untreated controls to obtain the log reduction in titers. The values are representative of those from two independent experiments, and error bars represent standard errors of the means.
Obatoclax inhibits virus entry.
Having ruled out any virucidal activity of OLX, the compound was tested in the previously described SFV entry assay (21) using the SFV-ts9 temperature-sensitive mutant. SFV-ts9 has a single point mutation in nonstructural protein 2 (nsP2; G389R) that results in multiple enzymatic defects in the N-terminal region of the protein and prevents replication at the elevated temperature of 39°C (22, 23). SFV-ts9 was coupled with an Rluc reporter (SFV-ts9–Rluc) and used to measure signals only from the initial RNA genomes that entered and were translated by the cellular machinery as a readout for viral entry. BHK-21 cells were infected with SFV-ts9–Rluc at 39°C and an MOI of 50 PFU/cell for 3 h and were either untreated or treated at the same time with 0.1% DMSO, 0.5 μM OLX, 25 μM chlorpromazine (a known entry inhibitor which causes defects in clathrin-mediated endocytosis) (24), or 50 nM bafilomycin A1 (an inhibitor of vacuolar ATPase [vATPase] that leads to an increase in the endosomal pH), which were present throughout the experiment (25, 26). OLX reduced the luciferase signals to almost negligible levels, similar to the signals achieved with bafilomycin A1, while chlorpromazine reduced the signals to ∼25% of the signals achieved with the untreated control (Fig. 3A). A time-of-addition assay was also performed with SFV-ts9. Pretreatment of cells with OLX for 120 min, 60 min, as well as 15 min prior to infection (treatment was discontinued postinfection) was sufficient to reduce the luciferase signals to a large extent. On the other hand, OLX inhibited virus entry to a lesser extent when it was added at 60 min p.i. than when it was added at the same time or 15 min p.i. When it was added at 120 min p.i., the inhibition was abolished (Fig. 3B). This set of results indicates that OLX, like bafilomycin A1, is an efficient inhibitor of SFV entry and the entry defect happens rapidly during 15 min of treatment of the cells.
FIG 3.

Obatoclax inhibits SFV entry. The results of entry assays with temperature-sensitive mutant SFV-ts9 are shown. BHK-21 cells were infected with SFV-ts9–Rluc at an MOI of 50 PFU/cell for 3 h at 39°C. At 3 h p.i., the cells were lysed and luciferase levels were measured. Values are expressed in relative luciferase units, which is the percentage of luciferase units compared to the number of luciferase units for untreated infected samples. Luciferase signals were measured from quadruplicate wells, and data are presented as means ± SEMs (n = 2). (A) Cells were treated with infection medium or infection medium containing 0.1% DMSO (solvent control), 0.5 μM OLX, 25 μM chlorpromazine, or 50 nM bafilomycin A1. (B) (Top) Schema for the experiment, where BHK-21 cells were treated with 0.5 μM OLX at different time points before and after infection. (Bottom) Luciferase activity measured after the treatment schedules shown in the top panel, identified by letters a to g, as compared to the untreated control set at 100%.
Obatoclax disrupts the low-pH environment of acidic organelles.
Due to the significant effect of OLX on virus entry, similar to that of bafilomycin A1, we investigated if the acidic environment of endosomal or lysosomal organelles is affected by OLX. The autofluorescent nature of OLX (emission peak, 490 nm; absorbance peak, 550 nm) (19) precluded any reliable conclusions from experiments using fluorescent pH-sensitive dyes from being made (data not shown). Instead, we used the vital stain neutral red for protonation and incorporation into acidic compartments, like lysosomes. The degree of incorporation is inversely proportional to the lysosomal pH (27). A neutral red retention assay was performed where HOS cells were loaded with 2.3 mM neutral red for 3 h in medium without any phenol red and then treated with 0.1% DMSO or 0.5 μM OLX. Treated cells were imaged under a light microscope at intervals of 0, 5, 15, and 30 min posttreatment (p.t.). For OLX-treated cells, the neutral red that had accumulated in acidic organelles could be observed at 0 min p.t., and the staining was marginally reduced at 5 min p.t. However, at 15 min p.t. and later, at 30 min p.t., there was a drastic reduction in neutral red-positive organelles, suggesting neutralization of the endosomal acidic environment during OLX treatment. In comparison, neutral red staining in cells treated with 0.1% DMSO remained unaffected when they were imaged at 30 min p.t. (Fig. 4A).
FIG 4.
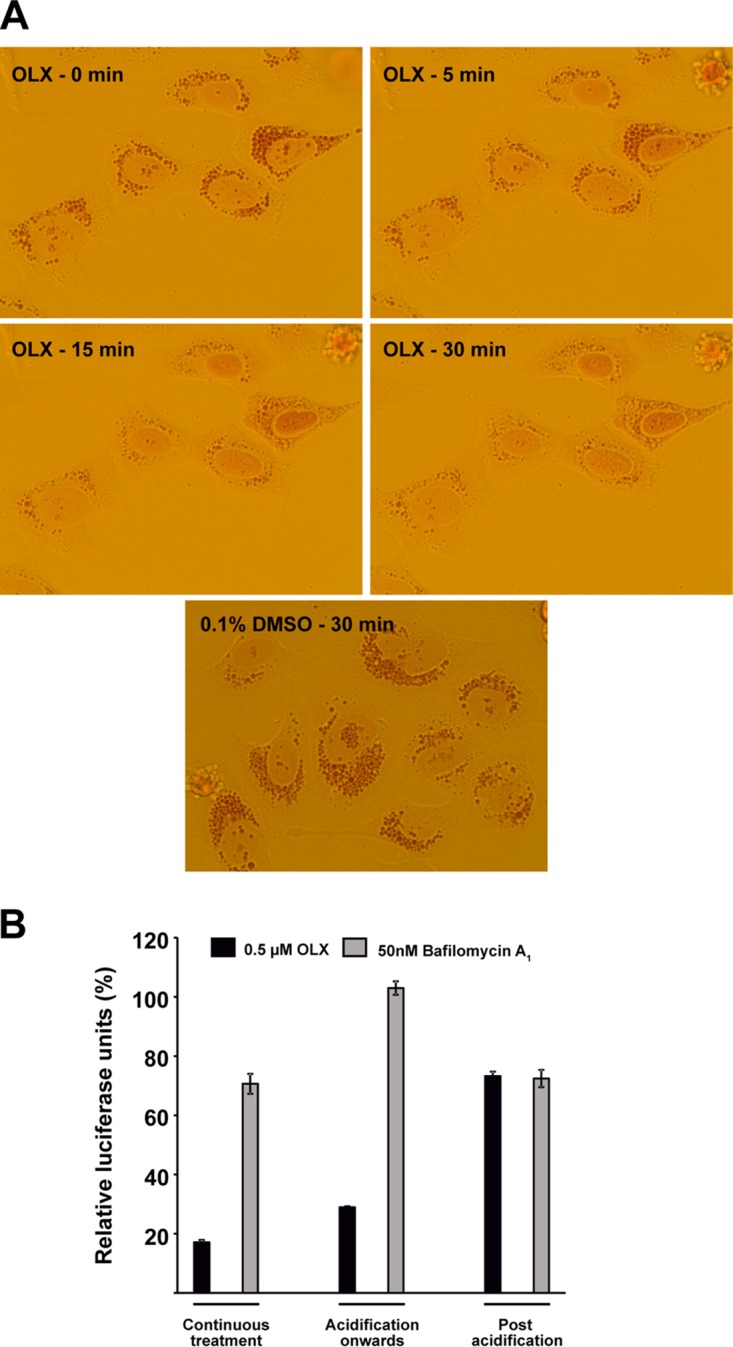
Obatoclax neutralizes the acidic environment of endocytic organelles. A neutral red retention assay was performed to assess the effect of OLX on endosomal acidification. (A) HOS cells were treated with 2.3 mM neutral red in cell culture medium devoid of pH indicator for 3 h, followed by treatment with 0.1% DMSO or 0.5 μM OLX. Images were captured at a ×40 magnification and the indicated times using a charge-coupled-device camera mounted on an Olympus CKX41 inverted light microscope and cellSens software. (B) Endosomal acidification bypass by inducing plasma membrane fusion. Prechilled BHK-21 cells were infected with SFV-ts9–Rluc at an MOI of 50 PFU/cell in neutral-buffered medium and allowed to adsorb and bind to the plasma membrane for 30 min at 4°C. Plasma membrane fusion was triggered by replacing the virus inoculum with acid-buffered medium for 20 min at 39°C, followed by replacement with neutral-buffered medium and further incubation for 3 h at 39°C. Infected cells were treated with 0.1% DMSO, 0.5 μM OLX, or 50 nM bafilomycin A1 according to the indicated schedules. At the end of the incubation period, cells were lysed and luciferase levels were measured. Values are expressed in relative luciferase units, which is the percentage of luciferase units compared to the number of luciferase units for the DMSO-treated controls for each treatment schedule. Luciferase signals were measured from triplicate wells, and data are presented as means ± SEMs (n = 3).
For the alphaviruses, viral fusion at the plasma membrane can be triggered by acidification of the extracellular medium (28). We tested if the inhibitory effect of OLX could be rescued by bypassing the endosomal pH requirement. BHK-21 cells were prechilled on ice for 10 min, followed by addition of the viral inoculum (SFV-ts9–Rluc at an MOI of 50 PFU/cell), and virus was allowed to bind to the plasma membrane for 30 min at 4°C. The viral inoculum was then aspirated, and cell-bound virus was pulsed with buffer at pH 5.5 for 15 min at 39°C. The acidic buffer was then replaced with neutral-buffered medium, and the cells were incubated for 3 h at 39°C and then processed for the luciferase assay. Three different treatment schedules were used, where 0.5 μM OLX or 50 nM bafilomycin A1 was present throughout the course of the experiment, added to the acidic buffer and being present thereafter, or added only after the acidification step. For each treatment condition, appropriate DMSO-treated controls were present. The inhibition that was normally seen with bafilomycin A1 treatment (Fig. 3A) was rescued by plasma membrane fusion regardless of the treatment schedule used (Fig. 4B). On the other hand, the inhibition caused by OLX could be rescued to the same extent only when the drug was added after the acid pulse (Fig. 4B). Surprisingly, OLX addition to the acidic buffer itself caused some level of neutralization, visualized by a color change in the acidic medium, which contained phenol red as a pH indicator. Addition of OLX at 15 min p.i. was sufficient to inhibit viral entry in the previous experiment (Fig. 3B). These results indicate that OLX indeed induces rapid neutralization of the endosomal pH and this inhibitory effect of OLX can indeed be reversed by inducing viral fusion at the plasma membrane.
Isolation of partially OLX-resistant SFV mutants.
Next, we explored the possibility of isolating OLX-resistant mutants to further characterize the compound's mode of action. BHK-21 cells were infected with wild-type SFV at a low MOI of 0.01 PFU/cell for 16 h in the presence or absence of 0.5 μM OLX. These parameters were selected to allow sufficient time for escape mutants to arise. Viral titers were estimated after the first passage, and subsequent dilutions were adjusted to maintain approximately the same MOI of 0.01 PFU/cell in every passage. Titrations were performed after every fifth passage to check for phenotypic resistance, and subsequent passaging dilutions were again adjusted to ensure the same MOI (see the schematic diagram in Fig. 5A). Partial phenotypic resistance was seen after 30 rounds of passaging of SFV in the presence of OLX. Multiple viral clones from the suspected resistant population were plaque purified, cultured into fresh virus stocks, and tested once again for phenotypic resistance; an increase in titer in the presence of OLX compared to the titer of the wild type was observed. Two such resistant clones were subsequently sequenced, and four nonsynonymous mutations, which were found in both of the resistant clones, were observed. Two mutations were present in nonstructural protein 2 (nsP2; E46D and V601I), and two were present in the E1 fusion glycoprotein (L369I and S395R).
FIG 5.

Partially obatoclax-resistant SFV mutants. (A) Passaging scheme employed for the isolation of OLX-resistant SFV mutants in BHK-21 cells. P, passage. (B) BHK-21 cells were infected with wild-type SFV and the respective mutant viruses in the presence of 0.1% DMSO or 0.5 μM OLX. The viral titers in the respective samples were determined. Values are represented as the fold increment in the viral titers compared to the titer for the wild type for DMSO-treated and OLX-treated samples. Values are representative of those from two independent experiments, and data are presented as means ± SEMs (n = 2). (C) BHK-21 cells were infected with wild-type SFV-ts9–Rluc and SFV-ts9–Rluc isolates containing an E1 mutation at an MOI of 50 PFU/cell for 3 h at 39°C. At 3 h p.i., the cells were lysed and the luciferase levels were measured. Luciferase levels are expressed as the fold increment in luciferase signals compared to that for wild-type SFV-ts9–Rluc. Luciferase signals were measured from quadruplicate wells, and data are presented as means ± SEMs (n = 3). (D) Dose-response entry assay to evaluate the antiviral activity of OLX. BHK-21 cells were infected with SFV-ts9–Rluc and the corresponding E1 mutant viruses at an MOI of 50 PFU/cell for 3 h at 39°C in the presence of OLX at concentrations ranging from 0.002 μM to 30 μM. At 3 h p.i., the cells were lysed and luciferase levels were measured. Percent inhibition values were calculated on the basis of the luciferase signals from infected cells treated with 0.1% DMSO. The half-maximal threshold (the EC50) is marked with a dotted line. Assays were performed in triplicate wells. Data are presented as means ± SEMs (n = 2).
Recombinant viruses which contained only the nsP2 mutations (designated the EV mutant), only the E1 mutations (designated the LS mutant), or all four mutations (designated the EVLS mutant) were constructed. Since OLX presumably inhibits viral fusion by neutralizing the endosomal pH, we constructed additional recombinant viruses by inclusion of the E1 L369I mutation in one virus (designated the L mutant) and the S395R mutation in another virus (designated the S mutant). All these mutants were tested for phenotypic resistance using the same parameters, and their resistance profiles were compared with the resistance profile of the original resistant isolate. In DMSO-treated cells, all mutant viruses grew to higher titers (2- to 6-fold) than the wild type. However, in OLX-treated cells, the relative increment was more prominent (4- to 16-fold). The EV mutant as well as the LS mutant showed almost 10-fold increases in viral titers compared to that of wild-type SFV in the presence of OLX (Fig. 5B). Viruses with the combination of all four mutations (EVLS mutants) grew to approximately 15-fold higher titers, similar to the original resistant isolate (Fig. 5B). Unexpectedly, the L mutant also showed the same level of resistance as the original isolate. On the other hand, the S mutant showed only a 4-fold increase in titer (Fig. 5B). This result suggests that the L369I mutation in the E1 fusion protein is sufficient to confer partial resistance against OLX.
To further analyze the role of the mutations in the E1 fusion protein, we investigated their effect in the SFV-ts9 entry assay. Mutants in which all the combinations of the E1 mutations (LS, L, and S) were cloned into SFV-ts9–Rluc were generated. In an entry assay performed in the absence of OLX, we observed a more than 2-fold increase in the absolute luciferase signals for all three mutant combinations (LS, L, and S) compared to that for wild-type SFV-ts9–Rluc (Fig. 5C), suggesting that these mutations in the E1 protein were selected to confer an increased fusogenic ability. Next, a dose-response entry assay was performed with BHK-21 cells infected with these SFV-ts9–Rluc isolates at an MOI of 50 PFU/cell and treated with OLX in a 3-fold dilution series ranging from 30 μM to 0.002 μM for 3 h at 39°C. In this assay, the virus containing only the L369I mutation was less sensitive to OLX, with the EC50 for the virus with that mutation being 7-fold higher (0.5 ± 0.02 μM) than that for the wild type (0.07 ± 0.02 μM). The EC50s of OLX for the other two mutants, the S and LS mutants, were higher (0.12 ± 0.01 μM and 0.15 ± 0.03 μM, respectively) (Fig. 5D). Thus, results from the entry assay indicated that the L369I mutation alone could mediate enhanced viral entry, even in the presence of OLX, corroborating the previous result assessing viral titers (Fig. 5B).
Other drugs targeting the Bcl-2 family of proteins do not inhibit virus entry.
Next, we investigated if other compounds, like OLX, antagonizing the Bcl-2 family of antiapoptotic proteins could also inhibit virus entry. We used TW-37, which has activity against Bcl-2, Bcl-xL, and Mcl-1 (29), and another compound called venetoclax (also known as ABT-199), which is a selective inhibitor of Bcl-2, Bcl-xL, and Bcl-w but has no activity against Mcl-1 (30). These compounds were tested in the SFV-ts9 entry assay at a concentration 10-fold higher than the concentration of OLX (5 μM). The assay, where BHK-21 cells were infected with SFV-ts9–Rluc at an MOI of 50 PFU/cell for 3 h, also included in parallel samples treated with 0.5 μM OLX, 25 μM chlorpromazine, and 50 nM bafilomycin A1. Neither TW-32 nor ABT-199 inhibited the luciferase signals in this assay (Fig. 6A), whereas we were able to recapitulate the data obtained earlier and shown in Fig. 3A showing that OLX, chlorpromazine, and bafilomycin A1 significantly inhibited SFV-ts9 entry. We also assessed the ability of these Bcl-2 inhibitors to disrupt the endosomal acidic environment. Neither TW-37 nor ABT-199 affected neutral red retention even after exposure for 6 h (Fig. 6B). This set of results suggests that the entry block mediated by OLX through the neutralization of the endosomal pH is independent of its antagonism of the Bcl-2 family of proteins.
FIG 6.

Bcl-2 inhibitors do not affect virus entry or endosomal acidification. (A) BHK-21 cells were infected with SFV-ts9–Rluc at an MOI of 50 PFU/cell for 3 h at 39°C. Infected cells were untreated or treated with 0.1% DMSO (solvent control), 0.5 μM OLX, 25 μM chlorpromazine, 50 nM bafilomycin A1, 5 μM TW-37, or 5 μM ABT-199. At 3 h p.i., cells were lysed and luciferase levels were measured. Values are expressed in relative luciferase units, which is the percentage of luciferase units compared to the number of luciferase units for untreated infected samples. Luciferase signals were measured from quadruplicate wells, and data are presented as means ± SEMs (n = 2). (B) HOS cells were treated with 0.66 g/liter of neutral red in cell culture medium devoid of pH indicator for 3 h, followed by treatment with 0.1% DMSO, 50 nM bafilomycin A1, 5 μM TW-37, or 5 μM ABT-199. Images were captured at a ×40 magnification and the indicated times using a charge-coupled-device camera mounted on an Olympus CKX41 inverted light microscope and cellSens software.
The antiviral activity of obatoclax depends on the low-pH requirement of different viruses.
Finally, we assessed the effectiveness of OLX against other RNA viruses from different families. We tested the activity of OLX against West Nile virus (WNV) and yellow fever virus (YFV) (by infection of BHK-21 cells for 24 h, followed by plaque assay titration of viral particles in the supernatants of infected cells) as well as a novel stable reporter Zika virus (ZIKV) (by infection of Vero E6 cells for 72 h, followed by measurement of NanoLuc reporter activity in the lysates of infected cells). WNV, YFV, and ZIKV are members of the Flaviviridae family possessing a class II fusion protein and have been shown to require a low pH for membrane fusion (8). We also employed viruses from the Picornaviridae family that do not have a clear acidification requirement in their entry process, like coxsackievirus A9 (CV-A9) (31), echovirus 6 (E-6), and echovirus 7 (E-7) (for the infection protocol, refer to the Materials and Methods section) (32). WNV, YFV, and ZIKV were highly sensitive to OLX treatment, with the EC50s for these viruses being low (≤0.13 μM) and the selectivity indices being high (Table 2; Fig. S4). On the contrary, OLX showed >30-fold higher EC50s for the pH-insensitive picornaviruses CV-A9, E-6, and E-7 than for the flaviviruses, and the selectivity indices were marginal (Table 2; Fig. S5).
TABLE 2.
EC50s of obatoclax against pH-dependent and -independent viruses
| Virus | EC50a (μM) | CC50b (μM) | SIc |
|---|---|---|---|
| WNV | 0.10 ± 0.04 | >20 | >200 |
| YFV | <0.125 | >20 | >160 |
| ZIKV | 0.13 ± 0.01 | 6.6 ± 4.4 | 50.77 |
| CV-A9 | 2.95 ± 0.01 | 13.47 ± 0.04 | 4.57 |
| E-6 | >4 | 13.47 ± 0.04 | <3.37 |
| E-7 | 3.9 ± 0.32 | 13.47 ± 0.04 | 3.45 |
EC50, concentration causing 50% inhibition of viral replication.
CC50, concentration causing a 50% reduction in cell survival.
SI, selectivity index, which is the ratio of the CC50 to the EC50.
DISCUSSION
OLX is a synthetic indole bipyrrole derivative of bacterial prodigiosin and was developed as a BH3 domain mimetic that inhibits the Bcl-2 family of prosurvival proteins (19, 33). It has been in clinical trials for the treatment of a wide variety of cancers either as a monotherapeutic treatment or in combination with other anticancer compounds (34). OLX was found to be effective against IAV at submicromolar concentrations (EC50 = 0.014 μM) and was proposed to inhibit IAV entry or internalization by targeting Mcl-1 (20). However, we show here for the first time that the antiviral property of OLX is its ability to rapidly neutralize the pH in acidic endosomes or lysosomes. This low-pH environment is necessary for the fusion or uncoating of viruses from diverse families and is thus considered a broad-spectrum host target for antiviral compounds. Indeed, OLX also inhibited other pH-dependent viruses of the Flaviviridae family at submicromolar concentrations, whereas viruses like CV-A9, which utilizes nonacidic multivesicular bodies for entry (31), and other pH-independent viruses, like E-6 and E-7, were unaffected by OLX treatment (Table 2). Also, it was shown earlier that OLX could not inhibit or prevent virus-induced cytotoxicity caused by a low-pH-independent enveloped virus like measles virus (20).
OLX was able to efficiently inhibit three different Old World alphaviruses (Fig. 1A), suggesting that the compound inhibits a common feature required by these viruses. This inhibition was also evident under high-MOI conditions, where virus particle production (Fig. 1B), viral protein expression (Fig. 1C), and RNA synthesis (Fig. 1D) were concomitantly impeded. Next, time-of-addition experiments with SFV were used to highlight the predominant effect of OLX on the early phase of the viral infectious cycle (Fig. 2C), with similar results being obtained for CHIKV (Fig. 2D). These results were corroborated by the results of an entry assay using the temperature-sensitive SFV-ts9–Rluc virus as a readout for virus entry. In that assay, OLX almost completely blocked the luciferase signals emanating only from the translation of viral genomes that had entered the host cells (Fig. 3A). A time-of-addition assay with SFV-ts9 showed that the effect was maximal when cells were pretreated with OLX. The inhibition was prevalent even when OLX was added at 15 min p.i. and corresponds well with the fusion time scale for both SFV (35) and CHIKV (36).
OLX brought about the rapid neutralization of the acidic environment in endosomes/lysosomes within 15 to 30 min of exposure (Fig. 4). Indeed, three recent reports have provided independent corroboration of our findings and showed that OLX not only increases the lysosomal pH but also accumulates in the lysosomes and causes a loss of lysosomal function (37–39). The fact that OLX is a weak base and the likelihood that it is protonated and accumulates in lysosomes, similar to another lysomotrophic agent, chloroquine, suggest a possible mechanism for the rapid elevation in the lysosomal pH (38). Additionally, Champa et al. have suggested that OLX could mediate the exchange of Cl− and HCO3− ions between the cytoplasm and the lysosome, which would also cause a rapid elevation in the lysosomal pH (37). Interestingly, addition of as little as 0.5 μM OLX to the acidic medium during endosomal bypass elevated the pH and resulted in only a minimal rescue, whereas addition of OLX after plasma membrane fusion resulted in lower levels of inhibition, similar to the findings observed with bafilomycin A1 (Fig. 4B). This serves to underline the mildly basic nature of OLX.
Consistent with what other groups have observed (37), the ability of OLX to neutralize acidic organelles is independent of its ability to inhibit the Bcl-2 family of prosurvival proteins. Another pan-Bcl-2 inhibitor, TW-37, as well as a selective antagonist of Bcl-2, ABT-199, could neither block virus entry (Fig. 6A) nor disrupt neutral red staining of acidic organelles, even when they were used to treat cells for 6 h at concentrations 10-fold higher than the concentration of OLX (Fig. 6B). OLX induced lysosome clustering in some cell types (39), but in some others it affected the lysosomal pH without causing the obvious destruction of these organelles (37). Nevertheless, the precise mechanism by which OLX brings about such a swift loss in lysosomal acidity remains to be determined.
The barrier for resistance development may be high when a host process essential for the viral life cycle is targeted. The fusion of viral and endosomal membranes, triggered by low-pH-induced conformational changes in the fusion protein, is one such critical step in the viral entry process. In this work, it took 30 rounds of passaging of SFV in the presence of OLX to obtain only partially resistant mutants. The resistant mutants grew to titers more than 1 log higher than the titer of wild-type SFV in the presence of OLX, but they did not reach titers as high as those of wild-type virus in untreated cells. The mutations L369I (in E1 domain III) and S395R (in the E1 stem region) were found in the E1 membrane fusion protein, providing indirect evidence that OLX affects virus membrane fusion-related processes. The L369I mutation alone was sufficient to confer the resistance phenotype both when viral titers in the presence of OLX were assessed (Fig. 5B) and also in a dose-response entry assay (Fig. 5D). Even though mutants with all three combinations of the E1 mutations appeared to have an increased fusogenic potential (Fig. 5C), the presence of the S395R mutation seemed to dampen the effect of the L369I mutation, as the LS mutant showed only an 8-fold increase in viral titers in the presence of OLX (Fig. 5B) and the EC50 of OLX for the LS mutant was increased only 2-fold compared to that for wild-type SFV-ts9–Rluc (Fig. 5D). This suggests that there could be possible interactions between these two residues (L369 and S395) in the E1 fusion protein. Further phenotypic analysis is required to fully elucidate the functionality of these residues. Interestingly, a 10-fold increase in the titer of the EV nsP2 mutant was also found when it was treated with OLX (Fig. 5B). Earlier, serial passage of CHIKV for only 7 rounds in HeLa cells was enough for cell culture adaptation, resulting in increased fitness and resistance to a cocktail of antiviral mutagens (40). This was also seen with hepatitis C virus, where increased viral fitness led to reduced drug sensitivity (41). It is therefore possible that although the chief mode of action of OLX is the neutralization of the endolysosomal acidic environment, additional fitness mutations might have emerged during the selection process.
The EC50s of OLX for CHIKV in the different cell lines tested were in the range of 0.04 to 0.15 μM. When OLX was given as 3-h infusions in clinical studies, plasma OLX concentrations reached a maximum of ∼0.4 μM (42, 43), suggesting that an effective therapeutic dose for prophylaxis against CHIKV and treatment of CHIKV infection can be envisaged. Chloroquine, a known lysomotrophic agent which has anti-CHIKV activity at concentrations (range, 5 to 20 μM) higher than those of OLX in vitro (11), did not demonstrate a selective advantage over a nonsteroidal anti-inflammatory drug, but the possibility that it has therapeutic efficacy was not ruled out (44). Further studies need to be performed in a suitable mouse model of CHIKV infection to ascertain the antiviral potential of OLX. Additionally, given the effectiveness of OLX against WNV, YFV, and ZIKV, it would be interesting to investigate if OLX is effective in animal models of these diseases (especially Zika fever), as well as against DENV, which affects millions of individuals every year. In conclusion, OLX is a promising compound for repurposing as an antiviral candidate and is effective against several pathogenic viruses at submicromolar concentrations.
MATERIALS AND METHODS
Cells and inhibitors.
Baby hamster kidney (BHK-21) and human osteosarcoma (HOS) cells were grown as described earlier (45, 46). Human hepatoma (Huh 7.5) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco) supplemented with nonessential amino acids, penicillin, streptomycin, and l-glutamine (Gibco). Vero E6 African green monkey kidney cells were cultured in Iscove's modified Dulbecco's medium (IMDM) containing 10% FBS. Human epithelial lung carcinoma (A549) cells (ATCC) were grown in DMEM containing 10% FBS supplemented with gentamicin. All cells were maintained at 37°C with 5% CO2. Obatoclax (Selleckchem), chlorpromazine (Calbiochem), bafilomycin A1 (Sigma), TW-37 (Selleckchem), ABT-199 (Selleckchem) were dissolved in dimethyl sulfoxide (DMSO) and used at the concentrations indicated above.
Viruses.
The infectious cDNA clones of wild-type SFV (47) and SINV (48) used in this study have been described earlier. Wild-type CHIKV (strain LR2006 OPY1) was generated from the infectious clone SP6-ICRES (21). SFV-Rluc and CHIKV-Rluc were derived from the respective wild-type clones, where the Renilla reniformis luciferase gene (Rluc) is inserted within the nsP3-coding region (21). The method used to generate SFV-ts9–Rluc, which has a point mutation in nsP2 and which carries the Rluc reporter, has been described earlier (21). SFV mutants were generated by subcloning the respective regions (between the SacI and XbaI sites in the pUC18 vector for the nsP2 mutations and two PsiI sites in the pUC57 vector for the E1 mutations) from plasmid pCMV-SFV4 (49) and performing site-directed mutagenesis using a QuikChange Lightning multi-site-directed mutagenesis kit (Agilent). The synthesized clones were sequenced and cloned back into the original pCMV-SFV4 vector. All alphavirus stocks were produced in BHK-21 cells, and viral titers were quantified by conventional plaque assay titration (21).
The details of the construction and properties of Zika virus (ZIKV) clone ZIKV-UbiNanoLuc derived from infectious cDNA (icDNA) will be described elsewhere. Briefly, the icDNA of ZIKV was obtained by assembly of synthetic cDNA fragments (GenScript, USA) corresponding to the sequence of the ZIKV Asian genotype, isolated in Brazil in 2015 (isolate BeH819015); the sequences of the 5′ and 3′ untranslated regions (UTRs), not fully resolved in the published sequence of the isolate (GI|975885966), were completed on the basis of the sequence of ZIKV isolate PE243/2015 from Brazil (GI|1026288139). Full-length cDNA, placed under the control of the bacteriophage SP6 RNA polymerase promoter, was assembled in the pCC1BAC vector by use of a CopyControl cloning kit (Epicentre, USA). Insertion of the sequence encoding the NanoLuc reporter was carried out by a method similar to that previously described for the icDNA clone of DENV (50). ZIKV-UbiNanoLuc was rescued in Vero cells that had been transfected with capped transcripts generated by SP6 RNA polymerase; the titers of the rescued virus were determined by plaque titration in Vero cells. ZIKV-UbiNanoLuc was subsequently passaged 5 times at a multiplicity of infection (MOI) of 0.1 PFU/cell; NanoLuc activities in infected cells were measured at different passages using a Promega Renilla luciferase assay system and were found to be constant, indicating that the obtained virus stock was genetically stable. West Nile virus (WNV; isolate NY 2000-crow3356) was grown on BHK-21/J cells and titers were determined as described previously for yellow fever virus (YFV) (51). Infectious cDNA clone 17D of YFV has been described earlier (52). The prototype picornaviruses used in this study included coxsackievirus A9 (CV-A9; GenBank accession number D00627), echovirus 6 (E-6; GenBank accession number AY302558), and echovirus 7 (E-7; GenBank accession number AY036578).
SDS-PAGE and Western blotting.
Analysis of CHIKV protein expression was performed as described earlier (45). Briefly, BHK-21 cells were infected with wild-type CHIKV in the format described above for the assay whose results are presented in Fig. 1C. Solubilized cell lysates were separated on a 10% SDS-polyacrylamide gel and blotted onto a Hybond enhanced chemiluminescence nitrocellulose membrane (GE Healthcare). Primary staining was performed using rabbit anti-CHIKV nsP1, nsP3, and capsid antibodies (all of which were prepared in-house), mouse anti-CHIKV E2 monoclonal antibody 3E4 (53), and mouse antiactin (Sigma). Alexa Fluor 680-conjugated anti-rabbit (Invitrogen) and IRDye800-conjugated anti-mouse (LI-COR Biosciences) antibodies were used for secondary staining, and the blots were scanned using an Odyssey infrared imaging system (LI-COR Biosciences).
Northern blotting.
BHK-21 cells seeded on 6-well plates were infected with wild-type CHIKV in the presence of serial dilutions of OLX as described above for the assay whose results are presented in Fig. 1D. At 6 h p.i., total RNA was isolated from the respective wells using the TRIsure reagent (Bioline) according to the manufacturer's instructions. CHIKV genomic and subgenomic RNAs were detected using a digoxigenin (DIG)-labeled RNA probe designed against the 3′ UTR of the viral genome, as described earlier (54).
Antiviral activity assay.
Ninety-six-well white-bottom culture plates (PerkinElmer) were seeded with BHK-21 cells, HOS cells, or Huh 7.5 cells. On the next day, the cells were infected with SFV-Rluc or CHIKV-Rluc at the MOIs indicated in the Results in the presence of OLX at concentrations ranging from 0.007 μM to 100 μM. After the respective incubation times (see the Results section), the cell culture supernatant was discarded, the cells were lysed, and the luciferase signals were measured using a Renilla luciferase assay system (Promega) and detected with a Varioskan Flash multimode reader (Thermo Fisher Scientific) according to the manufacturers' instructions.
Cell viability assays.
BHK-21, HOS, or Huh 7.5 cells seeded on 96-well white-bottom culture plates (PerkinElmer) were treated with OLX at various concentrations ranging from 0.007 μM to 100 μM. After incubation, the cell culture supernatant was removed, the cells were washed with phosphate-buffered saline (PBS), and cell viability was measured using the CellTiter-Glo reagent (Promega), which measures cellular ATP levels from metabolically active cells. For infections with flaviviruses, cell viability was measured in parallel using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma). Briefly, BHK-21 cells were treated with OLX at concentrations ranging from 20 μM to 0.016 μM. MTT was added to the supernatant at a final concentration of 200 μg/ml, and incubation was continued at 37°C for 90 min. After the cells were fixed with 4% formaldehyde, the cells were dissolved in isopropanol and colorimetric measurement was performed at an optical density of 562 nm. For infections with picornaviruses, cell viability was measured in parallel using a cell counting kit-8 (CCK-8; Dojindo) according to the manufacturer's instructions.
Endosomal bypass assay.
BHK-21 cells seeded on 96-well plate culture plates were prechilled on ice for 10 min, followed by addition of virus inoculum (SFV-ts9–Rluc) in neutral-buffered medium (minimal essential medium containing 0.2% bovine serum albumin [BSA], 20 mM HEPES buffer [pH 7.2], and 1% l-glutamine). The virus in the inoculum was allowed to adsorb and bind to the plasma membrane for 30 min at 4°C. The viral inoculum was then discarded, followed by induction of plasma membrane fusion by addition of acidic medium (minimal essential medium containing 0.2% BSA, 10 mM morpholine-ethane sulfonic acid [pH 5.5] and 1% GlutaMAX-1 [Invitrogen]) and incubation for 15 min at 39°C. The acidic medium was then replaced with neutral-buffered medium and incubated further for 3 h at 39°C, followed by cell lysis and analysis of luciferase signals as described above.
Plaque purification of resistant mutants.
BHK-21 cells seeded on 6-well plates were infected with the suspected resistant stocks in a 10-fold serial dilution format in the presence of 0.5 μM OLX. After 1 h of adsorption, the virus inoculum was discarded, the cells were washed with PBS, and overlay medium containing minimal essential medium without phenol red, 1% Bacto agar, 4% FBS, nonessential amino acids, penicillin, streptomycin, and l-glutamine was added (4 ml/well). At 48 h p.i., overlay medium containing neutral red stain (82.5 mg/liter) was added to enable the visualization of plaques. At 72 h p.i., plugs of plaques from wells with the highest dilution showing visible plaques were purified, and the purified plaques were stored at −80°C. All media contained 0.5 μM OLX to maintain a constant selection pressure.
Sequencing.
Viral RNA from phenotypically resistant plaque-purified stocks was isolated using a viral RNA minikit (Qiagen). cDNAs were synthesized using random primers and the viral RNA as a template with a high-capacity cDNA reverse transcription kit (Applied Biosystems). PCR products spanning the entire SFV genome were generated using the cDNAs as the templates and sequenced by standard Sanger sequencing. The sequenced contigs were assembled using BioEdit software (v7.2.5), and the sequence was compared to the genomic sequence of wild-type strain SFV4.
Picornavirus infection assays.
A549 cells were seeded on 96-well plates (PerkinElmer) with cell culture medium and incubated for 24 h. The cells were washed twice with phosphate-buffered saline, and OLX was added to the cells at concentrations ranging from 0.05 μM to 4 μM for 30 min. The cells were infected with the viruses in five replicates for 1 h on ice. This was followed by three washes with medium and addition of DMEM supplemented with 1% FBS with the same concentrations of OLX. At 6 h p.i., the cells were fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and stained with virus type-specific antibodies (from laboratory collections). Virus staining was visualized with secondary Alexa Fluor 488 antibodies and an Evos FL Auto microscope (Thermo Fisher). Infection efficiencies were analyzed with BioImageXD software (55).
Data and statistical analysis.
Half-maximal (50%) effective and cytotoxic concentrations (EC50 and CC50, respectively) were determined using GraphPad Prism software by generating antiviral and cell survival curves. Statistical analyses were done using Microsoft Excel software. Statistical significance was determined using a one-way analysis of variance (ANOVA) test. P values of less than 0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Margaret Kielian (Albert Einstein College of Medicine) for advice with the neutral red retention assay and for critical comments on the manuscript. We thank Janett Wieseler for excellent technical assistance and Tania Quirin for help with data analysis. We acknowledge the Drug Discovery and Chemical Biology Network for providing access to screening instrumentation. We also thank Bastian Thaa (University of Leipzig) for critical comments on the manuscript.
This work was funded by the Academy of Finland (grant 265997 to T.A.), the European Regional Development Fund through the Centre of Excellence in Molecular Cell Engineering, Estonia (grant 2014-2020.4.01.15-0013 to A.M.), the Estonian Research Council (grant IUT 20-27 to A.M.), and the European Union (AIROPico, FP7-PEOPLE-2013-IAPP grant no. 612308 to P.S.). F.S.V. was a fellow of the Integrative Life Sciences doctoral program. The Drug Discovery and Chemical Biology Network is funded by Biocenter Finland.
The funders had no role in the experimental design, data analysis and interpretation of the results, or the decision to submit this work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02227-16.
REFERENCES
- 1.Gould EA, Coutard B, Malet H, Morin B, Jamal S, Weaver S, Gorbalenya A, Moureau G, Baronti C, Delogu I, Forrester N, Khasnatinov M, Gritsun T, de Lamballerie X, Canard B. 2010. Understanding the alphaviruses: recent research on important emerging pathogens and progress towards their control. Antiviral Res 87:111–124. doi: 10.1016/j.antiviral.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver SC, Forrester NL. 2015. Chikungunya: evolutionary history and recent epidemic spread. Antiviral Res 120:32–39. doi: 10.1016/j.antiviral.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 3.Ahola T, Courderc T, Ng LF, Hallengard D, Powers A, Lecuit M, Esteban M, Merits A, Roques P, Liljestrom P. 2015. Therapeutics and vaccines against chikungunya virus. Vector Borne Zoonotic Dis 15:250–257. doi: 10.1089/vbz.2014.1681. [DOI] [PubMed] [Google Scholar]
- 4.Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825. doi: 10.3390/v2040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Duijl-Richter MK, Hoornweg TE, Rodenhuis-Zybert IA, Smit JM. 2015. Early events in chikungunya virus infection—from virus cell binding to membrane fusion. Viruses 7:3647–3674. doi: 10.3390/v7072792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vigant F, Santos NC, Lee B. 2015. Broad-spectrum antivirals against viral fusion. Nat Rev Microbiol 13:426–437. doi: 10.1038/nrmicro3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kielian M. 2014. Mechanisms of virus membrane fusion proteins. Annu Rev Virol 1:171–189. doi: 10.1146/annurev-virology-031413-085521. [DOI] [PubMed] [Google Scholar]
- 8.Smit JM, Moesker B, Rodenhuis-Zybert I, Wilschut J. 2011. Flavivirus cell entry and membrane fusion. Viruses 3:160–171. doi: 10.3390/v3020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suomalainen M, Greber UF. 2013. Uncoating of non-enveloped viruses. Curr Opin Virol 3:27–33. doi: 10.1016/j.coviro.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr Top Microbiol Immunol 343:43–89. doi: 10.1007/82_2010_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan M, Santhosh SR, Tiwari M, Lakshmana Rao PV, Parida M. 2010. Assessment of in vitro prophylactic and therapeutic efficacy of chloroquine against chikungunya virus in Vero cells. J Med Virol 82:817–824. doi: 10.1002/jmv.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, Seidah NG, Nichol ST. 2005. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2:69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC, Green CE, Iyer LV, Dilks HH, Davey RA, Kolokoltsov AA, Carrion R Jr, Patterson JL, Bavari S, Panchal RG, Warren TK, Wells JB, Moos WH, Burke RL, Tanga MJ. 2013. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One 8:e60579. doi: 10.1371/journal.pone.0060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelnabi R, Neyts J, Delang L. 2015. Towards antivirals against chikungunya virus. Antiviral Res 121:59–68. doi: 10.1016/j.antiviral.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savarino A, Di Trani L, Donatelli I, Cauda R, Cassone A. 2006. New insights into the antiviral effects of chloroquine. Lancet Infect Dis 6:67–69. doi: 10.1016/S1473-3099(06)70361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farias KJ, Machado PR, Muniz JA, Imbeloni AA, da Fonseca BA. 2015. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol 28:161–169. doi: 10.1089/vim.2014.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurgeit A, McDowell R, Moese S, Meldrum E, Schwendener R, Greber UF. 2012. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog 8:e1002976. doi: 10.1371/journal.ppat.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang YM, Lu JW, Lin CC, Chin YF, Wu TY, Lin LI, Lai ZZ, Kuo SC, Ho YJ. 2016. Antiviral activities of niclosamide and nitazoxanide against chikungunya virus entry and transmission. Antiviral Res 135:81–90. doi: 10.1016/j.antiviral.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. 2007. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A 104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J, Yadav B, Aittokallio T, Kaukinen P, Ahola T, Kuivanen S, Vapalahti O, Kantele A, Tynell J, Julkunen I, Kallio-Kokko H, Paavilainen H, Hukkanen V, Elliott RM, De Brabander JK, Saelens X, Kainov DE. 2012. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza A virus infection. J Biol Chem 287:35324–35332. doi: 10.1074/jbc.M112.392142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pohjala L, Utt A, Varjak M, Lulla A, Merits A, Ahola T, Tammela P. 2011. Inhibitors of alphavirus entry and replication identified with a stable chikungunya replicon cell line and virus-based assays. PLoS One 6:e28923. doi: 10.1371/journal.pone.0028923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balistreri G, Caldentey J, Kaariainen L, Ahola T. 2007. Enzymatic defects of the nsP2 proteins of Semliki Forest virus temperature-sensitive mutants. J Virol 81:2849–2860. doi: 10.1128/JVI.02078-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lulla V, Merits A, Sarin P, Kaariainen L, Keranen S, Ahola T. 2006. Identification of mutations causing temperature-sensitive defects in Semliki Forest virus RNA synthesis. J Virol 80:3108–3111. doi: 10.1128/JVI.80.6.3108-3111.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LH, Rothberg KG, Anderson RG. 1993. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol 123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowman EJ, Siebers A, Altendorf K. 1988. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A 85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266:17707–17712. [PubMed] [Google Scholar]
- 27.Seglen PO. 1983. Inhibitors of lysosomal function. Methods Enzymol 96:737–764. doi: 10.1016/S0076-6879(83)96063-9. [DOI] [PubMed] [Google Scholar]
- 28.White J, Kartenbeck J, Helenius A. 1980. Fusion of Semliki Forest virus with the plasma membrane can be induced by low pH. J Cell Biol 87:264–272. doi: 10.1083/jcb.87.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, Nikolovska-Coleska Z, Wang S, Al-Katib A. 2007. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res 13:2226–2235. doi: 10.1158/1078-0432.CCR-06-1574. [DOI] [PubMed] [Google Scholar]
- 30.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. 2013. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 31.Huttunen M, Waris M, Kajander R, Hyypia T, Marjomaki V. 2014. Coxsackievirus A9 infects cells via nonacidic multivesicular bodies. J Virol 88:5138–5151. doi: 10.1128/JVI.03275-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim C, Bergelson JM. 2012. Echovirus 7 entry into polarized intestinal epithelial cells requires clathrin and Rab7. mBio 3:e00304-11. doi: 10.1128/mBio.00304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Besbes S, Mirshahi M, Pocard M, Billard C. 2015. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 6:12862–12871. doi: 10.18632/oncotarget.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joudeh J, Claxton D. 2012. Obatoclax mesylate: pharmacology and potential for therapy of hematological neoplasms. Expert Opin Invest Drugs 21:363–373. doi: 10.1517/13543784.2012.652302. [DOI] [PubMed] [Google Scholar]
- 35.Vonderheit A, Helenius A. 2005. Rab7 associates with early endosomes to mediate sorting and transport of Semliki Forest virus to late endosomes. PLoS Biol 3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoornweg TE, van Duijl-Richter MK, Ayala Nunez NV, Albulescu IC, van Hemert MJ, Smit JM. 2016. Dynamics of chikungunya virus cell entry unraveled by single-virus tracking in living cells. J Virol 90:4745–4756. doi: 10.1128/JVI.03184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Champa D, Orlacchio A, Patel B, Ranieri M, Shemetov AA, Verkhusha VV, Cuervo AM, Di Cristofano A. 2016. Obatoclax kills anaplastic thyroid cancer cells by inducing lysosome neutralization and necrosis. Oncotarget 7:34453–34471. doi: 10.18632/oncotarget.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamelos VA, Fisher N, Bamrah H, Voisey C, Price JC, Farrell WE, Redman CW, Richardson A. 2016. The BH3 mimetic obatoclax accumulates in lysosomes and causes their alkalinization. PLoS One 11:e0150696. doi: 10.1371/journal.pone.0150696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu L, Wu WK, Gu C, Zhong D, Zhao X, Kong Y, Lin Q, Chan MT, Zhou Z, Liu S. 2016. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget 7:14693–14707. doi: 10.18632/oncotarget.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coffey LL, Vignuzzi M. 2011. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J Virol 85:1025–1035. doi: 10.1128/JVI.01918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheldon J, Beach NM, Moreno E, Gallego I, Pineiro D, Martinez-Salas E, Gregori J, Quer J, Esteban JI, Rice CM, Domingo E, Perales C. 2014. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J Virol 88:12098–12111. doi: 10.1128/JVI.01860-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goard CA, Schimmer AD. 2013. An evidence-based review of obatoclax mesylate in the treatment of hematological malignancies. Core Evid 8:15–26. doi: 10.2147/CE.S42568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hwang JJ, Kuruvilla J, Mendelson D, Pishvaian MJ, Deeken JF, Siu LL, Berger MS, Viallet J, Marshall JL. 2010. Phase I dose finding studies of obatoclax (GX15-070), a small molecule pan-BCL-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin Cancer Res 16:4038–4045. doi: 10.1158/1078-0432.CCR-10-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chopra A, Saluja M, Venugopalan A. 2014. Effectiveness of chloroquine and inflammatory cytokine response in patients with early persistent musculoskeletal pain and arthritis following chikungunya virus infection. Arthritis Rheumatol 66:319–326. doi: 10.1002/art.38221. [DOI] [PubMed] [Google Scholar]
- 45.Varghese FS, Kaukinen P, Glasker S, Bespalov M, Hanski L, Wennerberg K, Kummerer BM, Ahola T. 2016. Discovery of berberine, abamectin and ivermectin as antivirals against chikungunya and other alphaviruses. Antiviral Res 126:117–124. doi: 10.1016/j.antiviral.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Thaa B, Biasiotto R, Eng Neuvonen KM, Gotte B, Rheinemann L, Mutso M, Utt A, Varghese F, Balistreri G, Merits A, Ahola T, McInerney GM. 2015. Differential phosphatidylinositol-3-kinase-Akt-mTOR activation by Semliki Forest and chikungunya viruses is dependent on nsP3 and connected to replication complex internalization. J Virol 89:11420–11437. doi: 10.1128/JVI.01579-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liljestrom P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol 65:4107–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice CM, Levis R, Strauss JH, Huang HV. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J Virol 61:3809–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ulper L, Sarand I, Rausalu K, Merits A. 2008. Construction, properties, and potential application of infectious plasmids containing Semliki Forest virus full-length cDNA with an inserted intron. J Virol Methods 148:265–270. doi: 10.1016/j.jviromet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoggins JW, Dorner M, Feulner M, Imanaka N, Murphy MY, Ploss A, Rice CM. 2012. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc Natl Acad Sci U S A 109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kummerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76:4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bredenbeek PJ, Kooi EA, Lindenbach B, Huijkman N, Rice CM, Spaan WJ. 2003. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J Gen Virol 84:1261–1268. doi: 10.1099/vir.0.18860-0. [DOI] [PubMed] [Google Scholar]
- 53.Brehin AC, Rubrecht L, Navarro-Sanchez ME, Marechal V, Frenkiel MP, Lapalud P, Laune D, Sall AA, Despres P. 2008. Production and characterization of mouse monoclonal antibodies reactive to chikungunya envelope E2 glycoprotein. Virology 371:185–195. doi: 10.1016/j.virol.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 54.Utt A, Quirin T, Saul S, Hellstrom K, Ahola T, Merits A. 2016. Versatile trans-replication systems for chikungunya virus allow functional analysis and tagging of every replicase protein. PLoS One 11:e0151616. doi: 10.1371/journal.pone.0151616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kankaanpaa P, Paavolainen L, Tiitta S, Karjalainen M, Paivarinne J, Nieminen J, Marjomaki V, Heino J, White DJ. 2012. BioImageXD: an open, general-purpose and high-throughput image-processing platform. Nat Methods 9:683–689. doi: 10.1038/nmeth.2047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.