

ABSTRACT
Current treatments for chronic Chagas cardiomyopathy, a disease with high mortality rates and caused by the protozoan Trypanosoma cruzi, are unsatisfactory. Myocardial inflammation, including endothelial activation, is responsible for the structural and functional damage seen in the chronic phase. The clinical efficacy of benznidazole could be improved by decreasing chronic inflammation. Statins, which have anti-inflammatory properties, may improve the action of benznidazole. Here, the action of simvastatin in a murine model of chronic Chagas cardiomyopathy and the link with the production of the proresolving eicosanoid 15-epi-lipoxin A4, produced by 5-lipoxygenase, are evaluated. Simvastatin decreased the expression of the adhesion molecules E-selectin, intracellular adhesion molecule type 1 (ICAM-1), and vascular cell adhesion molecule type 1 (VCAM-1) in T. cruzi-infected mice. However, when this drug was administered to 5-lipoxygenase-deficient mice, the anti-inflammatory effect was not observed unless exogenous 15-epi-lipoxin A4 was administered. Thus, in chronic Chagas disease, 5-epi-lipoxin A4 induced by simvastatin treatment could improve the pathophysiological condition of patients by increasing the trypanocidal action of benznidazole.
KEYWORDS: 15-epi-lipoxin A4, 5-lipoxygenase, CAM, Chagas disease, chronic cardiomyopathy, simvastatin
INTRODUCTION
Chagas disease, or American trypanosomiasis, is an ailment produced by a flagellated protozoan, Trypanosoma cruzi. This parasite is transmitted via blood transfusions, via transplanted organs, vertically from mother to fetus, or through an insect vector, the hematophagous Triatoma infestans. Chagas disease is a malady afflicting 7 million people in 21 countries in Latin America where the disease is endemic, and its prevalence is increasing in countries where it is not endemic, due to migration (1). Chagas disease has the second-highest disease burden among tropical diseases (2) and causes approximately 7,000 deaths annually, which is more than are caused by malaria in the Americas (1, 2). The main strategies for controlling Chagas disease include the control of vectors, systematic screening of blood donors in all countries of endemicity, detection and treatment of congenital transmission, and treatment of infected children and acute cases (1). However, control programs are discontinuous and current therapies are limited due to their low efficacy (1). Clinically, Chagas disease presents an acute phase, which is diagnosed in less than 10% of the cases, based on the absence or presence of mild symptoms. The clinical course in most cases is a spontaneous recovery (3) but without the elimination of the parasite. The chronic phase may be asymptomatic (indeterminate), lasts 10 to 30 years, and is characterized by positive serological results and some degree of cardiac involvement with minimal, but persistent, inflammation. However, in approximately 30% of patients, the disease progresses to symptomatic forms involving the esophagus, colon, or heart (4). Symptoms and physical signs of chronic Chagas cardiomyopathy (CCC) include heart failure, cardiac arrhythmias, arterial or venous thromboembolism, and sudden death (5, 6).
Diverse evidence supports four main pathogenic mechanisms underlying CCC: (i) parasite-dependent myocardial damage, (ii) immune-mediated myocardial injury, (iii) cardiac dysautonomia, and (iv) microvascular abnormalities and ischemia causing zonal microinfarcts (7). During T. cruzi infections, endothelial cells (EC) change toward an active, prothrombotic, and vasoconstrictive state. EC activation occurs early in inflammatory processes, leading to vascular dysfunction and injury. These are crucial events associated with acute and chronic inflammatory conditions, including sepsis, atherosclerosis (8), and as shown by us, Chagas disease (9). EC activation starts when glycoproteins on the surface of the parasite bind to Toll-like receptor 4 (TLR4). This interaction triggers an intracellular signaling cascade involving the activation of the nuclear factor kappa B (NF-κB) (10, 11). This factor participates in the expression of cytokines and endothelial cell adhesion molecules (ECAMs), including intracellular adhesion molecule type 1 (ICAM-1), vascular cell adhesion molecule type 1 (VCAM-1), and E-selectin. These ECAMs are markers of EC activation because they affect permeability to macromolecules, surface protein expression, and leukocyte recruitment, and they promote platelet aggregation and thrombin formation.
Currently, treatments for Chagas disease are based on benznidazole (12). This drug is effective and well tolerated in children and during the acute phase; however, its clinical efficacy is inadequate for treating the chronic form of the illness due to its low efficacy, association with severe adverse events, and the fact that it does not modify the course of the disease (13). Thus, it is necessary to search for new strategies to improve the clinical efficacy of benznidazole and to directly modify the pathogenic mechanisms involved in the progression of CCC.
The aspirin-triggered lipoxin (ATL) 15-epi-lipoxin A4 (15-epi-LXA4) is a lipid in the group of anti-inflammatory proresolving molecules. It is produced by a metabolic switch of COX-2 induced by the acetylation of its active site, driving arachidonic acid metabolism toward the production of 15(R)-hydroxyeicosatetraenoic acid [15(R)-HETE], a substrate of 5-lipoxygenase (5-LO) (14). Cholesterol-lowering statins, as part of their pleiotropic effects, produce the same metabolic switch in COX-2 by prompting the nitrosylation of the enzyme (15). Recently, our group reported that aspirin decreases EC activation and myocardial inflammation in a T. cruzi-infected murine model via the production of 15-epi-LXA4 (16). In addition, simvastatin and benznidazole prevent EC activation in human umbilical vein endothelial cells (HUVECs), decreasing ECAM expression on the cell surface, an effect mediated by 15-epi-LXA4 (17). However, the role of simvastatin in endothelial activation and its link with 15-epi-LXA4 in vivo remain to be studied. Here, we provide evidence that helps clarify these issues.
RESULTS
Chronic model of Chagas disease in mice.
Figure 1 shows the progression of parasitemia and mortality in BALB/c and Sv/129 mice infected with T. cruzi strain Dm28c. As expected, parasitemia lasted approximately 25 days, with the peak of infection occurring at day 20 postinfection (dpi) (Fig. 1A). Parasitemia levels were similar in BALB/c and Sv/129 WT mice; however, parasitemia was significantly lower in the 5-LO−/− mice (P < 0.001). Moreover, despite significant parasitemia, survival rates were high for all mice at the end of the experiment, independent of their genetic background. Thus, T. cruzi infections were established during the first 30 dpi, generating a low-mortality model of murine Chagas disease.
FIG 1.

Parasitemia (A) and survival (B) progression in BALB/c and Sv/129 wild-type (WT) or 5-lipoxygenase (5-LO−/−) knockout mice. Bars in panel A correspond to the standard deviations from three independent experiments. ***, P < 0.001.
Effect of simvastatin on cardiac inflammation and endothelial activation in chagasic mice.
The effect of simvastatin on endothelial activation in a chronic model of Chagas disease was determined by treating BALB/c mice with simvastatin for 20 days, beginning on the 30th dpi. At 90 dpi, T. cruzi infections produced an intense endothelial activation, as demonstrated by increased staining for E-selectin, ICAM-1, and VCAM-1 (Fig. 2). Treatment with 40 mg/kg of body weight/day simvastatin, a dose previously reported for mice (18), induced a significant decrease in the expression of these three ECAMs (Fig. 2), with E-selectin expression undetectable via immunohistochemistry staining. Benznidazole produced a similar effect, as did the combination of benznidazole and a low dose of simvastatin.
FIG 2.

Representative photographs of immunohistochemically stained cardiac tissue from BALB/c mice infected with Dm28c Trypanosoma cruzi and treated with simvastatin or benznidazole at the indicated doses. Insets show healthy noninfected murine cardiac tissue. Sim, simvastatin; Bz, benznidazole.
Considering that simvastatin modulates inflammation, cardiac tissue was evaluated for inflammatory cell infiltration and fibrosis at 90 dpi. Figure 3A (“infected” panel) shows representative images of hematoxylin-and-eosin stained cardiac tissue from BALB/c mice demonstrating chronic myocarditis with extensive inflammatory infiltration. In treated mice, the recovery was significant, reaching states similar to those of healthy hearts. There were no differences among the different treatments, including the combination of a low dose of simvastatin with benznidazole. Similarly, the development of fibrosis was significant in the infected model (Fig. 3B, infected panel), as revealed by picrosirius red staining. This finding agrees with the results presented in Fig. 2.
FIG 3.

Representative photographs of hematoxylin and eosin-stained (A) or picrosirius red-stained (B) cardiac tissue from BALB/c mice infected with Dm28c Trypanosoma cruzi and treated with simvastatin or benznidazole at the indicated doses. Insets show healthy noninfected tissues. Sim, simvastatin; Bz, benznidazole.
15-Epi-lipoxin A4 mediates the effect of simvastatin on cardiac endothelial activation.
It has been previously reported that the anti-inflammatory effect of statins could be related to the induction of the production of 15-epi-lipoxin A4. To determine if the effects observed in BALB/c mice were mediated by 15-epi-LXA4, Sv/129 mice with the 5-LO gene knocked out were chronically infected with T. cruzi, and the effects of simvastatin and 15-epi-LXA4 on cardiac endothelial activation, inflammation, and fibrosis were compared.
Levels of 15-epi-LXA4 in serum were determined 80 days after T. cruzi infection in Sv/129 mice (Fig. 4). As expected, the production of 15-epi-LXA4 in wild-type (WT) mice was increased by simvastatin; however, in 5-LO−/− mice, this effect was not observed. Thus, in mice with T. cruzi infections, simvastatin induced the production of 15-epi-LXA4 via 5-LO activity, whereas 15-epi-LXA4 was detected in 5-LO−/− mice only when it was administered exogenously, reaching levels similar to those obtained in WT animals. T. cruzi infections or benznidazole treatment did not induce the production of this COX2/5-LO-derived eicosanoid.
FIG 4.
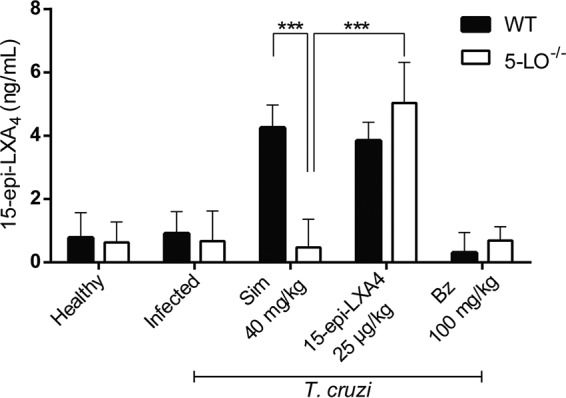
Levels of 15-epi-lipoxin A4 in sera of Sv/129 mice infected with Trypanosoma cruzi. ELISAs were performed at 80 dpi. Bars correspond to the standard deviations for three independent experiments. ***, P < 0.001.
The effect of 15-epi-LXA4 on ECAM expression in chagasic Sv/129 mice was also studied. Figure 5 shows representative immunohistochemical images for E-selectin, ICAM-1, and VCAM-1 from the hearts of T. cruzi-infected mice at 80 dpi. Endothelial activation for the three ECAMs analyzed is evident in the infected animals. Simvastatin in WT mice blunted this activation, but it did not have an effect in 5-LO−/− mice. When 15-epi-lipoxin A4 was administered exogenously, the expression of ECAMs, particularly E-selectin and ICAM-1, was almost abolished. The same pattern was observed for the levels of the soluble fractions of the ECAMs in plasma (Fig. 5B). Likewise, simvastatin decreased the inflammatory response and fibrosis in the hearts of Sv/129 mice (Fig. 6A and B, respectively). Although there were no significant differences between the results for WT and 5-LO−/− mice, there was a trend indicating a lesser effect in the knockout (KO) mice. However, although cardiac inflammation and fibrosis were expected in the group of mice without 5-LO, the response was at least similar to that of infected, untreated WT mice, with only a slight increase observed in inflammatory infiltration and fibrosis in the KO mice. Nevertheless, exogenous 15-epi-LXA4 did produce a significant reduction in inflammatory infiltration and fibrosis, with values reaching levels similar to those of healthy hearts in both WT and KO mice.
FIG 5.

(A) Representative images of cardiac tissue immunohistochemically stained for E-selectin, ICAM-1, and VCAM-1 from Sv/129 wild-type or 5-LO KO mice infected with Dm28c Trypanosoma cruzi and treated with simvastatin or 15-epi-lipoxin A4 at the indicated doses. (B) Levels of soluble E-selectin, ICAM-1, and VCAM-1 in serum. Insets show healthy noninfected murine cardiac tissue.
FIG 6.

Representative photographs of hematoxylin and eosin-stained (A) or picrosirius red-stained (B) cardiac tissue from Sv/129 mice infected with Dm28c Trypanosoma cruzi and treated with simvastatin or 15-epi-lipoxin A4 at the indicated doses. Insets show healthy noninfected murine cardiac tissue. *, P < 0.05.
It is possible that the effect of simvastatin and 15-epi-LXA4 on endothelial activation, cellular infiltration, and fibrosis could be related to a direct trypanocidal effect of simvastatin. However, as shown in Fig. 7, parasites persisted in cardiac tissue at the end of the experiment despite treatment, although they were present in a lower proportion than in the untreated infected controls. The presence of benznidazole may account for the greater clearance of parasites in mice treated with this drug due to its intrinsic trypanocidal activity. In any case, these results indicate the persistence of the parasite in the cardiac tissue of Sv/129 5-LO−/− mice treated with simvastatin (Fig. 7B).
FIG 7.

Parasite load in heart tissue of mice chronically infected with Trypanosoma cruzi and euthanized at 90 (A) or 80 (B) dpi. Bars correspond to the standard deviations for three independent experiments. **, P < 0.01; ***, P < 0.001.
DISCUSSION
A central issue related to CCC is the persistence of an inflammatory state characteristic of myocarditis that is responsible for permanent structural damage to the myocardium. This damage is ultimately responsible for the development of heart failure, arrhythmias, and death in chagasic patients. Current trypanocidal therapies are not able to stop or reverse the damage, although they can eradicate the parasite (13). Thus, it is necessary to provide alternate therapeutic strategies that allow host mechanisms to be modified and alter the course of the disease, including preventing organ deterioration, increasing benznidazole efficacy, and improving the quality of life and survival of CCC patients. Simvastatin is an attractive option to help accomplish this because it is approved, has a known safety profile, and could be quickly incorporated into conventional therapies for Chagas disease patients, without expensive and long-lasting clinical studies.
The murine model of CCC used in this work has already been validated (9). Additionally, it was successfully replicated in the Sv/129 mouse strain, which has not previously reported. While the progression of parasitemia in 5-LO KO mice was slightly lower than in WT mice, no significant mortality was observed (Fig. 1B), and parasite persistence in cardiac tissue was similar in WT and KO mice (Fig. 7B).
It is likely that if 15-epi-LXA4 measurements had been performed at 50 dpi, the levels of this eicosanoid would have been higher. Nevertheless, it is important to note that 15-epi-LXA4 was still detectable 30 days after ending its administration. Thus, a sustained effect of simvastatin can be attained, even if its administration is suspended early. However, although the presence of amastigote nests was not observed in the histological sections, the parasite was detected via quantitative PCR (qPCR) analysis.
It has been reported that statins alter the natural course of other parasitic diseases such as cerebral malaria and intestinal toxoplasmosis (19, 20). In addition, there is evidence that simvastatin decreases cardiac inflammation and fibrosis in dogs chronically infected with T. cruzi (21). The present work showed that simvastatin decreases endothelial activation in vivo, a key factor in the pathogenesis of Chagas cardiomyopathy due to its role in the generation of microthrombi, focal ischemia, and lymphocyte recruitment. In the present work, the most significant effect was on tissue-bound and soluble E-selectin and ICAM-1, as demonstrated previously (10, 22). Inflammation and fibrosis were also decreased in both BALB/c and Sv/129 mice.
Interestingly, the combination of 5 mg/kg simvastatin and 30 mg/kg benznidazole showed an effect similar to that of each of the drugs used separately at doses of 40 and 100 mg/kg, respectively. Although this is not a definite synergistic effect, it is a remarkable finding, since the dose of benznidazole in the combination was reduced by two-thirds, reducing inflammation and fibrosis and also the parasitic load of cardiac tissue. Possibly, the antiparasitic activity of benznidazole did not allow a full synergistic effect to be observed.
Furthermore, the most significant findings were observed when simvastatin was administered to Sv/129 5-LO−/− mice because in that setting, endothelial activation was unaffected, and only the administration of 15-epi-LXA4 restored the anti-inflammatory effect, although not completely, as we expected (Fig. 5). Thus, simvastatin exerts, at least partially, an effect related to the generation of 15-epi-LXA4. Lipoxins decrease endothelial dysfunction in cerebral malaria, toxoplasmosis, and Chagas disease (23). However, the link between statins and 15-epi-LXA4 has not been previously established in parasitic infections, especially in CCC. This study provides evidence that further supports the use of anti-inflammatories as an adjuvant in CCC therapies. It is possible that the anti-inflammatory effect of 15-epi-lipoxin A4 enables the immune system to clear parasites from cardiac tissue. It should be noted that simvastatin is not as efficacious as benznidazole in eradicating T. cruzi parasites from cardiac tissue. Although statins have trypanocidal effects in vitro, the proresolutory processes initiated by simvastatin-induced 15-epi-lipoxin A4 production enables the immune system to eliminate the parasites, probably because chronic inflammation is one of the evasive mechanisms used by the parasites.
Finally, from our results it is possible to state that in this murine model of Chagas heart disease, the presence of simvastatin improves benznidazole activity.
MATERIALS AND METHODS
Animals.
Eight-week-old, 20- to 25-g male BALB/c and sV/129 wild-type (WT) or 5-LO-deficient (5-LO−/−) mice were obtained from the Animal Facilities at the Faculty of Medicine, University of Chile, and Biological Sciences Institute of Minas Gerais Federal University (Belo Horizonte, Brazil) (24). The animals were maintained in a controlled environment at a constant temperature and under a 12-h day/night cycle, with food and water available ad libitum. Animal handling and experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health, USA (25). Supervising protocols were approved by the Institutional Bioethics Committee of the Faculty of Medicine, University of Chile (Protocol CBA# 0528 FMUCH, associated with the FONDECYT-Chile grant number 1130189).
Parasites and infection protocol.
Mice were intraperitoneally inoculated with 1,000 blood trypomastigotes of T. cruzi strain Dm28c. After randomization, animals were distributed in groups of six (BALB/c) or four (Sv/129) individuals; each group received a different treatment. At 5 dpi, direct microscopic visualization of circulating trypomastigotes in peripheral blood samples was used to confirm T. cruzi infection (26, 27). At 80 and 90 dpi, animals (Sv/129 and BALB/c, respectively) were euthanized with 150 mg/kg ketamine (Drag Pharma Investec, Santiago, Chile) and 30 mg/kg of xylazine (Laboratorios Alfasan, Buenos Aires, Argentina), and blood and hearts were extracted. The hearts were halved, with one half fixed in 4% formaldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.3) and the other half in 95% ethanol for histological and qPCR analyses, respectively.
Drug treatment protocol.
BALB/c mice were treated with 40 mg/kg/day simvastatin, 100 mg/kg/day benznidazole, or a combination of 5 mg/kg/day simvastatin and 30 mg/kg/day benznidazole. Sv/129 WT and 5-LO−/− mice received 40 mg/kg/day simvastatin, 100 mg/kg/day benznidazole, or 25 μg/kg/day 15-epi-LXA4 for 20 days starting at 30 dpi. Drugs were suspended in 0.5% aqueous methylcellulose and administered via oral gavage once daily. Mortality rates among the groups were not significantly different from mortality rates in the infected controls (P > 0.05; Kaplan-Meier analysis).
Histology.
Formaldehyde-fixed heart tissues were dehydrated with 50 to 100% ethanol, clarified with xylol, paraffin embedded, and sectioned in 5-μm slices for hematoxylin and eosin staining to observe inflammation or picrosirius red staining to examine cardiac fibrosis. Five representative, nonconsecutive microscopic fields from each sample were photographed (Nikon CoolPix 4500 camera coupled to a Zeiss Axioplan 2 light microscope). Inflammation was determined using ImageJ software according to the parameters previously reported (16, 28).
Immunohistochemistry.
Cell adhesion molecules were assessed via standard immunoperoxidase techniques (29) using specific anti-E-selectin (5 μg/ml), anti-VCAM-1 (1:1,000), and anti-ICAM-1 (2.5 μg/ml) antibodies (BD Biomedical). Primary antibodies were applied according to the supplier's instructions. Immunostaining was performed using a horseradish peroxidase-labeled streptavidin-biotin kit and diaminobenzidine as the chromogen in accordance with the manufacturer's directions (RTU-Vectastain kit; Vector Laboratories, USA). Heart sections were counterstained with Mayer's hematoxylin (Dako) and mounted with Entellan (Merck). Immunohistochemical controls were performed by replacing the primary antibodies with PBS. All controls were negative. Sections were examined via light microscopy (Zeiss Axioplan 2), and images were captured using a coupled camera (CoolPix 4500; Nikon).
Levels of soluble cell adhesion molecules in serum.
Soluble ICAM-1 (sICAM-1), E-selectin (sE-selectin), and VCAM-1 (sVCAM-1) were assayed from the sera of Sv/129 and BALB/c mice euthanized at 80 or 90 dpi, respectively. Assays were performed using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's protocols (mouse sICAM-1, sE-selectin, and sVCAM-1 Quantikine kits; R&D Systems, USA).
Real-time PCR.
Heart samples from T. cruzi-infected BALB/c and Sv/129 mice, treated as described above, were homogenized, and DNA was isolated using the Wizard Genomic DNA purification kit (Promega, USA) according to the manufacturer's instructions. DNA was quantified based on measurements of absorbance at 280 nm using a Varioskan spectrophotometer (Thermo Scientific, USA). qPCR analyses were performed using SYBR green PCR master mix (Thermo Fisher Scientific, USA). DNA amplification was performed using the primers S35 (5′-AAATAATGTACGGGGAGATGCATGA-3′) and S36 (5′-GGGTTCGATTGGGGTTGGTGT-3′), which are specific to T. cruzi minicircles. TNFa-5241 (5′-TCCCTCTCATCAGTTCTATGGCCCA-3′) and TNFa-5411 (5′-CAGCAAGCATCTATGCACTTAGACCCC-3′) primers, which amplify a 170-bp sequence of the Mus musculus tumor necrosis factor alpha (TNF-α) gene, were used as a loading control (26, 27). PCR amplifications were carried out on the 7300 real-time PCR system (Applied Biosystems, USA) as described previously (16).
Statistical analysis.
For all experiments, statistical significance was established at P values of <0.05. Data represent the means ± standard deviations (SD) from at least three independent experiments. All statistical analyses were performed using GraphPad Prism (5.0) software. One- and two-way analysis of variance (ANOVA) (with Tukey's posttests) or Student's t tests were performed as appropriate. For the survival analysis, a log rank test was performed.
REFERENCES
- 1.WHO. 2015. Investing to overcome the global impact of neglected tropical diseases: third WHO report on neglected tropical diseases. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.WHO. Estimates for 2000–2012. Global Health Observatory Data Repository, World Health Organization, Geneva, Switzerland: http://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html Accessed 30 December 2016. [Google Scholar]
- 3.Bern C. 2015. Chagas' disease. N Engl J Med 373:456–466. doi: 10.1056/NEJMra1410150. [DOI] [PubMed] [Google Scholar]
- 4.Rassi A Jr, Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 5.Rassi A Jr, Rassi A, Marcondes de Rezende J. 2012. American trypanosomiasis (chagas disease). Infect Dis Clin North Am 26:275–291. doi: 10.1016/j.idc.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Apt Baruch W. 2013. Parasitología humana. McGraw-Hill Interamericana, Mexico City, Mexico. [Google Scholar]
- 7.Teixeira AR, Hecht MM, Guimaro MC, Sousa AO, Nitz N. 2011. Pathogenesis of Chagas' disease: parasite persistence and autoimmunity. Clin Microbiol Rev 24:592–630. doi: 10.1128/CMR.00063-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spoladore R, Fisicaro A, Faccini A, Camici PG. 2014. Coronary microvascular dysfunction in primary cardiomyopathies. Heart 100:806–813. doi: 10.1136/heartjnl-2013-304291. [DOI] [PubMed] [Google Scholar]
- 9.Molina-Berrios A, Campos-Estrada C, Lapier M, Duaso J, Kemmerling U, Galanti N, Ferreira J, Morello A, Lopez-Munoz R, Maya JD. 2013. Protection of vascular endothelium by aspirin in a murine model of chronic chagas' disease. Parasitol Res 112:2731–2739. doi: 10.1007/s00436-013-3444-x. [DOI] [PubMed] [Google Scholar]
- 10.Yang J, Huang C, Yang J, Jiang H, Ding J. 2010. Statins attenuate high mobility group box-1 protein induced vascular endothelial activation: a key role for TLR4/NF-kappab signaling pathway. Mol Cell Biochem 345:189–195. doi: 10.1007/s11010-010-0572-9. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, Wittner M, Tanowitz HB. 1999. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappab and induces vascular adhesion molecule expression. Infect Immun 67:5434–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanowitz HB, Machado FS, Spray DC, Friedman JM, Weiss OS, Lora JN, Nagajyothi J, Moraes DN, Garg NJ, Nunes MC, Ribeiro AL. 2015. Developments in the management of chagas cardiomyopathy. Expert Rev Cardiovasc Ther 13:1393–1409. doi: 10.1586/14779072.2015.1103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr, Rosas F, Villena E, Quiroz R, Bonilla R, Britto C, Guhl F, Velazquez E, Bonilla L, Meeks B, Rao-Melacini P, Pogue J, Mattos A, Lazdins J, Rassi A, Connolly SJ, Yusuf S, BENEFIT Investigators. 2015. Randomized trial of benznidazole for chronic chagas' cardiomyopathy. N Engl J Med 373:1295–1306. doi: 10.1056/NEJMoa1507574. [DOI] [PubMed] [Google Scholar]
- 14.Chiang N, Bermudez EA, Ridker PM, Hurwitz S, Serhan CN. 2004. Aspirin triggers antiinflammatory 15-epi-lipoxin a4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci U S A 101:15178–15183. doi: 10.1073/pnas.0405445101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birnbaum Y, Ye Y, Lin Y, Freeberg SY, Huang MH, Perez-Polo JR, Uretsky BF. 2007. Aspirin augments 15-epi-lipoxin a4 production by lipopolysaccharide, but blocks the pioglitazone and atorvastatin induction of 15-epi-lipoxin a4 in the rat heart. Prostaglandins Other Lipid Mediat 83:89–98. doi: 10.1016/j.prostaglandins.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Molina-Berrios A, Campos-Estrada C, Henriquez N, Faundez M, Torres G, Castillo C, Escanilla S, Kemmerling U, Morello A, Lopez-Munoz RA, Maya JD. 2013. Protective role of acetylsalicylic acid in experimental Trypanosoma cruzi infection: evidence of a 15-epi-lipoxin a(4)-mediated effect. PLoS Negl Trop Dis 7:e2173. doi: 10.1371/journal.pntd.0002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campos-Estrada C, Liempi A, Gonzalez-Herrera F, Lapier M, Kemmerling U, Pesce B, Ferreira J, Lopez-Munoz R, Maya JD. 2015. Simvastatin and benznidazole-mediated prevention of Trypanosoma cruzi-induced endothelial activation: role of 15-epi-lipoxin A4 in the action of simvastatin. PLoS Negl Trop Dis 9:e0003770. doi: 10.1371/journal.pntd.0003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhi WH, Zeng YY, Lu ZH, Qu WJ, Chen WX, Chen L, Chen L. 2014. Simvastatin exerts antiamnesic effect in abeta25-35 -injected mice. CNS Neurosci Ther 20:218–226. doi: 10.1111/cns.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reis PA, Estato V, da Silva TI, d'Avila JC, Siqueira LD, Assis EF, Bozza PT, Bozza FA, Tibirica EV, Zimmerman GA, Castro-Faria-Neto HC. 2012. Statins decrease neuroinflammation and prevent cognitive impairment after cerebral malaria. PLoS Pathog 8:e1003099. doi: 10.1371/journal.ppat.1003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bereswill S, Munoz M, Fischer A, Plickert R, Haag LM, Otto B, Kuhl AA, Loddenkemper C, Gobel UB, Heimesaat MM. 2010. Anti-inflammatory effects of resveratrol, curcumin and simvastatin in acute small intestinal inflammation. PLoS One 5:e15099. doi: 10.1371/journal.pone.0015099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melo L, Caldas IS, Azevedo MA, Goncalves KR, da Silva do Nascimento AF, Figueiredo VP, de Figueiredo Diniz L, de Lima WG, Torres RM, Bahia MT, Talvani A. 2011. Low doses of simvastatin therapy ameliorate cardiac inflammatory remodeling in Trypanosoma cruzi-infected dogs. Am J Trop Med Hyg 84:325–331. doi: 10.4269/ajtmh.2011.10-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang JC, Huang F, Wu CJ, Chen YC, Lu TH, Hsieh CH. 2012. Simvastatin reduces vcam-1 expression in human umbilical vein endothelial cells exposed to lipopolysaccharide. Inflamm Res 61:485–491. doi: 10.1007/s00011-012-0435-9. [DOI] [PubMed] [Google Scholar]
- 23.Russell CD, Schwarze J. 2014. The role of pro-resolution lipid mediators in infectious disease. Immunology 141:166–173. doi: 10.1111/imm.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fahel JS, de Souza MB, Gomes MT, Corsetti PP, Carvalho NB, Marinho FA, de Almeida LA, Caliari MV, Machado FS, Oliveira SC. 2015. 5-Lipoxygenase negatively regulates Th1 response during Brucella abortus infection in mice. Infect Immun 83:1210–1216. doi: 10.1128/IAI.02592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Research Council, Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 26.Bustamante JM, Presti MS, Rivarola HW, Fernandez AR, Enders JE, Fretes RE, Paglini-Oliva P. 2007. Treatment with benznidazole or thioridazine in the chronic phase of experimental chagas disease improves cardiopathy. Int J Antimicrob Agents 29:733–737. doi: 10.1016/j.ijantimicag.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Yanagisawa M, Kisanuki YY, Jelicks LA, Chandra M, Factor SM, Wittner M, Weiss LM, Pestell RG, Shtutin V, Shirani J, Tanowitz HB. 2002. Role of cardiac myocyte-derived endothelin-1 in chagasic cardiomyopathy: molecular genetic evidence. Clin Sci (Lond) 103(Suppl 48):263S–266S. doi: 10.1042/CS103S263S. [DOI] [PubMed] [Google Scholar]
- 28.Boopathy AV, Martinez MD, Smith AW, Brown ME, Garcia AJ, Davis ME. 2015. Intramyocardial delivery of notch ligand-containing hydrogels improves cardiac function and angiogenesis following infarction. Tissue Eng Part A 21:2315–2322. doi: 10.1089/ten.tea.2014.0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molina-Berrios A, Campos-Estrada C, Lapier M, Duaso J, Kemmerling U, Galanti N, Leiva M, Ferreira J, Lopez-Munoz R, Maya JD. 2013. Benznidazole prevents endothelial damage in an experimental model of chagas disease. Acta Trop 127:6–13. doi: 10.1016/j.actatropica.2013.03.006. [DOI] [PubMed] [Google Scholar]