

ABSTRACT
Combination therapies are standard for management of human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infections; however, no such therapies are established for human hepatitis B virus (HBV). Recently, we identified several promising inhibitors of HBV RNase H (here simply RNase H) activity that have significant activity against viral replication in vitro. Here, we investigated the in vitro antiviral efficacy of combinations of two RNase H inhibitors with the current anti-HBV drug nucleoside analog lamivudine, with HAP12, an experimental core protein allosteric modulator, and with each other. Anti-HBV activities of the compounds were tested in a HepG2-derived cell line by monitoring intracellular core particle DNA levels, and cytotoxicity was assessed by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. The antiviral efficiencies of the drug combinations were evaluated using the median-effect equation derived from the mass-action law principle and combination index theorem of Chou and Talalay. We found that combinations of two RNase H inhibitors from different chemical classes were synergistic with lamivudine against HBV DNA synthesis. Significant synergism was also observed for the combination of the two RNase H inhibitors. Combinations of RNase H inhibitors with HAP12 had additive antiviral effects. Enhanced cytotoxicity was not observed in the combination experiments. Because of these synergistic and additive effects, the antiviral activity of combinations of RNase H inhibitors with drugs that act by two different mechanisms and with each other can be achieved by administering the compounds in combination at doses below the respective single drug doses.
KEYWORDS: RNase H, antiviral combination, hepatitis B virus, synergy, viral replication
INTRODUCTION
Hepatitis B virus (HBV) is one of the main causes of chronic liver disease worldwide. Two hundred forty million to 350 million people worldwide are chronically infected with HBV (1, 2) and are at risk for developing end-stage liver disease and hepatocellular carcinoma (3).
HBV is a hepatotropic enveloped DNA virus that replicates by reverse transcription via an RNA intermediate (4). The viral genome consists of a 3.2-kb partially double-stranded relaxed circular DNA (rcDNA). The rcDNA is repaired to a nuclear episomal covalently closed circular DNA (cccDNA) that is the template for viral transcription. Reverse transcription from the viral pregenomic RNA (pgRNA) template occurs inside viral core particles, leading to the formation of minus-strand HBV DNA covalently linked to the viral polymerase, concomitant degradation of pgRNA by the viral RNase H activity of the viral polymerase, and then synthesis of plus-strand DNA. Mature core particles are then either transported back into the nucleus, where the rcDNA is converted to cccDNA, or enveloped to form virions and secreted from the cell noncytolytically.
The most common antiviral regimen for HBV is treatment with nucleos(t)ide analogs (NAs). Five NAs are approved for treatment chronic HBV infection: lamivudine (LAM), telbivudine, entecavir, adefovir, and tenofovir (5). They inhibit reverse transcription of HBV DNA because their incorporation into the elongating DNA strand leads to chain termination. Potent inhibitors such as entecavir and tenofovir also target the priming step of reverse transcription and can lower viremia to undetectable levels (6). However, NA monotherapies very rarely eradicate the virus (7, 8). In addition, the newer, more potent NAs are extremely costly, prohibiting their use in many parts of the world with high HBV endemicity, such as much of Asia and sub-Saharan Africa (9).
Anti-HBV monotherapy is unlikely to be sufficient for the eradication of HBV infection in the majority of patients. Combination therapy has been successful against human immunodeficiency virus (HIV) and hepatitis C virus (HCV) (10, 11). Drugs used in combination therapy should have additive or synergistic activity to increase efficacy and delay or prevent the development of drug resistance and should have no added toxicity (12). However, clinical studies addressing NA combination therapy for HBV have provided little evidence for clinical benefit of this approach for HBV so far (13, 14).
The multifunctional HBV polymerase is the only enzyme encoded by the virus. Its reverse transcriptase (RT) domain synthesizes the viral DNA, and its RNase H domain hydrolyzes the RNA strand within the RNA/DNA hybrids that are generated during reverse transcription to enable synthesis of double-stranded DNA. Both activities are necessary for viral replication; however, the clinically available direct-acting anti-HBV drugs target the HBV RT, whereas RNase H inhibitors are yet to be developed. Therefore, the RNase H is an attractive target for new anti-HBV drugs that may be used in combination with the NAs to increase effectiveness of treatment (15, 16). Recently, we identified several promising compounds that inhibit both HBV RNase H activity and viral DNA synthesis (17–20). In addition to RNase H inhibitors, several other new agents to inhibit viral replication that target different critical steps in the HBV replication cycle are being developed. These agents include host-targeted antivirals (HTAs) and direct-acting antivirals (DAAs) (21). Among DAAs, multiple core protein allosteric modulators (CPAMs) (22) are being developed, including heteroaryldihidropyrimidines (HAPs). CPAMs affect HBV production by inappropriately activating core protein assembly in vitro and in vivo to yield empty or aberrant capsids (23–26).
New anti-HBV agents should not only inhibit HBV replication at nontoxic concentrations but also work in combination with the existing anti-HBV drugs. Here, we tested two novel RNase H inhibitors from different chemotypes (Fig. 1A) in combination with an existing anti-HBV drug, an experimental CPAM, and each other to evaluate whether HBV RNase H combination treatment results in improved efficiency against HBV replication. Lamivudine was selected as a representative NA because it employs the same chain-terminating mechanism as the other approved NAs. The developmental CPAM HAP12 was chosen because it is known to enhance the rate of core protein assembly and preferentially stabilizes noncapsid polymers of core protein (24, 27). We also assessed the effects of the combinations on cellular toxicity to guide the development of novel combination therapies.
FIG 1.
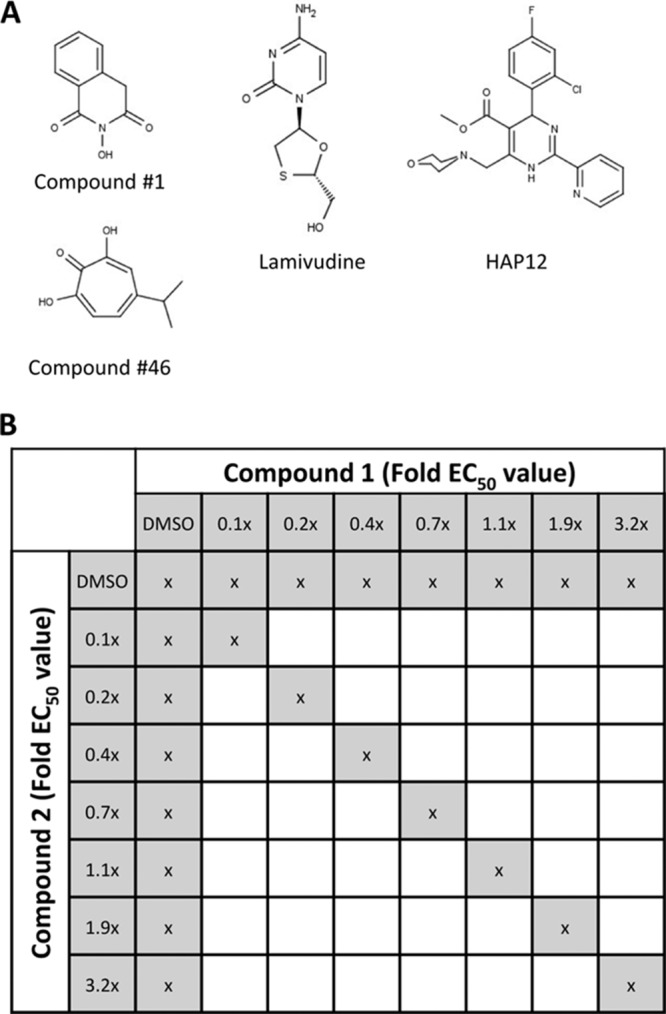
Compounds and study design. (A) HBV inhibitors used in this study. (B) Matrix of compound concentrations used during two-compound antiviral combination experiments. During each two-drug combination assay, individual compounds (1 and 2) were tested alone at seven concentrations from 0.1 to 3.2 times their respective EC50s. To test the combination activity of compounds 1 and 2, seven combination doses were administered to the cells as indicated to ensure constant compounds concentration ratio though the experiment. x, addition of compounds or DMSO.
RESULTS
Anti-HBV efficacy and cytotoxicity of individual compounds.
Anti-HBV activity of the individual compounds was determined in HepDES19 cells. HepDES19 cells are HepG2 hepatoblastoma cells with a tetracycline-repressible expression cassette for a replication-competent HBV genotype D genome (28) that express high levels of HBV. We determined anti-HBV activity of the compounds by treating cells replicating HBV following release of tetracycline suppression with the test compounds and quantifying the HBV minus- and plus-polarity DNA strands in core particles by quantitative PCR (qPCR) (19, 20). Two HBV RNase H inhibitors that we previously identified from different chemical scaffolds were selected for this study: #1 (19) and #46 (20) (Fig. 1). Compound #1 is an N-hydroxyisoquinolinedione, and #46 is the natural product β-thujaplicinol, an α-hydroxytropolone. Compound #1 had a 50% effective inhibitory concentration (EC50) of 4.2 ± 1.4 μM against plus-polarity DNA and did not suppress minus-polarity DNA (Table 1). Compound #46 inhibited plus-strand HBV DNA synthesis with an EC50 of 1.0 ± 0.6 μM. Minus-strand HBV DNA was suppressed at significantly higher concentrations (EC50 = 49 ± 3 μM) (Table 1). Thus, we observed the expected preferential inhibition of plus-polarity DNA strand synthesis by the RNase H inhibitors. We also determined EC50s for LAM and HAP12 (Table 1; Fig. 2) as well as their 50% cytotoxic concentrations (CC50s) in this cell system. As expected, HAP12 inhibited HBV plus- and minus-strand DNAs with similar efficiencies, with EC50s of approximately 0.3 μM (Table 1; Fig. 2), as it acts prior to DNA replication. LAM efficiently inhibited HBV minus-strand DNA (EC50 = 10 μM), and also plus-strand DNA with higher efficiency (EC50 = 0.4 μM) because the minus-strand DNA is the template for plus-strand DNA. The cytotoxicity of compounds was also tested in HepDES19 cells. LAM and HAP12 showed no cytotoxic effects at the highest tested concentration of 100 μM (Table 1). CC50 values for #1 and #46 were 75 μM and 25 μM, respectively (Table 1).
TABLE 1.
HBV replication inhibition efficiency and cytotoxicity of RNaseH inhibitors lamivudine and HAP12
FIG 2.
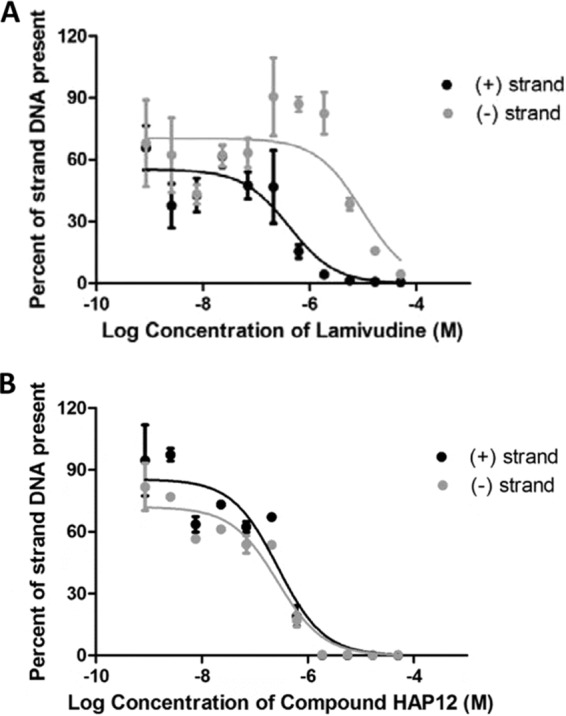
Efficacy of lamivudine (A) and HAP12 (B) against HBV replication. Inhibition of HBV replication was measured relative to DMSO-treated cells. EC50 values were calculated for both the plus and minus polarity DNA strands. The curves and the EC50 values are the means ± 1 standard deviation from two independent experiments.
Efficacy of RNase H inhibitors plus lamivudine.
For each compound pair, two to four independent combination experiments (Fig. 1B) were performed, and viral DNA accumulation was quantified by strand-preferential qPCR. To analyze anti-HBV efficacy of compound combinations, we employed the method of Chou and Talalay (29, 30). The combination index (CI) derived for mixed inhibitors was used to identify additivity (CI = 1), synergy (CI < 1), and antagonism (CI > 1). We first determined the effect on HBV replication of combinations of RNase H inhibitors #46 and #1 with LAM. The results of a representative experiment are shown in Fig. 3A. Isobolograms were constructed for the doses of LAM and #46 with 50%, 75%, and 90% HBV replication inhibition efficiency, referred to as fraction affected (Fa) of 0.5, 0.75, and 0.9, respectively. The combination of #46 and LAM was potently synergistic against HBV, as the experimental combination data points on the isobolograms fall to the lower left of the lines representing the predicted additive effect at each different efficiency level (Fig. 3A). The quantitative parameters of the combination experiments of #46 and #1 with LAM are given in Table 2. In all combination experiments, the linear correlation coefficient (r) for the median dose-effect plots was greater than 0.92, indicating good conformity to the median-effect principle of the mass-action law. The combinations of LAM with #46 or #1 consistently produced average CIs of <1 at efficiency levels from 50% to 95% (Table 2), indicating synergy. Average weighted CI values that emphasize values of high efficiency for LAM plus #46 and LAM plus #1 were 0.7 ± 0.1 and 0.5 ± 0.3, respectively. These values are significantly different by a one-sample t test from the expected CI of 1.0, with P values of 0.018 and 0.033, respectively, at a significance level of 0.05.
FIG 3.
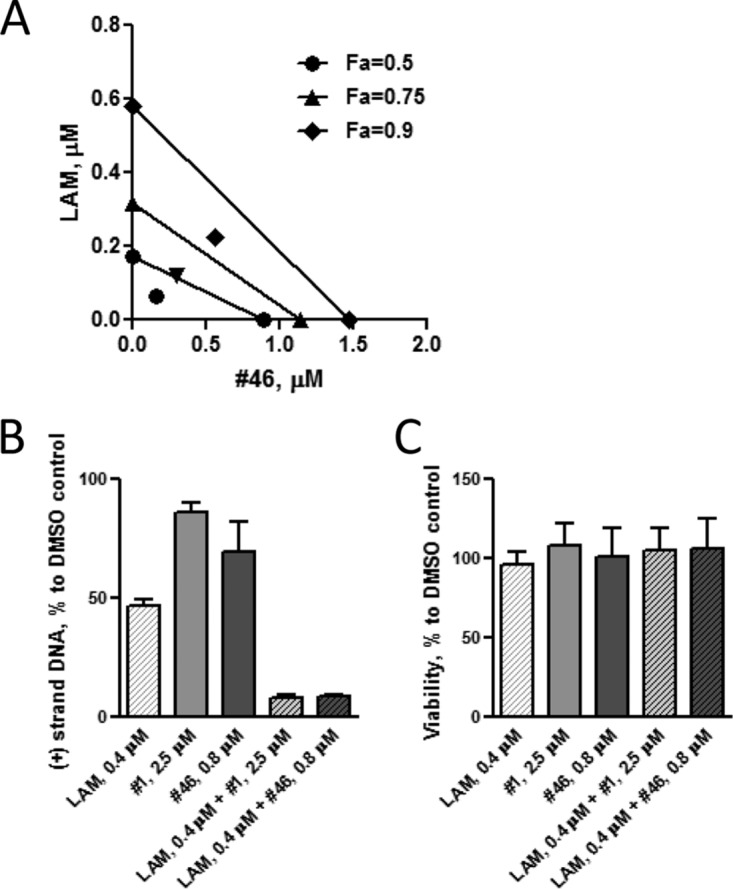
Anti-HBV efficacy of compound combinations. (A) Representative isobolograms of a #46-and-LAM combination experiment. Isobolograms at efficiency doses (referred to as fraction affected [Fa]) of 50%, 75%, and 90% are shown. The actual doses of #46 and LAM are plotted on the x and y axes, respectively. The points on the axes are the doses of each compound necessary to generate the given Fa value. The line drawn between the points on the axes corresponds to the possible combination of doses that are needed to generate the same Fa value, indicating the expected additive effect for the compound combination. The experimental combination data points for #46 and LAM fall on the lower left of the line at each efficiency level, indicating synergistic effects. A dose reduction for the LAM-and-#46 combination is also apparent at all three Fa values. (B) Inhibition of HBV plus-strand DNA by combinations of LAM and #1 or #46. HepDES19 cells replicating HBV were treated with the indicated compounds at concentrations calculated to inhibit HBV replication at 95% efficiency in combination. HBV core DNA was purified, the amount of plus-strand DNA was quantified by qPCR, and the amount of DNA for each treatment condition is shown as a percentage of the DMSO control; error bars are ±1 standard deviation. (C) Cytotoxic effect of LAM and #46 and #1. HepDES19 cells were exposed to the compounds for 3 days at concentrations that inhibit HBV replication at 95% efficiency in combination. Cell viability as a percentage of the DMSO control was assayed by MTS assay; error bars indicate ±1 standard deviation.
TABLE 2.
Combination effects of RNaseH inhibitors, lamivudine, and HAP12 on HBV replication inhibitione
| Compound combination | Combination ratio | ra | CIb values at inhibition of: |
Weightedc CI values | DRId at 95% inhibition |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| 50% | 75% | 90% | 95% | LAM | #1 | #46 | ||||
| LAM + #1 | 1:10 | 0.94 ± 0.05 | 0.70 ± 0.37 | 0.51 ± 0.31 | 0.41 ± 0.29 | 0.36 ± 0.29 | 0.50 ± 0.27 | 6.1 ± 0.8 | 2.0 ± 0.2 | |
| LAM + #46 | 1:3 | 0.96 ± 0.01 | 0.75 ± 0.16 | 0.70 ± 0.06 | 0.69 ± 0.15 | 0.70 ± 0.23 | 0.70 ± 0.13 | 3.3 ± 0.9 | 2.3 ± 0.5 | |
| #1 + #46 | 4:1 | 0.92 ± 0.09 | 0.85 ± 0.32 | 0.49 ± 0.03 | 0.34 ± 0.17 | 0.28 ± 0.22 | 0.40 ± 0.12 | 3.5 ± 0.7 | 9.5 ± 0.3 | |
| HAP12 + #1 | 1:15 | 0.93 ± 0.05 | 1.12 ± 0.25 | 1.05 ± 0.19 | 1.01 ± 0.15 | 0.98 ± 0.12 | 1.01 ± 0.15 | |||
| HAP12 + #46 | 1:4 | 0.92 ± 0.02 | 0.86 ± 0.02 | 0.83 ± 0.05 | 0.88 ± 0.02 | 0.95 ± 0.02 | 0.90 ± 0.01 | |||
r, linear correlation coefficient of the median-effect plot.
CI (combination index) was calculated by the CI equation of Chou and Talalay (29). CI < 1, CI = 1, and CI > 1 indicate synergism, additive effect, and antagonism, respectively.
The weighted CI is calculated as follows: CIwt = (CI50 + 2CI75 + 3CI90 + 4CI95)/10.
DRI, dose-reduction index, calculated by comparing the doses required to reach 95% of HBV replication inhibition when using the compound as a single agent and in combination.
r, CI, and DRI values shown are means ± 1 standard deviation.
A major objective of having synergistic drug combination is to reduce the doses of the drugs used, thereby reducing the toxicity while maintaining efficacy. The median-effect principle and the combination index theorem can be used to predict dose reduction for drugs during combination treatment (31). Therefore, we calculated the dose-reducing index (DRI), a measure of the degree to which the dose of each agent in a synergistic combination may be reduced to achieve a given effect level compared with the doses of each agent alone (31). A DRI of >1 is beneficial, and the greater the DRI value, the greater the dose reduction. The average DRI for compound #1 was 2 and for LAM was 6 at 95% HBV replication inhibition efficiency (Table 2), indicating that the concentrations of LAM and #1 necessary to inhibit HBV replication by 95% could be decreased 2- and 6-fold, respectively, when the compounds are used in combination. DRIs for compound #46 and LAM were similar, being 2 and 3, respectively (Table 2).
To confirm these calculated DRIs for LAM and RNase H inhibitor combinations, we measured the inhibition activity of LAM with #1 or #46 singly or in combination at the suboptimal reduced doses of LAM (0.4 μM) with #1 (2.5 μM) or #46 (0.8 μM) as calculated by the Chou-Talalay method to inhibit HBV replication by 95% when used in combination (Fig. 3B). As expected, #1 and #46 as single agents only slightly inhibited HBV DNA accumulation (14% ± 3.2% and 30% ± 10%, respectively). LAM alone had an inhibitory effect of 53% ± 2.1%. As predicted, the combination of LAM with #1 and #46 resulted in more than 90% of inhibition, 91.5% ± 0.7% and 91.4% ± 0.5%, respectively. Since the doses of #46, #1, and LAM used in combination experiments were significantly less than their respective 50% cytotoxic concentrations, cellular toxicity was not expected. However, to ensure that unpredicted combinatorial toxicities did not contribute to the antiviral synergistic effects for #46, #1, and LAM, we assayed the cytotoxic effects of #46, #1, and LAM combinations at concentrations that inhibit HBV plus-strand DNA synthesis by 95% (Fig. 3C). No significant cytotoxic effects were observed for any of the cultures treated with one or two compounds, indicating that the observed synergistic effect of LAM and #46 and #1 on HBV DNA synthesis was not due to the increased overall cellular toxicity.
Efficacy of RNase H inhibitors plus HAP12.
Combinations of HAP12 with #46 or with #1 were evaluated by the same method. They produced CIs of approximately 1 at all indicated levels of efficiency, and the weighted CIs for HAP12 were 0.9 ± 0.01 with #46 (P = 0.03) and 1.01 ± 0.15 with #1 (P = 0.86) (Table 2). These data imply additive anti-HBV effects for HAP12 when used with #1 and either additive or slightly synergistic interactions when used with #46.
Efficacy of RNase H inhibitor combinations.
We next tested the two RNase H inhibitors in combination. CI values of #1 and #46 at efficiency levels from 50% to 95% were all below 1 (from 0.85 ± 0.32 to 0.28 ± 0.22), and the weighted CI of 0.40 ± 0.12 was significantly less than 1.0 (P = 0.04), indicating that the two RNase H inhibitors were synergistic (Table 2). The average DRI for 95% of efficiency to inhibit HBV replication was 3.5 for compound #1 and 9.5 for compound #46, indicating that compounds #1 and #46 could be lowered 3- or 9-fold when given as combination therapy.
Loewe additivity model.
A variety of different methods have been developed for analysis of combination experiments (32, 33). Therefore, the antiviral effects of the compound combinations were also analyzed by Loewe additivity models using the Chalice Analyzer software (Table 3). Data analyses demonstrated that each of the LAM+#1, LAM+#46, and #1+#46 combinations had positive values for Loewe excess volume and synergy score, indicating synergistic interactions. The combinations of HAP12 with RNase H inhibitors also had positive values for Loewe excess volume and synergy score; however, because of high standard deviations, these data are considered insignificant, indicating an additive effect. Thus, analyses on the basis of the Loewe additivity model confirmed the results of analyses by the Chou-Talalay method for all tested combinations.
TABLE 3.
Loewe additivity model analyses of anti-HBV compound combinationsa
| Compound combination | Loewe excess volb | Synergy score |
|---|---|---|
| LAM + #1 | 0.49 ± 0.33 | 0.99 ± 0.28 |
| LAM + #46 | 0.46 ± 0.23 | 0.75 ± 0.35 |
| #1 + #46 | 0.49 ± 0.30 | 1.34 ± 0.16 |
| HAP12 + #1 | 0.24 ± 0.24 | 0.53 ± 0.49 |
| HAP12 + #46 | 0.31 ± 0.15 | 0.30 ± 0.29 |
Results are means ± 1 standard deviation.
Positive values indicate synergistic combinations; negative values would indicate antagonism.
Cytotoxic effects of combinatorial compound treatment.
Combination therapy can modulate cytotoxicity as well as efficiency. Therefore, we determined the effects of LAM and HAP12 on CC50 values of #46 and #1 by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. LAM and HAP12 each have CC50 values above 100 μM, the highest dose tested. Therefore, we measured CC50 values for #46 and #1 alone and in the presence of LAM and HAP12 at 100 μM. The CC50 value for #46 alone was 47 ± 12 μM, which was not significantly different from the CC50 value of 42 ± 8 μM for #46 in the presence of 100 μM LAM (P = 0.59; Student's t test) (Fig. 4A) or from the 34 ± 8 μM CC50 value for #46 in the presence of 100 μM HAP12 (P = 0.22) (Fig. 4B). Similarly, the CC50 value for compound #1 was more than 100 μM under all treatment conditions, alone or in combination with 100 μM LAM (Fig. 4C) or 100 μM HAP12 (not shown). These data exclude the possibility that the observed anti-HBV activities of these compound combinations were a result of cytotoxicity and/or cytostatic effects of the compounds.
FIG 4.

Cytotoxicity associated with combining anti-HBV agents. Cell viability was assayed by MTS assay. (A) Effect of LAM on #46 CC50. HepDES19 cells were exposed to #46 alone or in combination with 100 μM LAM. (B) Effect of HAP12 on #46 CC50. HepDES19 cells were exposed to #46 alone or in combination with 100 μM HAP12. (C) Effect of LAM on #1 CC50. HepDES19 cells were exposed to #1 alone or in combination with 100 μM LAM. (D) Combined cytotoxicity of #1 and #46. HepDES19 cells were seeded into a 96-well plate and exposed to #1 and #46 alone or in combination at a range of equimolar concentrations.
To determine the cytotoxic effects of combinations of two RNase H inhibitors, HepDES19 cells were treated with both #1 and #46 simultaneously or independently across a range of concentrations and analyzed for viability by the MTS assay (Fig. 4D). Combining #1 and #46 did not increase overall cytotoxicity, with cytotoxicity being determined by the more-toxic compound (#46) (total, 111 ± 3 μM, which corresponds to the #46 part being 56 ± 1 μM, as #1 and #46 were used in a 1:1 combination ratio). This is not significantly different from the CC50 for compound #46 alone (P = 0.36). Analysis using the method of Chou-Talalay (29) showed that the weighted cytotoxicity CI for #1 and #46 was 1.2. Thus, #1 and #46 have additive or slightly antagonistic effects on cytotoxicity.
DISCUSSION
Current HBV therapies attenuate chronic hepatitis B but rarely cure chronic HBV infections. This warrants a search for effective multidrug cocktails that target multiple steps in viral life cycle. Combination therapy is essential to management and cure of chronic viral infections, including HIV and HCV, and it is expected that combination therapy will provide similar benefits for patients with hepatitis B (15, 34). In addition, combination anti-HBV therapy is expected to achieve longer-term suppression of HBV replication and consequently viral load, with shorter treatment regimens than currently available. This should reduce side effects through reducing the dose of each drug used and also reduce the risk of resistant mutants developing.
Currently, there is no standard therapy that includes drug combination for treatment of patients with HBV infection. A few controlled clinical studies addressing NAs or NA plus interferon combination therapies against HBV have been undertaken, but there is little evidence of the clinical benefit of this approach (14, 35). Therefore, establishing whether combination therapy with newer agents provides increased benefit over monotherapy remains an urgent task.
Here, we hypothesized that treatment combining NAs with RNase H inhibitors could have additive or synergistic effects because these antiviral agents will be independently targeted to the HBV polymerase and RNase H catalytic centers. Our results indicate that combining an approved anti-HBV NA, LAM, with two different RNase H inhibitors, #46 and #1, produces greater in vitro antiviral effects on HBV DNA synthesis than any of the agents used alone (Fig. 5). This is similar to the synergistic effect of combining of β-thujaplicinol (compound #46) and calanolide A, a nonnucleoside HIV RT inhibitor, that was described against the HIV RT polymerase activity in vitro (36). Another study of influenza virus endonuclease inhibition by compound #1 demonstrated that compound #1 was competitive with RNA but noncompetitive with nucleoside triphosphates (NTPs) (37), further supporting the possibility of simultaneous targeting of two active sites of a viral polymerase by combination of NA and RNase H inhibitors.
FIG 5.

Sites of action of antiviral agents used in this study and their combined effects on HBV replication. The NAs and RNase H inhibitors act inside core particles and have synergistic effects on HBV replication inhibition. Core particle assembly modulators (e.g., HAP12) inhibit the formation of mature replication-competent HBV core particles, thus reducing the overall rate of HBV replication. The combination of RNase H inhibitors with core particle assembly modulators has an additive effect on HBV replication inhibition. Domains of HBV polymerase, i.e., terminal protein domain (TP), reverse transcriptase domain (RT), and RNase H domain (RH), and the RNA (black)/DNA (gray) heteroduplex are indicated inside core particles.
The molecular mechanism for the observed synergy between LAM and RNase H inhibitors is unclear at present. LAM is a cytidine analog that inhibits DNA polymerase function by terminating chain elongation and therefore is unlikely to have any direct inhibitory effect on the HBV RNase H. Similar to HIV RNase H, the active site of HBV RNase H is believed to contain two essential Mg2+ ions. Compounds #46 and #1 represent two different scaffold types that are both known HIV RNase H active-site metal binders (36, 38, 39). However, HBV RNase H inhibitors might indirectly affect HBV polymerase activity, thus synergistically contributing to the HBV polymerase inhibition by LAM. Binding of RNase H inhibitors to the RNase H active site may induce conformational changes that could distort HBV RT polymerase catalytic site. This could cause reduced interaction of HBV polymerase with the RNA/DNA template and/or steric effects that reduce HBV polymerase activity. Also, binding of RNase H inhibitors to the HBV RT RNase H domain might reduce/restrict the mobility of enzyme, thus slowing down or preventing DNA translocation and thereby inhibit elongation of the nascent viral DNA. There is no experimental evidence at present to support these mechanisms for HBV RNase H inhibitors. However, such phenomena are established for other drugs inhibiting divalent metal-dependent enzymes. For example, nonnucleoside reverse transcriptase inhibitors of HIV RT bind to a site distinct from its catalytic center and induce conformational changes that affect the polymerase activity (40–42). Furthermore, mutational studies with duck hepatitis B virus (DHBV), a common model for HBV, suggest that the RNase H domain of DHBV polymerase may contribute to multiple steps of viral genome replication, such as RNA encapsidation and minus-strand DNA synthesis (43, 44). Substitution mutations of the putative catalytic residues (45) as well as other charged residues in the RNase H domain (46) also affect HBV polymerase activity. In addition, HBV RNase H inhibitors might directly affect DNA strand elongation activity of HBV polymerase through chelating and/or altering the coordination geometry of the divalent metal cations in the HBV RT active site because HBV polymerase activity also requires divalent metal ions for catalysis (47).
Overall, the results presented here on the synergistic action of NA and RNase H inhibitors support the possibility of convergent therapy (i.e., using multiple compounds that target the same stage of viral replication) for future efficient treatment of chronic HBV infections. The aim would be to fully inhibit the HBV polymerase's multiple activities by directing two or more drugs at the same viral target and thus preventing viral replication (Fig. 5).
We also examined the combined effects of two RNase H inhibitors, #1 and #46, on HBV DNA accumulation. Surprisingly, in view of the fact that both compounds appear to act on the same site in HBV RNase H, these two compounds synergistically suppressed HBV DNA synthesis. Several possible mechanisms could explain this synergism. First, the enhanced inhibition of HBV DNA accumulation by #1 and #46 combination may simply reflect higher levels of RNase H inhibition with a two-compound regimen. Even when both RNase H inhibitors are present, the total intracellular availability of active compounds may be insufficient to saturate all HBV RNase H active sites. It is also possible that the access of RNase H inhibitors to the HBV polymerase within viral core particles might be limited. Addition of two RNase H inhibitors simultaneously would increase the probability of RNase H binding and inhibition by these compounds. Second, synergy could result from effects other than RNase H inhibition, although we have no data supporting this possibility. A precedent for synergistic interactions for two compounds that target a single active site exists. Combinations of the NAs LAM and penciclovir are synergistic against HBV in HepG2 2.2.15 cells (48), as well as against duck HBV in primary duck hepatocytes (49). A more recent study of in vitro combinations of tenofovir with different NAs (LAM, entecavir, telbivudine, and adefovir) in HepAD38 cells also showed additive to slight synergistic anti-HBV effects (50).
The challenge of combination therapy is to achieve a maximal antiviral effect with minimal toxicity. We did not observe any evidence of enhanced cytotoxicity during HBV replication inhibition assays at the highest tested combination doses. In addition, the cytotoxicity experiment to determine the effects of the highest possible concentration of LAM under our experimental conditions on CC50 of #46 and #1 demonstrated a lack of synergistic/additive in vitro cytotoxicity for the LAM and #46 and #1 combinations. Similarly, we did not find any evidence of synergistic toxicity of compounds #1 and #46. Together, our results demonstrate the feasibility of a strategy of combining two different of RNase H inhibitors in an anti-HBV convergent combination therapy (Fig. 5).
Divergent therapy involves a combination of antiviral agents targeting different stages of viral replication. To evaluate the potential for the divergent therapy against HBV, we focused on a combination of two classes of compounds: RNase H and core protein allosteric modulators (Fig. 5). We found additive antihepadnaviral activity of combining new developmental agents: RNase H inhibitors and core protein allosteric modulators. Thus, HBV cores that escape the action of these inhibitor(s) because of incomplete effectivity could be later targeted by RNase H inhibitor(s) at a later stage of the replication cycle. As with the RNase H inhibitors and LAM, we found no evidence for synergistic/additive in vitro cytotoxicity for HAP12 plus #1 or #46.
HBV is a genetically diverse virus, with 8 to 10 genotypes that differ by ≥8% at the nucleotide level (51). Our studies were done with a genotype D isolate, but it is probable that our conclusions will apply to other genotypes as well for two reasons. First, the catalytic center of the RNase H is likely to be highly conserved due to selective pressures to maintain its essential function. Second, we recently evaluated the activity of 18 variant HBV RNase H sequences from genotypes B, C, and D (51). Although the basal RNase H activity varied substantially among the variant enzymes, they were equivalently sensitive to compounds #1 and #46.
In conclusion, this study provides strong evidence that targeting multiple hepatitis B viral targets can improve the efficacy of molecularly targeted therapies without exacerbating cytotoxicity. It therefore opens one path toward developing rational combination strategies for treating chronic HBV infections.
MATERIALS AND METHODS
Compounds.
Compounds (Fig. 1A) were acquired commercially or were synthesized as described previously. LAM ([(-)-β-l-2′-3′ dideoxythiacytidine]) was purchased from Sigma, compound #1 (2-hydroxyisoquinoline-1,3(2H,4H)-dione) was obtained from Toronto Research Chemicals, and #46 (β-thujaplicinol) was acquired from the NCI Developmental Therapeutics Program. HAP12 was synthesized as previously described (24). All compounds were dissolved in dimethyl sulfoxide (DMSO; Sigma) at 10 mM and stored at −80°C.
Cell culture.
HepDES19 is a HepG2-derived stably transformed cell line that supports the tetracycline-inducible (tet-off) replication of HBV genome (28). HepDES19 cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle medium (DMEM)/F12 medium (Fisher) supplemented with 100 U of penicillin/ml, 10 μg of streptomycin/ml, and 10% fetal bovine serum (Fisher) in the presence of 1 μg/ml of tetracycline.
Determination of EC50 for individual compounds.
The 50% effective inhibitory concentration (EC50) for the individual compounds was determined by plating HepDES19 cells at 0.3 × 106 cells/well in 12-well plates in medium without tetracycline to induce DNA replication. Forty-eight hours later, cells were treated with medium containing antiviral compounds or DMSO as a vehicle control and without tetracycline. Two days later, cells were lyzed, and supernatants were collected and treated for 60 min at 37°C with Micrococcal Nuclease (New England BioLabs) (final concentration, 2 U/μl) in the presence of 10 mM CaCl2 to digest nonencapsidated nucleic acids (18). Encapsidated HBV DNA was then extracted using the Qiagen QIAamp cador Pathogen minikit (Qiagen). Quantitative PCR (qPCR) was performed with a strand-preferential assay with TaqMan reagents as described previously (19). The amounts of plus- and minus-polarity DNA strands were calculated as percentages relative to the quantity of DNA in DMSO-treated cells. The EC50s were calculated with GraphPad Prism software using a nonlinear regression log(inhibitor) versus response algorithm.
Combination experiments.
Each combination experiment included 24 samples, seven doses of each compound alone, three DMSO-only wells, and seven compound combinations. Of the seven concentrations tested for each compound, the middle dose was equal to 1.1 times the EC50, two higher doses (corresponding to 1.9 and 3.2 times the EC50), and four lower doses (corresponding to 0.1, 0.2, 0.4, and 0.7 times the EC50) were also tested (Fig. 1B). Compound treatment, DNA extraction, and viral DNA quantification were performed as described above. The data were analyzed using the method developed by Chou and Talalay (29, 31, 52), which allows quantitative determination of drug interactions, where combination indexes (CI) of<1, 1, and >1 indicate synergism, additive effect, and antagonism, respectively. The dose-reduction index (DRI) was determined by comparing the ratio of the doses required to reach a given degree of HBV replication inhibition for a single compound and for each compound in the combination. The CompuSyn program was used to calculate CIs and DRIs (ComboSyn, Inc.). The weighted CI value was calculated as follows: CIwt = (CI50 + 2CI75 + 3CI90 + 4CI95)/10 (31). A one-sample t test was used to determine the statistical significance of the calculated CI values from the expected value of 1.0. The antiviral effects of compound combinations were also assessed using the Loewe additivity model with Chalice Analyzer (Horizon Discovery). The activity of compound combinations was numerically evaluated using Loewe excess volume, which is positive for synergistic combinations and negative for antagonism, and synergy score, which is always positive and provides an additional prioritization favoring combinations whose synergy occurs at a high effect level (Horizon Discovery).
Cytotoxicity.
To assess cytotoxic effects of HBV inhibitor combinations, HepDES19 cells were seeded into 96-well plates at a density of 1 × 104 cells/well and exposed to compounds with a treatment schedule identical to that used for the HBV replication inhibition assays. Each RNase H inhibitor was tested at a range of concentrations alone and in combination with LAM or HAP12 at the highest doses available. Following compound treatment, cytotoxicity was assessed by MTS assay. Cytotoxicity was calculated as the percentage of cytotoxicity in compound-treated cells relative to cytotoxicity determined in DMSO-treated cells. The 50% effective cytotoxic concentration (CC50) values were calculated with GraphPad Prism software using the nonlinear regression log(inhibitor) versus response algorithm. Student's t test was used to compare cell viability among the single- and combination-treated cultures.
ACKNOWLEDGMENTS
We thank Stephen Locarnini for helpful conversations.
J.E.T. is an inventor on patents covering use of HBV RNase H inhibitors against HBV. Some of this intellectual property has been licensed to Arbutus Biopharma, Inc., and Arbutus Biopharma provides research support to the Tavis lab.
This work was supported by NIH grant R01 AI104494 to J.E.T. and an ancillary study within the Hepatitis B Virus Research Network (U01 DK082871, Adrian Di Bisceglie, P.I.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Trepo C, Chan HL, Lok A. 2014. Hepatitis B virus infection. Lancet 384:2053–2063. doi: 10.1016/S0140-6736(14)60220-8. [DOI] [PubMed] [Google Scholar]
- 3.Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med 350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 4.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 5.Lok AS, McMahon BJ, Brown RS Jr, Wong JB, Ahmed AT, Farah W, Almasri J, Alahdab F, Benkhadra K, Mouchli MA, Singh S, Mohamed EA, Abu Dabrh AM, Prokop LJ, Wang Z, Murad MH, Mohammed K. 2016. Antiviral therapy for chronic hepatitis B viral infection in adults: a systematic review and meta-analysis. Hepatology 63:284–306. doi: 10.1002/hep.28280. [DOI] [PubMed] [Google Scholar]
- 6.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2:e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, Kann M, Knolle PA, Benkirane M, Durantel D, Michel ML, Autran B, Cosset FL, Strick-Marchand H, Trepo C, Kao JH, Carrat F, Lacombe K, Schinazi RF, Barre-Sinoussi F, Delfraissy JF, Zoulim F. 2015. Towards an HBV cure: state-of-the-art and unresolved questions—report of the ANRS workshop on HBV cure. Gut 64:1314–1326. doi: 10.1136/gutjnl-2014-308943. [DOI] [PubMed] [Google Scholar]
- 8.Tong S, Revill P. 2016. Overview of hepatitis B viral replication and genetic variability. J Hepatol 64:S4–S16. doi: 10.1016/j.jhep.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perrillo RP, Marcellin P. 2010. Effect of newer oral antiviral agents on future therapy of chronic hepatitis B. Antivir Ther 15:13–22. doi: 10.3851/IMP1482. [DOI] [PubMed] [Google Scholar]
- 10.Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Brau N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P. 2014. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 370:1889–1898. doi: 10.1056/NEJMoa1402454. [DOI] [PubMed] [Google Scholar]
- 11.Arends JE, Kracht PA, Hoepelman AI. 2016. Performance of hepatitis C virus (HCV) direct-acting antivirals in clinical trials and daily practice. Clin Microbiol Infect 22:846–852. doi: 10.1016/j.cmi.2016.05.027. [DOI] [PubMed] [Google Scholar]
- 12.Lewin S, Walters T, Locarnini S. 2002. Hepatitis B treatment: rational combination chemotherapy based on viral kinetic and animal model studies. Antiviral Res 55:381–396. doi: 10.1016/S0166-3542(02)00071-2. [DOI] [PubMed] [Google Scholar]
- 13.Lai CL, Leung N, Teo EK, Tong M, Wong F, Hann HW, Han S, Poynard T, Myers M, Chao G, Lloyd D, Brown NA. 2005. A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology 129:528–536. [DOI] [PubMed] [Google Scholar]
- 14.Sung JJ, Lai JY, Zeuzem S, Chow WC, Heathcote EJ, Perrillo RP, Brosgart CL, Woessner MA, Scott SA, Gray DF, Gardner SD. 2008. Lamivudine compared with lamivudine and adefovir dipivoxil for the treatment of HBeAg-positive chronic hepatitis B. J Hepatol 48:728–735. doi: 10.1016/j.jhep.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 15.Tavis JE, Gehring AJ, Hu Y. 2013. How further suppression of virus replication could improve current HBV treatment. Expert Rev Anti Infect Ther 11:755–757. doi: 10.1586/14787210.2013.814846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tavis JE, Lomonosova E. 2015. The hepatitis B virus ribonuclease H as a drug target. Antiviral Res 118:132–138. doi: 10.1016/j.antiviral.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. 2013. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog 9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Y, Cheng X, Cao F, Huang A, Tavis JE. 2013. beta-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res 99:221–229. doi: 10.1016/j.antiviral.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Cai CW, Lomonosova E, Moran EA, Cheng X, Patel KB, Bailly F, Cotelle P, Meyers MJ, Tavis JE. 2014. Hepatitis B virus replication is blocked by a 2-hydroxyisoquinoline-1,3(2H,4H)-dione (HID) inhibitor of the viral ribonuclease H activity. Antiviral Res 108:48–55. doi: 10.1016/j.antiviral.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D'Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE. 2015. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob Agents Chemother 59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Block TM, Rawat S, Brosgart CL. 2015. Chronic hepatitis B: a wave of new therapies on the horizon. Antiviral Res 121:69–81. doi: 10.1016/j.antiviral.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. 2007. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res 76:168–177. doi: 10.1016/j.antiviral.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 24.Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A. 2008. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J Virol 82:10262–10270. doi: 10.1128/JVI.01360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol 5:1125–1136. doi: 10.1021/cb100275b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Chirapu SR, Finn MG, Zlotnick A. 2013. Phase diagrams map the properties of antiviral agents directed against hepatitis B virus core assembly. Antimicrob Agents Chemother 57:1505–1508. doi: 10.1128/AAC.01766-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC, Talalay P. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC, Talalay P. 1981. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem 115:207–216. [DOI] [PubMed] [Google Scholar]
- 31.Chou TC. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 32.Greco WR, Bravo G, Parsons JC. 1995. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 47:331–385. [PubMed] [Google Scholar]
- 33.Koizumi Y, Iwami S. 2014. Mathematical modeling of multi-drugs therapy: a challenge for determining the optimal combinations of antiviral drugs. Theor Biol Med Model 11:41. doi: 10.1186/1742-4682-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. 2013. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res 98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong GL, Wong VW, Chan HL. 2014. Combination therapy of interferon and nucleotide/nucleoside analogues for chronic hepatitis B. J Viral Hepat 21:825–834. doi: 10.1111/jvh.12341. [DOI] [PubMed] [Google Scholar]
- 36.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. 2005. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res 33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkes KE, Ermert P, Fassler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. 2003. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J Med Chem 46:1153–1164. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]
- 38.Hang JQ, Rajendran S, Yang Y, Li Y, In PW, Overton H, Parkes KE, Cammack N, Martin JA, Klumpp K. 2004. Activity of the isolated HIV RNase H domain and specific inhibition by N-hydroxyimides. Biochem Biophys Res Commun 317:321–329. doi: 10.1016/j.bbrc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 39.Billamboz M, Bailly F, Barreca ML, De Luca L, Mouscadet JF, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. 2008. Design, synthesis, and biological evaluation of a series of 2-hydroxyisoquinoline-1,3(2H,4H)-diones as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. J Med Chem 51:7717–7730. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- 40.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. 1992. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 41.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. 1995. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol 2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 42.Hsiou Y, Ding J, Das K, Clark AD Jr, Hughes SH, Arnold E. 1996. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 4:853–860. doi: 10.1016/S0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Robinson WS, Marion PL. 1992. Naturally occurring point mutation in the C terminus of the polymerase gene prevents duck hepatitis B virus RNA packaging. J Virol 66:1282–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J Virol 68:5232–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Marion PL. 1996. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. J Virol 70:6151–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ko C, Shin YC, Park WJ, Kim S, Kim J, Ryu WS. 2014. Residues Arg703, Asp777, and Arg781 of the RNase H domain of hepatitis B virus polymerase are critical for viral DNA synthesis. J Virol 88:154–163. doi: 10.1128/JVI.01916-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vashishtha AK, Wang J, Konigsberg WH. 2016. Different divalent cations alter the kinetics and fidelity of DNA polymerases. J Biol Chem 291:20869–20875. doi: 10.1074/jbc.R116.742494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korba BE. 1996. In vitro evaluation of combination therapies against hepatitis B virus replication. Antiviral Res 29:49–51. doi: 10.1016/0166-3542(95)00915-9. [DOI] [PubMed] [Google Scholar]
- 49.Colledge D, Locarnini S, Shaw T. 1997. Synergistic inhibition of hepadnaviral replication by lamivudine in combination with penciclovir in vitro. Hepatology 26:216–225. [DOI] [PubMed] [Google Scholar]
- 50.Zhu Y, Curtis M, Qi X, Miller MD, Borroto-Esoda K. 2009. Anti-hepatitis B virus activity in vitro of combinations of tenofovir with nucleoside/nucleotide analogues. Antivir Chem Chemother 19:165–176. doi: 10.1177/095632020901900404. [DOI] [PubMed] [Google Scholar]
- 51.Lu G, Villa JA, Donlin MJ, Edwards TC, Cheng X, Heier RF, Meyers MJ, Tavis JE. 2016. Hepatitis B virus genetic diversity has minimal impact on sensitivity of the viral ribonuclease H to inhibitors. Antiviral Res 135:24–30. doi: 10.1016/j.antiviral.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou TC. 1976. Derivation and properties of Michaelis-Menten type and Hill type equations for reference ligands. J Theor Biol 59:253–276. doi: 10.1016/0022-5193(76)90169-7. [DOI] [PubMed] [Google Scholar]