

Abstract
An enantioselective total synthesis of (+)-amphirionin-4 has been accomplished in a convergent manner. The synthesis features an efficient enzymatic lipase resolution to access the tetrahydrofuranol core in optically-active form. The functionalized tetrahydrofuran derivative was synthesized via an oxocarbenium ion-mediated highly diastereoselective syn-allylation reaction. The polyene side chain was synthesized using Stille coupling reactions. Nozaki-Hiyama-Kishi coupling was utilized to construct the C-8 stereocenter and complete the synthesis of (+)-amphirionin-4.
Graphical Abstract
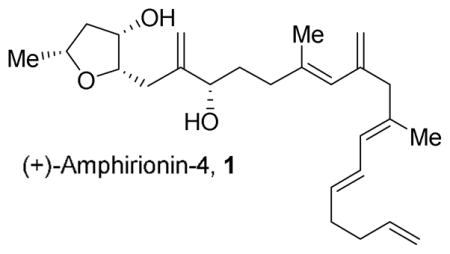
Marine dinoflagellates of the genus Amphidiuium species are rich sources of bioactive polyketide-like natural products with intriguing biological properties.1,2 Recently, Tsuda and co-workers isolated a number of polyketides, amphirionins-2, -4 and -5 from the dinoflagellate Amphidiuium species.3,4,5 Among these, the linear polyke-tide (+)-amphirionin-4 (1, Figure 1) was isolated from the Amphidiuium KCA09051 strain in the benthic sea sand collected off the Iriomote Island. This compound exhibited exceptionally potent proliferation-promoting activity (95% promotion) on murine bone marrow stomal ST-2 cells at 0.1 ng/mL concentration. Interestingly, it did not show proliferation promotion when administered to MC3T3-E1 and NIH3TC cells.4
Figure 1.

Retrosynthetic analysis of (+)-amphirionin-4.
The structure of (+)-amphirionin-4 was elucidated by Tsuda and co-workers using extensive NMR analysis and the absolute configuration of the C4 and C8 hydroxyl groups was determined by Mosher ester analysis.4 Recently, Britton and co-workers have reported the first synthesis of amphirionin-4.6 However, the specific rotation of the synthetic compound was opposite to that reported for natural amphirionin-4.4,6 Considering the intense proliferation-promotion activity of (+)-amphirionin-4 in ST-2 cells, its structural features, and its potential medicinal application, we sought to develop a convergent and concise enantioselective synthesis of (+)-amphirionin-4. Herein, we now report a route that proceeds in 9 linear steps, starting from a readily available racemic butyrolactone.
Our retrosynthesis of (+)-amphirionin-4 is shown in Figure 1. We planned to utilize a Nozaki-Hiyama-Kishi (NHK) 7,8 reaction similar to that reported by Britton and co-workers6 to assemble amphirionin-4 from vinyl iodide 2 and polyene aldehyde 3 at a late stage in the synthesis. The functionalized tetrahydrofuran ring 2 would be constructed from optically active γ-lactone 4 via a cis-selective allylation of the corresponding oxocarbenium ion derived from lactone 4. Optically active lactone 4 would be readily synthesized via acid-catalyzed condensation of pyruvic acid and acetaldehyde, followed by hydrogenation and lipase-catalyzed optical resolution of racemic lactone. This will provide rapid access to both enantiomers for structure-activity relationship studies of amphirionin-4. The polyene side chain 3 would be constructed by Julia-Kocienski olefination of the aldehyde derived from allylic alcohol 5.9 The requisite aldehyde precursor would be synthesized by iterative Stille cross-coupling reactions with appropriately protected vinyl iodide 6, and tributylstannanes 7 and 8.
Our synthesis of functionalized tetrahydrofuran derivatives is shown in Scheme 1. Racemic lactone 4 was synthesized on gram scale by acid-catalyzed condensation of pyruvic acid 9 and acetaldehyde, followed by hydrogenation of the resulting unsaturated lactone over 10 % Pd-C in EtOAc.10,11 Racemic lactone 4 was then subjected to enzymatic resolution utilizing PS-30 in vinyl acetate at 23 °C for 5 h.12,13 While the racemic lactone was obtained in low yield, it can be readily prepared in gram quantity and the current resolution protocol provided optically active alcohol (+)-4 and acetate derivative 10 in 47% and 50% yields, respectively, with high optical purity. Acetate 10 was saponified with aqueous sodium hydroxide in methanol to provide optically active alcohol (−)-4 in 82 % yield.14 The depicted absolute stereochemistry of lactones (+)-4 and (−)-4 was assigned based upon Kazlauskas’ model15 and comparison with the reported specific rotations.16 Optically active lactone (−)-4 was protected as the benzyl ether (>90% ee by chiral HPLC analysis of this benzyl derivative). Dibal-H reduction of the protected lactone provided the corresponding lactol, which was treated with acetic anhydride and pyridine in the presence of DMAP to provide acetate 11 in excellent yield. The hydroxyl group of lactone (−)-4 was also protected as the TBS ether. Dibal-H reduction of that lactone provided a mixture of lactols, which were converted to the acetate 12. The reaction of acetate 11, bearing a C2 benzyl ether, with allyltrimethylsilane in the presence of SnBr4 in CH2Cl2 at −78 °C to 23 °C provided the allyl derivatives 13 and 14 as a 3:1 mixture of diastereomers by 1H-NMR analysis. The corresponding reaction of acetate 12 containing a C2 silyl ether with allyltrimethylsilane under similar conditions furnished allylation products 15 and 16 in 80% yield. However, the diastereomeric ratio of 15 and 16 improved to 10:1 (by 1H NMR analysis). The relative stereochemistry of alkenes 13, 14, and 15 was assigned by 1H NMR NOESY analysis.
Scheme 1.

Synthesis of substituted tetrahydrofurans.
The observed cis-diastereoselectivity is consistent with a C2-benzyloxy substituted tetrahydrofuranyl substrate examined by Woerpel and co-workers.17 It appears that substituents at the C4-position, as in acetate 11, did not influence overall cis-diastereoselectivity. The observed stereochemical outcome can be rationalized using similar steric and electronic arguments to those proposed by Woerpel and co-workers.17 Presumably, the oxocarbenium ion intermediate 17 is preferred over 18 due to pseudoequatorial orientation of the C-2 alkoxy group. The σC-H orbital at C2 maximizes electron donation to the adjacent vacant orbital of the oxocarbenium ion. The bulky C2 siloxy group further enhances the pseudoequatorial orientation, and therefore the overall cis-selectivity.
The desired allyl derivative 15 was converted to the corresponding vinyl iodide as shown in Scheme 2. Oxidative cleavage of alkene 15 using PhI(OAc)2, NMO and catalytic OsO4 provided the corresponding aldehyde in 80% yield.18 Alkynylation of the resulting aldehyde with the Ohira-Bestmann reagent19,20 and MeONa in a mixture of MeOH and THF gave alkyne 20 in 70% yield. Hydrosilylation of the resulting alkyne 20 using Trost’s procedure21 with ([CpRu(MeCN)3]PF6) and triethylsilane followed by iododesilylation22 with NIS and 2,6-lutidine in hexafluoroisopropanol furnished vinyl iodide 2 in 80% yield.
Scheme 2.

Synthesis of vinyl iodide 2.
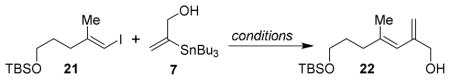
Synthesis of the side chain 5 is shown in Scheme 3. A zirconium-catalyzed carboalumination of 4-pentyne-1-ol 20 followed by reaction with iodine provided the corresponding vinyl iodide. TBS protection of the alcohol provided the vinyl iodide 21 in 91% yield.23 We then investigated the Stille coupling of vinyl iodide 21 with the known24 tributylstannane 7 under a variety of conditions. The results are summarized in Table 1. Reactions with Pd(MeCN)2Cl2 (10 or 20 mol%) in DMF at 23 °C resulted in 30% yield (entries 1 and 2). Catalyst loading was then increased to 30 mol% and reaction temperature was increased to 50 °C for 96 h. This resulted in low yield of coupling product 22 (entry 3). Coupling with Pd2(dba)3 (30 mol%) at 23 °C in a mixture of DIPEA/NMP also resulted in low yield of product (25 %, entry 4). We believe that these low yields can be attributed to the disubstituted vinylstannane, due to a slow transmetallation step and a competing cine substitution.25 In an attempt to enhance the transmetallation step, we decided to use cuprous chloride as a promoter in the Stille-coupling reactions as described by Corey and co-workers.25 Modified Stille-coupling of vinyl iodide 21 and tributylstannane 7 in the presence of CuCl and LiCl at 23 °C for 2 h furnished the desired diene 22 in excellent (90%) yield. Acetylation of diene 22 furnished the corresponding allyl acetate in 95% yield. Stille-coupling26 of the resulting allyl acetate with the known27 hydroxystannane 8 using Pd(dba)2 and LiCl in DMF at 50 °C afforded the coupling product in 96% yield. MnO2 oxidation of the resulting allylic alcohol furnished the aldehyde 23 in 80% yield.
Scheme 3.

Synthesis of aldehyde 23.
Table 1.
Stille-coupling reactions to access diene 22.
| ||||||
|---|---|---|---|---|---|---|
| entry | catalyst & additive | mol (%) | solvent | time (h) | yield (%) | |
| 1 | Pd(MeCN)2Cl2 | 10 | DMF | 24 | 30 | |
| 2 | Pd(MeCN)2Cl2 | 20 | DMF | 24 | 30 | |
| 3 | Pd(MeCN)2Cl2 | 30 | DMF | 96 | 28 | |
| 4 | Pd2dba3 | 30 | DIPEA, NMP | 24 | 25 | |
| 5 | Pd(PPh3)4 CuCl, LiCl |
10 | DMSO | 2 | 90 | |
Our synthesis of the polyene derivative 3 is shown in Scheme 4. Sulfone 26 was prepared in good yield by the reaction of 4-penten-1-ol 24 with 1-phenyl-1H-tetrazole-5-thiol 25, followed by oxidation using catalytic ammonium molybdate and hydrogen peroxide. Julia-Kocienski olefination9 between aldehyde 23 and sulfone 26 gave the polyene 27 in 89 % yield and only E-isomer was formed (by 1H-NMR analysis). Removal of the TBS group followed by DMP oxidation of the resulting alcohol provided aldehyde 3 in good yield.
Scheme 4.

Synthesis of aldehyde 3.
The final synthesis of (+)-amphirionin-4 is outlined in Scheme 5. NHK coupling6 between aldehyde 3 and vinyl iodide 2 was carried out in DMF at 0°C to 23 °C for 24 h. This provided TBS-protected amphirionin-4 28 as the major diastereomer (4:1) in 65 % yield. The diastereomers were separated by silica gel chromatography. Deprotection of the TBS group using 70% HF, 30% pyridine in the presence of excess pyridine furnished synthetic (+)-amphirionin-4 (1) in 98% yield. The 1H and 13C NMR of our synthetic amphirionin-4 {[α]D23 +6.4 (c 0.08, CHCl3)} are identical to the reported spectra for the natural (+)-amphirionin-4{Lit.4 [α]D 20 +6 (c 0.29, CHCl3)}, thus confirming the absolute configuration of our synthetic material.
Scheme 5.

Synthesis of (+)-Amphirionin-4 (1).
In summary, we have achieved an enantioselective synthesis of (+)-amphirionin-4 (1). The synthetic route is convergent and readily scalable. The longest linear path was 9 steps from readily available racemic butyrolactone. The synthesis featured a lipase resolution of a racemic lactone to give highly optically pure isomers. Other key reactions include a highly diastereoselective syn-allylation reaction, an efficient Stille coupling to construct the polyene side chain and a NHK coupling to establish the C8 allylic alcohol diastereoselectively. Our synthesis will provide rapid access to (+)-amphirionin-4 (1) and structural variants for biological studies. Further investigations of structural and biological studies are in progress.
Supplementary Material
Figure 2.

Stereochemical analysis for cis-allylation.
Acknowledgments
Financial support of this work was provided by the National Institutes of Health and Purdue University.
Footnotes
Experimental procedures and 1H- and 13C- NMR spectra are available for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kobayashi J, Tsuda M. Nat Prod. 2004;21:77–93. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]; (b) Kobayashi J, Kubota T. J Nat Prod. 2007;70:451–460. doi: 10.1021/np0605844. [DOI] [PubMed] [Google Scholar]
- 2.Blunt JW, Copp BR, Hu W-P, Munro MHG, North-cote PT, Prinsep MR. Nat Prod Rep. 2008;25:35–94. doi: 10.1039/b701534h. [DOI] [PubMed] [Google Scholar]
- 3.Kumagai K, Minamida M, Akakabe M, Tsuda M, Konishi Y, Tominaga A, Tsuda M, Fukushi E, Kawabata J. Bioorg Med Chem Lett. 2015;25:635–638. doi: 10.1016/j.bmcl.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Minamida M, Kumagai K, Ulanova D, Akakabe M, Konishi Y, Tominaga A, Tanaka H, Fukushi E, Kawabata J, Masuda A, Tsuda M. Org Lett. 2014;16:4858–4861. doi: 10.1021/ol5023504. [DOI] [PubMed] [Google Scholar]
- 5.Akakabe M, Kumagai K, Tsuda M, Konishi Y, Tominaga A, Tsuda M, Fukushi E, Kawabata J. Tetrahedron Lett. 2014;55:3491–3494. [Google Scholar]
- 6.Holmes M, Kwon D, Taron M, Britton R. Org Lett. 2015;17:3868–3871. doi: 10.1021/acs.orglett.5b01844. [DOI] [PubMed] [Google Scholar]
- 7.Takai K, Kimura K, Kuroda T, Hiyama T, Nozaki H. Tetrahedron Lett. 1983;24:5281–5284. [Google Scholar]
- 8.Jin H, Uenishi J, Christ WJ, Kishi Y. J Am Chem Soc. 1986;108:5644–5646. [Google Scholar]
- 9.Blakemore PR, Cole WJ, Kocienski P, Morley A. Synlett. 1998;1:26–28. [Google Scholar]
- 10.MaGee D, Mallais TC, Mayo PDM, Strunz GM. Tetrahedron. 2006;62:4153–4161. [Google Scholar]
- 11.Kang HY, Ji Y, Yu YK, Yu JY, Lee Y, Lee SJ. Bull Kor Chem Soc. 2003;24:1819–1826. [Google Scholar]
- 12.Bianchi D, Cesti P, Battistel E. J Org Chem. 1988;53:5531–34. [Google Scholar]
- 13.Ghosh AK, Chen Y. Tetrahedron Lett. 1995;36:505–508. doi: 10.1016/0040-4039(94)02296-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enders D, Sun H, Leusink FR. Tetrahedron. 1999;55:6129–6138. [Google Scholar]
- 15.Bornscheuer UT, Kazlauskas RJ. Hydrolases in Organic Synthesis. Wiley-VCH; Weinheim, Germany: 1999. [Google Scholar]
- 16.Ahrens H, Paetow M, Hoppe D. Tetrahedron Lett. 1992;33:5327–5330. [Google Scholar]
- 17.Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. J Am Chem Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- 18.Nicolaou KC, Adsool VA, Hale CRH. Org Lett. 2010;12:1552–1555. doi: 10.1021/ol100290a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohira S. Synth Commun. 1989;19:561–564. [Google Scholar]
- 20.Muller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996;521–522 [Google Scholar]
- 21.Trost BM, Ball ZT. J Am Chem Soc. 2005;127:17644–17655. doi: 10.1021/ja0528580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llardi EA, Stivala CE, Zakarian A. Org Lett. 2008;10:1727–1730. doi: 10.1021/ol800341z. [DOI] [PubMed] [Google Scholar]
- 23.Zheng YF, Oehlschlager AC, Hartman PG. J Org Chem. 1994;59:5803–5809. [Google Scholar]
- 24.Bellina F, Carpita A, Santis MD, Rossi R. Tetrahedron. 1994;50:4853–4872. [Google Scholar]
- 25.Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600–7605. [Google Scholar]
- 26.Valle LD, Stille JK, Hegedus LS. J Org Chem. 1990;55:3019–3023. [Google Scholar]
- 27.Chellat MF, Proust N, Lauer MG, Stambuli JP. Org Lett. 2011;13:3246–3249. doi: 10.1021/ol201183f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.