

Graphical abstract
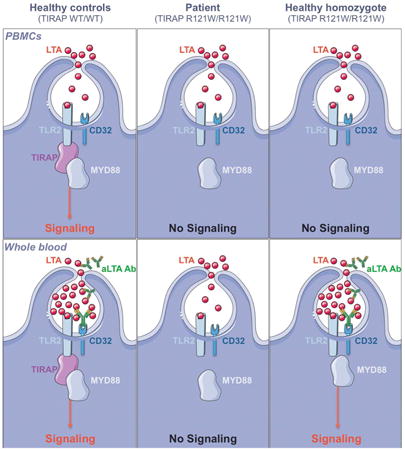
Adaptive immunity can compensate for a defect in the innate immune response, as seen with humans lacking a key TLR adaptor who can mount an antibody response to a bacterial pathogen.
Summary
The molecular basis of the incomplete penetrance of monogenic disorders is unclear. We describe here eight related individuals with autosomal recessive TIRAP deficiency. Life-threatening staphylococcal disease occurred during childhood in the proband, but not in the other seven homozygotes. Responses to all TLR1/2, TLR2/6, and TLR4 agonists were impaired in the fibroblasts and leukocytes of all TIRAP-deficient individuals. However, the whole-blood response to the TLR2/6 agonist staphylococcal lipoteichoic acid (LTA) was abolished only in the index case, the only family member lacking LTA-specific Abs. This defective response was reversed in the patient, but not in IRAK-4-deficient individuals, by anti-LTA mAb. Anti-LTA mAb also rescued the macrophage response in mice lacking TIRAP, but not TLR2 or MyD88. Thus, acquired anti-LTA Abs rescue TLR2-dependent immunity to staphylococcal LTA in individuals with inherited TIRAP deficiency, accounting for incomplete penetrance. Combined TIRAP and anti-LTA Ab deficiencies underlie staphylococcal disease in this patient.
Introduction
Staphylococcus aureus is a Gram-positive bacterium that colonizes the skin and nostrils of most healthy individuals (Li et al., 2015; Mulcahy and McLoughlin, 2016). It is also a common cause of human diseases, ranging from minor cutaneous infections to invasive hematogenous infections, as observed in children and young adults with necrotizing staphylococcal pneumonitis (Gillet et al., 2002). Most severe staphylococcal diseases remain unexplained. Various acquired immunodeficiencies, including those resulting from infection with human immunodeficiency virus, hemodialysis, chemotherapy, intravenous drug use, central catheters and vascular lines, increase the likelihood of severe staphylococcal disease (Lowy, 1998). The pathogenesis of invasive staphylococcal disease is complex in such settings, often involving neutropenia, skin breaches, or both. Alternatively, severe staphylococcal disease may result from various single-gene inborn errors of immunity, including disorders of phagocytes in particular (Zhang et al., 2015). Disorders of the TLR and IL-1R pathway, inborn errors of NF-κB, severe congenital neutropenia, chronic granulomatous disease, leukocyte adhesion deficiency, and autosomal dominant hyper IgE syndrome have all been implicated in severe staphylococcal disease (Bousfiha et al., 2015). All these defects affect phagocytes, albeit in different ways. Clinical features differ between patients with staphylococcal disease, reflecting the diversity of the underlying inborn errors.
Autosomal recessive MyD88 and IRAK-4 deficiencies selectively impair signaling via the TLR and IL-1R pathway (Picard et al., 2003; von Bernuth et al., 2008). More than 80 patients with these deficiencies have been diagnosed worldwide (reviewed in (Picard et al., 2010)). These patients are susceptible to invasive bacterial infections, particularly those caused by Streptococcus pneumoniae, but also those caused by S. aureus. They also develop superficial staphylococcal infections (Picard et al., 2011). Leukocytes from patients with deficiencies of IRAK-4 or MyD88 do not respond to activation via IL-1Rs or TLRs other than TLR3, which signals via TRIF but not MyD88, and TLR4, which signals via TRIF or MyD88. The molecular and cellular basis of staphylococcal disease in these patients remains unknown. We investigated this aspect, by testing the hypothesis that severe staphylococcal disease in otherwise healthy children may be caused by mutations in other genes of the TLR and IL-1R pathway (Casanova, 2015b).
Results
Homozygous TIRAP R121W mutation in a child with severe staphylococcal infection
We investigated a nine-year-old girl admitted to the intensive care unit with pneumonia and sepsis caused by Panton Valentine Leukocidin (PVL)-producing S. aureus, at the age of three months (P1, V.1, Figure 1A). She was born to consanguineous parents of Pakistani descent living in the United Kingdom. The presumed complete genetic tree was assessed by inferring relationships with King software based on genome-wide SNP genotyping (Manichaikul et al., 2010) (Figure 1A and Figure S1A). Known genetic etiologies of PIDs associated with life-threatening staphylococcal diseases were excluded by whole-exome sequencing (WES) and Sanger sequencing. We searched for rare variants (MAF<1%), and identified a homozygous substitution, c.361C>T, in exon 5 of TIRAP, which encodes a TIR domain-containing adapter of TLR2 and TLR4 (Fitzgerald et al., 2001; Horng et al., 2001; Yamamoto et al., 2002a). We confirmed the TIRAP mutation by Sanger sequencing of P1's genomic DNA extracted from whole blood cells (WBC), Epstein-Barr virus-immortalized B lymphocytes (EBV-B cells), and SV40-immortalized fibroblasts (SV40 fibroblasts) (Figure 1B). This missense variant results in the replacement of the arginine residue in position 121 of the TIR domain of TIRAP with a tryptophan residue (R121W) (Figure 1C). The R121 residue of TIRAP has been strongly conserved throughout evolution (Figure 1D). It is also conserved in 21 of the other 26 human molecules containing a TIR domain identified to date (e.g. R196 in MyD88, Figure S1B).
Figure 1. The TIRAP R121W mutation is a rare mutation affecting the TIR domain of TIRAP, with a modest impact at the mRNA and protein levels.
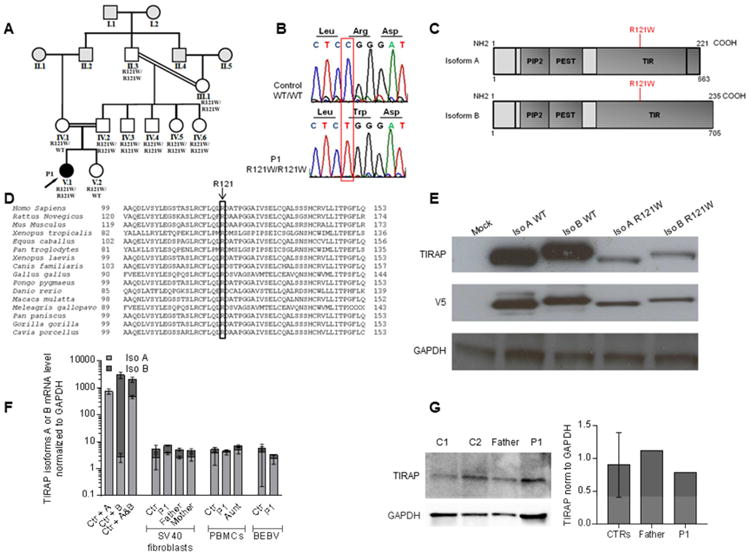
A. Pedigree of the kindred, with allele segregation. Each generation is identified with a roman numeral (from I to V), and each individual with an Arabic numeral (from left to right). The patient (P1) with clinical infectious disease is shown in black and indicated with an arrow. The relationships between individuals in gray were extrapolated according to the estimated kinship coefficients (Figure S1).
B. Electrophoregram of TIRAP genomic DNA sequences from a healthy control and the patient (V.1).
C. Schematic representation of TIRAP, showing the PIP2 domain, the PEST domain, the TIR domain and the coding exons. The R121W mutation in the TIR domain is indicated in red.
D. Conservation of the R121 amino acid in the TIR domain of TIRAP in 16 species.
E. HEK 293 T cells were transfected for 48 h with either TIRAP isoforms (A and B), in the WT form or with the R121W mutation. Western blot was performed on whole-cell extracts with a mouse monoclonal anti-TIRAP antibody and an anti-V5 tag antibody (n=3).
F. Reverse transcription-quantitative PCR was performed to determine the levels of the transcripts encoding TIRAP isoforms A and B in SV40 fibroblasts (from five controls, P1, P1's father and mother), PBMCs (from 9 controls, P1, and P1's aunt) and EBV-B cells (from 5 controls and P1). SV40 fibroblasts stably transfected with the cDNA encoding either isoform A or B were used as controls to test probe specificity. TIRAP mRNA levels encoding isoform A or B are expressed relative to GAPDH mRNA levels (2ΔΔC(t) method). For each probe, the results were normalized relative to one of the controls, for which the value had been set to 1. Error bars show SD (n=2).
G. TIRAP protein levels in PBMCs from two controls, P1's father and P1, as determined by western blotting with an anti-TIRAP mouse mAb. Image J software was used for quantification (n=1). Error bars show SD
In silico analysis with CADD and MSC predicted that the R121W variant would be deleterious (Itan et al., 2016; Kircher et al., 2014). The TIRAP gene is well conserved in the human population, with a neutrality index of 0.2346 (McDonald and Kreitman, 1991) and a GDI of 5.46 (Itan et al., 2015). No other rare or common variants of TIRAP were found in P1. No rare bi-allelic mutations were found in other genes of the TIR pathway, including those encoding individual TLRs and the other three TLR adaptors (TRAM, SARM and TRIF). Moreover, no other candidate variant was identified in the linkage regions defined by parametric multipoint linkage analysis (methods section) or among the other 107 non-synonymous rare homozygous variants found in P1 (Table S1). The patient was therefore homozygous for a potentially deleterious rare allele of TIRAP.
Homozygosity for the rare R121W variant in seven healthy relatives
This variant was not detected in 140 Pakistani individuals from the CEPH-HGD panel. Eight heterozygous, but no homozygous individuals were reported among 57,085 individuals from the Exome Aggregation Consortium (ExAc) database (minor allele frequency (MAF)=0.00007). This variant was detected, in the heterozygous state, in three of 3,033 individuals (6,066 chromosomes) in our in-house exome database. This patient was therefore homozygous for a rare TIRAP missense allele, with a MAF of less than 1/10,000. Surprisingly, however, seven relatives of P1 were also found to be homozygous for the R121W TIRAP mutation, including the proband's father (aged 33 years), four of the father's siblings (aged 20 to 28 years) and both his parents (aged 53 and 54 years) (Figure 1A). None of these individuals carried TIRAP polymorphisms. The proband's mother and younger sister were heterozygous for the R121W allele. None of these relatives, whether heterozygous or homozygous for the TIRAP allele, had ever suffered from any severe infection, including staphylococcal infections in particular. Thus, eight individuals from a consanguineous Pakistani kindred, including a child with severe staphylococcal infection and seven asymptomatic adults, were homozygous for a rare and probably deleterious TIRAP allele. These data suggested that P1 had autosomal recessive TIRAP deficiency, with incomplete penetrance for severe staphylococcal disease. We therefore investigated whether the eight individuals had TIRAP deficiency, and the mechanisms underlying incomplete clinical penetrance.
Normal mRNA and protein levels for the R121W TIRAP allele
Human TIRAP encodes two isoforms (Figure 1C) generated by four different transcripts. Two transcripts (NM_001039661.1 and NM_001318777) encode isoform A, which contains 221 amino acids (aa), (23.8 kDa), whereas the other two transcripts (NM_001318776 and NM_148910) encode isoform B, which contains 235 aa (25.3 kDa). The R121W mutation is located in exon 5 of reference transcript (encoding a sequence present in all isoforms). We assessed the impact of the mutation, by inserting the WT and R121W alleles into pCDNA3.1-V5 vectors encoding a C-terminally tagged version of each of the isoforms of TIRAP. Following the transient transfection of HEK 293T cells, the R121W allele yielded levels of both isoforms (A and B) about 50% lower than those obtained with the WT allele (Figure 1E). This suggested that both R121W TIRAP isoforms were somewhat less stable than the WT isoforms, at least when tagged molecules were overproduced in HEK 293T cells. We then evaluated TIRAP mRNA levels for both isoforms by RT-qPCR and RT-PCR in SV40 fibroblasts from P1, her father (R121W/W), and her mother (R121W/WT), and by RT-qPCR in EBV-B cells from P1, and PBMCs from P1 and her aunt (R121W/W) (Figure 1F and Figure S1C and S1D). P1, her father, mother, and aunt had mRNA levels within the normal range for the controls, with transcripts encoding isoform A predominating in the three cell types tested, and in all individuals tested. We were unable to detect the endogenous expression of either of the two TIRAP isoforms, by western blotting (WB), in SV40 fibroblasts from healthy controls, whereas TIRAP protein levels in control EBV-B cells varied considerably between individuals, consistent with the variability of mRNA levels between controls (data not shown, Figure 1F). In PBMCs from healthy controls only a 24 kDa protein, probably corresponding to isoform A, was detected, consistent with RT-qPCR results (Figure 1G). Homozygous family members had normal levels of this isoform of TIRAP in PBMCs. Overall, these data suggested that the R121W mutation had no detectable impact on mRNA or protein levels in circulating leukocytes.
Impaired subcellular localization of TIRAP R121W isoforms
We assessed the subcellular distribution of the mutant TIRAP molecules. Fluorescence microscopy of HEK 293T cells transiently transfected with a pcDNA3.1 vector encoding the WT form of TIRAP V5-tagged and labeled with Alexa Fluor 488-conjugated anti-V5 antibodies showed WT TIRAP to be present in precise small dots in the cytoplasm, whereas transfection with the R121W TIRAP construct resulted in diffuse cytoplasmic staining (Figure 2A). For the quantification of this subcellular localization phenotype, we cotransfected cells with constructs encoding V5-tagged WT TIRAP and either myc-tagged WT or R121W TIRAP, and then assessed the colocalization of the encoded proteins by determining Pearson's correlation coefficient (PCC) (Figure 2B). WT TIRAP had a PCC of 0.85 for colocalization with itself, whereas its PCC for colocalization with R121W was 0.25 (p < 0.0001). The R121W mutation therefore impaired the subcellular localization of TIRAP. We then investigated whether this cellular localization defect had an impact on the colocalization of TIRAP with its partner, MyD88. We obtained a PCC of 0.6 for the colocalization of WT TIRAP and MyD88, whereas the PCC for the colocalization of R121W TIRAP and MyD88 was 0.2 (Figure 2C) (p < 0.0001). These data suggest that the R121W mutation impairs TIRAP trafficking within cells, thereby reducing the accessibility of this molecule to MyD88.
Figure 2. The R121W mutation impairs the cellular localization of TIRAP and interaction with its partners MyD88 and TLR2.
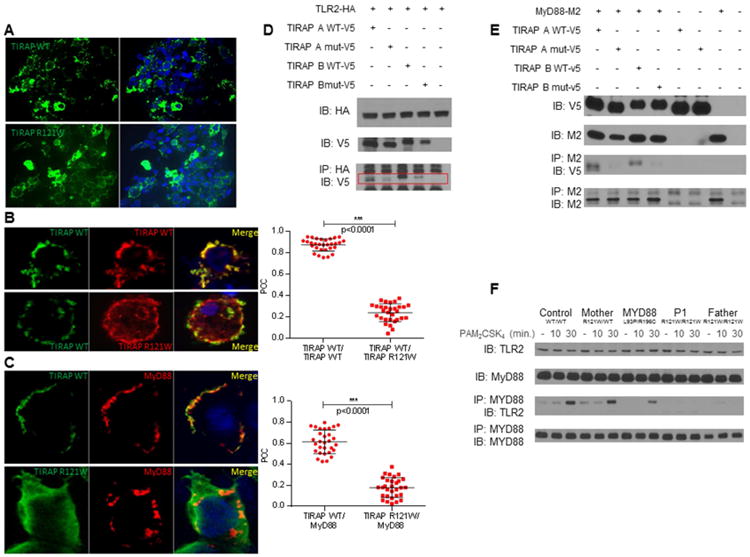
A. HEK 293T cells were transfected with a construct encoding a V5-tagged WT or mutant isoform A of TIRAP for 36 h and then labeled with an Alexa Fluor 488-conjugated anti-V5 antibody. Analyses were carried out by ApoTome fluorescence microscopy. The images shown are representative of three independent experiments and correspond to >95% of the images obtained (30 images were acquired per experiment).
B. Cells were cotransfected with constructs encoding TIRAP WT-V5 and either myc-tagged WT TIRAP or myc-tagged R121W TIRAP in (B) or cotransfected with constructs encoding myc-tagged MyD88 and either V5-tagged WT TIRAP or R121W TIRAP in (C). V5-tagged proteins were labeled with Alexa Fluor 488-conjugated anti-V5 antibodies and myc-tagged proteins were labeled with Alexa Fluor 555-conjugated anti-myc antibodies. Colocalization analysis was performed with Image J software, by calculating Pearson's correlation coefficient (PCC). Three independent experiments were performed and 30 cells were analyzed in each experiment. Error bars show the SD.
D. HEK 293T cells were cotransfected with constructs encoding both the TIRAP A and B isoforms with a V5 tag, WT or mutated, and with a construct encoding HA-tagged TLR2 (D) or and with a construct encoding M2-tagged MyD88 (E). HA or M2 immunoprecipitations and V5 immunoblotting were then performed on total cell extracts (n=2).
F. Co-immunoprecipitation of TLR2 and MyD88 with or without PAM2 stimulation for 10 and 30 min, in SV40 fibroblasts from a control (WT/WT), the P1's mother (R121W/WT), a MyD88-mutated patient (MyD88 L93P/R196C), and P1 and her father (both R121W/W) (n=2).
Impaired interaction of the TIRAP R121W isoforms with TLR2 and MyD88
Following stimulation, TLR2 or TLR4 recruits TIRAP, which recruits MyD88 to the receptor complex through TIR domain interactions (Horng et al., 2001; Yamamoto et al., 2002b). The TIR domain interactions between MyD88 and TLR2 or TLR4 have been characterized in structural studies (Nada et al., 2012; Ohnishi et al., 2009). Four residues of MyD88 have been identified as essential for this interaction, including R196, which is mutated (R196C) in three MyD88-deficient patients (von Bernuth et al., 2008). The R196 residue of MyD88 corresponds to the R121 residue of TIRAP (Figure S1B) (Valkov et al., 2011). Like the R196C mutation of MyD88, the R121W mutation of TIRAP seems to have at most a modest impact on protein levels (Figure 1G). The R121W mutation may impair the interaction of TIRAP with MyD88, thereby preventing downstream signaling and the activation of target gene transcription. The R121W mutation may also impair TIRAP interaction with TLR2 and TLR4 (Lin et al., 2012). We tested these hypotheses, by first performing in vitro co-immunoprecipitation assays in HEK 293T cells transfected with the WT and R121W TIRAP isoforms A and B, together with TLR2 or MyD88. Both WT TIRAP isoforms interacted with TLR2, whereas this interaction was strongly impaired when HEK 293T cells were transfected with the R121W TIRAP isoforms (Figure 2D). Similarly, both WT TIRAP isoforms interacted with MyD88, whereas this interaction was strongly impaired when HEK 293T cells were transfected with either R121W TIRAP isoform (Figure 2E). We also assessed TLR2/MyD88 co-immunoprecipitation after PAM2CSK4 (PAM2) stimulation, in SV40 fibroblasts from healthy controls, P1's mother (heterozygous for TIRAP), and P1 and her father, who are both homozygous for the mutant form of TIRAP (Figure 2F). The co-immunoprecipitation of TLR2 and MyD88 was observed upon TLR2 stimulation in both control and R121W-heterozygous fibroblasts, but not in R121W-homozygous fibroblasts, whereas MyD88-mutant fibroblasts (L93P/R196C) displayed an intermediate pattern. These data suggest that the R121W mutation disrupts the interaction of both TIRAP isoforms with their TLR2 (and presumably TLR4) and MyD88 partners. These data, therefore, suggest that individuals homozygous for R121W have a complete functional deficiency of TIRAP.
The R121W TIRAP allele is loss-of-function
The transient transfection of SV40 fibroblasts from P1 and her father with a vector encoding WT TIRAP isoform A or B induced IL-6 production to levels similar to those in transfected control cells (Figure 3A). With this high background, the increase in response to TLR2 or TLR4 stimulation was only modest, in both cell types (data not shown). By contrast, the transfection of these SV40 fibroblasts with constructs encoding either of the R121W TIRAP isoforms had no effect (Figure 3A). Overexpression of the WT MyD88 allele in SV40 fibroblasts from the control, P1, her father or a MyD88-deficient patient led to the constitutive activation of IL-6 production, but the overproduction of WT TIRAP in MyD88-deficient fibroblasts did not. Moreover, the stable transfection of fibroblasts from P1 or her father with lentiviral particles encoding WT TIRAP isoform A or B, or both, rescued constitutive IL-6 production, whereas no such rescue was observed after stable transfection with the R121W TIRAP construct or the empty vector (Figure S2A). Finally, the overproduction of each TIRAP WT isoform in HEK 293T cells, as already shown for TIRAP isoform A, induced constitutive activation of the NF-κB pathway and IL-6 production (Figure 3B and (Horng et al., 2001)), whereas no such activation was observed following the overproduction of TIRAP R121W isoforms. Thus, the R121W TIRAP allele is loss-of-function, at least for constitutive and MyD88-dependent IL-6 induction following overexpression in fibroblasts.
Figure 3. The TIRAP R121W allele is loss of function.
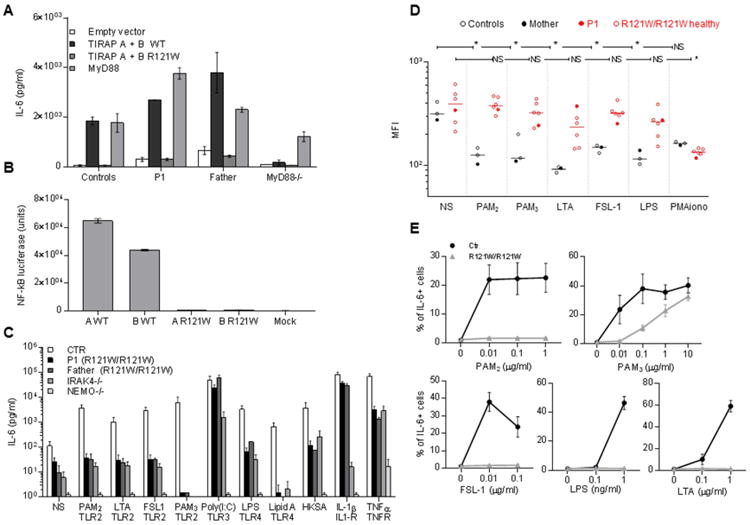
A. IL-6 induction in SV40 fibroblasts from a healthy control, P1, her father, and MyD88-deficient cells, upon transfection with WT or mutant TIRAP isoforms. IL-6 production was measured by ELISA in cells transfected with an empty vector, WT or mutant isoforms A and B, or in MyD88-deficient cells transfected with WT MyD88 (n=3). Error bars show SEM.
B. NF-κB luciferase assay in HEK 293T cells stably expressing a NF-κB-driven luciferase reporter plasmid upon transfection with WT or R121W TIRAP isoform A or B, (n=3). Error bars show SEM.
C. SV40 fibroblasts from healthy controls, P1, her father, an IRAK-4-deficient patient and NEMO-deficient fibroblasts were stimulated with TLR1/2 (PAM3), TLR2/6 (PAM2, purified LTA-SA, FSL-1), TLR3 (poly(I:C)) and TLR4 (LPS and lipid A) ligands, in addition to heat-killed S. aureus (HKSA) IL-1β (IL-1R) and TNF-α (TNF-αR). IL-6 production was assessed by ELISA in supernatants collected 24 h after stimulation and (n=3). Error bars show SEM.
D. Granulocyte L-selectin shedding assessed in several family members (III. 1, IV. 1-5 and V.1) after 1 h of stimulation with PAM2, PAM3, FSL-1, purified LTA, LPS and PMA as an activation control. Mean fluorescence intensity was determined by flow cytometry after the extracellular staining of CD62L. * p values < 0.05 (unpaired t-test) (n=1).
E. PBMCs from several family members (III. 1, IV. 1 to 5 and V.1) were stimulated with PAM2, FSL-1, LTA, PAM3 and LPS for 3 h, subjected to anti-CD3, anti-CD19 and anti-CD14 extracellular staining and IL-6 intracellular staining. The proportion of CD14+ cells positive for IL-6 is shown. 10,000 CD14+ cells were acquired. Each data point represents the mean of all individuals ± SEM.
Impaired responses via TLR2 and TLR4 in fibroblasts homozygous for R121W
TLR2 heterodimerizes with TLR1 for the recognition of triacylated lipopeptides (Jin et al., 2007), which are present in most Gram-positive bacteria, including S. aureus (Kurokawa et al., 2009), and with TLR6 for the recognition of diacylated lipopeptides, also present in S. aureus (Buwitt-Beckmann et al., 2006). It also polymerizes with TLR6 and CD36 for the recognition of staphylococcal lipoteichoic acid (LTA) from most Gram-positive bacteria, including S. aureus (Jimenez-Dalmaroni et al., 2009), as reviewed by (Zahringer et al., 2008). TLR4 recognizes LPS from Gram-negative bacteria (Poltorak et al., 1998). Macrophages and dendritic cells from TIRAP-deficient mice display abolished responses to the stimulation of TLR1/2, TLR2/6, and TLR4, and splenocyte proliferation in response to LPS is impaired (Horng et al., 2002; Yamamoto et al., 2002a). Human SV40 fibroblasts express TLR2 at their surface (data not shown). We thus assessed their cytokine production following stimulation with TLR2 and TLR4 agonists (PAM2 and FSL-1, two TLR2/6 synthetic agonists; purified LTA from S. aureus, another TLR2/6 agonist; PAM3CSK4 (PAM3), a synthetic TLR1/2 agonist; LPS and synthetic lipid A, two TLR4 agonists). We detected cytokine production by control fibroblasts upon stimulation by all these agonists. The response to PAM2, was both TLR2- and TIRAP-dependent, as demonstrated by the inhibition of IL-6 induction by siRNAs specific for TLR2 and TIRAP (Figure S2B), respectively, but not by scrambled siRNA. The responses to LPS and lipid A were also TLR4- and TIRAP-dependent (data not shown). Little or no IL-6 induction was observed in response to PAM2, LTA, FSL-1, PAM3, LPS, lipid A, and heat-killed S. aureus (HKSA), in SV40 fibroblasts from P1 and her father (both R121WW), or in IRAK-4- and NEMO-deficient cells (Figure 3C). However, unlike IRAK-4- and NEMO-deficient cells, SV40 fibroblasts from P1 and her father responded normally to IL-1β. All SV40 fibroblasts other than NEMO-deficient cells responded to TNF-α and poly(I:C) (a TLR3 agonist in human fibroblasts) (Zhang et al., 2007). These data suggest that homozygosity for the R121W allele abolishes TLR1/2 and TLR2/6 responses and strongly impairs TLR4 responses in human fibroblasts, consistent with the findings reported for TIRAP-deficient mouse macrophages and dendritic cells (Horng et al., 2002; Yamamoto et al., 2002a).
Impairment of phagocyte responses via TLR2 and TLR4 in R121W TIRAP homozygotes
We investigated P1's leukocyte responses to TLR2 and TLR4 agonists. CD62L (L-selectin) is a transmembrane protein cleaved by the metalloproteinase TACE upon TLR stimulation (Peschon et al., 1998). CD62L shedding in response to the stimulation of all TLRs other than TLR3 is abolished in granulocytes from IRAK-4- and MyD88-deficient patients (von Bernuth et al., 2006).. The granulocytes from six R121W/W individuals tested displayed impaired CD62L shedding in response to 1 h of stimulation with the TLR2 (PAM2, FSL-1, LTA, and PAM3) and TLR4 agonists (LPS) tested, when compared with two healthy controls and P1's heterozygous mother, but their response to PMA was normal (Figure 3D). In addition, the stimulation of PBMCs with various doses of PAM2, FSL-1, LTA, PAM3, and LPS for 3 h showed no intracellular labeling for IL-6 and TNF-α in CD14+ cells, particularly at low agonist concentrations (Figure 3E and Figure S2C). Similarly, transcriptome analysis after the stimulation of P1's whole blood for 2 h suggested that the induction of gene expression in response to PAM2, FSL-1, or LTA was abolished and that the induction of gene expression in response to PAM3 or LPS was impaired but not abolished (Figure S2D). These data suggest that granulocytes from R121W TIRAP homozygotes do not respond to the stimulation of TLR1/2, TLR2/6, and TLR4, whereas their monocytes do not respond to TLR2/6 and respond poorly to TLR1/2 and TLR4.
Residual responses to high concentrations of TLR agonists in monocytes homozygous for R121W TIRAP
After 3 h of stimulation, the responses of monocytes from R121W-homozygous PBMCs to low doses (10 ng/ml) of PAM3 were abolished, but the levels of production of both IL-6 and TNF-α in response to agonist stimulation at concentrations above 1 μg/ml were similar to those of healthy controls, suggesting the existence of TIRAP-independent responses to high concentrations of PAM3 and, possibly, other TLR2 agonists, at least in some cell types (Figure 3E and Figure S2C). An effect of contaminants is unlikely, as the agonist used was synthetic. Moreover, low levels of TNF-α induction were observed in response to LPS stimulation in CD14+ cells from the R121W-homozygous PBMCs tested. After 48 h of stimulation, IL-6 production in response to PAM3 (1 μg/ml) was normal in the PBMCs of all homozygous individuals tested, whereas it was profoundly impaired but not abolished in response to LPS (1 ng/ml) and lipid A (1 μg/ml); responses to PAM2 (1 μg/ml), FSL-1 (100 ng/ml), and LTA (1 μg/ml) were abolished (Figure 4A and Figure S3A). These results confirm the deleterious effect of the R121W allele on TLR2 and TLR4 responses in leukocytes. However, the normal induction of IL-6 production observed in R121W/W monocytes in response to TLR1/2 stimulation with PAM3, at late time points and high concentrations of agonist (and, to a lesser extent, in response to stimulation with LPS and lipid A) further suggests that TIRAP may be redundant in such circumstances, as we ruled out the possibility of the mutant TIRAP protein having residual activity. Alternatively, PAM3 and LPS/lipid A may also be recognized independently of TLR2 and TLR4, respectively. In the absence of TLR2- or TLR4-deficient subjects, given that the R121W TIRAP allele is loss-of-function, we can conclude that these late responses to high concentrations of TLR agonists were probably either TIRAP- or TLR-independent.
Figure 4. The lack of anti-LTA Abs in P1's plasma accounts for the defective whole-blood response to LTA.
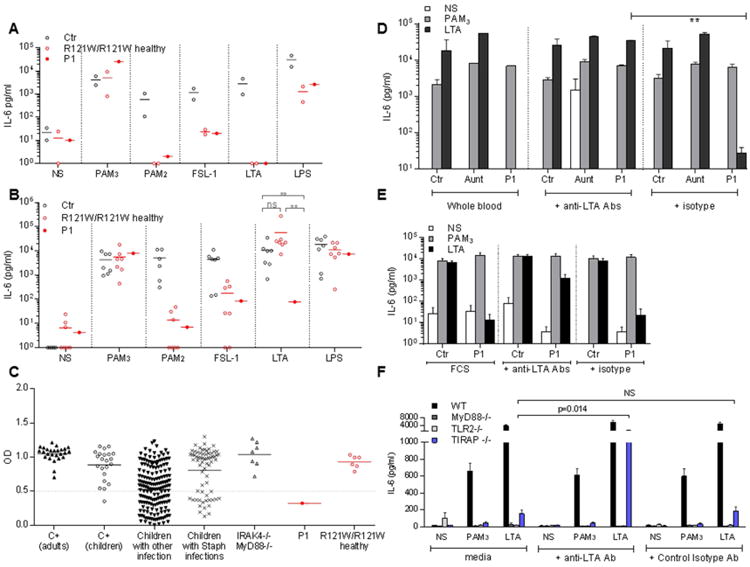
A. IL-6 production by PBMCs from controls, P1, her father (IV.2) and aunt (IV.5) upon stimulation with PAM3, PAM2 FSL-1, LTA and LPS for 48 h. (n=2)
B. IL-6 production by whole blood in II.3, III.1, IV.1 to 6 and V.1 after 48 h of stimulation with PAM3, PAM2, FSL-1, LTA, and LPS. The assay was performed several times and each point represents the mean for one individual. Whole blood from P1 was tested five times for FSL-1 stimulation, six for LTA, seven for PAM3and LTA and eight times for PAM2. Whole blood from other individuals was tested once (II.1), twice (III.1, IV.2, IV.3, IV.4, IV.5) or three times (IV.6) for each stimulus.
C. Tests for the presence of specific anti-LTA antibodies in plasma detected by ELISA are shown for a 1/100 dilution, tests were also performed at dilutions 1/20, 1/100, 1/500 (not shown).
D. Complementation of the defective IL-6 production in response to LTA stimulation in whole blood from the patient, by the addition of anti-LTA mAb (pagibaximab) (0.5 or 1 mg/ml). Comparison with the isotype control (0.5 or 1 mg/ml; palivizumab, anti-RSV). (n=2) (p=0.001, unpaired t-test). Error bars show SEM.
E. Complementation of impaired IL-6 production in response to LTA stimulation in PBMCs from P1 by the addition of anti-LTA mAbs. IL-6 production by control or P1 PBMCs after 48 h of stimulation with PAM3 or LTA in the presence of 10% FCS, 1 mg/ml exogenous isotype control mAbs or 1 mg/ml anti-LTA mAbs. Error bars show SEM (n=4).
F. Peritoneal macrophages from WT B6, Myd88-/-, Tiraptor/tor and TLR2lan/lan mice (n>3) were stimulated with PAM3 or LTA or left unstimulated and complemented with 0.5 mg/ml anti-LTA Abs (pagibaximab) or isotype control (anti-RSV, palivizumab). IL-6 production was assessed by ELISA. Error bars show SEM.
Impaired whole-blood response to LTA and lack of anti-LTA Abs in P1
In all individuals homozygous for the TIRAP R121W allele tested, IL-6 production by whole blood in response to 48 h of TLR2/6 stimulation with PAM2 and FLS-1 was impaired, whereas normal levels of IL-6 were produced in response to TLR1/2 stimulation with PAM3 and TLR4 stimulation with LPS and lipid A (Figure 4B and Figure S3A). However, the induction of IL-6 via TLR2/6 in response to LTA was reproducibly abolished in the whole blood of P1, whereas a normal response was observed for the other individuals homozygous for R121W TIRAP tested (Figure 4B). We investigated this striking difference in cellular phenotype, which we considered to be of potential clinical relevance as only P1 suffered from staphylococcal disease. The leukocyte response to LTA response is mediated principally by TLR2/6 heterodimers acting in concert with CD36 (Jimenez-Dalmaroni et al., 2009), but anti-LTA Abs have been shown to enhance this response through the recognition of their invariant IgG domain by CD32 (Bunk et al., 2010). The response to LTA was impaired in PBMCs, granulocytes, and monocytes from all R121W homozygotes (including P1), but normal in the whole blood of all these individuals other than P1. We therefore used enzyme-linked immunosorbent assays (ELISA) to test for the presence of circulating anti-LTA Abs in the plasma of individuals from this kindred. P1 was the only member of this kindred for whom anti-LTA IgG Abs were reproducibly undetectable in 10 plasma samples taken between the ages of four months and nine years. No maternal anti-LTA antibodies were detected in the plasma of P1 during the infectious episode. By contrast, the six relatives homozygous for R121W TIRAP tested and P1's mother (R121W/WT) had high titers of anti-LTA Abs (Figure 4C). P1 had high titers of Abs directed against H. influenzae, pneumococcal polysaccharides, tetanus, diphtheria toxins, and 68 other staphylococcal antigens, such as PVL and hemolysin-α (Figure S3B and Table S2). P1 was unique among the eight R121W homozygotes in three ways: her whole blood did not respond to LTA, she had no LTA-specific Abs in her plasma, and she suffered from invasive staphylococcal disease.
Rescue of the whole-blood LTA response in P1 with LTA-specific mAbs
We investigated whether the lack of anti-LTA IgG was responsible for the lack of whole-blood responses to LTA, by testing 26 healthy adults, 22 healthy children, 246 sick children (67 with invasive staphylococcal infections and 179 with other infections), seven patients with MyD88 or IRAK-4 deficiency (all of whom presented staphylococcal disease), and 10 patients with severe PVL+ S. aureus pneumonia for the presence of specific anti-LTA Abs (Figure 4C). All healthy adults and IRAK-4- and MyD88-deficient patients, most of the children (62%), whether healthy (96%), with S. aureus infections (72%) or with other types of infections (54%), and up to 60% of the patients with severe PVL+ S. aureus pneumonia, had high levels of serum anti-LTA Abs. Three of this last group of patients had a normal whole-blood response to LTA stimulation, one to eight months after infection (data not shown). Thus, the age at which serum anti-LTA IgG antibodies appear varies in the population, even after staphylococcal disease, consistent with previous reports (Wergeland et al., 1989). Our results also indicate that the absence of anti-LTA IgG does not influence the whole-blood response to LTA in TIRAP-expressing individuals (data not shown). IL-6 production in response to LTA stimulation was restored by incubating P1's PBMCs with LTA-responsive control plasma, suggesting that circulating anti-LTA Abs may rescue TIRAP deficiency (Figure S3C). We tested this hypothesis, by adding a chimeric mouse anti-LTA mAb to whole blood from healthy controls, P1, her aunt, and an IRAK-4-deficient patient, or to PBMCs from healthy controls and P1 (Figure 4D and 4E and Figure S4A). This restored IL-6 production in response to LTA stimulation in whole blood and PBMCs from P1, whereas an isotypic control mAb (directed against RSV) had no effect. Monocytes were the principal producers of IL-6 upon LTA stimulation in the presence of anti-LTA mAb (Figure S4B) (Bunk et al., 2010). The specific lack of anti-LTA Abs in the plasma of P1 therefore accounts for the abolition of IL-6 induction in the whole blood of this particular R121W homozygote in response to LTA stimulation via TLR2/6. The only R121W TIRAP homozygote to display catastrophic staphylococcal disease (P1) mounted no antibody response to LTA and her whole blood was unable to respond to LTA. The LTA-specific mAb did not rescue the response of IRAK-4-deficient cells, suggesting that the response of these cells is mediated by the canonical TLR2-MyD88 pathway. However, no TLR2- and MyD88-deficient individuals were available for study.
Anti-LTA mAb restores the LTA response in TIRAP-deficient mouse macrophages
We tested this model in mice. Like MyD88- and TLR2-deficient human macrophages, TIRAP-deficient mouse macrophages do not respond to TLR2 stimulation (Horng et al., 2001; Yamamoto et al., 2002a). We investigated whether the addition of exogenous anti-LTA antibodies restored the response to TLR stimulation. We first determined whether such Abs were present in mouse sera. No anti-LTA Abs were detected by ELISA in the 15 mice used for the study (data not shown). Mouse peritoneal macrophages from WT B6, Tirap-/-, Myd88-/- and Tlr2-/- mice (n>3) were then treated with LTA or low doses of PAM3 for 24 h, and the production of IL-6 and TNF-α was assessed in the presence and absence of exogenous anti-LTA or isotype control Abs. As in cells from P1, anti-LTA Abs restored the production of IL-6 and TNF-α in response to LTA, but not PAM3, in TIRAP-deficient mouse macrophages (Figure 4F and Figure S4C), whereas no such restoration was observed with the isotype control mAb. MyD88- and TLR2-deficient macrophages did not respond in this way to the addition of anti-LTA mAb. These findings unequivocally demonstrate that the TIRAP-independent mechanism compensating for TIRAP deficiency in the presence of anti-LTA Abs in mice, and, by inference, in humans, is dependent not only on IRAK4 but also on both TLR2 and MyD88. These data establish a plausible mechanism for the occurrence of staphylococcal disease in P1 but not in any of the other seven TIRAP-deficient individuals. The lack of anti-LTA IgG results in a failure to compensate for TIRAP deficiency, preventing leukocyte responses to staphylococcal LTA via TLR2/6, thereby accounting for invasive staphylococcal disease.
Discussion
We describe here a large family with autosomal recessive TIRAP deficiency. Eight family members are homozygous for the rare TIRAP R121W mutation affecting the TIR domain. TIRAP has been shown to play a crucial role in the TLR2- and TLR4-mediated and MyD88-dependent pathways in mice, as an adapter between these two TLRs and MyD88. Our data show that the R121W TIRAP allele is loss-of-function, as opposed to hypomorphic, because it cannot interact with upstream TLRs and downstream MyD88. TIRAP-deficient mouse macrophages display impaired activation of NF-κB in response to the stimulation of TLR2 and TLR4 by MALP2, PGN (peptidoglycans) (TLR2/6), BLP (bacterial lipopeptides) (TLR1/2) and LPS, respectively. LPS-induced splenocyte proliferation and cytokine production are also impaired in TIRAP-deficient mice, which are resistant to LPS-induced septic shock (Horng et al., 2002; Yamamoto et al., 2002a). Consistently, fibroblasts, granulocytes, and monocytes from TIRAP-deficient humans do not respond to TLR2/6 agonists PAM2, FSL-1, and LTA. However, residual activation of the TLR2 pathway was recently reported in mouse TIRAP-deficient macrophages, in response to high concentrations of PAM3 (TLR1/2) and MALP2 (TLR2/6) (Kenny et al., 2009). Moreover, prolongation or enhancement of the interaction between mouse TLR2 and its agonists PAM2 (TLR2/6) and PAM3 (TLR1/2) can induce TIRAP-independent responses, whereas MyD88 remains strictly required for all TLR2-dependent responses (Cole et al., 2010). These findings suggest that the response of R121W/W monocytes to high concentrations of or prolonged stimulation with PAM3 may be due to TIRAP-independent responses mediated via TLR1/2 and MyD88. Likewise, residual responses to LPS/lipid A in the patients may be TLR4-and MyD88-dependent. Our study thus provides the first comprehensive description of inherited TIRAP deficiency in humans, highlighting the essential role of this protein for TLR2/6 signaling. Our results also confirm that TIRAP is less critical than MyD88 and IRAK-4 for TLR1/2 and TLR4 responses, as it can be bypassed by increasing agonist concentration or the duration of stimulation.
The mouse TLR2 pathway plays an important role in host defense against S. aureus (Takeuchi et al., 2000). Humans with inherited IRAK-4 and MyD88 deficiencies are also prone to staphylococcal disease (reviewed in (Picard et al., 2010)), indicating that the human TIR signaling pathway is essential for protective immunity to S. aureus. The specific contribution of individual human TLRs and IL-1Rs has remained unclear (Casanova et al., 2011). We show that homozygosity for the loss-of-function R121W allele of TIRAP severely impairs, but does not abolish responses to TLR1/2 and TLR4 stimulation, whereas it abolishes responses to TLR2/6 stimulation. Impaired responses to TLR2 are much more likely than impaired responses to TLR4 to have contributed to the development of staphylococcal disease in P1, as S. aureus is a Gram-positive bacterium that contains both TLR1/2 and TLR2/6 agonists, including LTA in particular, but does not produce LPS (Lowy, 1998). However, the TIRAP mutation alone is not sufficient to account for P1's staphylococcal disease, as none of the other seven affected relatives had had any serious staphylococcal infections, despite frequent exposure to these commensal bacteria. The bacterial load in the initial inoculum, or the nature of the strain may influence the ability of TIRAP-deficient individuals to mount a protective immune response. It may therefore be relevant that P1 was infected with a PVL-positive strain of S. aureus; these relatively rare staphylococci are known to be more pathogenic in humans (Gillet et al., 2002). The observed clinical differences may also reflect the young age at which P1 encountered this pathogenic strain of S. aureus. The proband has been doing well for the last nine years, perhaps partly due to her robust Ab response to all staphylococcal antigens tested other than LTA. She is, however, on antibiotic prophylaxis (without IgG substitution, which would provide LTA-specific Abs, Figure S4D), making it impossible to draw firm conclusions concerning the efficacy of her current immune response to staphylococci.
A more likely hypothesis is based on the observation that P1 was the only TIRAP-deficient individual in the kindred to display a complete lack of whole-blood TLR2/6-dependent responses to staphylococcal LTA. This lack of response was causally related to the specific absence of circulating Abs against LTA in P1, as the phenotype was not only specific to this patient and absent from the other seven TIRAP-deficient relatives, but also complemented by the addition of LTA-specific mAbs to cells from P1. Monocytes are responsible for the response of TIRAP-deficient whole blood and PBMC to LTA in the presence of LTA-specific Abs. Moreover, TIRAP-deficient mouse macrophages responded to LTA via TLR2 in the presence of LTA-specific Abs. This Ab defect may, again, reflect the young age of P1, as LTA Abs first appear during childhood and their levels increase into adolescence, with considerable inter-individual variability (Dryla et al., 2005; Wergeland et al., 1989). If this is, indeed, the case, then the lack of staphylococcal disease during childhood in the other R121W homozygotes may have resulted from the staphylococci encountered being insufficiently virulent to cause disease, an earlier Ab response to LTA (triggered by any Gram-positive bacterium), or both. The normal levels of anti-LTA Abs in IRAK-4- and MyD88-deficient children (and the six TIRAP-deficient individuals other than P1 tested) indicate that a deficiency of TLR signaling pathways does not account for the lack of anti-LTA Ab production in P1. Furthermore, IRAK-4-deficient patients lack IL-6 production by whole blood in response to LTA (Figure S4A), despite presenting high titers of endogenous anti-LTA Abs, and even in the presence of exogenous anti-LTA mAb, indicating that the response to LTA mediated by anti-LTA Ab is TIRAP-independent but IRAK-4-dependent. Moreover, the LTA responses of mouse macrophages lacking MyD88 or TLR2 are not rescued by LTA-specific Abs, unlike those of TIRAP-deficient macrophages. The TIRAP-independent pathway rescued by LTA-specific Abs is therefore also TLR2- and MyD88-dependent. This rescue is probably mediated by the Fc receptor CD32 (Bunk et al., 2010), although CD32-deficient humans and mice were not tested.
The sustained lack of anti-LTA Abs in P1 is probably intrinsic and remains unexplained. However, this is not a unique feature, as approximately half the children under the age of nine years with no history of staphylococcal disease tested here had no detectable anti-LTA Abs. P1's serum was not tested before disease onset, but the observed Ab defect is unlikely to be a consequence of staphylococcal disease, as infection should have triggered the Ab response, and other children with staphylococcal disease (even with clinical phenotypes identical to that of this patient) produce such Abs. This defect is not the sole cause of the staphylococcal disease observed in P1, as children with various forms of Ab deficiency, including agammaglobulinemia, are not prone to such staphylococcal disease (Conley et al., 2009). The combined effects of a lack of anti-LTA Abs and the inherited TIRAP deficiency (i.e. of an acquired adaptive deficiency and an inborn innate deficiency) therefore underlie staphylococcal disease in this particular patient (Casanova et al., 2014). In the other individuals, IgG-dependent, adaptive immunity to LTA operates as a somatic modifier, selectively rescuing the inborn error of TIRAP, which disrupts TLR2-, TIRAP-, and MyD88-dependent innate immunity to LTA and staphylococci. This specific observation does not explain why staphylococcal (and pneumococcal) diseases become less frequent from adolescence onward in patients with inherited MyD88 or IRAK-4 deficiencies, whose cells do not respond to LTA in the presence of LTA-specific Abs. However, it suggests that other mechanisms of acquired immunity may be at work. In these patients, adaptive immunity is more likely to compensate (via pathways other than IRAK-4-MyD88) than to rescue (via the IRAK-4-MyD88 pathway itself) innate immunity. Nevertheless, our findings show that the clinical penetrance of an inborn error of innate immunity may be determined by adaptive immunity. This finding is consistent with twin studies in humans, which have revealed heritability to be lower for adaptive immunity than for innate immunity (Brodin et al., 2015; Casanova, 2015a). The somatic diversity of adaptive immune responses can compensate or even rescue inborn errors of innate immunity.
STAR Methods
1) Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to the lead contact author Jean-Laurent Casanova (St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, New York, USA; jean-laurent.casanova@rockefeller.edu)
2) Experimental Model and Subject Details
Clinical samples
PBMCs and blood experiments were performed using fresh blood collected in heparinized tubes from healthy volunteer obtained from French National Blood Service (The Etablissement Français du Sang), the patient and her family, as well as from one IRAK4-/- patient.
All plasma studies were conducted using archival plasma samples from 26 healthy adults, 22 healthy children, 67 children with invasive staphylococcal infections, 179 children with other type of infections), 7 patients with MyD88 or IRAK-4 deficiency and 10 patients with severe PVL+ S. aureus pneumonia
The experiments described here were conducted in accordance with local, national and international regulations and were approved by the French Ethics committee (ID-RCB: 2010-A00650-39), ANSM, (French National Agency for Medicines and Health Products Safety; B100713-40) and by the French Ministry of Research (IE-2010-547). Informed consent was obtained from all patients or their families, in the case of minors, in accordance with the World Medical Association, the Helsinki Declaration, and EU directives.
Cell lines
Primary human fibroblasts were grown from skin biopsy specimens and immortalized with SV40 T antigen (SV40 fibroblasts). They were cultured in DMEM (Gibco BRL, Invitrogen, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco BRL, Invitrogen, USA). All cells were grown at 37°C, under an atmosphere containing 5% CO2. The IRAK-4- and MyD88-deficient fibroblasts have been described elsewhere (Picard et al., 2003; von Bernuth et al., 2008). NEMO-deficient fibroblasts were derived from a female fetus with a completely skewed pattern of X inactivation expressing the del4-10 NEMO allele.
Mice
MyD88-/-mice was purchased from The Jackson Laboratory. Tiraptor/tor and Tlr2lan/lan mice were generated on a pure C57BL/6J background by ENU mutagenesis and are described at http://mutagenetix.utsouthwestern.edu. Animals were housed in an animal facility on a 12 h/12 hr light/dark cycle with free access to food and water. All experimental procedures using mice were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Southwestern Medical Center, and were conducted in accordance with institutionally approved protocols and guidelines for animal care and use.
All the mice were maintained at the University of Texas Southwestern Medical Center in accordance with institutionally approved protocols.
3) Method Details
Case report
We investigated a six-year-old girl of Pakistani origin, born full term, to second-degree consanguineous parents living in the United Kingdom (P1, V.1, Supplementary Figure S1). She received four injections of a pentavalent vaccine against Haemophilus influenzae type b, Diphtheria, Bordetella pertussis, tetanus and poliovirus at the ages of 2, 4, 6 and 36 months. She also received three injections of a seven-valent conjugate vaccine against pneumococcus (Prevenar®) at the ages of 2, 4 and 13 months and three injections of a conjugate meningococcus C vaccine at the ages of 3, 4 and 12 months. She received two injections of MMR vaccine. At the age of three months, she presented an acute respiratory illness leading to respiratory failure. PVL-producing S. aureus was identified as the cause of pneumonia and sepsis. On admission to the intensive care unit, the patient's C-reactive protein concentration was 52 mg/l (0 – 6), peaking at 180 mg/l 24 hours later. The patient displayed a marked acidosis, with a pH of 6.9. Conventional respiratory and circulatory support with inotropes failed and she required seven days of extracorporeal membrane oxygenation. Her staphylococcal infection was treated with a combination of intravenous clindamycin and linezolid. She made a complete recovery from this life-threatening illness and has remained well, with no further infective episodes, but on prophylactic cotrimoxazole treatment, over the last four years. Routine immunological results for phagocytes, complement, Ig (IgM 0.49 g/l, IgG 7.56 g/l, IgA 0.31 g/l at 3 months), antibody production, and for T and B cells were normal.
Genetic analysis
Human genomic DNA was isolated from polymorphonuclear (PMN) cells obtained from freshly collected whole blood by centrifugation on a Ficoll-Paque Plus gradient (GE Healthcare, GE Healthcare, Saclay, France), peripheral blood mononuclear cells (PBMCs) or cell lines (EBV-transformed B cells or SV40-immortalized fibroblasts). The cells were lysed by incubation overnight at 37°C in extraction buffer (10 mM Tris, pH 7.4, 1 mM EDTA, 0.5% SDS, and 10 mg/ml proteinase K). DNA was isolated by phenol/chloroform extraction, precipitated in ethanol and resuspended in 10 mM Tris, pH 7.4, 1 mM EDTA. Human RNA was isolated from SV40-transformed fibroblasts with Trizol reagent (Invitrogen Life Technologies, Paisley, UK), according to the manufacturer's instructions. RNA was reverse-transcribed with Superscript II reverse transcriptase (Invitrogen Life Technologies, Paisley, UK). PCR amplification was performed with the AmpliTaq DNA polymerase (Applied Biosystems, California, USA) and the GeneAmp PCR system 9700 (Applied Biosystem, California, USA). We amplified the full-length cDNA and the three coding exons of TIRAP separately. The conditions and primer sequences used for PCR amplification of the coding exons, including the flanking intron regions or cDNA sequences, are available upon request. The PCR amplicons were verified by electrophoresis in a 1% agarose gel and purified by centrifugation through Sephadex G-50 Superfine resin (GE Healthcare, Saclay, France) on multiscreen MAHV-N45 filter plates (Millipore, Molshein, France). The PCR products were sequenced by dideoxyribonucleotide termination methods, with the BigDye Terminator kit v3.1 (Applied Biosystem, California, USA) and appropriate primers (also available upon demand). Sequencing products were also purified by centrifugation through Sephadex G-50 Superfine resin and analyzed on an ABI Prism 3700 (Applied Biosystem, California, USA). Sequence files and chromatograms were analyzed with Genalys Software v3.3 from CNG, France (Takahashi et al., 2003) and Sequence Scanner Software v1.0 (Applied Biosystem, California, USA).
Exome sequencing
Massively parallel sequencing: Genomic DNA extracted from the patient's whole blood was sheared with a Covaris S2 ultrasonicator (Covaris). An adaptor-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome sequencing was performed with the Agilent Sureselect All Exons kit V4 of the Agilent Sureselect all exons kit V4+UTR (Table S3). We generated 2×100 base-pair (bp) paired-end reads on an Illumina HiSeq 2000.
Sequence alignment, variant calling, and annotation
The sequences were aligned with the human genome reference sequence (hg19 build), with BWA aligner (Li and Durbin, 2009). Downstream processing was carried out with the Genome Analysis Toolkit (GATK)(McKenna et al.), SAMtools(Li and Durbin, 2009), and Picard Tools (http://picard.sourceforge.net). Substitution calls were made with a GATK Unified Genotyper, whereas indel calls were made with a GATK IndelGenotyperV2. All calls with a read coverage ≤ 2× and a Phred-scaled SNP quality ≤ 20 were filtered out. All the variants were annotated with the GATK Genomic Annotator.
Genotyping and linkage analysis
Five members of this family (the proband, her parents, and her paternal grandparents) were genotyped with the Affymetrix Genome-wide SNP 6.0 array. Five other members (two uncles, two aunts and the proband's sister) were genotyped with the Affymetrix Human Mapping 250K Nsp Array. Genotype calling was achieved with Affymetrix Power Tools (http://www.affymetrix.com/partners_programs/programs/developer/tools/powertools.affx), for all 10 family members. We discarded monomorphic SNPs, SNPs with a call rate lower than 100% and SNPs presenting more than one Mendelian inconsistency within the family. SNPs were further filtered with population-based filters. We then used 117,517 high-quality SNP markers for linkage analysis. Parametric multipoint linkage analysis was carried out with Merlin software (Abecasis et al., 2002; Purcell et al., 2007). The family founders and HapMap CEU trios were used to estimate allele frequencies and to define linkage clusters, with an r2 threshold of 0.4.
Due to the high degree of inbreeding in this family and regarding lack of precise information about consanguinity loops, we performed different linkage analyses based on different models:
- A first analysis assuming autosomal-recessive inheritance with complete penetrance (homozygosity mapping), with a single loop of consanguinity at the level of the proband's parents;
- A second analysis assuming autosomal-recessive inheritance with complete penetrance (homozygosity mapping), with two loops of consanguinity at the levels of the proband's parents and grandparents.
Each of these two analyses were performed twice, with the disease status of healthy individuals homozygous for the TIRAP mutation taken as ‘healthy’ or ‘unknown’. Regions linked with an ‘unknown’ disease status included TIRAP as a candidate gene. This gene was not included in regions linked to a disease status of ‘healthy’. The proband and her relatives (father, grandparents, aunt and uncles) carried the same haplotype for TIRAP. We used a combination of this analysis and exome sequence to search for potential modifier genes.
Kinship calculation
Kinship coefficients (the probability that two alleles sampled at random from two individuals are identical by state) were calculated with KING robust (Manichaikul et al., 2010).
Quantitative PCR
Total RNA was extracted from 0.5million EBV-B cells or 0.5 SV-40 fibroblasts or 0.1 million of PBMCs, with the ZR RNA MicroPrepZymo (ZR1061, Zymo research, California, USA), according to the manufacturer's instructions. RNA was reverse-transcribed with Superscript II reverse transcriptase (Life Technologies, California, USA). Real-time quantitative PCR (qPCR) was performed using 2 different probes (TaqMan Gene Expression Assays Life Technologies custom probes for isoform A and B, sequences available in the key resource table), with an initial denaturation at 95°C for 20 s, followed by 40 cycles of denaturation at 95°C for 3 s and annealing/extension at 60°C for 30 s. mRNA levels are expressed relative to GUS mRNA levels, as determined by the 2-DDC(t) method. Error bars were calculated from at least two independent experiments.
Cell lysis, immunoprecipitation, and immunoblotting
Cells were lysed in a lysis buffer containing 30 mM Tris-HCl pH 7.5, 120 mM NaCl, 2 mM KCl, 1% Triton X-100, and 2 mM EDTA supplemented with protease inhibitor cocktail (cOmplete, Roche) and phosphatase inhibitor cocktail (PhoStop, Roche). Laemmli buffer supplemented with DTT was added to clear supernatants.
For immunoprecipitation, cells were lysed in lysis buffer (0.5% Triton X-100, 20 mM HEPES (pH 7.4), 150 mM NaCl, 12.5 mM b-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM dithiothreitol, 1 mM sodium orthovanadate, 2 mM EGTA, 20 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride). Cell extracts were incubated overnight at 4°C with 1 ug of the appropriate antibody and 20 μl of protein G Sepharose beads (prewashed and resuspended in lysis buffer at a 1:1 ratio). The beads were washed four times with lysis buffer.
Samples were separated by SDS–PAGE and the proteins were transferred onto a PVDF membrane. The membrane was blocked by incubation in TBS supplemented with 0.1% Tween 20 and 5% skimmed milk powder, and incubated with the primary antibody, followed by the appropriate horseradish peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized by enhanced chemiluminescence.
TIRAP cloning, overexpression and cell complementation
Complementary DNA was generated from RNA extracted from SV40-fibroblasts and the full-length A and B isoforms of TIRAP were amplified by PCR and inserted into a pCDNA3 vector with a C-terminal V5 tag. Site-directed mutagenesis was performed on the cDNAs for both isoforms, to introduce the R121W mutation. The primers used are available upon request. Sequencing confirmed that the mutation was incorporated, with no other alteration to the construct.
HEK293T cells were transfected with 1ug of each plasmid in 6cm petri dish with calcium phosphate (Thermofisher) according to manufacturer instructions. 36 hours later, cells were collected and lysed according to protocol above.
TIRAP-deficient and MyD88-deficient SV40-fibroblasts were transfected with 100 ng of empty pCDNA3 vector, pCDNA3-TIRAP-V5 vectors encoding isoform A WT, isoform B WT, isoform A with the R121W mutation, isoform B with the mutation, or pCDNA3-MyD88-V5, with the LTX transfection kit (Invitrogen, SARL, France), used according to the manufacturer's instructions. Twenty-four hours later, cells were stimulated by incubation with various ligands for 24 hours. The supernatants were harvested and IL-6 production was assessed by ELISA.
Immunostaining
HEK 293T cells stably transfected with a TLR2 construct (Invivogen) were plated and transfected with 1 μg in total of plasmids encoding the WT or mutant TIRAP, or MyD88, in IBIDI eight-well microslides. They were stained 36 hours after transfection, washed in 0.01% Tween in PBS, fixed by incubation with 4% PFA for 30 minutes and permeabilized by incubation with 0.3% Triton for 30 minutes on ice. Cells were then blocked by incubation in 3% BSA and 10% goat serum for 1 hour and stained with 1/500 anti-V5 (mouse) Ab or 1/500 anti-myc (rabbit) (both from Invitrogen) for 1 hour. Secondary antibodies were then added. The cells were incubated for one hour in the dark and images were captured with an ApoTome microscope (Carl-Zeiss). Pearson's correlation coefficients were calculated with Image J software (JacoP plugin).
SV-40 stimulation and cytokine determinations by ELISA
Twenty-four hours before stimulation, 1×105 SV40 fibroblasts were plated in 1 ml of DMEM supplemented with 2% FBS per well in a 24-well plate. Cells were either left unstimulated or were stimulated for 24 hours with different ligands: 10 ng/ml IL-1β (R&D Systems), 20 ng/ml TNF-α (R&D Systems), 25 μg/ml poly(I:C) (polyinosinic-polycytidylic acid from GE Healthcare) and TLR2 ligands: 1 μg/ml PAM2CSK4, 1 μg/ml PAM3CSK4, 1 μg/ml purified LTA-SA, 100 ng/ml FSL-1 (all from Invivogen), 1μg/ml of LPS (from Sigma)
Twenty-four hours later, supernatants were collected and IL-6 levels were assessed by ELISA, with the Sanquin kit, used according to the manufacturer's instructions. Samples and antibodies were diluted in 1 × HPE dilution buffer (M1940 Sanquin). Milk was used for blocking, and antibody binding was detected with streptavidin-conjugated horseradish peroxidase (M2032, Sanquin) and the TMB microwell peroxidase substrate (50-76-00 KPL). Finally, the reaction was stopped by adding 1.8 M H2SO4. Optical density was determined with an MRX microplate reader (Thermolab System).
CD62L shedding
We stimulated 100 μl of fresh blood by incubation for one hour with various TLR agonists, at 37°C, under an atmosphere containing 5% CO2. After activation, the red blood cells were lysed by incubation in extraction buffer (10 mM Tris, pH 7.4, 1 mM EDTA, 0.5% SDS) for 5 minutes and washed twice in PBS. Cells were then labeled with anti-CD62L (BD) antibody and captured by flow cytometry. The analysis was performed on granulocytes gated according to size and granulosity (FCS/SSC settings).
Whole-blood activation
Blood samples were collected in tubes containing heparin and were tested, generally after 24 hours of transport at room temperature. Blood was diluted 1:2 in RPMI 1640 medium (Gibco BRL, Invitrogen, USA). Aliquots of diluted blood were dispensed into a 48-well plate and incubated at 37°C, under an atmosphere containing 5% CO2, under different sets of conditions: with medium alone (non-stimulated), with agonists of different receptors (20 ng/ml IL-1β (R&D Systems), 10 ng/ml TNF-α (R&D Systems), 25 μg/ml poly(I:C) (polyinosinic-polycytidylic acid from GE Healthcare) and TLR2 ligands: 1 ug/ml PAM2CSK4, 1 ug/ml PAM3CSK4, 1 μg/ml purified LTA-SA, 100 ng/ml FSL-1, 500 ug/ml of lipidA (all from Invivogen), 1 ng/ml of LPS Salmonella enterica serotype minnesota Re 595 (Sigma) or with 10-7 M PMA/ 10-5 M ionomycin (as positive control of stimulation) in presence of 10ug/ml of polymixin B. Supernatants were collected after 48 hours and stored at -20°C until their use for cytokine determinations by ELISA. Exogenous monoclonal anti-LTA Ab (Pagibaximab) was obtained through collaboration with Biosynexus Incorporated (Gaithersburg, MD, USA) and palivizumab (Synagis®, Abbott) was used as an isotypic control. These mAb were used at 0.5 or 1mg/ml.
Flow cytometry
PBMCs were cultured in DMEM supplemented with 10% FBS at a density of 2 × 106 cells/ml. They were stimulated for 3 hours with TLR agonists or left unstimulated. Concomitantly to stimulation, protein transport was blocked with GolgiPlug (BD Biosciences). Cells were labeled with anti-CD14 (BD), anti-CD19 (BD) and anti-CD3 antibodies (Miltenyi Biotec). They were fixed and permeabilized (BD, 554722) and then stained with antibodies against IL-6 (BD) or TNF-α (BD). PBMCs were captured by flow cytometry with a BD Canto II machine and FACS Diva software. The data were analyzed with FACS Diva or FlowJo software (Tree Star).
Detection of plasmatic antibodies
ELISA plates were coated with 10 μg/ml of purified LTA from S. aureus (Invivogen) by incubation for 1 h at 37°C, and blocked by incubation with 5% milk in PBS for 1 h at room temperature. Plasma isolated from fresh blood collected into heparin was diluted 1/20, 1/100 and 1/500 and incubated on the plate for 1 h at room temperature. Anti-LTA antibodies were detected with peroxidase-conjugated goat anti-human IgG antibodies and the TMB microwell peroxidase substrate (50-76-00 KPL). The reaction was stopped by adding H2SO4 (1.8 M). Optical density was determined with an MRX microplate reader (Thermolab Systems).
For IVIgs a starting dilution at 10 mg/ml was made to get closer from human plasma concentration of Igs. Then, OD was assessed at the following dilutions 1/20, 1/100, 1/500, 1/2500, 1/7500, 1/22500.
Luminex analyses for antibodies detection
IgG levels against 70 recombinant S. aureus proteins were measured in serum samples using a bead-based flow cytometry technique (xMAP®; Luminex® Corporation, Austin, TX, USA), as previously described (den Reijer et al., 2016). All measurements were taken in duplicate, and the median fluorescence inten-sities (MFIs), a semiquantitative measure of antibody levels, were averaged. Duplicate measurements for which the coefficient of variation was larger than 25 % were excluded from further analysis. All measurements were corrected for non-specific background signal by subtracting the MFIs of control beads not coupled to any protein.
Monocytes negative selection
PBMCs were collected and washed four times at 300× g for 10 min with RPMI 1640. Monocytes were isolated from PBMCs by negative magnetic isolation (Miltenyi Biotec, Germany). Briefly, PBMCs were incubated with FcR Blocking reagent (Miltenyi Biotec, Germany) and with a cocktail of biotinylated antibodies against CD3, CD7, CD16, CD19, CD56, CD123 and Glycophorin A for 10min on ice. Then cells were incubated with magnetic bead-conjugated anti-biotin antibodies for 15 min on ice and applied onto MACS LS columns. Cells passing the column without binding were collected as monocytes fraction, and the monocyte-depleted PBMCs were collected by eluting the bound cell fraction. Each fraction were added at a density of 250.000 cells/well in RPMI, 5%FBS. Prior to stimulation with PAM3 and LTA (performed according to the conditions described for PBMCs activation in a total volume of 250ul) monoclonal anti-LTA antibodies (pagibaximab, 1mg/ml) or isotypic control (palivizumab, anti-RSV, 1mg/ml) were added to each fraction. Il-6 production was assessed by ELISA after 24 hours stimulation.
Mouse macrophage stimulation
A total of 1×105 mouse peritoneal macrophages from wild type B6 or mutant mice (n≥3) (MyD88-/-, Tiraptor/tor, Tlr2lan/lan) were treated with 40ng/ml of PAM3CSK4 or 1ug/ml of purified LTA from S. aureus for 24 h and supplemented with 0.5mg/ml of either isotype control (anti-RSV antibody) or anti-LTA antibodies. Supernatants were collected and analyzed by mouse IL-6 and TNF ELISA.
4) Quantification and Statistical Analysis
Data are presented as the means ± SEM or SD (indicated in the legend for each figure). The significance of differences between groups was evaluated using t-test. The p value < 0.05 was considered significant.
Statistical parameters including the exact value of n, precision measures (mean ± s.e.m) and statistical significance are reported in the Figures and the Figure Legends when necessary. Data are judged to be statistically significant when p < 0.05 by one-tailed Student's T-Test, where appropriate. In figures, asterisks denote statistical significance (*, p < 0.05; **, p < 0.01). Pearson correlation coefficients were calculated with PRISM and statistical significance is based upon the assumption that values exhibit a Gaussian distribution. Statistical analysis was performed in Graph Pad PRISM 6 or 7.
Supplementary Material
Figure S1: The TIRAP R121W mutation is a rare mutation affecting the TIR domain of TIRAP, with a modest impact at the mRNA and protein levels, related to Figure 1
A. Familial relationship, within-family relationships estimated with KING. The x-axis represents the expected kinship coefficient for a non-consanguineous family and the y-axis represents the estimated kinship coefficient in this family. Each dot represents the kinship coefficient between two members of the family. Black dots represent an estimated kinship coefficient concordant with the expected kinship coefficient for this kind of relationship. Red dots represent an estimated kinship coefficient discordant with the expected kinship coefficient for this kind of relationship. Pedigree constructed according to the estimated kinship coefficients.
B. Alignment of the human TIRAP protein with other human TIR domain-containing molecules
C. TIRAP RT-PCR, ranging from 27 to 33 cycles, on RNA extracted from SV40 fibroblasts from P1 and her father, compared to the results for a healthy control. Primer specific for transcripts encoding isoform A or B were used.
D. Quantitative PCR was performed to measure the levels of the transcripts encoding TIRAP isoforms A and B in SV40 fibroblasts (from five controls, P1, P1's father and mother), PBMCs (from 9 controls, P1, and P1's aunt) and EBV-B cells (from 5 controls and P1). SV40 fibroblasts stably transfected with the cDNA encoding isoform A or B were used as controls to test probe specificity. The percentages of transcripts encoding isoform A or B were normalized relative to the total number of transcripts encoding isoforms A and B, set at 100%. Error bars represent the SD.
Figure S2 : The TIRAP R121W allele is loss of function, related to Figure 3
A. Complementation study in SV40 fibroblasts from P1 (up), her father (middle) and a control (bottom), after stable transfection with lentiviral particles encoding the WT or mutant A or B isoform of TIRAP, or both. Particles containing luciferase instead of TIRAP were used for mock transfection. IL-6 production was assessed by ELISA. Error bars represent the SEM.
B. IL-6 mRNA levels assessed by RT-qPCR and expressed relative to GUS mRNA levels by the 2ΔΔC(t) method, in SV40 fibroblasts from a healthy control. Cells were stimulated for 4 h with PAM2 or TNF-α, 24 h after transfection with siRNAs against TLR2 and TIRAP.
C. PBMCs from several family members (III.1, IV.1-5 and V.1) were stimulated with various doses of PAM2, PAM3, LTA, FSL-1, and LPS for 3 h, subjected to extracellular staining with anti-CD3, anti-CD19 and anti-CD14 antibodies and intracellular staining for TNF-α. The proportion of CD14+ cells positive for TNF-α is shown. Acquisition was performed on 10,000 CD14+ cells. Each data point represents the mean of all individuals ± SEM (related to Figure 3E).
D. Transcriptome analysis performed on whole blood after 3 h of stimulation with PAM2, FSL-1, LTA, PAM3, LPS or PMA ionomycin. The results for P1 and her mother (R121W/WT) are compared with those for one healthy control and one IRAK-4-deficient patient.
Figure S3: The lack of anti-LTA Abs in P1's plasma accounts for the defective whole-blood response to LTA, related to Figure 4
A. PBMCs and whole blood from healthy controls, P1 and her father were stimulated with lipid A (1 μg/ml), LPS (10 ng/ml) and PMA/ionomycin for 48 h. IL-6 production was assessed by ELISA. Error bars represent the SD.
B. Assessment, with Luminex technology, of antibodies against 68 staphyloccocal antigens, (only 6 are shown in the figure, all results are summarized in Table S2), in 6 different plasma samples from P1 (between the ages of 4 months and 9 years), 6 homozygous relatives (V.2, IV.1, IV.2, IV.4, IV.6 and III.1), 6 IRAK-4 or MyD88-deficient patients and 11 sex- and aged-matched controls. Bonferroni correction for multiple testing indicated that the patient did not have significantly lower levels of antibody than the controls for any of the antigens tested.
C. PBMCs from P1 and two controls were left unstimulated or were stimulated by PAM3 or LTA in the presence of fetal calf serum (FCS) or plasma from each individual. IL-6 production was assessed by ELISA. Error bars represent the SD.
Figure S4: The lack of anti-LTA Abs in P1's plasma accounts for the defective whole-blood response to LTA, related to Figure 4
A. IL-6 production by whole blood was assessed in a healthy control and an IRAK4-deficient patient after 48 h of stimulation with PAM3 or LTA, supplemented or not with 0.5 mg/ml control isotype antibody (anti-RSV, Synagis) or monoclonal anti-LTA (pagibaximab) antibody.
B. IL-6 production by control PBMCs, monocytes, and monocyte-depleted PBMCs after 24 h of stimulation with 1 μg/ml PAM3 or 1 μg/ml LTA in the presence of medium alone, or medium supplemented with 1 mg/ml anti-LTA mAb or isotype control mAb (anti-RSV). Representative of 3 experiments.
C. Peritoneal mouse macrophages from WT B6, Myd88-/-, Tiraptor/tor and TLR2lan/lan (n>3) were stimulated with PAM3 (40 ng/ml) or LTA (1 μg/ml) or left unstimulated and complemented with 0. 5mg/ml anti-LTA Ab (pagibaximab) or isotype control (anti-RSV, Synagis). TNF-α production was assessed by ELISA. Error bars represent the SEM.
D. Assessment, by ELISA, of levels of anti-LTA antibodies in 16 different batches of commercially available IVIG, with comparison to healthy controls (n=10) and P1. Error bars represent the SEM.
Table S1: Related to figure 1.
List of genes presenting a non-synonymous rare homozygous variation in P1 exome data.
Table S2: Related to figure 4.
IgG levels against 68 recombinant S. aureus proteins were measured in serum samples, by a bead-based flow cytometry technique, in 6 different plasma samples from the patient (obtained between the ages of 1 and 8 years), 5 homozygous relatives, 6 IRAK-4- or MyD88-deficient patients and 11 aged-matched controls. The table summarizes mean and SD values for each antigen in each plasma category. Bonferroni correction for multiple testing was performed and p values are indicated for significant differences from controls.
Table S3: Related to STAR Methods section
Metrics and coverage data for the three exomes sequenced
Highlights.
Inherited TIRAP deficiency impairs cellular responses to TLR2 and TLR4 stimulation
Human TIRAP is redundant for protective immunity against most microorganisms
Anti-LTA antibodies rescue TLR2-dependent responses to LTA in TIRAP-deficient cells
TIRAP deficiency causes staphylococcal disease in the absence of anti-LTA antibodies
Acknowledgments
We thank the patient and her family for participating in the study and all the members of both branches of the laboratory for discussions. We thank J Bustamante and SY Zhang for providing the plasma samples, S Fahy, J Flatot, M Courat, L Amar and Y Nemiroskaya for their assistance. We also thank R Desvaux and N Goudin for their help at the imaging platform. We thank Biosynexus Incorporated, Gaithersburg, MD USA for providing us with pagibaximab (anti-LTA Ab). The Laboratory of Human Genetics of Infectious Diseases is supported by INSERM, Paris Descartes University, the Imagine Institute, ANR-10-LABX-62-IBEID, ANR-2012-BSV3-0003-02, ANR-10-IAHU-01, the St. Giles Foundation, the National Center for Research Resources, and the National Center for Advancing Sciences (NCATS) grant number 8UL1TR000043 from the National Institutes of Health, and The Rockefeller University. This work was partly supported by the Innovative Medicines Initiative Joint Undertaking under grant agreement no. [115523], COMBACTE, from the European Union's FP7/2007-2013 and EFPIA. Laura Israel was supported by Fondation pour la Recherche Médicale. Carolina Prando was supported by the Thrasher Research Fund and Yuval Itan was supported by the AXA Research Fund.
Footnotes
Author contributions: None of the authors of this manuscript have a financial interest related to this work. L.I., A.P. and J.L.C. designed the study and wrote the manuscript. L.I., Y.W., K.B., E.D.M., Z.Z., M.C., T.L., E.L., L.L., L.Y., A.M. and S.W. performed the experiments. Y.W., K.B., E.D.M. equally contributed to the manuscript. V.P., A.B., A.A., C.Pr. and Y.I conducted exome, linkage and statistical analysis. P.D.A., L.E.W., M.L., and J.H. provided material from the patients and reagents. V.S.S, M.D., F.V., C.Pi., L.A., D.C., X.L., and B.B. provided expertise and feedback. J.L.C., A.P. and L.A secured funding. P.D.A conducted the medical follow-up, provided expertise and critical reading. All the authors critically reviewed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, et al. The 2015 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2015;35:727–738. doi: 10.1007/s10875-015-0198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015;160:37–47. doi: 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunk S, Sigel S, Metzdorf D, Sharif O, Triantafilou K, Triantafilou M, Hartung T, Knapp S, von Aulock S. Internalization and coreceptor expression are critical for TLR2-mediated recognition of lipoteichoic acid in human peripheral blood. J Immunol. 2010;185:3708–3717. doi: 10.4049/jimmunol.0901660. [DOI] [PubMed] [Google Scholar]
- Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ. TLR1- and TLR6-independent recognition of bacterial lipopeptides. J Biol Chem. 2006;281:9049–9057. doi: 10.1074/jbc.M512525200. [DOI] [PubMed] [Google Scholar]
- Casanova JL. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A. 2015a;112:E7118–7127. doi: 10.1073/pnas.1521644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A. 2015b;112:E7128–7137. doi: 10.1073/pnas.1521651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol. 2011;29:447–491. doi: 10.1146/annurev-immunol-030409-101335. [DOI] [PubMed] [Google Scholar]
- Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211:2137–2149. doi: 10.1084/jem.20140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole LE, Laird MH, Seekatz A, Santiago A, Jiang Z, Barry E, Shirey KA, Fitzgerald KA, Vogel SN. Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. J Leukoc Biol. 2010;87:275–281. doi: 10.1189/jlb.0909619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- den Reijer PM, Sandker M, Snijders SV, Tavakol M, Hendrickx AP, van Wamel WJ. Combining in vitro protein detection and in vivo antibody detection identifies potential vaccine targets against Staphylococcus aureus during osteomyelitis. Med Microbiol Immunol. 2016 doi: 10.1007/s00430-016-0476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryla A, Prustomersky S, Gelbmann D, Hanner M, Bettinger E, Kocsis B, Kustos T, Henics T, Meinke A, Nagy E. Comparison of antibody repertoires against Staphylococcus aureus in healthy individuals and in acutely infected patients. Clin Diagn Lab Immunol. 2005;12:387–398. doi: 10.1128/CDLI.12.3.387-398.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- Gillet Y, Issartel B, Vanhems P, Fournet JC, Lina G, Bes M, Vandenesch F, Piemont Y, Brousse N, Floret D, et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359:753–759. doi: 10.1016/S0140-6736(02)07877-7. [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13:109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Velez M, Scott E, Ciancanelli MJ, Lafaille FG, Markle JG, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A. 2015;112:13615–13620. doi: 10.1073/pnas.1518646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Dalmaroni MJ, Xiao N, Corper AL, Verdino P, Ainge GD, Larsen DS, Painter GF, Rudd PM, Dwek RA, Hoebe K, et al. Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PLoS One. 2009;4:e7411. doi: 10.1371/journal.pone.0007411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Kenny EF, Talbot S, Gong M, Golenbock DT, Bryant CE, O'Neill LA. MyD88 adaptor-like is not essential for TLR2 signaling and inhibits signaling by TLR3. J Immunol. 2009;183:3642–3651. doi: 10.4049/jimmunol.0901140. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Lee H, Roh KB, Asanuma M, Kim YS, Nakayama H, Shiratsuchi A, Choi Y, Takeuchi O, Kang HJ, et al. The Triacylated ATP Binding Cluster Transporter Substrate-binding Lipoprotein of Staphylococcus aureus Functions as a Native Ligand for Toll-like Receptor 2. J Biol Chem. 2009;284:8406–8411. doi: 10.1074/jbc.M809618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Peres AG, Damian AC, Madrenas J. Immunomodulation and Disease Tolerance to Staphylococcus aureus. Pathogens. 2015;4:793–815. doi: 10.3390/pathogens4040793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Lu J, Zhou W, Shen Y. Structural insights into TIR domain specificity of the bridging adaptor Mal in TLR4 signaling. PLoS One. 2012;7:e34202. doi: 10.1371/journal.pone.0034202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy ME, McLoughlin RM. Host-Bacterial Crosstalk Determines Staphylococcus aureus Nasal Colonization. Trends Microbiol. 2016;24:872–886. doi: 10.1016/j.tim.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Nada M, Ohnishi H, Tochio H, Kato Z, Kimura T, Kubota K, Yamamoto T, Kamatari YO, Tsutsumi N, Shirakawa M, et al. Molecular analysis of the binding mode of Toll/interleukin-1 receptor (TIR) domain proteins during TLR2 signaling. Mol Immunol. 2012;52:108–116. doi: 10.1016/j.molimm.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc Natl Acad Sci U S A. 2009;106:10260–10265. doi: 10.1073/pnas.0812956106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. 2011;24:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 2010;89:403–425. doi: 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Matsuda F, Margetic N, Lathrop M. Automated identification of single nucleotide polymorphisms from sequencing data. J Bioinform Comput Biol. 2003;1:253–265. doi: 10.1142/s021972000300006x. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- Valkov E, Stamp A, Dimaio F, Baker D, Verstak B, Roversi P, Kellie S, Sweet MJ, Mansell A, Gay NJ, et al. Crystal structure of Toll-like receptor adaptor MAL/TIRAP reveals the molecular basis for signal transduction and disease protection. Proc Natl Acad Sci U S A. 2011;108:14879–14884. doi: 10.1073/pnas.1104780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernuth H, Ku CL, Rodriguez-Gallego C, Zhang S, Garty BZ, Marodi L, Chapel H, Chrabieh M, Miller RL, Picard C, et al. A fast procedure for the detection of defects in Toll-like receptor signaling. Pediatrics. 2006;118:2498–2503. doi: 10.1542/peds.2006-1845. [DOI] [PubMed] [Google Scholar]
- von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, Chrabieh M, Mustapha IB, Ghandil P, Camcioglu Y, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–696. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wergeland HI, Haaheim LR, Natas OB, Wesenberg F, Oeding P. Antibodies to staphylococcal peptidoglycan and its peptide epitopes, teichoic acid, and lipoteichoic acid in sera from blood donors and patients with staphylococcal infections. J Clin Microbiol. 1989;27:1286–1291. doi: 10.1128/jcm.27.6.1286-1291.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002a;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002b;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- Zahringer U, Lindner B, Inamura S, Heine H, Alexander C. TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology. 2008;213:205–224. doi: 10.1016/j.imbio.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Su HC, Lenardo MJ. Genomics is rapidly advancing precision medicine for immunological disorders. Nat Immunol. 2015;16:1001–1004. doi: 10.1038/ni.3275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: The TIRAP R121W mutation is a rare mutation affecting the TIR domain of TIRAP, with a modest impact at the mRNA and protein levels, related to Figure 1
A. Familial relationship, within-family relationships estimated with KING. The x-axis represents the expected kinship coefficient for a non-consanguineous family and the y-axis represents the estimated kinship coefficient in this family. Each dot represents the kinship coefficient between two members of the family. Black dots represent an estimated kinship coefficient concordant with the expected kinship coefficient for this kind of relationship. Red dots represent an estimated kinship coefficient discordant with the expected kinship coefficient for this kind of relationship. Pedigree constructed according to the estimated kinship coefficients.
B. Alignment of the human TIRAP protein with other human TIR domain-containing molecules
C. TIRAP RT-PCR, ranging from 27 to 33 cycles, on RNA extracted from SV40 fibroblasts from P1 and her father, compared to the results for a healthy control. Primer specific for transcripts encoding isoform A or B were used.
D. Quantitative PCR was performed to measure the levels of the transcripts encoding TIRAP isoforms A and B in SV40 fibroblasts (from five controls, P1, P1's father and mother), PBMCs (from 9 controls, P1, and P1's aunt) and EBV-B cells (from 5 controls and P1). SV40 fibroblasts stably transfected with the cDNA encoding isoform A or B were used as controls to test probe specificity. The percentages of transcripts encoding isoform A or B were normalized relative to the total number of transcripts encoding isoforms A and B, set at 100%. Error bars represent the SD.
Figure S2 : The TIRAP R121W allele is loss of function, related to Figure 3
A. Complementation study in SV40 fibroblasts from P1 (up), her father (middle) and a control (bottom), after stable transfection with lentiviral particles encoding the WT or mutant A or B isoform of TIRAP, or both. Particles containing luciferase instead of TIRAP were used for mock transfection. IL-6 production was assessed by ELISA. Error bars represent the SEM.
B. IL-6 mRNA levels assessed by RT-qPCR and expressed relative to GUS mRNA levels by the 2ΔΔC(t) method, in SV40 fibroblasts from a healthy control. Cells were stimulated for 4 h with PAM2 or TNF-α, 24 h after transfection with siRNAs against TLR2 and TIRAP.
C. PBMCs from several family members (III.1, IV.1-5 and V.1) were stimulated with various doses of PAM2, PAM3, LTA, FSL-1, and LPS for 3 h, subjected to extracellular staining with anti-CD3, anti-CD19 and anti-CD14 antibodies and intracellular staining for TNF-α. The proportion of CD14+ cells positive for TNF-α is shown. Acquisition was performed on 10,000 CD14+ cells. Each data point represents the mean of all individuals ± SEM (related to Figure 3E).
D. Transcriptome analysis performed on whole blood after 3 h of stimulation with PAM2, FSL-1, LTA, PAM3, LPS or PMA ionomycin. The results for P1 and her mother (R121W/WT) are compared with those for one healthy control and one IRAK-4-deficient patient.
Figure S3: The lack of anti-LTA Abs in P1's plasma accounts for the defective whole-blood response to LTA, related to Figure 4
A. PBMCs and whole blood from healthy controls, P1 and her father were stimulated with lipid A (1 μg/ml), LPS (10 ng/ml) and PMA/ionomycin for 48 h. IL-6 production was assessed by ELISA. Error bars represent the SD.
B. Assessment, with Luminex technology, of antibodies against 68 staphyloccocal antigens, (only 6 are shown in the figure, all results are summarized in Table S2), in 6 different plasma samples from P1 (between the ages of 4 months and 9 years), 6 homozygous relatives (V.2, IV.1, IV.2, IV.4, IV.6 and III.1), 6 IRAK-4 or MyD88-deficient patients and 11 sex- and aged-matched controls. Bonferroni correction for multiple testing indicated that the patient did not have significantly lower levels of antibody than the controls for any of the antigens tested.
C. PBMCs from P1 and two controls were left unstimulated or were stimulated by PAM3 or LTA in the presence of fetal calf serum (FCS) or plasma from each individual. IL-6 production was assessed by ELISA. Error bars represent the SD.
Figure S4: The lack of anti-LTA Abs in P1's plasma accounts for the defective whole-blood response to LTA, related to Figure 4
A. IL-6 production by whole blood was assessed in a healthy control and an IRAK4-deficient patient after 48 h of stimulation with PAM3 or LTA, supplemented or not with 0.5 mg/ml control isotype antibody (anti-RSV, Synagis) or monoclonal anti-LTA (pagibaximab) antibody.
B. IL-6 production by control PBMCs, monocytes, and monocyte-depleted PBMCs after 24 h of stimulation with 1 μg/ml PAM3 or 1 μg/ml LTA in the presence of medium alone, or medium supplemented with 1 mg/ml anti-LTA mAb or isotype control mAb (anti-RSV). Representative of 3 experiments.
C. Peritoneal mouse macrophages from WT B6, Myd88-/-, Tiraptor/tor and TLR2lan/lan (n>3) were stimulated with PAM3 (40 ng/ml) or LTA (1 μg/ml) or left unstimulated and complemented with 0. 5mg/ml anti-LTA Ab (pagibaximab) or isotype control (anti-RSV, Synagis). TNF-α production was assessed by ELISA. Error bars represent the SEM.
D. Assessment, by ELISA, of levels of anti-LTA antibodies in 16 different batches of commercially available IVIG, with comparison to healthy controls (n=10) and P1. Error bars represent the SEM.
Table S1: Related to figure 1.
List of genes presenting a non-synonymous rare homozygous variation in P1 exome data.
Table S2: Related to figure 4.
IgG levels against 68 recombinant S. aureus proteins were measured in serum samples, by a bead-based flow cytometry technique, in 6 different plasma samples from the patient (obtained between the ages of 1 and 8 years), 5 homozygous relatives, 6 IRAK-4- or MyD88-deficient patients and 11 aged-matched controls. The table summarizes mean and SD values for each antigen in each plasma category. Bonferroni correction for multiple testing was performed and p values are indicated for significant differences from controls.
Table S3: Related to STAR Methods section
Metrics and coverage data for the three exomes sequenced