

Abstract
The c-Jun N-terminal kinase (JNK)/stress activated protein kinase is preferentially activated by stress stimuli. Growth factors, particularly ligands for G protein-coupled receptors, usually induce only modest JNK activation, although they may trigger marked activation of the related extracellular signal-regulated kinase. In the present study, we demonstrated that homozygous disruption of glycogen synthase kinase 3β (GSK-3β) dramatically sensitized mouse embryonic fibroblasts (MEFs) to JNK activation induced by lysophosphatidic acid (LPA) and sphingosine-1-phosphate, two prototype ligands for G protein-coupled receptors. To a lesser degree, a lack of GSK-3β also potentiated JNK activation in response to epidermal growth factor. In contrast, the absence of GSK-3β decreased UV light-induced JNK activation. The increased JNK activation induced by LPA in GSK-3β null MEFs was insufficient to trigger apoptotic cell death or growth inhibition. Instead, the increased JNK activation observed in GSK-3β−/− MEFs was associated with an increased proliferative response to LPA, which was reduced by the inhibition of JNK. Ectopic expression of GSK-3β in GSK-3β-negative MEFs restrained LPA-triggered JNK phosphorylation and induced a concomitant decrease in the mitogenic response to LPA compatible with GSK-3β through the inhibition of JNK activation, thus limiting LPA-induced cell proliferation. Mutation analysis indicated that GSK-3β kinase activity was required for GSK-3β to optimally inhibit LPA-stimulated JNK activation. Thus GSK-3β serves as a physiological switch to specifically repress JNK activation in response to LPA, sphingosine-1-phosphate, or the epidermal growth factor. These results reveal a novel role for GSK-3β in signal transduction and cellular responses to growth factors.
Glycogen synthase kinase 3 (GSK-3)1 is an evolutionarily conserved and ubiquitously expressed serine/threonine kinase (1). In contrast to many other protein kinases, GSK-3 is constitutively active in intact cells (1–3). Phosphorylation of human GSK-3β at tyrosine 216 is critical for efficient kinase activity (1). GSK-3 kinase activity is inhibited through the phosphorylation of serine 9 of GSK-3β and serine 21 of GSK-3α by protein kinase A, protein kinase B, or protein kinase C (4–8). Therefore, GSK-3 activity can be down-regulated by environmental stimuli that are linked to the activation of multiple intracellular signaling pathways (4–8). Interestingly, the constitutive activity of GSK-3β suppresses cellular proliferation and survival (1, 9, 10). Many growth factors down-regulate GSK-3β activity via inducing the phosphorylation of serine 9, an integral process of the proliferative/survival network (9–11).
GSK-3 was originally identified as a regulator of glycogen synthesis (1). Later studies implicated GSK-3 in multiple biological processes. GSK-3 phosphorylates a broad range of physiological substrates, including several transcription factors and the translational factor eIF2B (12, 13). GSK-3β is required for cell fate specification in Dictyostelium spp. and is a component of the Wnt signaling cascade required for Drosophila and Xenopus development (14–17). In mammalian cells, regulation of the Wnt signaling is achieved, at least in part, by intracellular levels of β -catenin (18). In the absence of Wnt signals, a large multimeric protein complex that consists minimally of the scaffold protein Axin, adenomatous polyposis coli, GSK-3β, and β -catenin is formed (18–20). GSK-3β phosphorylates β -catenin in the complex, targeting β -catenin for ubiquitination and the subsequent degradation by a proteasome pathway (19, 20). Mutation or deletion of the putative GSK-3β phosphorylation sites of β -catenin results in constitutively active, highly stable forms of β -catenin (21, 22). In GSK-3β −/− mouse embryonic fibroblasts, however, the cytoplasmic levels of β -catenin seem to be normal (23), indicating that GSK-3α or other kinases can act redundantly to fulfill this functionality in the absence of GSK-3β. Similarly, inhibition of GSK-3β activity by activation of the phosphatidylinositol 3-kinase protein kinase B/Akt pathway does not allow accumulation of cytoplasmic β -catenin or activation of β -catenin-TCF/LEF-1 (T cell-specific factor/lymphoid enhancer-binding factor 1)-mediated gene expression (24).
In addition to a role in modulation of free β -catenin levels, GSK-3β has been shown to negatively regulate Axin-mediated MEKK1 activation (25, 26). Specifically, MEKK1 binds to the scaffold protein Axin, and transfection/overexpression of Axin increases the activation of MEKK1 (25, 26). The effect of Axin on MEKK1 activation is compromised by co-expression of GSK-3β or casein kinase Ie, which is compatible with a model wherein GSK-3β or casein kinase Ie competes with MEKK1 for binding to Axin (26). Based upon this scenario, GSK-3β would function as a negative regulator of cellular MEKK1 activity (25, 26). However, a recent study demonstrated that overexpression of GSK-3β potentiated UV light-induced or tumor necrosis factor α -induced activation of the MEKK1-SEK-JNK pathway (27). Thus, a positive regulatory role for GSK-3β in stress-induced or proinflammatory cytokine-induced activation of the MEKK1-SEK-JNK pathway has been proposed (27).
In the present study, we demonstrated that homozygous disruption of GSK-3β dramatically sensitized mouse embryonic fibroblasts (MEFs) (23) to JNK activation induced by ligands for G protein-coupled receptors (GPCRs) such as lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) (28). To a lesser degree, the lack of GSK-3β also potentiated JNK activation induced by ligation of the epidermal growth factor (EGF) transmembrane tyrosine kinase receptor. However, UV light-triggered JNK activation was not enhanced in GSK-3β null MEFs. These results suggest that GSK-3β acts as a physiological switch to specifically repress growth factor-induced but not stress-induced JNK activation. The increased JNK activation observed in GSK-3β −/− MEFs correlated with an increase in the proliferative response to LPA, suggesting that GSK-3β mediated inhibition of JNK repressed growth factor-induced proliferation. Consistently, the introduction of GSK-3β into GSK-3β -negative MEFs limited LPA-triggered JNK activation with a concomitant decline in the proliferative response to LPA.
EXPERIMENTAL PROCEDURES
Reagents
LPA (oleoyl; 18:1) and S1P were purchased from Avanti Polar Lipids (Alabaster, AL) and Calbiochem, respectively. Before use, these phospholipids were dissolved in phosphate-buffered saline containing 0.5% fatty acid-free bovine serum albumin (Roche Applied Science). Indo-1-AM was obtained from Molecular Probes (Eugene, OR). EGF, fetal bovine serum, and LiCl were obtained from Sigma. Anti-phospho-ERK was purchased from Promega (Madison, WI). Anti-phos-pho-c-Jun (S63) and anti-phospho-JNK antibodies were purchased from Cell Signaling (Beverly, MA). Anti-GSK-3 monoclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-glyceraldehyde-3-phosphate dehydrogenase was from Ambion (Austin, TX). The JNK inhibitor SP600125 was purchased from Calbiochem and dissolved in dimethyl sulfoxide (Me2SO). All cell culture reagents were from Invitrogen.
Cells
The fibroblast cell lines Swiss 3T3 (from the American Type Culture Collection) and GSK-3β −/− and GSK-3β +/+ MEFs (23) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cell lines were frozen at early passages and used for <10 weeks in continuous culture.
Western Blot
Cells were lysed in ice-cold X-100 lysis buffer (1% Triton X-100, 50 mM Hepes, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 100 mM NaF, 10 mM sodium pyrophosphate, and proteinase inhibitor mixture (Roche Applied Science)) or lysed directly in SDS sample buffer (2% SDS, 62.5 mM Tris (pH 6.8), 10% glycerol, and 5% β -mercaptoethanol). Cell lysates were resolved by SDS/PAGE, transferred to Immobilon (poly(vinylidene difluoride)), and immunoblotted with antibodies following the protocols provided by the manufacturers. Immunocomplexes were visualized with an enhanced chemiluminescence detection kit (Amersham Biosciences) using horseradish peroxidase-conjugated secondary antibodies (Bio-Rad).
[3H]Thymidine Incorporation
GSK-3β −/− and GSK-3β +/+ MEFs were plated in 6-well plates and grown to subconfluence. After a brief starvation, the cells were washed and fed serum-free medium containing various concentrations of LPA. After 16 h, cells were pulse-labeled with [3H]thymidine (2 μCi/well) for the final 6 h. The cells were then washed twice with phosphate-buffered saline, twice with 5% (w/v) trichloroacetic acid, and once with 95% (v/v) ethanol. Trichloroacetic acid-insoluble material was dissolved in 0.2 M sodium hydroxide overnight and scintillation-counted for radioactivity.
Plasmids
To conditionally express GSK-3β in mammalian cells, we cloned GSK-3β into the tetracycline-off plasmid EC1214A (29). This inducible system combines the two required tetracycline-inducible expression elements (the tetracycline-repressive transcriptional activator protein tTA and the tTA-responsive promoter) into a single plasmid (29). HA-GSK-3β was excised from the plasmid pcDNA3-HA-GSK-3β (5) by partial digestion with BamHI. The resulting 1399-bp fragment was cloned into the BamHI site of EC1214A (29). The structure of the EC1214A-GSK-3β plasmid was confirmed by restriction enzyme digestion and DNA sequencing.
The GSK-3β mutants (GSK-3β -K85M and GSK-3β -Y216F) (26) were generated from pcDNA3-HA-GSK-3β by in vitro site-directed mutagenesis using the QuikChange™ site-directed mutagenesis kit (Invitrogen). The oligonucleotides used to create the GSK-3β -K85M mutant were 5′-CTGGTCGCCATCAtGAAAGTATTGCAGGACAAG-3′ and 5′-GTCCTGCAATACTTTCaTGATGGCGACCAGTTC-3′. The oligonucleotides used for the creation of the GSK-3′-Y216F mutant were 5′CCC-AATGTTTCGTtTATCTGTTCTCGGTACTATAG-3′ and 5′-CCGAGA-ACAGATAaACGAAACATTGGGTTC-3′. The lowercase letters indicate the bases that are mutated in the GSK-3′ mutant constructs.
Stable and Inducible Expression of GSK-3β
GSK-3β −/− MEFs were transfected with Lipofectamine 2000 (Invitrogen) and pcDNA3-HA-GSK-3β along with pcDNA3.1/GS (Invitrogen). To select stable clones, transfected cells were subcultured at a 1:30 dilution and grown in the presence of 300 μg/ml Zeocin™. Discrete Zeocin-resistant colonies were isolated by ring cloning and expanded sequentially in 24-well plates, 6-well plates, and 60-mm dishes. Positive clones that expressed GSK-3β were identified by immunoblotting with the antibody against GSK-3α/β.
For the re-expression of GSK-3β in GSK-3β −/− MEFs in an inducible manner, GSK-3β −/− cells were transfected with EC1214A-GSK-3β along with pcDNA3.1/GS (Invitrogen). Transfected cells were cultured in the presence of 2 μg/ml tetracycline to ensure a minimal leakage and in the presence of 300 μg/ml Zeocin for the selection of stable transfectants. Discrete Zeocin-resistant colonies were isolated by ring cloning and expanded sequentially in 24-well plates, 6-well plates, and 60-mm dishes. Inducible expression of GSK-3β in individual clones in the absence of tetracycline was examined by immunoblotting.
GSK-3β Retrovirus Constructs
To generate retroviral constructs to express wild type or mutant GSK-3β, the HA-GSK-3β, HA-GSK-3β -K85M, and HA-GSK-3β -Y216F constructs were excised from their corresponding pcDNA3 vectors by digestion with BamHI and then cloned into a Moloney murine leukemia retrovirus vector, LZRS-EGFP (30) (a gift of Dr. J. Chun, the Scripps Research Institute, La Jolla, CA). The complete inserts and the flanking regions of the final retroviral vectors were confirmed by automatic sequencing. These retroviral constructs contain an internal ribosome entry site sequence that permitted concomitant expression of the GSK-3β gene and the enhanced green fluorescent protein (GFP) gene from a single transcript driven by the 5′-long terminal repeat promoter (30).
Generation of GSK-3β Retrovirus and Infection of Cells
To generate a high titer of viral stocks, LZRS-EGFP-GSK-3β, LZRS-EGFP-GSK-3β -K85M, or LZRS-EGFP-GSK-3β -Y216F were transfected into the BOSC23 packaging line (American Type Culture Collection) (31) using Lipofectamine 2000 (12 μg of DNA per 100-mm dish). Approximately 60–72 h after the beginning of transfection, the supernatants were harvested, cleared by a brief spin, and used for infection or stored in aliquots at −80 °C for later use.
GSK-3β −/− MEFs in 6-well plates at ~60% confluence were incubated for 18 h with 1 ml of viral supernatants in the presence of Polybrene (6 μg/ml). The infected cells were then kept in complete medium for 48 h. The GFP-positive cells were isolated by flow cytometry and replated in culture. A high purity of sorted cells was indicated by the presence of >90% cells positive for GFP. The cells were starved overnight in serum-free medium prior to ligand stimulation.
UV Light Exposure
Cells were exposed to UV light irradiation as described previously (32). Briefly, sub-confluent Swiss 3T3, GSK-3β −/−, and GSK-3β +/+ cells were starved in serum-free medium and exposed to UV light irradiation (10, 20, 40, and 80 J/m2) using a UV light cross-linker (Stratagene). UV light irradiated cells were then re-fed serum-free medium and returned to the CO2 incubator for a period of 30 min before cell lysates were prepared and analyzed for JNK phosphorylation by Western blotting.
Cytoplasmic [Ca2+]i Assay
After starvation in serum-free medium for 24 h, GSK-3β+/+ and GSK-3β−/− MEFs were harvested and loaded with 1 μM Indo-1 AM in phosphate buffered saline for 30 min at 37 °C. Cells were washed in phosphate-buffered saline and resuspended at 2 × 106 cells/ml in a [Ca2+]i assay buffer (140 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 25 mM Hepes, and 10 mM glucose, pH 7.4). Cytoplasmic [Ca2+]i was determined at an excitation wavelength of 331 nm and an emission wavelength of 410 nm using a fluorescence spectrophotometer (Hitachi F-4000). Approximately 3 × 106 cells were used for [Ca2+]i determination in a stirred quartz cuvette kept at 37 °C.
RESULTS
JNK and ERK Are Differentially Regulated by GPCR Agonists
In normal fibroblasts such as Swiss 3T3 cells, EGF and particularly, agonists of GPCR such as LPA and S1P induced only marginal phosphorylation of JNK as compared with the cell stress mediated by UV light exposure. In contrast, treatment of cells with EGF or LPA resulted in marked phosphorylation of ERK, whereas UV light irradiation did not alter ERK phosphorylation in a detectable manner (Fig. 1). Additionally, LPA- and S1P-induced JNK activation demonstrated different kinetics from that of ERK activation. Peak activation of ERK occurred within 5 min of LPA or S1P addition and declined immediately afterward. In contrast, LPA- or S1P-induced phosphorylation of JNK peaked at later time points and persisted for at least 60 min after treatment with LPA or S1P.
Fig. 1. JNK and ERK are differentially regulated by growth factors.

Swiss 3T3 cells were starved in serum-free medium and incubated with LPA (1 μM), S1P (1 μM), or EGF (20 ng/ml). Cells were lysed in SDS sample buffer at 0, 5, 10, 30, and 60 min post-treatment and analyzed by immunoblotting for JNK and ERK phosphorylation using phospho-specific (p-Erk and p-JNK) antibodies. Starved Swiss 3T3 were subjected to UV light irradiation using an UV cross-linker at 10, 20, 40, and 80 J/m2 (J). UV light-irradiated cells were re-fed serum-free medium and returned to a CO2 incubator for 30 min before the cells were lysed and analyzed for JNK and ERK phosphorylation. Reprobing with anti-JNK1 antibody was included to show similar levels of loading among samples. For this and all other illustrations in the paper, similar results were obtained from three independent experiments.
Homozygous Disruption of GSK-3β Increases Growth Factor-induced JNK Phosphorylation
Growth factor-induced activation of the ERK and JNK pathways are mediated by interactive signaling cascades that bifurcate downstream of Ras (33, 34). The limited and delayed activation of JNK in response to LPA or S1P suggests that the signal leading to JNK activation is either intrinsically weak or restrained by other signaling molecules. Because, when overexpressed, GSK-3β inhibits activation of MEKK1, an upstream component of the JNK pathway (26), we examined whether GSK-3β could act as a negative regulator of LPA- or S1P-induced JNK activation.
Because GSK-3β is universally expressed and constitutively active in almost all cell types (35), overexpression of the kinase by transfection does not offer an appropriate approach to define the physiological role of GSK-3β in signaling processes. In fact, previous transfection/overexpression experiments have yielded controversial results concerning the role of GSK-3β in the modulation of MEKK1 activity (25–27). To clarify this issue, we took advantage of GSK-3β null MEFs and wild type MEFs to study the effect of GSK-3β on cellular JNK activation. In GSK-3β−/− MEFs, LPA and S1P induced much higher and more prolonged phosphorylation of JNK than in GSK-3β+/+ MEFs (Fig. 2A) or Swiss 3T3 cells (see Fig. 1). EGF also induced higher levels of JNK phosphorylation in GSK-3β−/− cells than in GSK-3β+/+ cells, although the effect was not as dramatic as that of LPA or S1P (Fig. 2, A and B).
Fig. 2. Homozygous disruption of GSK-3β sensitizes cells to JNK phosphorylation induced by LPA, S1P, or EGF.

GSK-3β knock-out (GSK-3β−/−) and wild type MEFs (GSK-3β+/+) were starved in a serum-free medium and stimulated with 1 μM LPA (A), 1 μM S1P (B), or 20 ng/ml EGF (A) for the indicated periods of time (minutes). The stimulated cells were lysed in SDS sample buffer and analyzed by immunoblotting for JNK and ERK phosphorylation using the corresponding phospho-specific (p-Erk and p-JNK) antibodies. Reprobing with an anti-JNK1 antibody was included to show equal loading among samples. The absence of GSK-3β in the knock-out MEFs was confirmed by reprobing with an anti-GSK-3α/β antibody (B).
In contrast, the absence of GSK-3β did not increase LPA, S1P, or EGF-induced ERK activation. Both the magnitude and the duration of ERK phosphorylation were similar between GSK-3β+/+ and GSK-3β−/− cells, indicating a specific role for GSK-3β in restraining growth factor-stimulated JNK but not ERK phosphorylation. Similarly, the absence of GSK-3β in GSK-3β−/− cells did not alter LPA-induced phosphorylation/ activation of p38 MAPK (data not shown). The specificity of the effect of GSK-3β on growth factor-mediated JNK activation is further supported by the observation that the absence of GSK-3β did not increase UV light-mediated JNK activation. In fact, UV light-triggered JNK phosphorylation was lower in GSK-3β null cells compared with that in the wild type cells (Fig. 3). Similarly, TNFα induced stronger JNK phosphorylation in wild type MEFs than in GSK-3β−/− MEFs (data not shown). These results indicate that, in contrast to the inhibitory effect on growth factor-induced JNK activation, GSK-3β positively regulates stress-mediated and pro-inflammatory cytokine-mediated JNK activation.
Fig. 3. Lack of GSK-3β does not enhance UV-induced JNK phosphorylation.

GSK-3β−/− and GSK-3β+/+ MEFs were subjected to UV light radiation at the indicated doses (J, J/m2) using a UV light cross-linker as described under “Experimental Procedures.” Following UV light exposure, the cells were re-fed serum-free medium and returned to a CO2 incubator for 30 min. Cells were then lysed in SDS sample buffer and analyzed for JNK phosphorylation by immunoblotting with a JNK phospho-specific (p-JNK) antibody. Reprobing with anti-JNK1 antibody was included to confirm comparable levels of loading among samples.
The differential activation of JNK by LPA in GSK-3β−/− and GSK-3β+/+ cells could be due to clonal variation in LPA or S1P receptor expression or action in the two established MEF lines. To address this possibility, we assessed the ability of LPA to increase intracellular calcium changes, an early signaling event downstream of LPA receptor activation (28, 30). LPA induced similar dose- and time-dependent increases in [Ca2+]i in GSK-3β−/− and GSK-3β+/+ cells (data not shown).
Inhibition of GSK-3β Activity by LiCl Potentiates LPA-induced JNK Activation
To further evaluate the role of GSK-3β kinase activity in the inhibition of JNK activation by LPA, we assessed the impact of inhibiting GSK-3β activity on LPA- induced JNK activation. To determine whether the inhibition of GSK-3β activity could mimic the phenotype of GSK-3β−/− cells, we treated GSK-3β+/+ cells with various concentrations of LiCl, a specific inhibitor of GSK-3 (35, 36), for 75 min before stimulation with LPA. As shown in Fig. 4, pre-treatment of cells with LiCl enhanced LPA-mediated JNK phosphorylation in a dose-dependent manner, which is compatible with GSK-3β kinase activity playing a critical role in limiting LPA-induced JNK phosphorylation.
Fig. 4. Inhibition of GSK-3β activity by LiCl potentiates LPA-induced JNK phosphorylation.
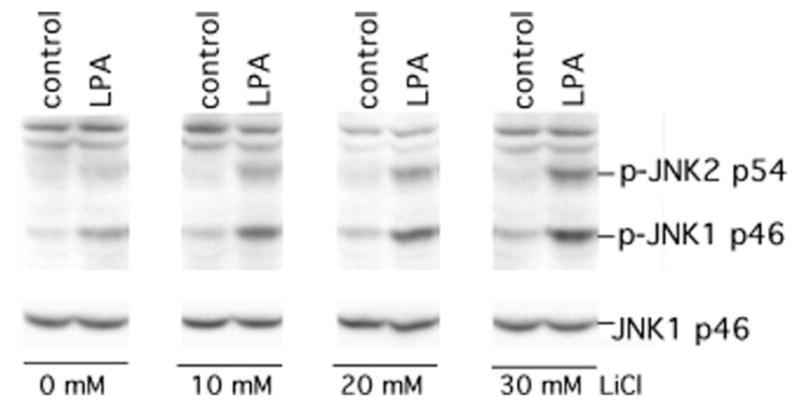
GSK-3β+/+ MEFs were starved and stimulated without (control) or with LPA (1 μM) for 30 min in the presence of lithium chloride at the indicated concentrations. LiCl was added to medium 75 min before LPA stimulation. Cell lysates were prepared and analyzed for JNK phosphorylation with a JNK phospho-specific (p-JNK) antibody. Reprobing with anti-JNK1 antibody was included to confirm comparable levels of loading among samples.
Increased JNK Phosphorylation Correlates with Increased Proliferative Response to LPA
Under most circumstances, strong or sustained activation of JNK is linked to apoptotic cell death or inhibition of cell proliferation (37–39). To determine the biological significance of the effects of GSK-3β on JNK activation, we compared the survival and proliferative responses to LPA between GSK-3β+/+ and GSK-3β−/− cells. We have demonstrated previously that LPA is a potent growth and survival factor in murine fibroblasts (40). In serum-free conditions, <10% of GSK-3β+/+ and GSK-3β−/− cells underwent apoptosis (Fig. 5A). Treatment of cells with LPA reduced the apoptotic rate to 1–2% in both lines, indicating that LPA protects both wild type and GSK-3β-negative cells from serum deprivation-induced apoptosis. The results suggest that the increased JNK activation by LPA was not sufficient to trigger the apoptotic process in GSK-3β−/− cells. This suggestion is consistent with the observation that LPA-induced JNK activation in GSK-3β −/− cells was only modest compared with the marked JNK activation induced by UV light (Fig. 1) and other death stimuli (32, 41).
Fig. 5. Increased JNK activation in GSK-3β−/− cells does not induce apoptosis and is associated with an increase in mitogenic response to LPA.

A, GSK-3β−/− and GSK-3β+/+ cells were starved and incubated without or with LPA (10 μM). After 24 h, cells (including floating and adherent cells) were harvested, fixed, and stained via terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL) using ApopTag (Chemicon International). The percentages of TUNEL-stained positive apoptotic cells were determined by flow cytometry. The results are presented as means ± S.D. of duplicate data from three independent experiments. B, the mitogenic activity of LPA in GSK-3β−/− and GSK-3β+/+ cells was measured by [3H]thymidine incorporation as described under “Experimental Procedures.” The data were presented as fold increases (means ± S.D. of triplicate assays) with the activity in unstimulated control cells defined as one.
Interestingly, LPA stimulated significantly higher levels of [3H]thymidine incorporation in GSK-3β−/− cells than in GSK-3β+/+ cells as demonstrated in Fig. 5B, an observation compatible with a modest increase in JNK activation acting in concert with other proliferative signaling pathways to augment cell proliferation in LPA-stimulated conditions.
Re-expression of GSK-3β Desensitizes Cells to LPA-induced JNK Activation and Proliferative Response to LPA
To confirm the ability of GSK-3β to restrain growth factor-induced JNK activation, we re-expressed GSK-3β in GSK-3β−/− cells by transfection with the GSK-3β expression vector pcDNA3-HA-GSK-3β. Two stable clones (G28 and G60) expressing GSK-3β at levels lower than that of wild type MEFs were selected (Fig 6A, top). Although these cells tended to lose GSK-3β expression with passage for an undefined reason, we used early passages, which retained GSK-3β, to examine the effect of GSK-3β on LPA-induced JNK activation. As shown in Fig. 6A (bottom), LPA stimulated marked phosphorylation of JNK in the GSK-3β-negative control clone (C9), similarly to the parental GSK-3β−/− cells. However, LPA-induced JNK phosphorylation in the two GSK-3β expressing clones was dramatically reduced. JNK phosphorylation induced by LPA was inversely correlated with GSK-3β expression levels in the two stable clones (Fig. 6A), confirming that the presence of GSK-3β inhibited LPA-induced JNK phosphorylation.
Fig. 6. GSK-3β inhibits LPA-induced JNK phosphorylation.

A, the effect of stable re-expression of GSK-3β. GSK-3β −/− cells were co-transfected with pcDNA3-HA-GSK-3β and pcDNA3.1/GS (containing the Zeocin resistance gene). Stable clones (G28 and G60) that expressed HA-GSK-3β were identified by immunoblotting with an antibody against GSK-3α/β and GSK-3β, with wild type MEFs included as a positive control (top). G28, G60, and empty vector control clone C9 were starved and treated with LPA (1 μM) for the indicated periods of time (min) (bottom). Cell lysates were prepared and analyzed by immunoblotting for JNK phosphorylation with a JNK phospho-specific (p-JNK) antibody (bottom). B, effect of the inducible expression of GSK-3β. The EC1214A-GSK3β inducible system involves a single vector carrying both the tetracycline-responsive transcriptional activator (tTA) and a tetracycline responsive element (TRE). The plasmid produces the tTA under the control of the cytomegalovirus promoter, and then tTA binds to TRE, leading to transcriptional activation of GSK3β in the absence of tetracycline. TK, thymidine kinase; BGH, bovine growth hormone; pA, poly(A). A stable clone with minimum expression of GSK-3β in the presence of tetracycline was identified and incubated in the presence (Tet 2 μg/ml) and absence of tetracycline (Tet 0 μg/ml) for 36 h. The cells were stimulated without or with LPA (1 μM) for 30 min and analyzed by immunoblotting for JNK phosphorylation with a JNK phospho-specific (p-JNK) antibody. Induction of GSK-3β expression in the inducible clone following removal of tetracycline was confirmed by immunoblotting with antibody against GSK-3α/β (bottom).
Because of the tendency to lose GSK-3β expression in constitutive stable clones, we utilized a tetracycline-regulatable expression system to conditionally express GSK-3β in GSK-3β−/− cells. This approach involves a single vector carrying the gene to be expressed and the tetracycline repressor (Fig. 6B, top) (29). In an identified inducible clone the expression of GSK-3β was rapidly induced upon the removal of tetracycline from the culture medium and could be reversibly repressed by the addition of tetracycline (Fig. 6B, bottom). LPA-induced JNK phosphorylation was significantly inhibited when the GSK-3β protein was induced by removal of tetracycline (Fig. 6B, bottom), further supporting an inhibitory role for GSK-3β in LPA-induced JNK activation.
To determine whether re-expression of GSK-3β compromises the mitogenic response to LPA, we developed a retroviral system to co-express GSK-3β and GFP in the knock-out MEFs. Highly enriched populations with >90% GFP-positive cells were isolated by flow cytometry. The cells were analyzed for JNK phosphorylation and cell cycle progression in response to LPA. As expected, the expression of GSK-3β nearly abolished LPA-induced JNK phosphorylation (data not shown). As indicated by propidium iodide staining, re-expression of GSK-3β decreased LPA-induced cell cycle progression with an accumulation of cells in G1 (72% in GSK-3β-positive cells versus 50% in GSK-3β-negative cells, p < 0.01) and a decrease of cells in S phase (14% in GSK-3β-positive cells versus 30% in GSK-3β-negative cells, p < 0.01). Furthermore, as indicated by cell counts, GSK-3β expression decreased LPA-induced increases in both cell proliferation and saturation density (p < 0.01) (Fig. 7B), confirming that GSK-3β inhibited LPA-induced cell proliferation. Consistent with this finding, the growth response to LPA in GSK-3β wild type MEFs was similar to that in GSK-3β infected cells (Fig. 7, A and B).
Fig. 7. Re-expression of GSK-3β inhibits LPA-induced cell proliferation.
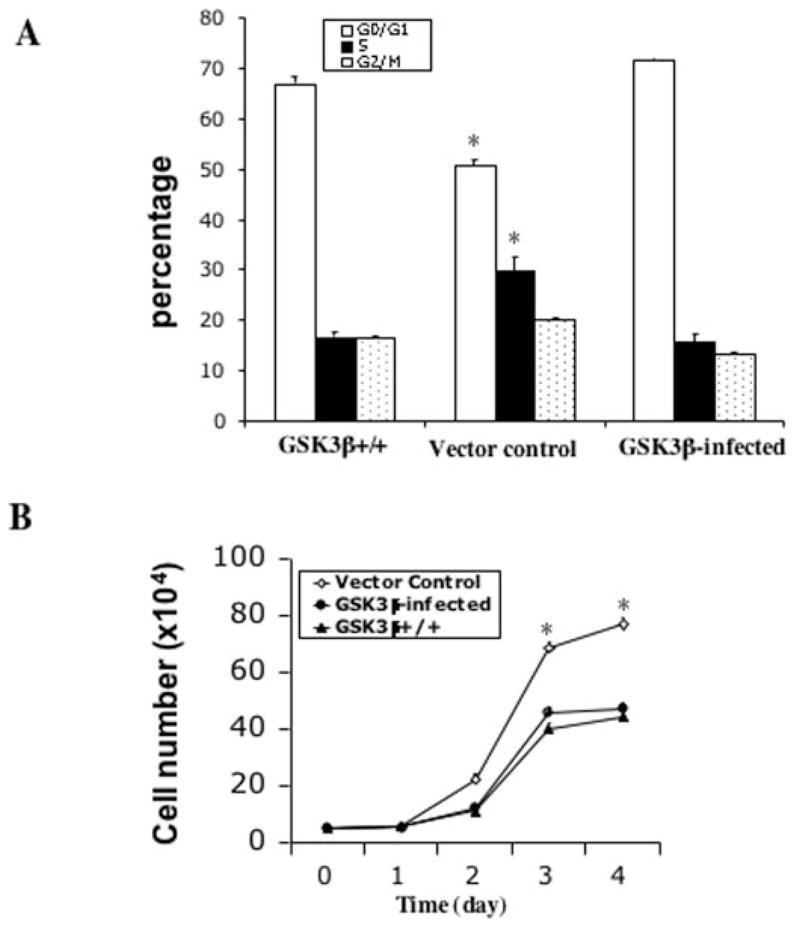
GSK-3β−/− cells were infected with the HA-GSK-3β retro-virus or the control virus as detailed under “Experimental Procedures.” GFP-positive cells from HA-GSK-3β retrovirus-infected cells (GSK-3β infected) and control virus-infected cells (Vector control) were sorted, replated, and cultured for growth response to LPA. A, cell cycle analysis of the GSK-3β-infected, vector control, and GSK-3β+/+ cells. The cells were starved for 16 h and incubated with 10 μM LPA for 24 h before collection by trypsinization. Cells were stained with propidium iodide for flow cytometric analysis of cell cycle progression. B, growth curves of the GSK-3β-infected, vector control, and GSK-3β+/+ cells. Cells were seeded in 6-well plates (5 × 104 cells/well). After attachment, cells were starved for 16 h and cultured in serum-free medium containing 10 μM LPA. Cell numbers were determined at 24-h intervals by counting in a hemocytometer. For both panel A and panel B data are presented as means ± S.D. of duplicate assays from three independent experiments. Statistical differences between GSK-3β-infected cells and vector control cells are indicated by an asterisk (p < 0.01).
To verify the importance of JNK activation in the increased cell proliferation in GSK3β−/− MEFs, we examined the effect of the specific JNK inhibitor SP600125 on the growth response to LPA in the GSK3β-negative cells. LPA-induced c-Jun phosphorylation was inhibited by incubation of the cells with SP600125 in a dose-dependent manner (Fig. 8A). SP600125 also dose-dependently inhibited LPA-induced increases in cell number and saturation density (p < 0.01) (Fig. 8B), indicating that JNK activation is necessary for the increased growth response to LPA in GSK-3β-negative cells.
Fig. 8. Inhibition of JNK activation suppresses the increased proliferative response to LPA in GSK3β-negative cells.

A, the effect of the JNK inhibitor SP600125 on LPA-induced c-Jun phosphorylation. GSK-3β-negative cells were starved and incubated with SP600125 at the indicated concentrations. One hour later the cells were stimulated with 10 μM LPA for 30 min. Cells were lysed in SDS sample buffer and analyzed by immunoblotting for c-Jun phosphorylation using phospho-specific (p-c-Jun) antibodies. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B, the effect of the JNK inhibitor on LPA-mediated cell proliferation. GSK3β-negative MEFs were prepared and plated as described for Fig. 7B. After attachment and starvation, cells were incubated with LPA (10 μM) in the presence of the indicated concentration of SP600125 that was added 1 h before the addition of LPA. Cell numbers were determined at 24-h intervals by counting in a hemocytometer. The data are presented as means ± S.D. of duplicate assays from three independent experiments. Statistical differences between cells treated with vehicle (Me2SO) and SP600125 are indicated by an asterisk (p < 0.01).
GSK-3β Kinase Activity Is Required for Inhibition of LPA-stimulated JNK Activation
We next examined whether the kinase activity of GSK-3β was required for its ability to inhibit growth factor-induced JNK activation. To this end, we made a kinase-dead mutant of GSK-3β (K85M) and a hypomorphic mutant (Y216F) with markedly decreased kinase activity (1, 26, 42) by site-directed mutagenesis. The K85M mutant also lacks the ability to bind to the scaffold protein Axin, whereas the Y216F mutant retains the Axin binding ability, thus allowing interaction with MEKK1 (1, 26, 42).
The wild type and mutant forms of GSK-3β were expressed in GSK-3β−/− MEFs using recombinant retroviruses as described above. Highly enriched populations of GFP-positive cells were isolated by flow cytometry. The kinase dead K85M mutant failed to suppress LPA-mediated JNK phosphorylation (Fig. 9) suggesting the importance of the GSK-3β kinase activity in the process. Consistent with this suggestion, expression of the hypomorphic mutant (Y216F) of GSK-3β partially decreased LPA-induced JNK phosphorylation compared with wild type GSK-3β (Fig. 9). Because the Y216F mutant is fully capable of binding with Axin (26, 42), the partial effect of the mutant on LPA-induced JNK phosphorylation suggests that the kinase activity, rather than the interaction with Axin or MEKK1, is critical for GSK-3β inhibition of LPA-induced JNK activation.
Fig. 9. GSK-3β inhibition of LPA-induced JNK activation requires kinase activity.

Wild type and mutant (Y216F and K85M) forms of GSK-3β were introduced into GSK-3β−/− cells by retrovirus-mediated gene transfer as detailed under “Experimental Procedures.” Enriched populations of GFP-positive cells were isolated by flow cytometry and replated in culture. After starvation in serum-free medium, the cells were incubated with LPA (1 and 10 μM) for 30 min before being lysed in SDS sample buffer and analyzed by immunoblotting for JNK phosphorylation with a JNK phospho-specific (p-JNK) antibody. Reprobing with an anti-GSK-3α/β antibody was included to serve as a loading control and to verify re-expression of GSK-3β in retrovirus-transduced cells. Similar results were obtained in three independent experiments.
DISCUSSION
In mammalian cells, JNK is most strongly activated by UV light exposure, heat shock, inhibition of cellular protein synthesis, and other stress-related conditions (32, 39, 41). Growth factors, especially ligands for GPCRs, usually induce only modest JNK activity, although they may trigger marked activation of the related ERK (33, 39, 42, 43). The differential effects of growth factors on JNK and ERK activity suggest that the existence of an as yet unidentified mechanism that specifically restrains JNK but not ERK activation upon the stimulation of GPCR. The present study demonstrates that GSK-3β limits JNK activation in response to the growth factors LPA, S1P, and EGF. In contrast, GSK-3β positively regulates UV light- and TNFα-mediated JNK activation.
In GSK-3β−/− MEFs, as compared with wild type MEFs, a markedly increased degree of JNK phosphorylation was induced by LPA, S1P, and EGF. In contrast, the absence of GSK-3β did not potentiate LPA, S1P, or EGF-induced activation of ERK and p38 mitogen-activated protein kinase, two related members of the mitogen-activated protein kinase family. We confirmed the inhibitory effect of GSK-3β and a requirement for its kinase activity on growth factor-mediated JNK activation by blocking GSK-3β kinase activity with LiCl in wild type cells and by re-expression of GSK-3β in the knock-out cells. Ectopic expression of GSK-3β in GSK-3β −/− cells reversed the increase in JNK phosphorylation induced by LPA, indicating that increased JNK phosphorylation in LPA-treated GSK-3β−/− MEFs was due to the lack of GSK-3β rather than a clonal variation between the two established MEF lines.
Kim et al. reported recently that GSK-3β activates MEKK1 and promotes UV light- and TNFα-induced MEKK1-SEK-JNK activation (27). This finding predicts that homozygous deletion of GSK-3β would impair UV light- or TNFα-triggered JNK phosphorylation. In our study, we indeed observed that JNK phosphorylation induced by UV light exposure or TNFα was decreased in GSK-3β−/− MEFs (Fig. 3), suggesting that GSK-3β acts as a positive mediator of stress- and proinflammatory cytokine-induced JNK activation. However, the results of our present study indicate that GSK-3β has an opposite effect on growth factor-stimulated JNK activation. This finding is compatible with a previous observation that GSK-3β competes with MEKK1 for binding to the scaffold protein Axin (26). The interaction between MEKK1 and Axin appears to positively regulate MEKK1 activity (25, 26). GSK-3β may prevent binding of MEKK1 to Axin and therefore limit activation of the Axin-MEKK1-SEK-JNK pathway. However, the Y216F mutant, which is fully capable of binding to Axin, is compromised as compared with wild type GSK-3β in its ability to inhibit LPA-induced JNK phosphorylation. Furthermore, the inhibition of GSK-3 activity by LiCl in wild type cells sensitizes LPA-induced JNK phosphorylation. These results collectively suggest that a GSK-3β kinase activity-dependent mechanism is required for the full execution of the ability of GSK-3β to inhibit JNK activation. If GSK-3β binding to Axin and interruption of the Axin-MEKK1-SEK-JNK cascade are involved in the partial effect observed with the Y216F mutant of GSK-3β, it is clear that the interaction of GSK-3β with Axin is insufficient for GSK-3β to exert its optimal inhibitory effect on growth factor-induced JNK activation.
A wide range of biological outcomes has been attributed to cellular JNK activation. The majority of published evidence supports a role of JNK in conveying apoptotic responses when cells are exposed to cytokines, DNA damage, or anti-cancer therapies (37–39). These stress or death insults usually induce robust and sustained JNK activation (37–39). However, many studies support the requirement of basal and stimulated JNK activity for cell growth and survival (39, 44–48). The pro-proliferative action of JNK is consistent with the prominent role of c-Jun, the major substrate of JNK, and AP-1 transcriptional activity in promotion of cell cycle progression (39, 44–47). In GSK-3β null MEFs, the augmented JNK activation in response to LPA was not associated with increased apoptosis or growth inhibition. Instead, GSK-3β null cells demonstrated a more robust proliferative response to LPA. The inhibition of JNK activation by a pharmacological inhibitor decreased LPA-induced increases in cell proliferation and saturation density, further suggesting that the modestly increased JNK activation observed in GSK-3β null cells is required for the increased cell cycle progression induced by LPA. Thus, GSK-3β coordinately restricts growth factor-induced JNK phosphorylation and cell proliferation.
In summary, our present work identifies the ubiquitously expressed GSK-3β as an intracellular negative regulator of JNK activation induced by LPA, S1P, or EGF. Our results provide a new mechanism by which GSK-3β coordinates cellular responses to environmental stimuli.
Footnotes
This work was supported by the Lynne Cohen Foundation Ovarian Cancer Research Award (to X. F. and G. B. M.), Department of Defense Breast Cancer Research Program Grant DAMD17-03-1-0409 (to S. L.), National Institutes of Health Grants CA82716 and CA64602 (to G. B. M.), and National Institutes of Health Core Grant P30 CA016672 (to the M. D. Anderson Cancer Center).
The abbreviations used are: GSK, glycogen synthase kinase; EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; JNK, the c-Jun N-terminal kinase/stress activated protein kinase; LPA, lysophosphatidic acid; MEF, mouse embryonic fibroblast; MEKK, mito-gen-activated protein kinase/ERK kinase; SEK, stress-activated protein kinase/ERK kinase; S1P, sphingosine-1-phosphate; TNFα, tumor necrosis factor α.
References
- 1.Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stambolic V, Woodgett JR. Biochem J. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sutherland C, Leighton IA, Cohen P. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 5.Fang X, Yu S, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Proc Natl Acad Sci U S A. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heiden-reich KA. Mol Cell Biol. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB. Mol Cell Biol. 2002;22:2099–2110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goode N, Hughes K, Woodgett JR, Parker PJ. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- 9.Cui H, Meng Y, Bulleit RF. Brain Res Dev Brain Res. 1998;111:177–188. doi: 10.1016/s0165-3806(98)00136-9. [DOI] [PubMed] [Google Scholar]
- 10.Pap M, Cooper GM. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 11.Shin SY, Yoon SC, Kim YH, Kim YS, Lee YH. Exp Mol Med. 2002;34:444–450. doi: 10.1038/emm.2002.62. [DOI] [PubMed] [Google Scholar]
- 12.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Biochim Biophys Acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- 13.Welsh GI, Proud CG. Biochem J. 1993;294:625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harwood AJ, Plyte SE, Woodgett JP, Strutt H, Kay RR. Cell. 1995;80:139–148. doi: 10.1016/0092-8674(95)90458-1. [DOI] [PubMed] [Google Scholar]
- 15.He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez I, Itoh K, Sokol SY. Proc Natl Acad Sci U S A. 1995;92:8498–8502. doi: 10.1073/pnas.92.18.8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siegfried E, Chou TB, Perrimon N. Cell. 1992;71:1167–1179. doi: 10.1016/s0092-8674(05)80065-0. [DOI] [PubMed] [Google Scholar]
- 18.Hinoi T, Yamamoto H, Kishida M, Takada S, Kishida S, Kikuchi A. J Biol Chem. 2000;275:34399–34406. doi: 10.1074/jbc.M003997200. [DOI] [PubMed] [Google Scholar]
- 19.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. Proc Natl Acad Sci U S A. 1999;96:6273–6278. doi: 10.1073/pnas.96.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 22.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 23.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 24.Ding VW, Chen RH, McCormick F. J Biol Chem. 2000;275:32475–32481. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Neo SY, Wang X, Han J, Lin SC. J Biol Chem. 1999;274:35247–35254. doi: 10.1074/jbc.274.49.35247. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Qiu WJ, Liu DX, Neo SY, He X, Lin SC. J Biol Chem. 2001;276:32152–32159. doi: 10.1074/jbc.M104451200. [DOI] [PubMed] [Google Scholar]
- 27.Kim JW, Lee JE, Kim MJ, Cho EG, Cho SG, Choi EJ. J Biol Chem. 2003;278:13995–14001. doi: 10.1074/jbc.M300253200. [DOI] [PubMed] [Google Scholar]
- 28.Goetzl EJ, An S. Adv Exp Med Biol. 1999;469:259–264. doi: 10.1007/978-1-4615-4793-8_38. [DOI] [PubMed] [Google Scholar]
- 29.Xu HJ, Zhou Y, Ji W, Perng GS, Kruzelock R, Kong CT, Bast RC, Mills GB, Li J, Hu SX. Oncogene. 1997;15:2589–2596. doi: 10.1038/sj.onc.1201446. [DOI] [PubMed] [Google Scholar]
- 30.Ishii I, Contos JJ, Fukushima N, Chun J. Mol Pharmacol. 2000;58:895–902. doi: 10.1124/mol.58.5.895. [DOI] [PubMed] [Google Scholar]
- 31.Pear W, Scott M, Nolan GP. In: Methods in Molecular Medicine: Gene Therapy Protocols. Robbins P, editor. Humana Press; Totowa, NJ: 1997. pp. 41–57. [DOI] [PubMed] [Google Scholar]
- 32.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 33.Minden A, Lin A, Claret FX, Abo A, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 34.Scita G, Nordstrom J, Carbone R, Tenca P, Giardina G, Gutkind S, Bjarnegard M, Betsholtz C, Di Fiore PP. Nature. 1999;401:290–293. doi: 10.1038/45822. [DOI] [PubMed] [Google Scholar]
- 35.Doble BW, Woodgett JR. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryves WJ, Harwood AJ. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- 37.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 38.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 39.Davis RJ. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 40.Fang X, Yu S, LaPushin R, Lu Y, Furui T, Penn LZ, Stokoe D, Erickson JR, Bast RC, Jr, Mills GB. Biochem J. 2000;352:135–143. [PMC free article] [PubMed] [Google Scholar]
- 41.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki T, Maehama T, Yamamoto T, Takasuga S, Hoshino S, Nishina H, Hazeki O, Katada T. J Biochem. 1998;124:934–939. doi: 10.1093/oxfordjournals.jbchem.a022210. [DOI] [PubMed] [Google Scholar]
- 43.Castillo SS, Teegarden D. J Nutr. 2003;133:3343–3349. doi: 10.1093/jn/133.11.3343. [DOI] [PubMed] [Google Scholar]
- 44.Potapova O, Gorospe M, Bost F, Dean NM, Gaarde WA, Mercola D, Holbrook NJ. J Biol Chem. 2000;275:24767–24775. doi: 10.1074/jbc.M904591199. [DOI] [PubMed] [Google Scholar]
- 45.Bost F, McKay R, Bost M, Potapova O, Dean NM, Mercola D. Mol Cell Biol. 1999;19:1938–1949. doi: 10.1128/mcb.19.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 47.Du L, Lyle CS, Obey TB, Gaarde WA, Muir JA, Bennett BL, Chambers TC. J Biol Chem. 2004;279:11957–11966. doi: 10.1074/jbc.M304935200. [DOI] [PubMed] [Google Scholar]
- 48.Jacobs-Helber SM, Sawyer ST. Blood. 2004;104:696–703. doi: 10.1182/blood-2003-05-1754. [DOI] [PubMed] [Google Scholar]