

Abstract
The present article reports three clinical cases in order to elucidate the diversity of the pathophysiological mechanisms that underlie rheumatoid arthritis associated pulmonary hypertension. The condition's three major causes are: interstitial lung disease, vasculitis, and chronic thromboembolic disease, but it should be noted that the multiple pulmonary manifestations of rheumatoid arthritis, can all contribute to chronic lung disease or hypoxia. The first patient in this report suffered from moderate restriction due to fibrosis and was diagnosed with pulmonary hypertension during an episode of life threatening hypoxia. Early upfront combination therapy prevented intubation and reversed hypoxia to adequate levels. The second presented patient was a case of isolated pulmonary hypertension attributable to vasculopathy. The patient maintained normal lung volumes but low diffusion capacity and echocardiography dictated the need for right heart catheterization. Finally, the third patient presented severe functional limitation due to several manifestations of rheumatoid arthritis, but a past episode of acute pulmonary embolism was also reported although it had never been evaluated. Chronic thromboembolic disease was eventually proved to be one major cause of the patient's pulmonary hypertension. The importance of early identification of pulmonary hypertension in patients with rheumatoid arthritis is therefore emphasized, especially since multiple treatment options are available, symptoms can be treated, and right heart failure can be avoided.
Keywords: Rheumatoid arthritis, Pulmonary hypertension, Pulmonary vasodilators
Abbreviations: PH, Pulmonary hypertension; RA, rheumatoid arthritis; mPAP, mean pulmonary artery pressure; PAWP, pulmonary arterial wedge pressure; RICU, Respiratory Intensive Care Unit; DLCO, diffusing capacity for carbon monoxide; 6MWT, 6-min walking test; RVSP, right ventricular systolic pressure; CTD, connective tissue disease
1. Introduction
Pulmonary hypertension (PH) is a continuously deteriorating disease of the pulmonary vasculature, directly affecting quality of life and leading to right heart failure. Moreover, PH can appear as a complication of connective tissue diseases [1], resulting from autoimmunity, inflammation, and vascular changes, increasing the morbidity and mortality of the primary illness. In this article, we present three clinical cases in order to discuss the correlation between PH and rheumatoid arthritis (RA), a correlation not so frequent, albeit problematic in its early identification and management. Currently the prevalence of PH in RA can only be grossly estimated by echocardiographic data, and according to several studies, it ranges from 0.8% to 21–27.5% [2], [3]. Echocardiography is an important screening tool. However, the gold-standard diagnostic tool of PH remains right heart catheterization. Resting mean pulmonary artery pressure (mPAP) of at least 25 mm Hg and a pulmonary arterial wedge pressure (PAWP) of less than 15 mm Hg determines the diagnosis. We present three cases aimed to illustrate the diversity of the pathophysiological mechanisms that underlie RA associated PH and to emphasize the importance of a thorough evaluation. Early identification is crucial, especially since multiple treatment options are available, symptoms can be treated, and right heart failure can be avoided.
2. Case reports
2.1. Case 1
In September 2015, a 60 year-old woman with known history of rheumatoid arthritis (rheumatoid factor >20, cyclic citrullinated peptide antibodies >300, elevated c-reactive protein and erythrocyte sedimentation rate, synovitis in 8 small joints) and co-existing pulmonary fibrosis with chronic type 1 respiratory failure was urgently transferred to the Respiratory Intensive Care Unit (RICU) due to life-threatening hypoxia. Assessment of vital signs revealed a respiratory rate of 35 breaths/min, a heart rate of 110 bpm and a saturation of 85%, receiving maximal flow of oxygen through a non-rebreather mask. Blood gas analysis showed pH of 7.47, pO2 of 50 mm Hg, and PCO2 of 36 mm Hg. Physical examination revealed crackles on both lungs, a prominent pulmonic compound of the second heart sound, and leg edema. Electrocardiogram showed right axis deviation, pulmonary P waves, and rSR′ in V1. Hematological and biochemical tests showed: leukocytosis (16.02 k/μL −80% neutro), Hct at 49.9%, Hb of 16.6 g/dL, normal procalcitonin but elevated C-reactive protein of 3.250 mg/dl, normal troponin, and elevated b-type natriuretic peptide levels. Heart echocardiography revealed right heart enlargement, tricuspid valve regurgitation, and an estimated right ventricular systolic pressure (RVSP) of 68 mm Hg. The left heart maintained normal dimensions and systolic function, but the intraventricular septum was flattened forming a D-shaped left ventricle. The patient underwent pulmonary function tests indicating moderate restrictive lung disease with a FVC of 63% pred, TLC of 50.6% pred and a diffusing capacity for carbon monoxide (DLCO) of 19% pred. Thoracic high resolution CT revealed modest honeycombing, ground-glass opacities with fibrotic compound, traction bronchiectasis, and thickening of interlobular septa (illustrated in Image 1). A ventilation perfusion scan was urgently performed and showed normal perfusion. A right heart catheterization confirmed precapillary pulmonary hypertension [PAP (s/d/m):74/35/51 mm Hg, PAWP: 12 mm Hg, Right Atrial Pressure: 11 mm Hg, Cardiac Index: 1.6 L/min/m2, SvO2: 64%, PVR: 13 Wood units, SaO2: 89%]. Early upfront combination therapy was planned for the patient and she was immediately treated with inhaled iloprost six times a day and tb sildenafil 20 mg tqd combined with her previous treatment of iv furosemide and per os methylprednisolone. Her oxygenation status gradually improved. By Day 10, she was able to maintain a pO2 of 58 mm Hg with 5 lt/min through nasal cannula. She was discharged on Day 15 with further improvement of oxygenation. At the one-year follow up she was stable under oxygen therapy in functional class NYHA III [New York Heart Association class III].
Image 1.

Chest X-ray (right) showing right atrium enlargement and chest CT-scan (left) showing modest honeycombing, ground-glass opacities with fibrotic compound, traction bronchiectasis, and thickening of interlobular septa.
2.2. Case 2
In 2008, a 68-year old patient was presented with dyspnea on exertion and type 1 respiratory failure. The patient had seropositive rheumatoid arthritis (rheumatoid factor: 787, cyclic citrullinated peptide antibodies > 300, elevated c-reactive protein and erythrocyte sedimentation rate) with erosive arthritis in both upper (metacarpophalangeal joints, wrists, elbows) and lower extremities (metatarsophalangeal joints) and pulmonary fibrosis. The patient had received previous treatment with methotrexate plus methylprednisolone, leflunamide, cyclosporine, and abadacept, all unable to control his arthritis (DAS28 score = 5.92, high disease activity) and was treated with azathioprine and methylprednisolone at the time of presentation. He underwent chest X-ray showed enlargement of proximal pulmonary arteries. Lung function tests revealed slightly diminished total lung capacity (5.8 lt, 82.3% pred) and a greatly impaired DLCO (55%). The patient could only walk 150 m with desaturation of 10% in a 6-min walking test (6MWT). Chest CT revealed a reticulonodular pattern with honeycombing and mild lymphadenopathy. On echocardiography, the estimated RVSP was 105 mm Hg and there were also signs of left ventricular diastolic dysfunction. Right heart catheterization was performed after excluding acute pulmonary embolism and chronic thromboembolic disease. The measured mPAP was 63 mm Hg and the PAWP was 10 mm Hg. The patient started treatment with oxygen, ambrisentan, furosemide, and spironolactone, resulting in the improvement of symptoms, and exercise capacity. Seven years later he complained about deterioration in dyspnea on exertion. A chest CT scan revealed similar fibrotic lesions with preserved pulmonary volumes (TLC = 5.4 lt, 78.3% pred), while DLCO was further diminished (34.2% pred). He could only walk 70 m due to severe hypoxia. Echocardiography revealed an estimated RVSP of 120 mm Hg and a CT angiography was negative for pulmonary embolism. Deterioration was attributed to the progression of vasculopathy and therapy was intensified, by adding tadalafil and inhaled iloprost to the previous ambrisentan monotherapy. The patient was gradually stabilized with less dyspnea on exertion, smaller degree of desaturation, and improved functional capacity.
2.3. Case 3
The third case involves an 80-year old woman, ex-smoker, with seronegative rheumatoid arthritis (negative rheumatoid factor and cyclic citrullinated peptide antibodies, elevated c-reactive protein, synovitis in 10 small joints and 6 large joints) treated with azathioprine, and prednisolone, presented with dyspnea on exertion, and severe functional limitation. The patient has previously received various disease modifying antirheumatic drugs such as methotrexate, leflunomide and infliximab, which did not control effectively joint disease. The patient also reported treatment with acenocoumarol and long term oxygen therapy for five years after an episode of pulmonary embolism that was not further evaluated. On clinical examination, crackles in basal lung fields, leg edema, and kyphoscoliosis were identified. Blood gas analysis on 5 lt O2/min revealed a pO2 of 54 mm Hg. She could not walk more than 25 m in a 6MWT due to extreme dyspnea. Lung function tests revealed a mild obstructive-restrictive impairment and a moderately diminished DLCO. An echocardiogram was next performed, revealing diastolic dysfunction along with an elevated estimated RVSP of 85 mm Hg. On high resolution CT, ground glass lesions, mosaic pattern, and atelectasis were visible (as illustrated in Image 2), whereas on a CT-angiography there is no evidence of pulmonary embolism. Intravenous diuresis was initiated and slight improvement was observed. Due to the patient's history of pulmonary embolism and the mosaic pattern in HRCT, chronic thromboembolic pulmonary disease was suspected. A triplex sonogram of lower limb veins revealed indeed an old thrombus and post-thrombotic lesions, whereas a ventilation perfusion scan confirmed the diagnosis of chronic thromboembolic disease as it revealed multiple perfusion defects. Thrombophilia testing identified an elevated activated protein C resistance value. Finally, right heart catheterization with pulmonary angiography were performed, confirming pulmonary hypertension [mPAP: 38 mm Hg, PAWP: 12 mm Hg, PVR: 4.5 Wood units, CI: 3.4 L/min/m2, and SvO2: 58%]. The patient was judged inoperable due to distal disease and significant comorbidities. Medical treatment with riociguat was initiated. Her functional status is now NYHA II, she can walk more than 250 m in a 6MWT, and plasma BNP levels are reduced.
Image 2.
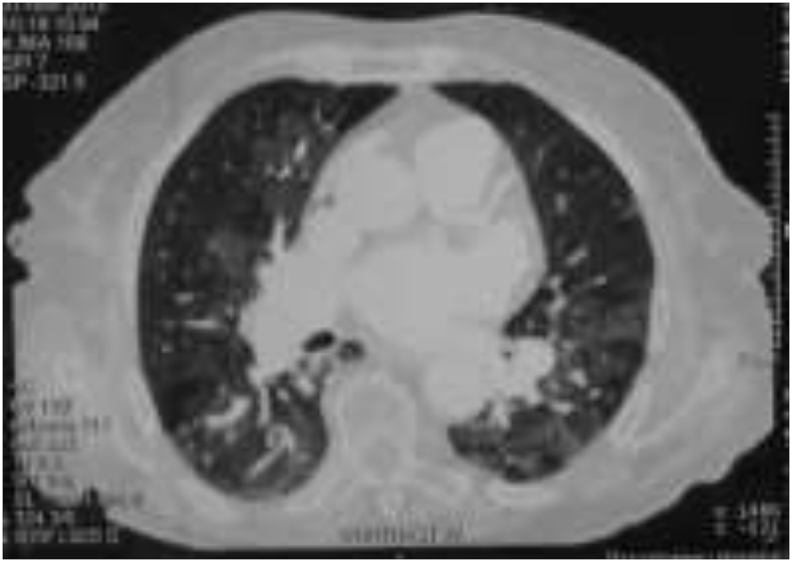
HRCT: ground glass lesions, mosaic pattern, and atelectasis.
3. Discussion
Prevalence of PAH in patients with CTDs as reported in several studies can range from 2.8% to 32%. The prevalence in patients with systemic sclerosis fluctuates from 3.6% to 32%. An important issue seems to be the inflation of PAH prevalence estimate when echocardiography is used for the diagnosis. In studies for systemic sclerosis based on right heart catheterization, PAH prevalence is 8.2%, whereas in studies based on echocardiography the prevalence estimate is 18%. In systemic lupus erythematosus patients and rheumatoid arthritis patients, echocardiographic data are only available and the prevalence ranges from 2.8% to 4.2% and from 21% to 27.5%, respectively [3]. It should be emphasized that the diagnosis of rheumatoid arthritis can occur in overlap with other connective tissue disease, such as scleroderma where the diagnosis of PAH is much more common. However, in this report we carefully selected patients with a definite diagnosis of RA according to the 2010 ACR/EULAR Classification Criteria for Rheumatoid Arthritis.
Rheumatoid arthritis can provoke several pulmonary manifestations. Parenchymal disease appears in 40–62% of cases as usual interstitial pneumonia [4], [5], but can also appear as nonspecific interstitial pneumonia, acute interstitial pneumonia or diffuse alveolar damage and organizing pneumonia [6]. Pleural disease is also common, involving pleural effusion, pneumothorax, bronchopleural fistula, and trapped lung syndrome. Another manifestation can be airway obstruction as the result of cricoarytenoid arthritis, bronchiectasis, follicular bronchiolitis, and obliterative bronchiolitis. Apart from parenchymal lesions, vascular disease can also appear, including pulmonary hypertension, and rheumatoid vasculitis.
Furthermore, the risk of venous thromboembolism appears to be increased probably due to prothrombotic effects of chronic inflammation [7], [8]. This diversity of manifestations complicates clinical presentation and respiratory symptoms are often masked as patients diminish their activity due to joint disease [6].
Treatment options include vasodilators of the pulmonary circulation targeting the reduction of pulmonary vascular resistance and augmentation of cardiac output and of mixed venous blood saturation. However, treatment with vasoactive agents in patients with lung fibrosis carries the risk of deteriorating hypoxemia due to the inhibition of hypoxic vasoconstriction in low ventilation/perfusion lung units. Frequent monitoring of vital signs and saturation is of great significance when a vasodilator is introduced to a patient's treatment. Furthermore, selective pulmonary vasodilation by inhalation of the vasoactive agent is an appealing option in order to circumvent the risk of aggravating ventilation/perfusion mismatch, which appears with systemic vasodilatory therapy.
The aforementioned data describe our strategy in the first case presented. For this critical PH patient, upfront combination therapy with inhaled iloprost and per os sildenafil was decided due to the presence of severe right ventricular dysfunction. This case was challenging, due to concomitant lung fibrosis. Discrimination between Pulmonary Arterial Hypertension associated with connective tissue disease (group 1) and PH caused by lung disease (group 3) was difficult. Specific drug therapy is highly indicated for group 1 patients. On the other hand, group 3 patients may benefit from targeted therapy, yet there is no evidence for long term effects and, thus, treatment is not approved. Some evidence exists for sildenafil use in idiopathic pulmonary fibrosis. This phosphodiesterase-5 inhibitor preferentially vasodilates well-ventilated lung units, and appears to ameliorate gas exchange and dyspnea in these patients [9].
The case was also a strong reminder of the importance of early identification of pulmonary hypertension as the patient received her diagnosis inside the RICU during severe respiratory failure. Immediate upfront combination therapy prevented the need for mechanical ventilation and its possible deleterious hemodynamic and infectious complications.
Pulmonary hypertension occurs usually within the context of interstitial lung disease, but cases of isolated pulmonary hypertension due to vasculopathy have also been reported [10]. The second case was an example of isolated PH in the absence of severe interstitial disease, classified as Pulmonary Arterial Hypertension (group 1). Pulmonary function tests of the patient only revealed severe impairment of the DLCO with preserved lung volumes. TLC values could not justify his severe pulmonary hypertension. Vasculopathy was rapidly progressing, resulting in severe functional impairment. This patient has also undergone several changes in his immunosuppressive therapy due to severe progression of erosive arthritis. The effect of immunosuppressive therapy for rheumatoid arthritis on the evolution of pulmonary hypertension appears controversial in the current literature [11], [12]. In general, they have a beneficial effect in controlling lung involvement in CTD, but none of them carries a specific indication for CTD-interstitial lung disease, and their effectiveness has not been properly studied [13].
The third case was significantly more complex. Rheumatoid arthritis, left heart diastolic dysfunction, kyphoscoliosis, and prior smoking history can justify functional impairment and CT findings. Indeed, they were attributed to the cumulative effect of multiple causes and further evaluation was not provided to an old, yet active, patient. The diagnosis of chronic thromboembolic disease is often delayed, as a discrete acute episode of pulmonary embolism is not clinically apparent in up to 62% of patients with proximal disease and 49%of those with distal disease. Speculation of such diagnosis is low, particularly, in a non specialized center [14].
4. Conclusions
Rheumatoid arthritis associated pulmonary hypertension can be attributed to interstitial lung disease, vascular disease, and chronic thromboembolic disease. Chronic inflammation reduces patient's functional capacity and conceals early symptoms of cardiovascular disease and pulmonary hypertension. Clinicians should suspect PH and pursue further diagnostic testing when a) symptoms are disproportionate to the severity of parenchymal lung disease, b) exercise limitation is disproportionate to lung volume abnormalities, and c) prominent arterial oxygen desaturation occurs during exercise. RHC is the gold standard method for the diagnosis of PH. Early diagnosis and treatment prevents right heart failure and significantly enhances quality of life.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- 1.Shahane A. Pulmonary hypertension in rheumatic diseases: epidemiology and pathogenesis. Rheumatol. Int. 2013;33(7):1655–1667. doi: 10.1007/s00296-012-2659-y. [DOI] [PubMed] [Google Scholar]
- 2.Dawson J.K., Goodson N.G., Graham D.R., Lynch M.P. Raised pulmonary artery pressures measured with Doppler echocardiography in rheumatoid arthritis patients. Rheumatology. 2000;39(12):1320–1325. doi: 10.1093/rheumatology/39.12.1320. [DOI] [PubMed] [Google Scholar]
- 3.Yang X., Mardekian J., Sanders K.N., Mychaskiw M.A., Thomas J. Prevalence of pulmonary arterial hypertension in patients with connective tissue diseases: a systematic review of the literature. ClinRheumatol. 2013;32:1519. doi: 10.1007/s10067-013-2307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lauretis A., Veeraraghavan S., Renzoni E. Review series: aspects of interstitial lung disease: connective tissue disease-associated interstitial lung disease: how does it differ from IPF? How should the clinical approach differ? Chron. Respir. Dis. 2011;8:53–82. doi: 10.1177/1479972310393758. [DOI] [PubMed] [Google Scholar]
- 5.Hallowell R.W., Horton M.R. Interstitial lung disease in patients with rheumatoid arthritis: spontaneous and drug induced. Drugs. 2014;74:443–450. doi: 10.1007/s40265-014-0190-z. [DOI] [PubMed] [Google Scholar]
- 6.Shaw M., Collins B.F., Ho L.A., Raghu G. Rheumatoid arthritis-associated lung disease. Eur. Respir. Rev. 2015;24(135):1–16. doi: 10.1183/09059180.00008014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung W.S., Peng C.L., Lin C.L. Rheumatoid arthritis increases the risk of deep venous thrombosis and pulmonary thromboembolism: a nationwide cohort study. Ann. Rheum. Dis. 2014;73:7. doi: 10.1136/annrheumdis-2013-203380. [DOI] [PubMed] [Google Scholar]
- 8.Bacani A.K., Gabriel S.E., Crowson C.S. Noncardiac vascular disease in rheumatoid arthritis: increase in venous thromboembolic events? Arthritis Rheum. 2012;64:9. doi: 10.1002/art.33322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The Idiopathic Pulmonary Fibrosis Clinical Research Network A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N. Engl. J. Med. 2010;363:620–628. doi: 10.1056/NEJMoa1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahler C.M., Colleselli D. Pulmonary arterial hypertension (PAH) in connective tissue diseases. Rheumatology. 2006;45:11–13. doi: 10.1093/rheumatology/kel291. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez O., Sitbon O., Jais X., Simonneau G., Humbert M. Immunosuppressive therapy in connective tissue diseases-associated pulmonary arterial hypertension. Chest. 2006;130:182–189. doi: 10.1378/chest.130.1.182. [DOI] [PubMed] [Google Scholar]
- 12.Behr J., Ryu J.H. Pulmonary hypertension in interstitial lung disease. Eur. Respir. J. 2008;31(6):1357–1367. doi: 10.1183/09031936.00171307. [DOI] [PubMed] [Google Scholar]
- 13.Meyer K.C., Bierach J. Immunosuppressive therapy for autoimmune lung diseases. Immunol. Allergy Clin. N. Am. 2012;32:633–669. doi: 10.1016/j.iac.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Condliffe R., Kiely D.G., Gibbs J.S. Improved outcomes in medically and surgically treated chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008;177:1122–1127. doi: 10.1164/rccm.200712-1841OC. [DOI] [PubMed] [Google Scholar]