

Abstract
OBJECTIVE: The present study aimed to explore the expression profiles of circular RNAs (circRNAs) in glioblastoma multiforme (GBM) in an attempt to identify potential core genes in the pathogenesis of this tumor. METHODS: Differentially expressed circRNAs were screened between tumor tissues from five GBM patients and five normal brain samples using Illumina Hiseq. Bioinformatics analysis was used to analyze their potential function. CircBRAF was further detected in different WHO grades glioma tissues and normal brain tissues. Kaplan-Meier curves and multivariate Cox's analysis were used to analyze the association between circBRAF expression level and prognosis of glioma patients. RESULTS: A total of 1411 differentially expressed circRNAs were identified in GBM patients including 206 upregulated circRNAs and 1205 downregulated circRNAs. Differential expression of circRNAs was closely associated with the biological process and molecular function. The downregulated circRNAs were mainly associated with ErbB and Neurotrophin signaling pathways. Moreover, the expression level of circBRAF in normal brain tissues was significantly higher than that in glioma tissues (P < .001). CircBRAF was significantly lower in glioma patients with high pathological grade (WHO III & IV) than those with low grade (WHO I & II) (P < .001). Cox analysis revealed that high circBRAF expression was an independent biomarker for predicting good progression-free survival and overall survival in glioma patients (HR = 0.413, 95% CI 0.201-0.849; HR = 0.299, 95% CI 0.135-0.661; respectively). CONCLUSION: The present study identified a profile of dysregulated circRNAs in GBM. Bioinformatics analysis showed that dysregulated circRNAs might be associated with tumorigenesis and development of GBM. In addition, circBRAF could severe as a biomarker for predicting pathological grade and prognosis in glioma patients.
Introduction
Glioma is the most common primary intraparenchymal tumor of the central nervous system (CNS). It can be classified into four grades as WHO I to IV according to the WHO criteria 2007 [1], among which glioblastoma multiforme (GBM) belonging to WHO grade IV is the most aggressive form with rapid recurrence. Currently, surgery, postoperative radiotherapy, and temozolomide-based chemotherapy are the mainstay treatments for GBM. However, the highly proliferative, aggressive, and invasive nature of GBM contributes to dismal prognosis, with a mean survival of only 14.6 months [2]. To better understand and improve the therapeutic efficacy, many studies have focused on the genome mutation and transcriptome instability in GBM [3], [4], [5].
CircRNAs are a group of non–protein-coding RNAs identified from some transcribed genes about 20 years ago [6], [7], [8]. Recent studies have demonstrated their widespread distribution and substantial presence by using high-throughput sequencing and novel computational approaches [9], [10], [11], suggesting that circRNAs are a family of naturally occurring endogenous ncRNAs with single-stranded covalently closed circular molecules with neither 5′-3′ polarity nor a polyadenylated tail [11]. CircRNAs have been characterized in several tumors and cell lines [12], [13], [14], [15], [16], [17]. Cumulative studies have indicated that some circRNAs play an important role in the pathogenesis and development of cancer. Circ_001569 can promote cell proliferation and invasion of colorectal cancer by sponging miR-145 and upregulating miR-145 functional targets E2F5, BAG4, and FMNL2 [18]. Another study also found that silencing of circHIPK3 could inhibit cell proliferation of several tumor cell lines such as Huh7, HCT-116, and HeLa [14]. A recent study found that circZNF292 was positively associated with human glioma tube formation [19].
Despite the findings of the existing studies suggesting that circRNAs play an important role in the development of many tumors, the expression profile and potential function of circRNAs in glioma remain unclear. The aim of the present study was to explore the expression profiles of circRNAs in GBM in an attempt to identify potential core genes in the pathogenesis of this CNS malignant tumor and to furthermore determine the association between circRNAs and the prognosis of glioma patients.
Material and Methods
Acquisition of the Clinical Specimens
Five GBM and five normal brain specimens were obtained from patients who underwent surgery at Changzheng Hospital (Shanghai, China). GBM was diagnosed according to the 2007 WHO classification of tumors of the CNS [1]. Another 68 Chinese glioma patients (24 female and 44 male; age: 50.18 ± 15.7 years) underwent first surgical resection in Changzheng Hospital between March 2012 and October 2015. All glioma patients did not receive chemotherapy or radiotherapy before surgery. The clinical, surgical, imaging, and pathologic records of the patients were reviewed retrospectively. The pathologic diagnosis was confirmed by two experienced pathologists independently, and histopathological grade was determined according to the 2007 WHO Classification of Tumors of the Central Nervous System [1]. Tumor recurrence and progression were defined as clinical and/or imaging progression on follow-up magnetic resonance imaging findings. All the surviving patients were followed up for at least 1 year on the outpatient basis at 3-month intervals for the first 6 months, then at 6-month intervals for the next 2 years, and annually for life thereafter. The study was performed in accordance with the 1964 Helsinki Declaration and subsequent amendments or comparable ethical standards. The study protocol was approved by the Ethic Committee of Changzheng Hospital affiliated to the Second Military Medical University (Shanghai, China), and informed consent was obtained from the surviving patients or family members of those who had died.
Sequencing Library Preparation and circRNA Sequencing
Total RNA was isolated from the specimens using RNAiso Plus reagent (TaKaRa, Japan) according to the manufacturer's protocol. The NanoDrop ND-1000 spectrophotometer (Agilent Inc., USA) was used to measure RNA quantification and quality. RNA integrity and gDNA contamination were tested by denaturing agarose gel electrophoresis. Sequencing library was determined by Agilent 2100 Bioanalyzer using the Agilent DNA 1000 chip kit (Agilent, part# 5067-1504, USA). Total RNA from each sample was used to prepare the circRNA sequencing library via the following steps: 1) 5 μg total RNA was pretreated to enrich circRNA using circRNA Enrichment Kit (Cloud-seqInc, USA); 2) RNA libraries were constructed by using pretreated RNAs with TruSeq Stranded Total RNA Library Prep Kit (Illumina, USA) according to the manufacturer's instructions; 3) the libraries were controlled qualitatively and quantitatively by using the BioAnalyzer 2100 system (Agilent Technologies, Inc., USA), denatured as single-stranded DNA molecules, captured on Illumina flow cells, amplified in situ as clusters, and finally sequenced for 150 cycles on Illumina HiSeq Sequencer according to the manufacturer's instructions.
RNA-seq Data Statistics and RNA Labeling
Compared with the classical linear transcripts, circRNAs were covalently closed. Circular RNA molecules were spliced at the canonical splice site. The back-splicing reads indirectly represented the expression quality of circRNAs. After image recognition and base recognition, raw sequencing reads were acquired from Illumina HiSeq Sequencer. The total number of reads in the samples was obtained for standardization and log2 conversion, and the number of standardized reads was compared between the samples. The raw reads and the human reference genome (hg19) were compared using STAR software [20], and the Ensemble (V70) GTF file was used as the guide. CircRNAs were detected using the DCC software [21]. Altogether 59,386 circRNAs candidates were detected in these samples. CircBase database and circ2Trait disease database were used to label the circRNAs.
Identification of Differentially Expressed circRNAs
After normalization of the raw reads, circRNAs belonging to at least two samples were chosen for further data analysis. Using the normalized number of reads, significantly differentially expressed circRNAs were identified through Volcano Plot filtering. Hierarchical clustering was performed using Heatmap2 in software R, and circRNAs with a fold change more than 2.0, P value less than .05, and false discovery rate (FDR) less than 0.05 were filtered.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
To validate the accuracy of the circRNAs-seq data, qRT-PCR was performed as a confirmatory method. cDNA of the tumor and normal samples was synthesized by reverse transcription using a Primescipt RT reagent kit with gRNA Eraser according to manufacturer's protocols (TaKaRa, Japan). The differentially expressed circRNAs were measured by qRT-PCR using SYBR premix ExTaq (TaKaRa, Japan) on Applied Biosystems StepOnePlus Real-Time PCR system. The primers of randomly selected 12-pair outward-facing circRNAs were shown in Table 1. The qRT-PCR condition was set at an initial denaturation step of 10 minutes at 95°C and 95°C for 10 seconds, 60°C for 60 seconds, 95°C for 10 seconds for a total of 40 cycles, with a final step heating slowly from 60°C to 99°C. The relative expression of each circRNA was calculated relative to GAPDH.
Table 1.
Primers and DNA Sequences Used in This Study
| cirRNA | Primer Sequence (5′-3′) |
|---|---|
| hsa:circ_0000199 | F:TCATTGCTTTCAGGGCTCTT |
| R:ATAGAAACGTGTGCGGTCCT | |
| hsa:circ_0002968 | F:GGAGCTCATGGATGCAAATC |
| R:TTGTTGTCACGCTTGCTTCT | |
| hsa:circ_0005603 | F:ATCAGTGGACAGCTTGTGGA |
| R:TCCTGATGACTCAATGCTGTG | |
| hsa:circ_0008345 | F:GGCCCAGAAGAAGGAATCAG |
| R:GCAATTTCCTGGTCCACTTG | |
| chr4:103,501,692–103,504,114+ | F:TGGAGTCTGGGAAGGATTTG |
| R:ATTTCCTCCCCTCCAGTCAC | |
| hsa:circ_0003586 | F:CCTTAACTATCCAAAAGCCAAA |
| R:TCAATATTTCCCGCAACCAC | |
| hsa:circ_0004872 | F:TGCAGATCCAGACCATGATCACA |
| R:TTCTCATGTCTGAAGCGCAGT | |
| hsa:circ_0006411 | F:TCTCGCCTCCCACACCAAA |
| R:TCCACACACGAGCTTCACC | |
| hsa:circ_0001566 | F:CTTTGTGGTATTAAACATCTGC |
| R:TCACATTTACTGTCGCTCA | |
| circBRAF | F:CTCCAGCTTGTATCACCAT |
| R:TCTTCATCTGCTGGTCGGAA | |
| chr19:5,761,476–5,761,630+ | F:TCACAGTTCCTCAGGCTTAACA |
| R:ATACACCATCCAGCGCCTTC | |
| chr18:107,887–108,499- | F:GCCCCTTTAGGCAGAGCTTA |
| R:CTACAGGGGGCTTGTGACAT | |
| BRAF | F:GAAGACCTCACAGTAAAAATAGGTGA |
| R: CCACAAAATGGATCCAGACA |
Bioinformatics Analysis of Differentially Expressed circRNAs
CircRNAs were generated from intron-derived RNAs and back-spliced exons. The parent linear mRNAs of differentially expressed circRNAs were studied with Gene Ontology (GO) (http://www.geneontology.org) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome,jp/kegg) databases. Based on GO and KEGG pathway analysis, differentially expressed circRNAs were annotated and their functions were presumpted.
Interactions between circRNAs and miRNAs
Interactions between differentially expressed circRNAs and miRNAs were investigated by using miRanda and Target Scan databases for predicting target miRNAs, whereby circRNAs that may function as miRNA sponges could be inferred. The network between circRNAs and miRNAs was also constructed by Cytoscape based on the binding sites of the differentially expressed circRNAs and miRNAs.
Validation of circBRAF Expression in Glioma Patients
GO and pathway analysis showed that circBRAF located in chr7:140,476,711-140,494,267 was a potential core gene that might be related to tumorigenesis of glioma. The relative expression of circBRAF and BRAF mRNA was validated by qRT-PCR in the five GBM and five normal brain samples. The relative expression of circBRAF was validated in 68 glioma species with different grades and 13 normal brain species by qRT-PCR as described above. Also, the association of circBRAF with prognosis of glioma patients was further evaluated.
Statistical Analysis
Chi-square test or Fisher's exact test was used to analyze relevant factors. The Kaplan-Meier method was used to estimate the survival probability. The difference in survival probability between groups was analyzed by log-rank tests. Multivariate analysis of prognosis factors was performed using a Cox regression analysis. For the stability of the equation, the histology grade was not included in the multivariate analysis. All statistical analyses were performed in SPSS 20.0 (SPSS, Inc., Chicago, IL). P < .05 was considered statistically significant.
Results
Differential Expression of circRNAs in Tumor Tissues from Glioma Patients
CircRNAs were sequenced in five GBM and five normal brain samples. The circRNA-seq reads of each sample are shown in Table 2. There were 12,048 circRNAs overlapped when compared to Rybak-Wolf's research on human brain [22]. Hierarchical clustering analysis clearly screened the differentially expressed circRNAs between these two groups (Figure 1). As shown in Figure 2A, a total of 1411 differentially expressed circRNAs (fold change ≧ 2.0, P < .05, FDR < 0.05) were identified in glioma patients by Volcano Plot, including 206 upregulated circRNAs and 1205 downregulated circRNAs. Scatter plot analysis made the variation of circRNAs expressions of two groups be visualized in these plots (Figure 2B).
Table 2.
Reads Statistics
| Sample | Raw Reads | Mapped Reads | circRNA Number |
|---|---|---|---|
| N1 | 63,504,910 | 54,253,992 | 6928 |
| N2 | 95,430,420 | 80,774,288 | 9112 |
| N3 | 97,418,358 | 80,320,270 | 7839 |
| N4 | 116,474,654 | 77,690,582 | 7418 |
| N5 | 73,894,064 | 48,289,650 | 7496 |
| T1 | 109,894,058 | 92,221,972 | 23,113 |
| T2 | 40,432,568 | 24,233,908 | 3139 |
| T3 | 66,533,768 | 53,711,986 | 9178 |
| T4 | 60,880,950 | 43,383,662 | 2733 |
| T5 | 65,515,164 | 41,651,528 | 4507 |
Figure 1.
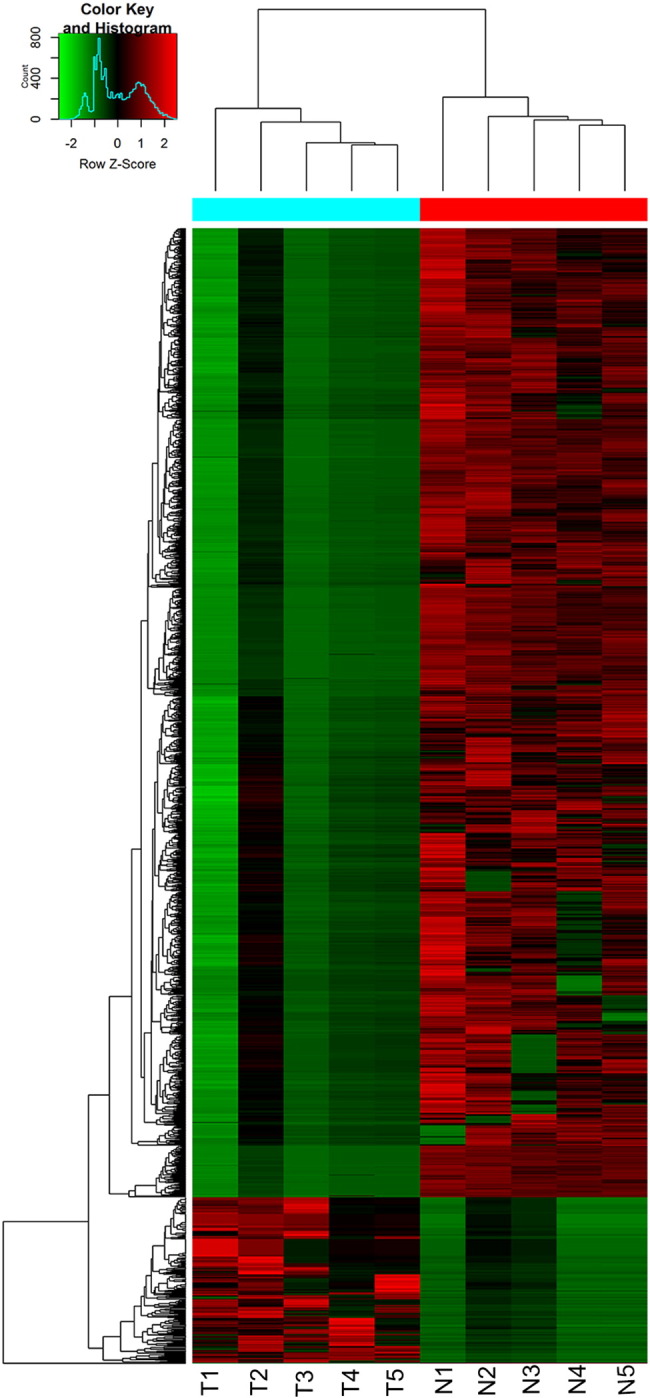
Hierarchical cluster analysis of circRNAs; unique circRNA expression profile of the 10 samples in red (upregulated expression) or green (downregulated expression) scale. The dendrogram shows the relationships among the expression levels of samples.
Figure 2.

Volcano plot filtering out the differentially expressed circRNA (red frame represented differentially expressed circRNA with fold change > 2, P < .05, FDR < 0.05 (A); scatter plot (red dots and blue dots represented highly abundant circRNAs and low-abundance circRNAs) (B); good correlation of results from circRNA-seq with RT-PCR (12 circRNAs) (C); GO annotation analysis of downregulated circRNAs with top 10 enrichment score (D) and molecular functions (E); GO annotation analysis of upregulated circRNAs with top 10 enrichment score (F) and molecular functions (G); KEGG pathway enrichment analysis of downregulated circRNAs with top 10 enrichment score (H).
Validation of the Accuracy of circRNA-seq Data by qRT-PCR
To verify the differential expression of the candidate circRNA, qRT-PCR was conducted in five GBM and five normal samples. We selected 12circRNAs (hsa:circ_0000199, hsa:circ_0002968, hsa:circ_0005603, hsa:circ_0008345, chr4:103,501,692–103,504,114+, hsa:circ_0003586, hsa:circ_0004872, hsa:circ_0006411, hsa:circ_0001566, chr19:5,761,476-5,761,630+, chr18:107,887-108,499−, circBRAF) according to the results of RNA sequencing. As shown in Figure 2C, the results obtained from RT-PCR were consistent with RNA sequencing data.
GO Analysis and KEGG Pathway Analysis of Differentially Expressed circRNA
GO analysis showed that down-regulated circRNAs were in favor of the enzyme binding, transferees activity, protein binding, organelle organization, and protein modification processes (Figure 2, D and E). As for upregulated circRNAs, the closely related GO terms were fertilization, G0 to G1 transition, manganese ion transport, sperm capacitation, store-operated calcium channel activity, and actin-dependent ATPase activity (Figure 2, F and G). KEGG pathway analysis showed no enrichment pathway in the up-regulated circRNAs, but 70 pathways were identified to be associated with downregulated circRNAs. And ErbB signaling pathway and Neurotrophin signaling pathway were the top enrichment pathways in downregulated circRNAs (Figure 2H).
Identification of circRNA-miRNA Network
Bioinformatics analysis showed that some circRNAs functioned as miRNA sponges in mammal cells. As mentioned above, miRanda and Target Scan databases were used to predict miRNAs binding sites in circRNAs. Based on the differentially expressed circRNAs and predicted miRNA binding sites, 56 differentially expressed circRNAs and 154 predicted miRNAs were used to construct the circRNA-miRNA network (Figure 3).
Figure 3.

circRNA-miRNA network of 56 differentially expressed circRNAs and 154 predicted miRNAs.
Expression Level of circBRAF in Glioma Patients
The demographic feature and clinical characteristics of 68 glioma patients were presented in Table 3. Forty-six cases were high-grade (WHO III, 12; WHO IV, 34), while 22 cases were low-grade glioma (WHO I, 6; WHO II, 16). The circRNA-seq data showed that circBRAF derived from BRAF gene exon 8-13 was significantly downregulated in tumor group. The distinct product of the expected size was amplified using outward-facing primers and confirmed by Sanger sequencing (Figure 4A). There were no significantly differences between circBRAF and BRAF mRNA expression in normal brain. However, circBRAF was significantly downregulated in GBM tissues than BRAF mRNA as indicated by qRT-PCR analysis (Figure 4B). Then, circBRAF was detected in 13 normal brain tissue specimens and 68 glioma specimens of different grades. The results showed that the relative expression level of circBRAF in the glioma tissues was significantly lower than normal brain tissues, and the relative expression level of circBRAF in high-grade glioma specimens was significantly lower than low-grade glioma specimens (P < .001) (Figure 4C). The correlations between circBRAF expression and other prognostic factors were shown in Table 4. The Kaplan-Meier analysis revealed a survival difference between high–circBRAF expression group and low–circBRAF expression group, suggesting that glioma patients with high circBRAF expression had significantly better progression-free survival (PFS) and overall survival (OS) than those with low circBRAF expression (P < .001 for both; Figure 4, D and E). Univariate analysis showed that high circBRAF expression, younger age, longer duration of symptoms, high preoperative Karnofsky Performance Status (KPS), low histologic grades, and single lobe lesion were closely associated with the longer PFS and OS (Table 5). In addition, multivariate Cox analysis revealed that high circBRAF expression was an independent factor for good PFS and OS in glioma patients (HR = 0.413, 95% CI 0.201-0.849; HR = 0.299, 95% CI 0.135-0.661; respectively) (Table 6). Taken together, the high expression of circBRAF represented a lower grade of glioma and a poorer prognosis.
Table 3.
Population Characteristics
| Age | |
|---|---|
| Min-max | 9-79 |
| Mean (years ± SD) | 50.18 ± 15.7 |
| Sex | |
| Male, n (%) | 44 (64.7%) |
| Female, n (%) | 24 (35.3%) |
| KPS score (preoperative) | |
| ≼70, n (%) | 7 (10.3%) |
| > 70, n (%) | 61 (89.7%) |
| No. of lesions | |
| Single, n (%) | 43 (63.2%) |
| Multiple, n (%) | 25 (36.8%) |
| Extent of surgery | |
| Gross total resection (GTR), n (%) | 46 (67.6%) |
| Subtotal resection (STR), n (%) | 22 (32.4%) |
| Histology grade (WHO grade) | |
| Low (I & II), n (%) | 22 (32.4%) |
| High (III & IV), n (%) | 46 (67.6%) |
| Adjuvant treatment | |
| Radiotherapy, n (%) | 50 (73.5%) |
| Chemotherapy, n (%) | 56 (82.4%) |
| Recurrence, n (%) | 39 (57.4%) |
| Death during follow-up, n (%) | 34 (50.0%) |
| Follow-up period (months) | |
| Range | 1-54 |
| Mean (months ± SD) | 20.60 ± 14.66 |
Figure 4.

The genomic loci of circBRAF in BRAF gene and the expression of circBRAF was validated by qRT-PCR followed by Sanger sequencing (arrows represent divergent primers binding to the genome region of circBRAF) (A); the relative expression of circBRAF and BRAF mRNA in five normal brain and five GBM tissues (B); the relative expression of circBRAF in different grades glioma and normal brain tissues (C); Kaplan-Meier PFS curves of glioma patients in correlation with circBRAF expression (D); Kaplan-Meier OS curves of glioma patients in correlation with circBRAF expression (E); **P value < .01.
Table 4.
Correlations between Relative Expression of circBRAF and Other Prognostic Factors
| Relative Expression of circBRAF (Number (% of Total)) |
||||
|---|---|---|---|---|
| Low | High | P Value | ||
| Sex | Male | 25 (36.8) | 19 (27.9) | .331 |
| Female | 10 (14.7) | 14 (20.6) | ||
| Age (years) | <18 | 1 (1.5) | 5 (7.4) | .011⁎ |
| 18-50 | 7 (10.3) | 14 (20.6) | ||
| ≽50 | 27 (39.7) | 14 (20.6) | ||
| Duration of symptoms (months) | <12 | 29 (42.6) | 24 (35.3) | .387 |
| ≽12 | 6 (8.8) | 9 (13.2) | ||
| KPS score | <80 | 30 (44.1) | 31 (45.6) | .429 |
| ≽80 | 5 (7.4) | 2 (2.9) | ||
| Histologic grade | Low | 1 (1.5) | 21 (30.9) | <.001⁎⁎ |
| High | 34 (50.0) | 12 (17.6) | ||
| Tumor size (cm) | <5 | 11 (16.4) | 10 (14.9) | 1.000 |
| ≽5 | 23 (34.3) | 23 (34.3) | ||
| No. of lesions | Single, n (%) | 19 (27.9) | 24 (35.3) | .137 |
| Multiple, n (%) | 16 (23.5) | 9 (13.2) | ||
| Extent of surgery | Gross total resection (GTR) | 24 (35.3) | 22 (32.4) | 1.000 |
| Subtotal resection (STR) | 11 (16.2) | 11 (16.2) | ||
| Radiotherapy | No | 10 (14.7) | 8 (11.8) | .786 |
| Yes | 25 (36.8) | 25 (36.8) | ||
| Chemotherapy | No | 5 (7.4) | 7 (10.3) | .534 |
| Yes | 30 (44.1) | 26 (38.2) | ||
| Recurrence | No | 9 (13.2) | 20 (29.4) | .007⁎ |
| Yes | 26 (38.2) | 13 (19.1) | ||
| Survival | Yes | 12 (17.6) | 22 (32.4) | .015⁎ |
| No | 23 (33.8) | 11 (16.2) | ||
Asterisks indicate statistical significance.
P ≼ .05.
P < .01.
Table 5.
Patient Characteristics and Univariate Analysis of Prognostic Factors Affecting PFS and OS.
| Factors | PFS |
OS |
|||||
|---|---|---|---|---|---|---|---|
| 1-year Rate (%) | 3-year Rate (%) | P Value | 1-year Rate (%) | 3-year Rate (%) | P Value | ||
| Sex | male/female | 56.8 vs. 54.2 | 41.8 vs. 38.7 | 0.932 | 62.1 vs. 87.0 | 33.2 vs. 56.8 | 0.079 |
| Age | ≽18/18–50/≽50(years) | 100 vs.76.2 vs.39.0 | 100 vs.47.5 vs.26.1 | 0.003⁎ | 100 vs.85.7 vs.58.5 | 100 vs.57.0 vs.23.2 | 0.003⁎ |
| Duration of symptoms | <12/≽12(months) | 49.1 vs. 80.0 | 36.1 vs. 70.0 | 0.010⁎ | 66.0 vs. 86.7 | 34.1 vs.68.9 | 0.041⁎ |
| KPS score | ≽70/<70 | 60.7 vs. 14.3 | 43.1 vs. 14.3 | 0.001⁎ | 73.1 vs. 42.9 | 46.1 vs.0 | 0.001⁎ |
| Histology grade | low/high | 95.5 vs. 37.0 | 83.9 vs. 18.8 | <0.001⁎⁎ | 100 vs. 56.5 | 88.4 vs.17.7 | <0.001⁎⁎ |
| Tumor size (cm) | <5 cm/≽5 cm | 57.1 vs. 56.5 | 51.4 vs. 35.6 | 0.420 | 71.4 vs. 71.7 | 43.7 vs. 40.4 | 0.382 |
| No. of lesions | single/multi | 65.1 vs. 40.0 | 50.8 vs. 17.5 | 0.015⁎ | 81.3 vs. 52.0 | 53.7 vs. 18.9 | 0.001⁎ |
| Extent of surgery | total/subtotal | 56.5 vs. 54.5 | 45.4 vs. 28.6 | 0.598 | 76.1 vs. 58.7 | 48.0 vs.27.5 | 0.270 |
| Radiotherapy | no/yes | 55.6 vs. 56.0 | 37.5 vs. 38.8 | 0.668 | 66.7 vs. 71.9 | 47.1 vs. 37.3 | 0.829 |
| Chemotherapy | no/yes | 58.3 vs. 55.4 | 50.0 vs. 36.3 | 0.694 | 75.0 vs. 69.6 | 54.7 vs. 37.3 | 0.634 |
| Relative expression of circBRAF | low/high | 34.3 vs. 78.8 | 21.9 vs. 58.9 | <0.001⁎⁎ | 51.4 vs. 74.1 | 23.7 vs. 59.7 | <0.001⁎⁎ |
Asterisks indicates statistical significance.
P ≼ 0.05.
P<0.01.
Table 6.
Multivariate Analysis of Prognostic Factors for PFS and OS
| Factors | FPS |
OS |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Value | HR | 95% CI | P Value | |
| Age | 2.203 | 1.143-4.244 | .018⁎ | 2.382 | 1.157-4.905 | .019⁎ |
| Duration of symptoms | – | – | .006⁎ | – | – | .82 |
| KPS score | 2.623 | 1.060-6.490 | .037⁎ | – | – | .092 |
| No. of lesions | – | – | .078 | 2.878 | 1.396-5.936 | .004⁎ |
| Relative expression of circBRAF | 0.413 | 0.201-0.849 | .016⁎ | 0.299 | 0.135-0.661 | .003⁎ |
Asterisks indicate statistical significance.
P ≼ .05.
Discussion
CircRNA, emerging as important regulator of tumorigenesis, has been found to be associated with the development of several different type of cancers. It has been established that dysregulation of circRNA could contribute to the initiation and promotion of tumors. To clarify the potential role of circRNA in pathogenesis of glioma, we identified circRNA expression signature in tumor tissue from GBM patients and conducted the function analysis. In the present study, a total of 1411 differentially expressed circRNAs were observed in tumor tissues from glioma patients including 206 upregulated circRNAs and 1205 downregulated circRNAs. GO analysis and KEGG pathway analysis revealed that the differentially expressed circRNAs participated in several biological processes and signaling pathways such as ErbB signaling pathway and Neurotrophin signaling pathway. Several studies have shown that BRAF plays an important role in the development and progression of cancers [23]. But few studies have reported circular transcriptomes produced from the BRAF gene. Interestingly, we found that circBRAF, one of the downregulated circRNAs, was significantly associated with pathological grade and prognosis of glioma patients.
The present study showed that there was a significant difference in circRNAs expression between the tumor and normal brain tissues and most circRNAs were downregulated in GBM tissues. In consistent with our results, several previous studies have reported that circRNAs were abundant in the brain [15], [24]. A similar phenomenon was also observed in progressive laryngeal cancer [12], breast cancer, colorectal cancer, gastric cancer, hepatocellular carcinoma, and prostate adenocarcinoma [14], [17]. CircRNAs were significantly downregulated in these tumors when compared with matched normal tissues. On the contrary, some previous reports found that circRNAs were significantly upregulated in bladder urothelial carcinoma and kidney clear cell carcinoma [14]. Although the accurate role of circRNAs in tumorigenesis and prognosis of tumors remained unclear, we speculated that the abnormal expression of circRNAs and its skewed distribution in tumors might be a widespread phenomenon.
Accumulating evidences showed that circRNAs played a key role in many biological processes, and some circRNAs are identified as RNA banding protein and mRNA magnet to directly translate protein [11], [25]. GO analysis showed that downregulated circRNAs were significantly associated with protein binding. Previous studies have found that circRNAs could bind and sequester RNA-binding proteins to form large RNA-protein complexes; these RNA-protein complexes might regulate the pool of RNA-binding proteins [26]. GO analysis also showed that of the closely GO terms related to upregulated circRNAs included G0 to G1 transition, manganese ion transport, store-operated calcium channel activity. Previous studies have found these biological processes may be involved in the development of glioma. For example, a previous study has found that cell cycle G0/G1 arrest could significantly affect glioma cell proliferation [27]. Also, some researchers have reported that Ca2+ might also be an important positive regulator of tumorigenesis in GBM in processes involving quiescence, maintenance, proliferation, or migration [28]. Based on our results, we hypothesized that upregulated circRNAs may regulate the G0/G1 transition or participate in the tumorigenesis of glioma by modulating Ca2+transport. These require investigators to confirm in the future research. KEGG pathway analysis showed that ErbB and neurotrophin signaling pathways were the top enrichment pathways in downregulated circRNAs. Several studies have showed that the ErbB signaling pathway was evolutionarily conserved [29], [30]. Blockade of ErbB signaling could inhibit the slow cycling JARID1B-positive cell population, which is critical for long-term maintenance of melanoma growth [30]. The ErbB signaling pathway could participate in metastasis of prostate cancer via regulation on epithelial-mesenchymal transition and stemness of prostate cancer cells [29]. That is to say, the ErbB signaling pathway may be closely related to proliferation, cell cycle, and differentiation of cancer cells. Currently, there are no studies focusing on the role of circRNAs in the ErbB pathway. The relationship between circRNAs and ErbB pathway needs to be further investigated. In most cases, neurotrophins have been shown to favor tumor development and progression. In glioma, neurotropins (NGF, BDNF, and NT-3) and their receptors (TrkA, TrkBTekC, and P75NTR) were detected in several lines of brain tumor–initiating cells. Furthermore, NGF, BDNF, and NT-3 were able to stimulate the proliferation of these tumor cells [31]. According to the results of KEGG analysis, downregulated circRNAs may be involved in the pathogenesis of glioma through regulation of neurotropins signaling pathway.
Recent functional research found that circRNAs could harbor miRNAs and function as miRNA sponges [32]. For instance, circCDR1as could harbor over 70 conserved binding sites for miR-7 and acts as a sponge for miR-7 to regulate the expression of human epidermal growth factor receptor, α-synuclein, and insulin receptor substrate-2 [32]. The murine circRNA circSry might harbor more than 10 miR-138 binding sites [13], [32]. A recent study showed that circHIPK3 could sponge 9 miRNAs with 18 potential binding sites, and it also could directly bind to miR-124 and inhibit miR-124 activity [14]. To date, few studies have focused on the interaction of circRNAs and miRNAs in GBM. Here, we established a circRNA-miRNA network of GBM based on our RNA-seq data. The circRNA-miRNA network was able to help us further understand the role of circRNA in the tumorigenesis of glioma patients.
Previous studies found that BRAF played an important role in numerous human tumors. BRAF gene could participate in various pathways including ErbB signaling pathway, MAPK signaling pathway, neurotrophin, and mTOR signaling pathway [30], [33]. Our results showed that circBRAF derived from BRAF was significantly downregulated in GBM tumor tissue compared with normal brain tissue. Based on the extended experiment of the relative expression of circBRAF in different grades of glioma, we found that the relative expression level of circBRAF was lower in glioma patients with high pathologic grade than those with low grade. In addition, we evaluated the association of circBRAF expression with prognosis of glioma patients. Univariate and multivariate analyses showed that high circBRAF expression was significantly associated with good PFS and OS in glioma patients. Although the special function and mechanisms of circBRAF were unclear, we believe that circBRAF may serve as a biological biomarker for diagnosis or prognosis in glioma.
In conclusion, the present study identified a profile of dysregulated circRNAs in GBM. Bioinformatics analysis showed that dysregulated circRNAs might be associated with tumorigenesis and development of GBM and circBRAF could severe as a biomarker for predicting pathological grade and prognosis in glioma patients. The present study could widen the horizon of gene research in glioma and lay a preliminary foundation for further research of circRNAs in glioma.
Conflicts of Interest
None.
Authors' Contributions
Junle Zhu, Jingliang Ye, and Lei Zhang designed and carried out the experiments, and wrote the manuscript. Junle Zhu and Jingliang Ye analyzed the sequencing data and developed analysis tools. Lili Xia, Hongkang Hu, and Heng Jiang analyzed experimental results. Zhiping Wan, Fei Sheng and Yan Ma assisted in obtaining samples and collected clinical information. Wen Li, Jun Qian and Chun Luo reviewed and edited the manuscript. All authors read and approved the manuscript.
Footnotes
Funding: This study was supported by grants from the National Key Basic Research Program (973 project) (2015CB554004) from the National Natural Science Foundation (81272213) of China and the Ministry of Science and Technology of China.
Contributor Information
Jun Qian, Email: qianjun19@126.com.
Chun Luo, Email: boyluochun@126.com.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koh Y, Park I, Sun C-H, Lee S, Yun H, Park C-K, Park S-H, Park J, Lee S-H. Detection of a distinctive genomic signature in rhabdoid glioblastoma, a rare disease entity identified by whole exome sequencing and whole transcriptome sequencing. Transl Oncol. 2015;8:279–287. doi: 10.1016/j.tranon.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch CJ, Lustig RA, Yang X-Y, Jenkins WT, Wolf RL, Martinez-Lage M, Desai A, Williams D, Evans S. Microvesicles as a biomarker for tumor progression versus treatment effect in radiation_temozomide-treated glioblastoma patients. Transl Oncol. 2014;7:752–758. doi: 10.1016/j.tranon.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myung JK, Hj C, Kim H, Park C-K, Lee SH, Choi SH, Park P, Yoon J, Park S-H. Prognosis of glioblastoma with oligodendroglioma component is associated with the IDH1 Mutation and MGMT methylation status. Transl Oncol. 2014;7:712–719. doi: 10.1016/j.tranon.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell. 1991;64:607–613. doi: 10.1016/0092-8674(91)90244-s. [DOI] [PubMed] [Google Scholar]
- 7.Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. 1993;7:155–160. doi: 10.1096/fasebj.7.1.7678559. [DOI] [PubMed] [Google Scholar]
- 8.Zaphiropoulos PG. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci U S A. 1996;93:6536–6541. doi: 10.1073/pnas.93.13.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32:453–461. doi: 10.1038/nbt.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 12.Xuan L, Qu L, Liu M, Zhou H, Wang P, Yu H, Wu T, Wang X, Li Q, Tian L. Circular RNA: a novel biomarker for progressive laryngeal cancer. Am J Transl Res. 2016;8:932–939. [PMC free article] [PubMed] [Google Scholar]
- 13.Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 16.Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777. doi: 10.1371/journal.pgen.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang M, Zhong Z, Lv M, Shu J, Tian Q, Chen J. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget. 2016 doi: 10.18632/oncotarget.9706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie H, Ren X, Xin S, Lan X, Lu G, Lin Y, Yang S, Zeng Z, Liao W, Ding YQ. Emerging roles of circRNA_001569 targeting miR-145 in the proliferation and invasion of colorectal cancer. Oncotarget. 2016;7:26680–26691. doi: 10.18632/oncotarget.8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang P, Qiu Z, Jiang Y, Dong L, Yang W, Gu C, Li G, Zhu Y. Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/beta-catenin signaling pathway. Oncotarget. 2016 doi: 10.18632/oncotarget.11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32:1094–1096. doi: 10.1093/bioinformatics/btv656. [DOI] [PubMed] [Google Scholar]
- 22.Rybak-Wolf A, Stottmeister C, Glazar P, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, Herzog M, Schreyer L, Papavasileiou P. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015:870–885. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Rizzo D, Ruggiero A, Amato M, Maurizi P, Riccardi R. BRAF and MEK inhibitors in pediatric glioma: new therapeutic strategies, new toxicities. Expert Opin Drug Metab Toxicol. 2016:1–9. doi: 10.1080/17425255.2016.1214710. [DOI] [PubMed] [Google Scholar]
- 24.Venø MT, Hansen TB, Venø ST, Clausen BH, Grebing M, Finsen B, Holm IE, Kjems J. Genome biol-spatio-temporal regulation of circular RNA expression during porcine embryonic brain. Genome Biol. 2015;16:245. doi: 10.1186/s13059-015-0801-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with Pre-mRNA splicing. Mol Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 26.Wilusz JE, Sharp PA. Molecular biology. A circuitous route to noncoding RNA. Science. 2013;340:440–441. doi: 10.1126/science.1238522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du W, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858. doi: 10.1093/nar/gkw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leclerc C, Haeich J, Aulestia FJ, Kilhoffer MC, Miller AL, Neant I, Webb SE, Schaeffer E, Junier MP, Chneiweiss H. Calcium signaling orchestrates glioblastoma development: facts and conjunctures. Biochim Biophys Acta. 2016;1863:1447–1459. doi: 10.1016/j.bbamcr.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Ren D, Guo W, Huang S, Wang Z, Li Q, Du H, Song L, Peng X. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int J Oncol. 2016;48:595–606. doi: 10.3892/ijo.2015.3270. [DOI] [PubMed] [Google Scholar]
- 30.Zhang K, Wong P, Salvaggio C, Salhi A, Osman I, Bedogni B. Synchronized targeting of notch and ERBB signaling suppresses melanoma tumor growth through inhibition of Notch1 and ERBB3. J Invest Dermatol. 2016;136:464–472. doi: 10.1016/j.jid.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forsyth PA, Krishna N, Lawn S, Valadez JG, Qu X, Fenstermacher DA, Fournier M, Potthast L, Chinnaiyan P, Gibney GT. p75 neurotrophin receptor cleavage by alpha- and gamma-secretases is required for neurotrophin-mediated proliferation of brain tumor-initiating cells. J Biol Chem. 2014;289:8067–8085. doi: 10.1074/jbc.M113.513762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 33.Kakkar A, Majumdar A, Kumar A, Tripathi M, Pathak P, Sharma MC, Suri V, Tandon V, Chandra SP, Sarkar C. Alterations in BRAF gene, and enhanced mTOR and MAPK signaling in dysembryoplastic neuroepithelial tumors (DNTs) Epilepsy Res. 2016;127:141–151. doi: 10.1016/j.eplepsyres.2016.08.028. [DOI] [PubMed] [Google Scholar]