

Abstract
Objective
To determine whether differentially methylated CpGs in synovium‐derived fibroblast‐like synoviocytes (FLS) of patients with rheumatoid arthritis (RA) were also differentially methylated in RA peripheral blood (PB) samples.
Methods
For this study, 371 genome‐wide DNA methylation profiles were measured using Illumina HumanMethylation450 BeadChips in PB samples from 63 patients with RA and 31 unaffected control subjects, specifically in the cell subsets of CD14+ monocytes, CD19+ B cells, CD4+ memory T cells, and CD4+ naive T cells.
Results
Of 5,532 hypermethylated FLS candidate CpGs, 1,056 were hypermethylated in CD4+ naive T cells from RA PB compared to control PB. In analyses of a second set of CpG candidates based on single‐nucleotide polymorphisms from a genome‐wide association study of RA, 1 significantly hypermethylated CpG in CD4+ memory T cells and 18 significant CpGs (6 hypomethylated, 12 hypermethylated) in CD4+ naive T cells were found. A prediction score based on the hypermethylated FLS candidates had an area under the curve of 0.73 for association with RA case status, which compared favorably to the association of RA with the HLA–DRB1 shared epitope risk allele and with a validated RA genetic risk score.
Conclusion
FLS‐representative DNA methylation signatures derived from the PB may prove to be valuable biomarkers for the risk of RA or for disease status.
Rheumatoid arthritis (RA) is a chronic inflammatory disease with the potential to cause substantial disability, primarily because of the erosive and deforming process that occurs in the joints. It is the most common systemic autoimmune disease, with a worldwide prevalence approaching 1% 1, 2. The etiology of RA is complex, with both genetic and nongenetic contributions. Findings from a rigorous assessment of RA heritability using twin studies suggested that 50–60% of the occurrence of RA in twins is explained by genetic effects 3. Approximately 50% of this genetic contribution can be explained by genes in the major histocompatibility complex (MHC) 3. In addition, at least 101 independent non‐MHC risk loci have been identified 4. Evidence of a role for environmental factors has also been presented, but currently, exposure to tobacco smoke is the only well‐established risk factor 5.
DNA methylation is an epigenetic modification resulting from the addition of a methyl group to a cytosine base at positions in the DNA sequence where a cytosine is followed by a guanine (“CpGs”), which can lead to altered expression of DNA. The process of DNA methylation is essential for proper mammalian development and other functions, and methylation patterns are affected by environmental changes. Methylation status is also influenced by the interaction between genetics and environment, and a growing number of human diseases have been associated with aberrant DNA methylation 6. Maintenance of DNA methylation is critical for the development and function of immune cells 6, 7.
Altered patterns of DNA methylation at CpG sites have been observed in individuals with RA. A 1990 study by Richardson et al found that global methylation of genomic DNA from T cells of RA patients was lower when compared to T cells of healthy controls 8. Altered methylation patterns have also been observed in small studies of specific genes in RA, including the promoter regions of IL6 using peripheral blood mononuclear cells (PBMCs) and DR3 (alternative name, TNFRSF25) using synovial fibroblasts 9, 10. Liu et al studied global DNA methylation among 129 Taiwanese individuals and found that those with RA had significantly lower levels of DNA methylation in PBMCs compared to healthy control PBMCs 11. Recently, Glossop et al identified ∼2,000 differentially methylated CpGs in both T lymphocytes and B lymphocytes between treatment‐naive patients with early RA and healthy individuals, and in a separate analysis, they found that DNA methylation profiles in synovial fluid–derived fibroblast‐like synoviocytes (FLS) had similarities with the profiles in tissue‐derived FLS 12, 13.
A recent investigation identified 15,220 differentially methylated CpG sites in synovium‐derived FLS between RA patients and either patients with osteoarthritis or normal controls, and these differences appear to distinguish RA cases from non‐RA controls (see ref. 14 and Rhead B: personal communication). These 15,220 FLS CpGs serve as the candidate sites for the current investigation. FLS in the synovial intimal lining of joints have key roles in the production of cytokines that perpetuate inflammation and in the production of proteases that contribute to cartilage destruction in RA 15. An overlap in the methylation pattern between FLS and peripheral blood (PB) cells could be indicative of disease‐associated biologic processes detectable in the periphery. Because samples of PB are easily accessible, such signatures may be useful biomarkers for the risk of RA or for disease status.
PATIENTS AND METHODS
Study design
Participants included 63 female patients with RA (ages ≥18 years with a diagnosis meeting the American College of Rheumatology 1987 revised criteria for RA 16, 17) and 31 female unaffected control subjects (locally based), all of whom reported having European ancestry. Table 1 summarizes the characteristics of our study population. All participants provided a PB sample for genotyping and measurement of methylation.
Table 1.
Characteristics of the study participants at the time of blood samplinga
| Patients with RA (n = 63) | Controls (n = 31) | |
|---|---|---|
| Seropositive (for RF or anti‐CCP), no. (%) | 57 (90) | – |
| Age, mean ± SD years | 56.4 ± 14.8 | 57.5 ± 16.5 |
| Smoking, no. (%) | ||
| Ever | 33 (52) | 13 (42) |
| Never | 30 (48) | 18 (58) |
| Current | 4 (6) | 1 (3) |
| Not current | 59 (94) | 30 (97) |
| Disease duration, mean ± SD years | 14.0 ± 10.5 | – |
| Erosive disease, no. (%) | ||
| Present | 39 (62) | – |
| Absent | 22 (35) | – |
| Missing | 2 (3) | – |
| Disease activity, mean ± SD CDAI | 10.1 ± 9.2 | – |
No significant differences (by Wilcoxon's rank sum test or chi‐square test) were observed between the groups. RA = rheumatoid arthritis; RF = rheumatoid factor; anti‐CCP = anti–cyclic citrullinated peptide; CDAI = Clinical Disease Activity Index (score range 0–76; data not available for 4 subjects).
Genotyping
Study participants were genotyped using Illumina HumanOmniExpress, HumanOmniExpressExome, or Human660W‐Quad BeadChips, which were read on an Illumina HiScan array scanner. Genotype results were merged using Plink software, version 1.07 18, and only single‐nucleotide polymorphisms (SNPs) that were assessed using all 3 chips were retained for analysis. SNPs with failed genotype calls in at least 10% of individuals, those with a minor allele frequency of lower than 1%, or those found to not be in Hardy‐Weinberg equilibrium (P ≤ 0.000001) in controls were removed from the analysis.
Ancestry
The EigenStrat program 19 was used to visualize ancestral clustering of the study population relative to individuals from 11 HapMap populations 20. As expected, our study population of individuals of self‐identified European ancestry clustered with the HapMap populations of Utah residents with ancestry from northern and western Europe and Tuscans in Italy. We excluded individuals with self‐reported non‐European ancestry, because of the potential for confounding (see Supplementary Figures 1–3 for the ancestral clustering of participants with self‐identified European ancestry, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract).
Cell sorting
Whole blood was collected from each subject into four 10‐ml EDTA collection tubes. PBMCs were isolated using a Ficoll‐Paque density gradient and stained with conjugated monoclonal antibodies against CD45 (fluorescein isothiocyanate conjugated), CD19 (phycoerythrin [PE] conjugated), CD45RA (PE–Cy7 conjugated) (all from BD PharMingen), CD3 (Brilliant Violet 421 conjugated), CD4 (CF594 conjugated) (both from BD Horizon), CD14 (allophycocyanin [APC] conjugated) (BD Biosciences), and CD27 (APC–eFlour 780 conjugated) (ebioscience). Cells were then stored overnight in buffer at 4°C and sorted the following day, on a BD Biosciences FACSAria fluorescence‐activated cell sorter (FACS). The following populations were gated for sorting following exclusion of debris and doublets: monocytes (CD45+CD14+), B cells (CD45+CD14 −CD3 −CD19+), naive CD4+ T cells (CD45+CD14 −CD19 −CD3+CD4+CD27+CD45RA+), and memory CD4+ T cells (CD45+CD14 −CD19 −CD3+CD4+CD45RA−). Cell counts and purity checks were performed after sorting, and then cells were stored frozen as a pellet at −80°C.
Validation of overnight cell storage
To enable DNA methylation profiling of a large number of FACS‐sorted samples, a protocol for storing blood samples overnight prior to sorting was established and validated. Whole blood was collected from a single individual into ten 10‐ml EDTA collection tubes. PBMCs were isolated and stained as described above, and then either sorted the same day or stored overnight in buffer at 4°C and sorted the following day. Paired DNA samples from the 2 time points were collected from all 4 cell types. All DNA samples were quantified using a Nanodrop spectrophotometer. All samples were subjected to bisulfite conversion on the same day and then assayed on Illumina 450k BeadChips simultaneously.
Methylation analyses
A total of 371 genome‐wide DNA methylation profiles were generated using the Illumina Infinium HumanMethylation450 BeadChip kit and read on an Illumina HiScan array scanner. A beta value, representing the ratio of the fluorescence intensity of the methylated probe to the fluorescence intensity overall (methylated plus unmethylated), was derived for each CpG site. We performed an extensive quality control process, in which Illumina GenomeStudio software was used to examine Jurkat cells as controls, to examine the between‐chip and within‐chip variations, and to evaluate replicate samples. All replicate samples had r2 values of >0.99, and Jurkat cell replicate samples showed r2 values of >0.98. Background signal was subtracted using the methylumi R package “noob” method 21, and the values in all samples were normalized with the use of the all sample mean normalization method 22, followed by beta‐mixture quantile normalization 23 to correct for type I and type II probe differences.
Multidimensional scaling (MDS) plots for each cell type, before and after background subtraction and normalization, were examined to assess for the presence of batch effects. The batch effects were found to be minimal, and were reduced following data normalization (for examples, see Supplementary Figures 4 and 5, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract). In these analyses, 286 CpG sites with low detection rates (read P > 0.05) in more than 20% of samples were removed, and 1 sample with low detection rates (read P > 0.05) in more than 20% of sites was removed. The following CpG sites were also removed from analysis: the 65 non‐CpG “rs” SNP probes included in the 450k BeadChip, the 30,969 sites with probes predicted to hybridize to more than one location in the genome after bisulfite conversion (cross‐reactive probes), as identified in a study by Chen et al 24, and the 28,355 sites with a known polymorphism at the site being measured (polymorphic CpGs), as identified by Chen et al 24, that were either present in our European‐ancestry population or present in Europeans in the 1,000 Genomes Project 24. The final data set used for analysis consisted of 428,232 CpG sites in 371 samples (94 CD14+ monocyte samples, 91 CD19+ B cell samples, 94 CD4+ memory T cell samples, and 92 CD4+ naive T cell samples).
An MDS plot of all 371 samples (Figure 1) showed that each of the 4 immune cell types clustered together, as expected based on their DNA methylation patterns. Differences in methylation among the different cell types were much larger than the differences between RA patients and controls within each cell type, as expected. There was greater scattering for B cells, which is reflective of the diversity of that cell type, as compared to monocytes and the T cell subpopulations examined in this study.
Figure 1.
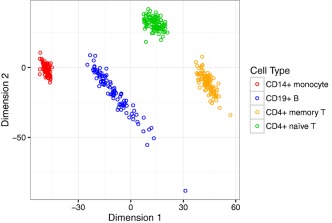
Multidimensional scaling (MDS) plot of DNA methylation profiles in CD14+ monocytes, CD19+ B cells, CD4+ memory T cells, and CD4+ naive T cells. This MDS plot of 371 samples (4 immune cell types each from patients with rheumatoid arthritis and unaffected control subjects) shows that samples cluster according to cell type, as expected.
Wilcoxon's rank sum tests
Four immune cell types were assayed for each individual sample: CD14+ monocytes, CD19+ B cells, CD4+ memory T cells, and CD4+ naive T cells. Consistent with the methylation differences seen in FLS, hypermethylation or hypomethylation of DNA in PB samples from RA patients relative to controls was evaluated separately for each immune cell type. For each of the hypermethylated candidate CpGs (n = 5,532) and hypomethylated candidate CpGs (n = 8,406) from the FLS study, we used a 1‐tailed Wilcoxon's rank sum test to assess differences in the median beta value between RA patients and controls. P values were adjusted using the Benjamini‐Hochberg method, to control for the false discovery rate (FDR) 25. We controlled the error rate for either 5,532 tests or 8,406 tests, depending on the candidate list. Methylation changes at a second set of 1,788 candidate CpG sites (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract) in 98 genes deemed likely to be important to RA biology (based on a recent genome‐wide association study [GWAS] meta‐analysis of >100,000 subjects [4]) were also evaluated, and an exploratory association analysis was conducted using all CpGs on the 450k BeadChip. For the GWAS candidate CpGs and the chip‐wide tests, we used a 2‐tailed Wilcoxon's rank sum test, controlling for 1,676 tests and 428,232 tests, respectively.
Principal components analysis (PCA)
In order to determine whether the proportions of cell subtypes in the sorted cells were confounding the results, we performed Reference‐Free Adjustment for Cell‐Type Composition (ReFACTor) analysis 26, in which a PCA is performed on a subset of sites that are informative with respect to the cell composition in the data. The ReFACTor method finds the most informative sites, in an unsupervised manner. To measure the potential confounding, we examined quantile–quantile (Q–Q) plots for each cell type for a standard epigenome‐wide association study (EWAS), using only the methylation sites with a mean methylation level in the range of 0.2–0.8, following the suggestion made by Liu et al to remove consistently methylated and consistently unmethylated probes when performing EWAS 27. Deflation was observed in the Q–Q plots of all cell types except CD4+ naive T cells, implying that power was deficient. To assess the expected Q–Q plot under the condition of power deficiency, we permuted the phenotype and repeated the EWAS analysis, and repeated this procedure 100 times for each cell type.
To determine whether a correction was required in the cell types, we used the genomic control lambda measurement of inflation 28. We considered the median lambda of the 100 EWAS executions as the expected lambda. The approach was to add individual ReFACTor principal components (PCs) to the analysis until the inflation was corrected with respect to the expected lambda 29. Only the values in the CD4+ naive cells were found to be inflated, and adjustment for the first ReFACTor PC (PC1) resulted in removal of this inflation, suggesting that a possible cell substructure is present in the CD4+ naive cells. The ReFACTor analysis was executed on the CD4+ naive T cell data with the parameter K = 2 (representing the number of assumed cell types for the ReFACTOR program). We added this first ReFACTor component, PC1, in logistic regression models to evaluate the results that were adjusted for confounding by cell substructure.
Logistic regression models
To evaluate possible confounding effects, logistic regression models of RA case status were carried out against each FLS CpG that was significant at q < 0.05 in the Wilcoxon's rank sum tests (1,056 models), adjusting for smoking, age, batch (date the plate was run), and PC1 calculated from the ReFACTor analysis (as described above), which aims to quantify cell substructure 26. Unadjusted models were compared to models adjusted for age only, models adjusted for ever having smoked only, models adjusted for batch only, models adjusted for ReFACTor PC1 only, models adjusted for age, smoking, and batch combined, and models adjusted for age, smoking, batch, and ReFACTor PC1 combined.
Receiver operating characteristic (ROC) curve analyses
ROC curve analysis was used to explore the potential for the FLS sites to serve as a biomarker for the RA disease process, as compared to the potential of a previously validated genetic risk score for RA 30, 31 and the presence or absence of HLA–DRB1 shared epitope alleles 32, 33. The hypermethylation score for each person was calculated by summing the beta values across the 1,056 significantly differentially methylated loci in the FLS. A continuous weighted genetic risk score was also calculated, based on the studies by Eyre et al 30 and Yarwood et al 31. The genetic risk score included 43 of the 45 non‐HLA SNPs (rs13397 and rs59466457 were missing), and it was calculated by multiplying the number of copies of risk alleles, using probability data from genome‐wide imputation, for each SNP by the natural logarithm of the odds ratio, as reported in the study by Eyre et al 30, and summing these values across the 43 SNPs for each person. Presence of the shared epitope was coded as a binary variable. Individuals with ≥1 copy of the following alleles were assigned a value of 1 for the shared epitope: HLA–DRB1*0101, *0102, *0401, *0404, *0405, *0408, or *1001 34. The pROC package in R was used to plot each of these variables as a predictor, with RA case status as the response variable 35.
To determine the influence of adjusting for potential confounders of the hypermethylation score, we created 2 additional hypermethylation scores. The first hypermethylation score was based on the 830 FLS sites that remained significant (P < 0.05) in the logistic regression models after adjustment for age, smoking, and batch. The second hypermethylation score was based on the 79 FLS sites that remained significant (P < 0.05) after adjustment for age, smoking, batch, and ReFACTor PC1.
Study approval
Written informed consent was provided by all participants prior to inclusion in this study, and the research was in compliance with the Declaration of Helsinki. Institutional Review Board approval was estabilished in place at the University of California, San Francisco, where the study subjects were recruited.
RESULTS
Lack of effect of overnight cell storage
The methylation profiles of isolated cell populations were not impacted by overnight storage of the cells (correlation between profiles derived from all paired samples was very high, at r2 > 0.997). Details are summarized in the Supplementary Text (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract).
Identification of sites of significant hypo‐ or hypermethylation among the candidate FLS CpGs
After the P values from Wilcoxon's rank sum tests were adjusted for multiple testing by controlling for the FDR (P values adjusted for multiple testing hereafter referred to as q values), 1,056 significantly hypermethylated CpG sites in CD4+ naive T cells were found to have a q value of <0.05 (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract). At this same threshold, there were no sites of significantly hypomethylated CpG candidates in CD4+ naive T cells, nor were there any significantly hypermethylated or hypomethylated FLS CpG candidates in any of the remaining cell types (CD14+ monocytes, CD19+ B cells, and CD4+ memory T cells). Results are summarized in Table 2.
Table 2.
Results of candidate fibroblast‐like synoviocyte CpG analysesa
| Cell type | CpGs with raw P < 0.05 | CpGs with absolute median methylation difference of >10%b | CpGs with absolute median methylation difference between 1% and 10%b | CpGs with FDR q < 0.05 | CpGs with FDR q < 0.05 and median methylation difference >1% |
|---|---|---|---|---|---|
| CD14+ monocytes | |||||
| Hypomethylated | 263 | 4 | 175 | 0 | 0 |
| Hypermethylated | 100 | 0 | 61 | 0 | 0 |
| CD19+ B cells | |||||
| Hypomethylated | 96 | 1 | 59 | 0 | 0 |
| Hypermethylated | 1,408 | 1 | 732 | 0 | 0 |
| CD4+ memory T cells | |||||
| Hypomethylated | 262 | 0 | 204 | 0 | 0 |
| Hypermethylated | 66 | 1 | 36 | 0 | 0 |
| CD4+ naive T cells | |||||
| Hypomethylated | 160 | 1 | 62 | 0 | 0 |
| Hypermethylated | 2,569 | 0 | 1,105 | 1,056 | 517 |
Wilcoxon's rank sum tests were carried out for each candidate CpG in each of the 4 cell types, with 1‐sided P values, according to whether the CpG was hypermethylated or hypomethylated in the original study. FDR = false discovery rate.
The values for absolute median methylation differences are among the CpGs with unadjusted P < 0.05.
Results of logistic regression analyses
Logistic regression analysis was conducted with RA case status as the outcome and methylation beta value as the predictor variable for each of the 1,056 FLS CpGs identified. Of these, 1,035 CpGs were significantly associated with RA case status (P < 0.05, by 1‐sided test) in the unadjusted model, while 830 CpGs remained significant when adjusting for age, smoking, and batch together, and 79 CpGs remained significant when adjusting for age, smoking, batch, and ReFACTor PC1. Results are summarized in Supplementary Table 2 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract), and the shifts in P values with each of the different models are visualized in Figure 2.
Figure 2.
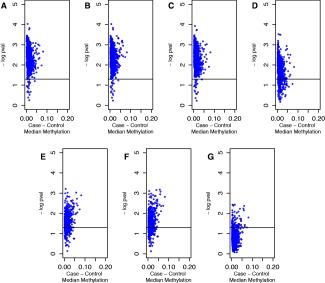
Plots of 1‐sided P values and differences in median methylation levels in fibroblast‐like synoviocyte CpGs between rheumatoid arthritis (RA) patients and unaffected controls in logistic regression models with RA case status as the outcome and methylation beta value as the predictor variable, after adjustment for covariates. CpGs in the models are those significant at q < 0.05 in Wilcoxon's rank sum tests. Models are adjusted for A, no covariates, B, age only, C, smoking only, D, batch only, E, Reference‐Free Adjustment for Cell‐Type Composition (ReFACTor) principal component 1 (PC1) only, F, age, smoking, and batch together, and G, age, smoking, batch, and ReFACTor PC1 together.
Comparison of methylation profiles to shared epitope status and genetic risk score as predictors of RA risk
The association of hypermethylation of CpG sites in CD4+ naive T cells with the risk of RA was compared to a weighted genetic risk score based on non‐HLA risk alleles, and also compared to the presence or absence of the HLA–DRB1 shared epitope, a major genetic risk factor for RA 36. The hypermethylation score and shared epitope models each performed similarly. Figure 3 shows the ROC curves for each of the 3 measures, and Table 3 summarizes the point estimates and 95% confidence intervals (95% CIs) for the area under the ROC curve (AUC) for association with RA case status in each model. The hypermethylation score had the largest AUC, at 72% (95% CI 61–83%). The shared epitope had an AUC of 66% (95% CI 56–76%), and the genetic risk score had an AUC of 51% (95% CI 38–63%). The AUC for the hypermethylation score based on the 830 CpGs found to be significant at P < 0.05 in the logistic regression model adjusted for age, smoking, and batch was 71.8% (95% CI 61.0–82.7%), which is similar to the hypermethylation score based on the CpGs found to be significant after Wilcoxon's rank sum test in unadjusted models. The AUC using only the 79 CpGs that were significant in the logistic regression model adjusted for age, smoking, batch, and ReFACTor PC1 was 80.7% (95% CI 71.3–90.1%). Results are summarized in Table 3.
Figure 3.
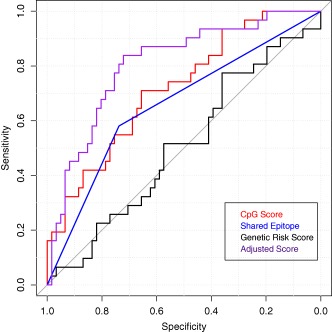
Receiver operating characteristic curves of the CpG hypermethylation score, the HLA–DRB1 shared epitope status, and the rheumatoid arthritis (RA) genetic risk score as predictors of RA case status. The hypermethylation score is the sum of the beta values across the 1,056 significant CD4+ naive T cell sites and is a measure of hypermethylation. Shared epitope is a binary variable taking on the value of 1 if a person has 1 or 2 copies of the shared epitope. Genetic risk score is a weighted score of 43 single‐nucleotide polymorphisms, a scoring method that has been validated in previous studies 30, 31. The adjusted hypermethylation score represents the sum of the 79 CpGs that were significant (P < 0.05) after adjustment for age, smoking, batch, and Reference‐Free Adjustment for Cell‐Type Composition principal component 1.
Table 3.
ROC curve analyses of predictors of association with RA case statusa
| Model | AUC (95% CI) |
|---|---|
| Hypermethylation score (1,056 CpG sites) | 72 (61–83) |
| Presence of the HLA–DRB1 shared epitope | 66 (56–76) |
| Genetic risk score | 51 (38–63) |
| Hypermethylation score (830 CpG sites; regression model adjusted for age, smoking, batch) | 72 (61–83) |
| Hypermethylation score (79 CpG sites; regression model adjusted for age, smoking, batch, ReFACTor PC1) | 81 (71–90) |
Analyses of the area under receiver operating characteristic (ROC) curve (AUC), expressed as a percentage, were carried out for a hypermethylation score based on the 1,056 CpG sites significant at q < 0.05 from the Wilcoxon's rank sum tests. This score was compared to shared epitope status (positive/negative) and a rheumatoid arthritis (RA) genetic risk score. Two other hypermethylation scores were constructed based on the 1,056 CpGs that remained significant (P < 0.05) in logistic regression models after adjustment for various covariates. 95% CI = 95% confidence interval; ReFACTor PC1 = Reference‐Free Adjustment for Cell‐Type Composition principal component 1.
Results for GWAS candidate CpGs
Among the GWAS set of candidate CpGs assessed by Wilcoxon's rank sum tests (1,676 CpGs), 1 CpG (hypermethylated) in CD4+ memory cells and 18 CpGs (6 hypomethylated, 12 hypermethylated) in CD4+ naive T cells were significantly associated (q < 0.05) with RA susceptibility. Results are summarized in Supplementary Table 4 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract). We also carried out logistic regression analysis using RA status as the outcome for each of the 18 CpGs that were differentially methylated in CD4+ naive T cells, with adjustments for various covariates. Results are summarized in Supplementary Table 5 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract).
Genome‐wide results
Results of the genome‐wide tests of differences in methylation are summarized in Supplementary Table 6 (available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract). No CpG sites were significantly differentially methylated after correction for multiple testing (adjusting the P value for 428,232 tests). Differences in global methylation were investigated by comparing the mean methylation levels in RA patients to those in unaffected controls (for results, see Supplementary Table 7, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39952/abstract). No significant differences in mean methylation levels were observed in any cell type.
DISCUSSION
In the present study, hypermethylated CpG sites previously identified in FLS of patients with RA relative to patients with osteoarthritis or healthy controls were also distinguished in CD4+ naive T cells from the PB of RA patients relative to healthy controls. Our results show that a disease‐associated signature can be observed in cells obtained from whole blood, which is more accessible than the synovial fluid for clinical or epidemiologic studies.
The results of our study extend the recent findings demonstrating that DNA methylation profiles in PBMCs differ between RA patients and controls 12. While Glossop et al observed differences in both B lymphocytes and T lymphocytes 12, most results from the present study were confined to CD4+ naive T cells. However, taken together, the combined findings increase the evidence that PB cells contain a DNA methylation signature that can distinguish patients with RA from unaffected controls. Furthermore, the DNA methylation profile differences detected in T cells of treatment‐naive patients in the study by Glossop et al 12 suggest that there are methylation changes important in RA that are not a consequence of medication or long disease duration.
The 1,056 differentially methylated candidate FLS CpGs associated with RA in this study were limited to the CD4+ naive T cell population. Most of the observed differences were small, with a difference in median beta value of <10% between RA patients and controls. Of the 1,056 sites, 517 had a methylation difference of >1% (results in Supplementary Table 1). These 517 sites resided in 357 genes as well as intergenic regions, and across all chromosomes. It is uncertain what effect size is biologically meaningful for DNA methylation. Some researchers impose a threshold of either 5% or 10% difference in methylation to consider the results relevant 37, while others include modest effect sizes 38. One recent study showed replicable methylation differences associated with smoking, ranging from 1.2% to 24% 39.
Although differences in this study were small, they were robust, surviving stringent multiple‐testing correction. A hypermethylation score constructed from the 1,056 significant CpG sites predicted RA case status with an AUC of 73%, and awaits validation in an independent data set. The hypermethylation score based on the 830 CpG sites with significance at P < 0.05 after adjustment for smoking, age, and batch in the logistic regression models had a similar AUC, of 71.8%, suggesting that the score was not strongly influenced by these covariates. The hypermethylation score calculated using the 79 CpG sites with significance at P < 0.05 after adjustment for smoking, age, batch, and ReFACTor PC1 in the logistic regression models had a slightly higher AUC, of 80.7%, suggesting that adjustment for possible cell substructure may improve the ability of our FLS CpG site score to serve as a biomarker for RA. Because DNA methylation was measured subsequent to the diagnosis of RA, we cannot determine with certainty whether the FLS methylation signature in the CD4+ naive T cells is predictive of an RA diagnosis or is a biomarker of the disease process.
One of the top 10 replicated sites (those with the most significant P values) in CD4+ naive T cells, cg21480173, was found in the gene TYK2, which has been associated with RA and other autoimmune diseases 40. The remaining 9 top hits were found in the following genes: PRKAR1B, ABCC4, COMT, CAI2, MCF2L, GALNT9, C7orf50, or non‐gene regions, which have not been previously associated with RA. These results demonstrate that novel genes related to RA may be discovered through DNA methylation analysis. We also observed differential methylation in CpG sites that reside in genes that have previously been associated with RA 4. For example, 2 of the CpGs reside in the promoter regions for both GATA3 and GATA3‐AS1 (cg17566118 and cg15852223), and both are hypomethylated in RA patients relative to controls. It is important to note that our results were not due to genetic variation or to genetic ancestry differences between RA patients and controls.
The lack of significant findings in cell types other than CD4+ naive T cells suggests that CD4+ naive T cells are particularly relevant to RA through epigenetic mechanisms involving DNA methylation. There is strong evidence from previous studies that aberrant T cell activation pathways are involved in the pathogenesis of RA, including in the naive T cell population, which has not yet participated in immune responses 41. CD4+ naive T cells from RA patients have been shown 1) to have premature senescence, 2) to be defective in up‐regulating telomerase due to deficiencies in the telomerase component human telomerase reverse transcriptase, 3) to have increased DNA damage load and increased rates of apoptosis, 4) to be unable to metabolize amounts of glucose equal to those metabolized by healthy control cells of the same age, and 5) to generate lower amounts of ATP 42, 43, 44, 45. Although our methylation findings need to be replicated, the striking findings in CD4+ naive T cells and the existing literature on abnormalities in this cell population in RA suggest that the methylation changes observed may be involved in disease pathogenesis.
However, it is also plausible that methylation changes are a response to the disease process itself or are a result of exposure to medications. Additional studies involving patients with early or preclinical disease will be required to determine at what point in the course of the disease process such differential methylation patterns occur. Longitudinal studies may also help elucidate why results from the present study support a hypermethylation signature in RA, in contrast to a signature of hypomethylation, which has been demonstrated in previous studies 8, 11. Hypermethylation may occur at a specific point along the course of RA, or may be specific to the FLS‐associated sites rather than the global methylome.
Results from logistic regression modeling suggest that although some variables are confounding the relationship between methylation and RA case status, evidence for association persists. Specifically, adjustments for age or smoking did not markedly impact the number of FLS CpGs that were significantly associated with RA at P < 0.05. Adjusting for batch or ReFACTor PC1 reduced the number of statistically significant CpGs by ∼200, but many remained statistically significant (841 in models adjusted for batch, 837 in models adjusted for ReFACTor PC1). Even when all 4 of these variables were controlled for in the models, 79 CpGs remained significant. Figure 2 visually represents the shifting of the P values across these regression models. Evidence for association also persisted in analyses of GWA‐identified candidate CpGs, even in fully adjusted models (results in Supplementary Table 5).
This study has many strengths. DNA methylation profiles were analyzed in 4 sorted cell types from 94 individuals, all of whom were women of European ancestry, which reduced the genetic heterogeneity of the study population. Examination of individual cell types from FACS‐sorted blood allowed us to measure methylation results with more confidence, rather than relying on whole blood and cell type proportions 46. Restriction of the study to female subjects eliminates the possibility of confounding by sex. Moreover, since RA affects women at a 3:1 ratio relative to men, results are generalizable to the group who experience the greatest disease burden.
Stringent quality control of the methylation data, as described in Patients and Methods, is another strength of this study. In addition to implementation of the standard quality control steps of background subtraction, normalization, and removal of sites with low quality scores, CpG sites with known SNPs in individuals of European ancestry at the cytosine or guanine being measured on the 450k BeadChip were removed, which is important because methylation measurements for CpG sites harboring SNPs are likely to simply reflect genetic polymorphism at that site rather than a true measure of methylation. We also removed from analysis CpG sites with cross‐reactive sequencing probes on the 450k BeadChip, i.e., probes that could hybridize to more than one location in the genome and reflect methylation at 2 different genomic locations rather than only the intended target site. Rigorous quality control measures increase confidence that the observed differential methylation is an accurate reflection of the disease biology, and not due to artifacts.
Both whole genome and whole methylome data were utilized in the present study. The whole genome data allowed us to determine the genetic ancestry of all participants. The original FLS study by Whitaker et al involved anonymous samples, and the authors did not have information on ethnicity or race 14. Therefore, it is possible that we are underestimating the overlap between FLS and CD4+ naive T cell sites if we are comparing different ethnicities in analyzing the CD4+ naive T cell and FLS samples. In addition, we were able to demonstrate that even after controlling for age, smoking, batch, and possible cell substructure (ReFACTor PC1), a number of FLS and GWA candidate sites remain significantly associated with RA.
This study also has limitations. We could not assess temporality between methylation and RA case status. Results may be confounded by case‐specific factors such as medication and inflammation. Indeed, other studies have observed associations between methylation and medications 47, 48; however, the case–control nature of the present study did not allow us to adjust for the effects of RA medications, since they were present only among RA cases.
Our findings are restricted to CpG sites that are represented on the 450k BeadChip. The BeadChip prioritized inclusion of features such as RefSeq genes, CpG islands, shores, and shelves, areas of the genome such as the MHC region, and sites known to be important in cancer 49, 50. Therefore, additional CpG sites relevant to RA may be missing. Furthermore, although our ROC analysis demonstrated that differential methylation of ∼1,000 CpGs in the PB has the potential to distinguish RA cases from controls, our hypermethylation score needs to be further tested as a predictor in an independent data set.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Criswell had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Shao, Link, Barcellos, Criswell.
Acquisition of data
Shao, H. L. Quach, D. Quach, Shah, Sinclair, Graf, Link, Harrison, Wang, Firestein, Barcellos, Criswell.
Analysis and interpretation of data
Rhead, Holingue, Cole, Link, Rahmani, Halperin, Firestein, Barcellos, Criswell.
Supporting information
Supplementary Figure 1. Eigenvector 1 and 2 from EIGENSTRAT in all samples.
Supplementary Figure 2. Eigenvector 2 and 3 from EIGENSTRAT in all samples.
Supplementary Figure 3. Eigenvector 1 and 2 from EIGENSTRAT in European ancestry samples.
Supplementary Figure 4. MDS of CD4+ naïve T cells before normalization.
Supplementary Figure 5. MDS of CD4+ naïve T cells after normalization.
Supplementary Table 2. Logistic Regression Results from Wilcoxon Rank Sum Test FLS Sites
Supplementary Table 4. Candidate GWAS CpG Results
Supplementary Table 5. Logistic Regression Results Using 18 Significant CpGs from Wilcoxon Rank Sum Test, GWA Sites
Supplementary Table 6. Genome‐wide Results.
Supplementary Table 7. Mean Methylation in RA Cases and Controls.
Supplementary Table 1. FLS Candidate Sites Replicated in CD4 Naïve Cells in Peripheral Blood of RA Cases.
Supplementary Table 3. Candidate CpGs from Genes Previously Associated with RA.
ACKNOWLEDGMENT
We thank Vladimir Chernitskiy for his assistance in data acquisition.
Supported by the Rheumatology Research Foundation (Within Our Reach grant and Health Professional Research preceptorship), the Arthritis Foundation, the Rosalind Russell/Ephraim P. Engleman Rheumatology Research Center, and the University of California–Stanford Arthritis Center of Excellence, which is funded in part by the Arthritis Foundation. The FLS study was funded by the Rheumatology Research Foundation, the Arthritis Foundation, and the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases grant 1R01‐AR‐065466 to Dr. Firestein).
REFERENCES
- 1. Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD, Tanasescu R. Extra‐articular manifestations in rheumatoid arthritis. Maedica (Buchar) 2010;5:286–91. [PMC free article] [PubMed] [Google Scholar]
- 2. Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther 2009;11:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 2000;43:30–7. [DOI] [PubMed] [Google Scholar]
- 4. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Källberg H, Ding B, Padyukov L, Bengtsson C, Rönnelid J, Klareskog L, et al. Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis 2011;70:508–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robertson KD. DNA methylation and human disease. Nat Rev Genet 2005;6:597–610. [DOI] [PubMed] [Google Scholar]
- 7. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity 2013;38:414–23. [DOI] [PubMed] [Google Scholar]
- 8. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 1990;33:1665–73. [DOI] [PubMed] [Google Scholar]
- 9. Takami N, Osawa K, Miura Y, Komai K, Taniguchi M, Shiraishi M, et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum 2006;54:779–87. [DOI] [PubMed] [Google Scholar]
- 10. Nile CJ, Read RC, Akil M, Duff GW, Wilson AG. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 2008;58:2686–93. [DOI] [PubMed] [Google Scholar]
- 11. Liu C, Fang T, Ou T, Wu C, Li R, Lin Y, et al. Global DNA methylation, DNMT1, and MBD2 in patients with rheumatoid arthritis. Immunol Lett 2011;135:96–9. [DOI] [PubMed] [Google Scholar]
- 12. Glossop JR, Emes RD, Nixon NB, Packham JC, Fryer AA, Mattey DL, et al. Genome‐wide profiling in treatment‐naive early rheumatoid arthritis reveals DNA methylome changes in T and B lymphocytes. Epigenomics 2016;8:209–24. [DOI] [PubMed] [Google Scholar]
- 13. Glossop JR, Haworth KE, Emes RD, Nixon NB, Packham JC, Dawes PT, et al. DNA methylation profiling of synovial fluid FLS in rheumatoid arthritis reveals changes common with tissue‐derived FLS. Epigenomics 2015;7:539–51. [DOI] [PubMed] [Google Scholar]
- 14. Whitaker JW, Shoemaker R, Boyle DL, Hillman J, Anderson D, Wang W, et al. An imprinted rheumatoid arthritis methylome signature reflects pathogenic phenotype. Genome Med 2013;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartok B, Firestein GS. Fibroblast‐like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 2010;233:233–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 17. Barton JL, Trupin L, Schillinger D, Gansky SA, Tonner C, Margaretten M, et al. Racial and ethnic disparities in disease activity and function among persons with rheumatoid arthritis from university‐affiliated clinics. Arthritis Care Res (Hoboken) 2011;63:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. International HapMap Consortium . The International HapMap Project. Nature 2003;426:789–96. [DOI] [PubMed] [Google Scholar]
- 21. Davis S, Du P, Bilke S, Triche T Jr, Bootwalla M. Methylumi: Handle Illumina methylation data. R package version 2.0. 2014.
- 22. Yousefi P, Huen K, Aguilar Schall R, Decker A, Elboudwarej E, Quach H, et al. Considerations for normalization of DNA methylation data by Illumina 450K BeadChip assay in population studies. Epigenetics 2013;8:1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez‐Cabrero D, et al. A β‐mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013;29:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross‐reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013;8:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995;57:289–300. [Google Scholar]
- 26. Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, et al. Sparse PCA corrects for cell type heterogeneity in epigenome‐wide association studies. Nat Methods 2016;13:443–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, et al. Epigenome‐wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013;31:142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic‐based association studies. Theor Popul Biol 2001;60:155–66. [DOI] [PubMed] [Google Scholar]
- 29. Zou J, Lippert C, Heckerman D, Aryee M, Listgarten J. Epigenome‐wide association studies without the need for cell‐type composition. Nat Methods 2014;11:309–11. [DOI] [PubMed] [Google Scholar]
- 30. Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, et al. High‐density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 2012;44:1336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yarwood A, Han B, Raychaudhuri S, Bowes J, Lunt M, Pappas DA, et al. A weighted genetic risk score using all known susceptibility variants to estimate rheumatoid arthritis risk. Ann Rheum Dis 2015;74:170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30:1205–13. [DOI] [PubMed] [Google Scholar]
- 33. Holoshitz J. The rheumatoid arthritis HLA‐DRB1 shared epitope. Curr Opin Rheumatol 2010;22:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 2012;44:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open‐source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Van der Helm‐van Mil AH, Verpoort KN, Breedveld FC, Huizinga TW, Toes RE, de Vries RR. The HLA–DRB1 shared epitope alleles are primarily a risk factor for anti–cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum 2006;54:1117–21. [DOI] [PubMed] [Google Scholar]
- 37. Stefansson OA, Moran S, Gomez A, Sayols S, Arribas‐Jorba C, Sandoval J, et al. A DNA methylation‐based definition of biologically distinct breast cancer subtypes. Mol Oncol 2015;9:555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tsai PC, Bell JT. Power and sample size estimation for epigenome‐wide association scans to detect differential DNA methylation. Int J Epidemiol 2015. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Georgiadis P, Hebels DG, Valavanis I, Liampa I, Bergdahl IA, Johansson A, et al. Omics for prediction of environmental health effects: blood leukocyte‐based cross‐omic profiling reliably predicts diseases associated with tobacco smoking. Sci Rep 2016;6:20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune‐mediated diseases. Nat Rev Genet 2013;14:661–73. [DOI] [PubMed] [Google Scholar]
- 41. Cope AP, Schulze‐Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol 2007;25 Supp 46:S4–S11. [PubMed] [Google Scholar]
- 42. Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A 2009;106:4360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev 2005;204:55–73. [DOI] [PubMed] [Google Scholar]
- 44. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med 2009;206:1435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 2013;210:2119–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012;7:e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Plant D, Wilson AG, Barton A. Genetic and epigenetic predictors of responsiveness to treatment in RA. Nat Rev Rheumatol 2014;10:329–37. [DOI] [PubMed] [Google Scholar]
- 48. Kim YI, Logan JW, Mason JB, Roubenoff R. DNA hypomethylation in inflammatory arthritis: reversal with methotrexate. J Lab Clin Med 1996;128:165–72. [DOI] [PubMed] [Google Scholar]
- 49. Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics 2011;98:288–95. [DOI] [PubMed] [Google Scholar]
- 50. J Sandoval, H Heyn, S Moran, J Serra-Musach, MA Pujana, M Bibikova, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011;6:692–702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Eigenvector 1 and 2 from EIGENSTRAT in all samples.
Supplementary Figure 2. Eigenvector 2 and 3 from EIGENSTRAT in all samples.
Supplementary Figure 3. Eigenvector 1 and 2 from EIGENSTRAT in European ancestry samples.
Supplementary Figure 4. MDS of CD4+ naïve T cells before normalization.
Supplementary Figure 5. MDS of CD4+ naïve T cells after normalization.
Supplementary Table 2. Logistic Regression Results from Wilcoxon Rank Sum Test FLS Sites
Supplementary Table 4. Candidate GWAS CpG Results
Supplementary Table 5. Logistic Regression Results Using 18 Significant CpGs from Wilcoxon Rank Sum Test, GWA Sites
Supplementary Table 6. Genome‐wide Results.
Supplementary Table 7. Mean Methylation in RA Cases and Controls.
Supplementary Table 1. FLS Candidate Sites Replicated in CD4 Naïve Cells in Peripheral Blood of RA Cases.
Supplementary Table 3. Candidate CpGs from Genes Previously Associated with RA.