

Abstract
Purpose
The aims of this study are to elucidate if molecular markers can be used to differentiate between the two main types of ameloblastoma (unicystic and solid/multicystic), and to determine whether a biologically ‘less-aggressive’ subtype exists.
Methods
A retrospective analysis of 33 solid/multicystic ameloblastomas and six unicystic ameloblastomas was completed using immunohistochemistry for five molecular markers: P16, P53, MMP-9, Survivin, and Ki-67. Tumors were graded as either negative or positive (mild, moderate, strong), and the results were related to both ameloblastoma subtypes and outcomes following treatment.
Results
Unicystic ameloblastomas were more likely to test strongly positive for P53 than solid/multicystic ameloblastomas (p < 0.05), whereas the latter were more likely to be negative for Survivin (p < 0.05). Solid/multicystic and Type 3 unicystic ameloblastomas that were positive for P16, but also negative for MMP-9 and Survivin, were less likely to recur than all other tumors (p < 0.05). The proliferation index of an ameloblastic carcinoma (11 %) was shown to be higher than benign ameloblastomas (4.5 %).
Conclusions
Immunohistochemistry can be valuable in lesions where histological sub-typing of an ameloblastoma is unclear. A biologically ‘less-aggressive’ subtype may exist, and hence further research into this area is required.
Keywords: P16, Markers, Diagnosis, Outcome, Ameloblastoma
Introduction
Ameloblastoma is an odontogenic neoplasm characterized by its invasive behavior and has a propensity to recur following treatment. Understanding its underlying cellular mechanisms and molecular markers can be considered important for a number of reasons [1]. Firstly, it may assist in the diagnosis and differentiation between odontogenic tumors and their subtypes. For example, radical surgery forms the mainstay of treatment for solid/multicystic ameloblastomas (SMA), whilst most unicystic ameloblastomas (UA) can be effectively managed with conservative therapy [2, 3]. Secondly, this information may identify a biologically ‘less-aggressive’ subtype where more conservative, rather than radical, therapy could be successfully introduced. Similarly, human papillomavirus (HPV) positive oropharyngeal squamous cell carcinomas (OPSCC) has an improved prognosis compared to their HPV-negative counterparts, and hence de-escalated treatment is currently being explored. Lastly, certain molecules integral to ameloblastoma development could possibly receive targeted therapy, resulting in prevention or delayed oncogenesis.
The aims of this study are to elucidate if molecular markers can be used to differentiate between the two main types of ameloblastoma (UA and SMA), and to determine whether a biologically ‘less-aggressive’ subtype exists.
Materials and Methods
Case Identification
Forty-nine cases of ameloblastoma were identified from the surgical and pathology databases of The Royal Melbourne Hospital from 2001 to 2012. Patient files and histological slides were assessed by an oral and maxillofacial pathologist and a senior surgical registrar, and the diagnosis of ameloblastoma was confirmed and then sub-typed according to the World Health Organization (WHO) classification [4]. A total of nine cases had inadequate pathological or clinical information, and were excluded from the study. Treatment methods were divided into conservative or radical. Conservative management included enucleation, curettage, and/or marsupialization, whereas radical treatment followed a standard protocol of tumor resection with a margin of 1–1.5 cm confirmed on specimen radiograph, frozen section, and histopathological examination. Outcomes were classified as either ‘recurrence’ or ‘no recurrence’.
Immunohistochemistry (IHC)
4 μm thick sections from formalin fixed paraffin embedded (FFPE) tissue blocks of the 40 ameloblastomas were tested with five antibodies at appropriate dilutions (Table 1). Sections were de-waxed through a series of xylene solutions (×3) followed by absolute ethanol (×2), 70 % ethanol and distilled water. Retrieval of antigen was conducted using a high pH retrieval solution, and IHC testing was completed by a Leica BOND-MAX™ automated IHC stainer and Bond™ Polymer Refine Detection system (Leica Biosystems, Melbourne, Australia). Controls (positive and negative) were analyzed by the same method (Table 1).
Table 1.
Antibodies and controls used for immunohistochemistry testing
| Antibody | Brand | Cat. no. | Dilution | Control |
|---|---|---|---|---|
| P16 | Roche CINtec (Arizona, USA) | 06594441001 | 1:3 | Basaloid SCC in lymphoid tissue |
| P53 | Novocastra (Leica Biosystems, Newcastle, UK) | NCL-L-P53-DO7 | 1:50 | Colon adenocarcinoma |
| MMP-9 | Epitomics (California, USA) | 1939-1 | 1:200 | Non-small cell lung carcinoma |
| Ki-67 | Dako Australia (Campbellfield, Victoria, Australia) | M724001 | 1:100 | Tonsillar tissue |
| Survivin | Dako Australia (Campbellfield, Victoria, Australia) | M362429 | 1:50 | Tonsillar tissue |
For each tumor, ten randomized areas were evaluated under high magnification (400×) for the number of tumor cells positive for P16, P53, MMP-9, and Survivin antibodies. A cell was deemed ‘positive’ if it displayed strong antibody uptake (intense staining) of its nucleus and/or cytoplasm. Negative tumor cells in the same areas were also counted, and a percentage calculated by dividing the number of positive cells by the number of total cells (1000) and the tumor was graded accordingly (Table 2) [5–7]. To reduce bias, tumors were also classified as overall negative (<25 %) and overall positive (25–100 %). Proliferating index (PI) was formulated using the number of tumor cells positive for Ki-67 in the ten areas.
Table 2.
| Grading | Positive tumor cells (%) |
|---|---|
| 0 = negative | <5 |
| 1 = weak positive | 5–24 |
| 2 = moderate positive | 25–50 |
| 3 = strong positive | >50 |
Statistical analysis was conducted using Minitab® Statistical Software (Pennsylvania, USA). Fisher’s exact tests were conducted using tumor subtypes, outcomes, and molecular markers, and statistical significance was determined by p < 0.05. The Melbourne Health Human Research Ethics Committee (Institutional Review Board) granted ethical approval for this study.
Results
Thirty-three cases of SMA, and 6 cases of UA were identified. An ameloblastic carcinoma was also tested but excluded from statistical analysis due to its distinctive tumor biology. Males were affected more than females (64:36 %), and the mandible was involved in 81.5 % of cases, most commonly in the posterior region (82 %). Patients were followed up for a mean of 51 months, and six patients suffered tumor recurrence.
Molecular Markers and Diagnosis
The IHC results are summarized in Fig. 1 and Table 3. There were no statistically significant differences between SMA and UA subtypes with P16 and MMP-9 stains. However, a statistically significant difference between these subtypes was seen in P53 and Survivin levels. UA tumors were more likely to be Strong Positive (Grade 3 or >50 % of positive cells) for P53 (p = 0.03), and SMA tumors were more likely to be negative (<5 % positive cells) for Survivin (p = 0.04).
Fig. 1.

Solid/multicystic and unicystic ameloblastomas with an overall positive grade for molecular markers (P16, MMP-9, Survivin, P53)
Table 3.
Immunohistochemistry results of the 33 SMA and 6 UA tumors
| Antigen and grading | UA | SMA |
|---|---|---|
| P16 | ||
| Negative | 0 | 1 |
| Weak | 0 | 5 |
| Moderate | 1 | 9 |
| Strong | 5 | 18 |
| MMP-9 | ||
| Negative | 3 | 12 |
| Weak | 1 | 9 |
| Moderate | 0 | 8 |
| Strong | 2 | 4 |
| Survivin | ||
| Negative* | 2 | 26 |
| Weak | 1 | 2 |
| Moderate | 1 | 2 |
| Strong | 2 | 3 |
| P53 | ||
| Negative | 1 | 12 |
| Weak | 0 | 5 |
| Moderate | 0 | 5 |
| Strong* | 5 | 11 |
* Statistical significance <0.05
Molecular Markers and Outcomes
Six recurrences were found in the SMA and Type 3 UA cases (37 cases total). Of the eight lesions treated conservatively, four recurred (50 %), compared to just two of the 29 treated with radical treatment (6.9 %) (p = 0.013). Further analysis did not reveal a significant association between individual IHC antibodies and outcomes, although ameloblastomas that were combined ‘P16-positive, MMP-9-negative, and Survivin-negative’ were unlikely to recur when compared to all other tumors (p = 0.01) (Table 4). Amongst those tumors that received radical treatment, this combination of IHC markers was not found to be statistically significant (p = 0.111). The ameloblastic carcinoma was excluded from the above analysis, however it was strongly positive for P53.
Table 4.
Molecular markers associated with a reduced recurrence rate in SMA and Type 3 UA tumors
| Combination of markers | No recurrence | Recurrence |
|---|---|---|
| P16 +ve, MMP-9 −ve, Survivin −ve* | 23 | 1 |
| Remaining tumors | 8 | 5 |
* Statistical significance <0.05
Proliferation Index (PI)
The PI for the SMA was similar to that of the UA (4.9 and 4.3 %), although the ameloblastic carcinoma had a notably higher PI (11.4 %) compared to its benign counterparts.
Discussion
The origins and molecular biology of the ameloblastoma is largely unknown despite its discovery over a century ago. Although this tumor has distinct histological features, definitive tumor diagnosis and sub-typing can be complex for a number of reasons including an inadequate tissue biopsy by the clinician, concurrent inflammation or ulceration, or difficulty differentiating normal tissue from a pathological lesion. IHC can assist where there is diagnostic uncertainty and this was demonstrated by our results.
Survivin is an inhibitor of apoptosis, and although it is found in many pathological lesions [8], there is limited evidence of an association with odontogenic tumors. One study found higher levels of Survivin mRNA in ameloblastomas than normal tooth germs using polymerase chain reaction (PCR) [9], although expression between the different subtypes was unclear. The results from our study indicate that the SMA has a statistically lower expression for Survivin (<5 % positive cells) than the UA, and thus this may be used as a distinguishing factor between these two subtypes. p53 is a tumor suppressor gene that is frequently altered in oncogenesis [10, 11], and its protein (P53) induces cell-cycle arrest (or apoptosis) when genomic damage is detected. P53 is undetectable in normal cellular levels, but can be recognized by IHC when it has a longer half-life through genetic mutation (Fig. 2). Elevated levels have been detected in several odontogenic tumors including the keratocystic odontogenic tumor (KCOT), ameloblastoma, and malignant ameloblastoma [10–13]. In particular, ameloblastomas have a higher level of P53 expression compared to normal tooth germs and ‘less-invasive’ odontogenic tumours, however, like Survivin the differences between ameloblastoma subtypes is still uncertain [14]. In our tumor group, UA significantly overexpressed P53 compared to the SMA, which may be explained by the high proportion of Type 3 UAs (mural invasion and proliferation). In lesions where it is difficult to diagnose a histological subtype (e.g. biopsy specimens of a cystic lesion), these results indicate a strongly positive result for P53 supports a UA rather than an SMA.
Fig. 2.
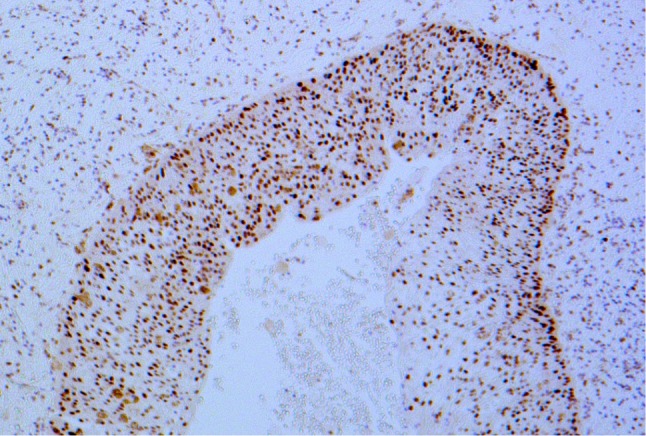
P53—strongly positive result for P53 in a unicystic ameloblastoma (×100 magnification)
Molecular markers may also have a role in the identification of a ‘less-aggressive’ tumor subtype that may be adequately treated with conservative, rather than radical, therapy. For example HPV-positive oropharyngeal squamous cell carcinoma (OPSCC) has a significantly improved prognosis compared to their HPV-negative counterparts [15, 16], and thus de-escalated treatment is currently being investigated for this group of patients. Over recent years P16 has been shown to be an accurate surrogate marker for HPV infection in OPSCC, however it can also act as an independent prognostic indicator in those cases that are P16-positive but HPV-negative [17]. In our group of tumors, SMA and Type 3 UA lesions had a statistically significant lower recurrence rate if they were a combined P16-positive, MMP 9-negative, and Survivin-negative, and thus this may indicate a biologically ‘less-aggressive’ subtype of ameloblastoma. Current recommendations advocate radical surgery for these lesions, and when this was taken into consideration statistical significance was not reached. However, understanding the underlying molecular basis to these markers supports the theory that a ‘less-aggressive’ ameloblastoma subtype may exist.
P16 (INK4a) protein inhibits cell cycle progression by inactivating cyclin-dependent kinases (CDKs) that phosphorylate the suppressor-suppressor protein pRB, thus leading to a deceleration of the cell cycle [6]. Loss of P16 function is thought to occur early in oncogenesis, and it has been found associated with many benign and malignant neoplasms including odontogenic tumors, and oral, pancreatic, and esophageal cancer [18, 19]. Kumamoto et al. [20] showed that there was no difference in P16 expression between ameloblastomas and normal tooth germs (Fig. 3), however the number of controls used in that study were limited (8 tooth germs). Artese et al. [6] evaluated 36 odontogenic tumors and found that amongst high risk tumors (e.g. SMA) there were more P16 positive cells on the periphery of the tumor compared to low risk tumors (including the UA and PA). In contrast our results indicate P16 levels were high in both SMA and UA tumors (81.8 and 100 % respectively), and no statistical difference was found between these two tumor groups (p = 0.564).
Fig. 3.
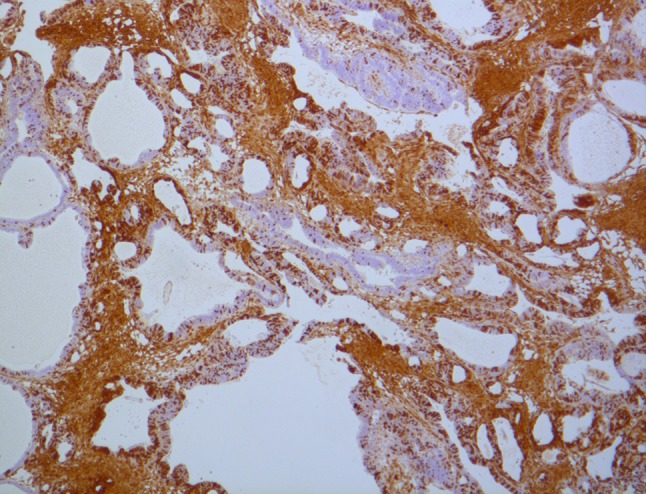
P16—strongly positive result for P16 in a solid/multicystic ameloblastoma (×40 magnification)
MMPs (matrix metalloproteinases) are proteolytic enzymes that assist in tumor invasion by degradation of the extracellular matrix [21].‘Invasive’ odontogenic tumors, such as ameloblastoma and odontogenic myxoma, have been shown to express MMPs-1, -2, and -9, thus assisting tumor progression [22–25]. MMPs are found in both tumor and stromal cells, and like P16 are mainly found on the periphery of the lesion [22, 23, 26]. Ameloblastomas have a higher expression of MMP-2 and -9 compared to normal tooth germs, but lower than that of malignant tumors such as the ameloblastic carcinoma [26, 27]. Zhang et al. suggested that by inhibition of MMPs could possibly suppress SMA invasion into local tissues, thus MMPs could be regarded a future treatment target [28, 29]. Our study results found similar levels of MMP expression in both the UA and SMA, with approximately 1/3 of lesions regarded as positive for MMP-9.
Ki-67 is a marker of tumor proliferation, as its binds to chromosomes during cell mitosis and is rapidly degraded after cellular division [30]. It is overexpressed in a number of proliferative lesions, including KCOT [12, 13, 31], ameloblastic carcinoma, and ameloblastic fibrosarcoma [27, 30]. Ameloblastomas have a variable, but relatively low, PI ranging between 2.8 % and 10 % [32–35] (Fig. 4). Ki-67 can be difficult to detect using IHC, especially in decalcified tissue samples where IHC staining can become unstable. It is overexpressed in both SMAs and UAs compared to both normal tooth germs and other odontogenic lesions, with the exception of the KCOT which consistently has a higher PI compared to the ameloblastoma [32, 36–38]. Certainly our results did not indicate a significant difference between SMA (4.9 %) and UA tumors (4.3 %), although the ameloblastic carcinoma showed a noticeably higher PI of 11.4 %. This supports other studies where the ameloblastic carcinoma has been shown to have a higher PI (17.2 %) than that of benign ameloblastomas (3.6 %) [27, 33, 34].
Fig. 4.
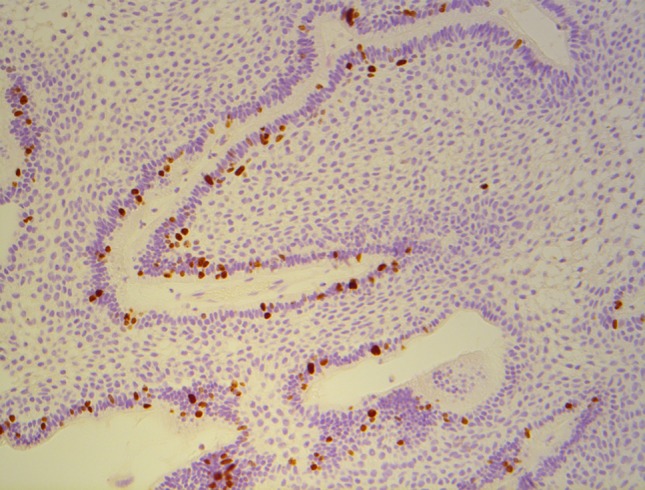
Ki-67—solid/multicystic ameloblastoma showing Ki-67 positive cells in a peripheral location (×100 magnification)
In conclusion, IHC for molecular markers can be valuable in the assessment of ameloblastoma. Those tumors strongly positive for P53 are more likely to represent a UA, whereas as negative test for Survivin supports an SMA. This information may provide assistance in cases where an ameloblastoma subtype is difficult to identify on histopathology alone (particularly biopsy specimens). Ameloblastomas that are positive for P16, and negative for MMP-9 and Survivin, may represent a biologically ‘less-aggressive’ tumor, and thus radical treatment could potentially be avoided. Further research is necessary to evaluate the true influence of different treatment methods in association with these markers. Finally our single case of ameloblastic carcinoma had a higher PI compared to its benign counterparts, and supports current literature in distinguishing this malignancy from the more common benign subtypes.
Acknowledgments
The authors would like to acknowledge the staff at the Anatomical and Pathology Laboratory at The Royal Melbourne Hospital. Special thanks to Associate Professor Nastri and Associate Professor Wiesenfeld for their assistance in this study. A research grant was kindly provided by the Australia and New Zealand Association of Oral and Maxillofacial Surgeons (ANZAOMS) Research and Education Foundation Inc. and Trust.
References
- 1.Kumamoto H. molecular pathology of odontogenic. J Oral Pathol Med. 2006;35:65–74. doi: 10.1111/j.1600-0714.2006.00380.x. [DOI] [PubMed] [Google Scholar]
- 2.Li T, Wu Y, Yu S, Yu G. Unicystic ameloblastoma: a clinicopathologic study of 33 Chinese patients. Am J Surg Pathol. 2000;24(10):1385–1392. doi: 10.1097/00000478-200010000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Singh TSC. Ameloblastoma: report on two cases and review of the literature. N Z Dent J. 2009;105(1):13–17. [PubMed] [Google Scholar]
- 4.Gardner D, Heikinheimo K, Shear M, Philipsen H, Coleman H (2005) Ameloblastoma. In: Barnes L, Eveson JW, Reichart P, Sidransky D (eds) World Health Organization classification of tumours: pathology and genetics of head and neck tumours. International Agency for Research on Cancer, Lyon
- 5.Artese L, Lessi G, Piattelli A, Rubini C, Goteri G, Pernotti V, Piccirilli M, Carinci F. p16 expression in odontogenic cysts. Dent Res J. 2008;5(2):61–64. doi: 10.1177/030089160809400513. [DOI] [PubMed] [Google Scholar]
- 6.Artese L, Piattelli A, Rubini C, Goteri G, Pernotti V, Lessi G, Piccirilli M, Carinci F. p16 expression and odontogenic tumours. Tumori. 2008;94:718–723. doi: 10.1177/030089160809400513. [DOI] [PubMed] [Google Scholar]
- 7.Mendelsohn AH, Lai CK, Shintaku IP, Elashoff DA, Dubinett SM, Abemayor E, St John MA. Histopathologic findings of HPV and p16 positive HNSCC. Laryngoscope. 2010;120(9):1788–1794. doi: 10.1002/lary.21044. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto T, Tanigawa N. The role of survivin as a new target of diagnosis and treatment in human cancer. Med Electron Microsc. 2001;34(4):207–212. doi: 10.1007/s007950100017. [DOI] [PubMed] [Google Scholar]
- 9.Kumamoto H, Ooya K. Expression of survivin and X chromosome-linked inhibitor of apoptosis protein in ameloblastomas. Virchows Arch. 2004;444(2):164–170. doi: 10.1007/s00428-003-0941-9. [DOI] [PubMed] [Google Scholar]
- 10.Kumamoto HIT, Ohki K, Takahashi N, Ooya K. p53 gene status and expression of p53, MDM2, and p14ARF proteins in ameloblastomas. J Oral Pathol Med. 2004;33:292–299. doi: 10.1111/j.0904-2512.2004.00044.x. [DOI] [PubMed] [Google Scholar]
- 11.Sharifi-Sistani N, Zartab H, Babakoohi S, Saghravanian N, Jamshidi S, Esmaili H, Mohtasham N, Zamanzadeh M, Abbaszadeh-Bidokhty H. Immunohistochemical comparison of the expression of p53 and MDM2 proteins in ameloblastomas and keratocystic odontogenic tumors. J Craniofac Surg. 2011;22(5):1652–1656. doi: 10.1097/SCS.0b013e31823188e9. [DOI] [PubMed] [Google Scholar]
- 12.Gadbail AR, Chaudhary M, Patil S, Gawande M. Actual Proliferating Index and p53 protein expression as prognostic marker in odontogenic cysts. Oral Dis. 2009;15(7):490–498. doi: 10.1111/j.1601-0825.2009.01590.x. [DOI] [PubMed] [Google Scholar]
- 13.Gurgel CA, Ramos EA, Azevedo RA, Sarmento VA, da Silva Carvalho AM, dos Santos JN. Expression of Ki-67, p53 and p63 proteins in keratocyst odontogenic tumours: an immunohistochemical study. J Mol Histol. 2008;39(3):311–316. doi: 10.1007/s10735-008-9167-0. [DOI] [PubMed] [Google Scholar]
- 14.Salehinejad JZ-MR, Saghafi S, Jafarian AH, Ghazi N, Rajaei AR, Marouzi P. Immunohistochemical detection of p53 and pCNA in ameloblastoma and adenomatoid odontogenic tumor. J Oral Sci. 2011;53(2):213–217. doi: 10.2334/josnusd.53.213. [DOI] [PubMed] [Google Scholar]
- 15.Chaturvedi AK. Epidemiology and clinical aspects of HPV in head and neck cancers. Head Neck Pathol. 2012;6(Suppl 1):S16–S24. doi: 10.1007/s12105-012-0377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ang K, Sturgis E. Human papillomavirus as a marker of the natural history and response to therapy of head and neck squamous cell carcinoma. Semin Radiat Oncol. 2012;22(2):128–142. doi: 10.1016/j.semradonc.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JS, Jr, Thorstad WL, Chernock RD, Haughey BH, Yip JH, Zhang Q, El-Mofty SK. p16 positive oropharyngeal squamous cell carcinoma:an entity with a favorable prognosis regardless of tumor HPV status. Am J Surg Pathol. 2010;34(8):1088–1096. doi: 10.1097/PAS.0b013e3181e84652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol. 2003;163(2):477–491. doi: 10.1016/S0002-9440(10)63677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi S, Shirasawa H, Sashiyama H, Kawahira H, Kaneko K, Asano T, Ochiai T. P16INK4a expression adenovirus vector to suppress pancreas cancer cell proliferation. Clin Cancer Res. 1999;5(12):4182–4185. [PubMed] [Google Scholar]
- 20.Kumamoto H, Kimi K, Ooya K. Detection of cell cycle-related factors in ameloblastomas. J Oral Pathol Med. 2001;30:309–315. doi: 10.1034/j.1600-0714.2001.300509.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhong Y, Guo W, Wang L, Chen X. Molecular markers of tumor invasiveness in ameloblastoma: an update. Ann Maxillofac Surg. 2011;1(2):145. doi: 10.4103/2231-0746.92780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribeiro BF, Iglesias DP, Nascimento GJ, Galvao HC, Medeiros AM, Freitas RA. Immunoexpression of MMPs-1, -2, and -9 in ameloblastoma and odontogenic adenomatoid tumor. Oral Dis. 2009;15(7):472–477. doi: 10.1111/j.1601-0825.2009.01575.x. [DOI] [PubMed] [Google Scholar]
- 23.Pinheiro JJVFV, Moretti AIS, Jorge AG, Jaeger RG. Local invasiveness of ameloblastoma. Role played by matrix metalloproteinases and proliferative activity. Histopathology. 2004;45:65–72. doi: 10.1111/j.1365-2559.2004.01902.x. [DOI] [PubMed] [Google Scholar]
- 24.Henriques AC, Vasconcelos MG, Galvao HC, de Souza LB, de Almeida Freitas R. Comparative analysis of the immunohistochemical expression of collagen IV, MMP-9, and TIMP-2 in odontogenic cysts and tumors. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(4):468–475. doi: 10.1016/j.tripleo.2011.05.033. [DOI] [PubMed] [Google Scholar]
- 25.Rosenthal EL, Matrisian LM. Matrix metalloproteases in head and neck cancer. Head Neck. 2006;28(7):639–648. doi: 10.1002/hed.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumamoto H, Yamauchi K, Yoshida M, Ooya K. Immunohistochemical detection of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in ameloblastomas. J Oral Pathol Med. 2003;32:114–120. doi: 10.1034/j.1600-0714.2003.00086.x. [DOI] [PubMed] [Google Scholar]
- 27.Yoon HJ, Jo BC, Shin WJ, Cho YA, Lee JI, Hong SP, Hong SD. Comparative immunohistochemical study of ameloblastoma and ameloblastic carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(6):767–776. doi: 10.1016/j.tripleo.2011.06.036. [DOI] [PubMed] [Google Scholar]
- 28.Zhang B, Zhang J, Huang HZ, Chen WL, Tao Q, Zeng DL, Zhang LT, Xu JH. Inhibition of ameloblastoma invasion in vitro and in vivo by inhibitor of metalloproteinase-2 activity. J Oral Pathol Med. 2009;38(9):731–736. doi: 10.1111/j.1600-0714.2009.00771.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang A, Zhang B, Huang H, Zhang L, Zeng D, Tao Q, Wang J, Pan C. Suppression of local invasion of ameloblastoma by inhibition of matrix metalloproteinase-2 in vitro. BMC Cancer. 2008;8:182. doi: 10.1186/1471-2407-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bologna-Molina R, Mosqueda-Taylor A, Lopez-Corella E, de Almeida OP, Carrasco-Daza D, Farfan-Morales JE, Molina-Frechero N, Damian-Matsumura P. Comparative expression of syndecan-1 and Ki-67 in peripheral and desmoplastic ameloblastomas and ameloblastic carcinoma. Pathol Int. 2009;59(4):229–233. doi: 10.1111/j.1440-1827.2009.02355.x. [DOI] [PubMed] [Google Scholar]
- 31.Slootweg PJ. p53 protein and Ki-67 reactivity in epithelial odontogenic lesions. An immunohistochemical study. J Oral Pathol Med. 1995;24(9):393–397. doi: 10.1111/j.1600-0714.1995.tb01207.x. [DOI] [PubMed] [Google Scholar]
- 32.Jaaskelainen KJK, Leivo I, Saloniemi I, Knuutila S, Heikinheimo K. Cell proliferation and chromosomal changes in human ameloblastoma. Cancer Genet Cytogenet. 2002;136:31–37. doi: 10.1016/S0165-4608(02)00512-5. [DOI] [PubMed] [Google Scholar]
- 33.Akrish S, Buchner A, Shoshani Y, Vered M, Dayan D. Ameloblastic carcinoma: report of a new case, literature review, and comparison to ameloblastoma. J Oral Maxillofac Surg. 2007;65(4):777–783. doi: 10.1016/j.joms.2005.11.116. [DOI] [PubMed] [Google Scholar]
- 34.Bello IO, Alanen K, Slootweg PJ, Salo T. Alpha-smooth muscle actin within epithelial islands is predictive of ameloblastic carcinoma. Oral Oncol. 2009;45(9):760–765. doi: 10.1016/j.oraloncology.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Sandra F, Mitsuyasu T, Nakamura N, Shiratsuchi Y, Ohishi M. Immunohistochemical evaluation of PCNA and Ki-67 in ameloblastoma. Oral Oncol. 2001;37(2):193–198. doi: 10.1016/S1368-8375(00)00079-8. [DOI] [PubMed] [Google Scholar]
- 36.Florescu A, Simionescu C, Ciurea R, Pitru A. P53, Bcl-2 and Ki67 immunoexpression in follicular solid ameloblastomas. Rom J Morphol Embryol. 2012;53(1):105–109. [PubMed] [Google Scholar]
- 37.Razavi S, Tabatabaie S, Hoseini A, Hoseini E, Khabazia A. A comparative immunohistochemical study of Ki-67 and Bcl-2 expression in solid ameloblastoma and adenomatoid odontogenic tumor. Dent Res J. 2012;9(2):192–197. doi: 10.4103/1735-3327.95235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soluk Tekkesin M, Mutlu S, Olgac V. Expressions of bax, bcl-2 and Ki-67 in odontogenic keratocysts (keratocystic odontogenic tumor) in comparison with ameloblastomas and radicular cysts. Turk Patoloji Derg. 2012;28(1):49–55. doi: 10.5146/tjpath.2012.01097. [DOI] [PubMed] [Google Scholar]