

Abstract
Clinically, non-invasive carcinomas are confined to the epithelial side of the basement membrane and are classified as benign, whereas invasive cancers invade through the basement membrane and thereby acquire the potential to metastasize. Recent findings suggest that, in addition to protease-mediated degradation and chemotaxis-stimulated migration, basement membrane invasion by malignant cells is significantly influenced by the stiffness of the associated interstitial extracellular matrix and the contractility of the tumor cells that is dictated in part by their oncogenic genotype. In this review, we highlight recent findings that illustrate unifying molecular mechanisms whereby these physical cues contribute to tissue fibrosis and malignancy in three epithelial organs: breast, pancreas, and liver. We also discuss the clinical implications of these findings and the biological properties and clinical challenges linked to the unique biology of each of these organs.
Keywords: Matrix rigidity, tumor invasion, breast cancer, pancreatic cancer, hepatocellular carcinoma
1. Introduction
One hallmark of epithelial cancers is the infiltration of malignant cells through the basement membrane, which heralds the difference between non-lethal neoplastic lesions and invasive cancers with associated high mortality. The basement membrane is an important histologically identifiable barrier between non-invasive (i.e., carcinoma in situ) and invasive forms of cancer. Identifying factors that induce and enable transformed cells to cross this barrier is an active area of investigation. Emerging evidence indicate that basement membrane invasion by malignant cells is substantially influenced by crosstalk between three inter-related factors: stromal stiffening, epithelial cytoskeletal contractility, and growth factor/cytokine signaling (Figure 1). Activation of stromal cells and immune cell infiltration lead to interstitial stromal stiffening in the tumor microenvironment, which stimulates the cytoskeletal contractility of transformed epithelial cells. Conversely, increased epithelial contractility induces greater stromal stiffening as well as formation of invadosomes and production of matrix metalloproteinases (MMPs) required for basement membrane invasion. Importantly, both stromal stiffening and epithelial contractility serve to amplify growth factor and cytokine signaling pathways that are implicated in promoting basement membrane invasion. In this review, we discuss recent data that illustrate these fundamental principles in three different epithelial organs: breast, pancreas, and liver. We highlight the clinical significance of understanding the molecular mechanisms that promote basement membrane invasion in the development of adenocarcinoma in these organs and recent scientific advances that shed light on this important checkpoint in cancer progression. Two unifying themes emerge from synthesis of the literature. First, matrix rigidity is an important risk factor and mediator of tumor progression, and second, oncogenic mutations and signaling pathways act in concert with the stromal microenvironment to promote tumor cell invasion through the basement membrane.
Figure 1.
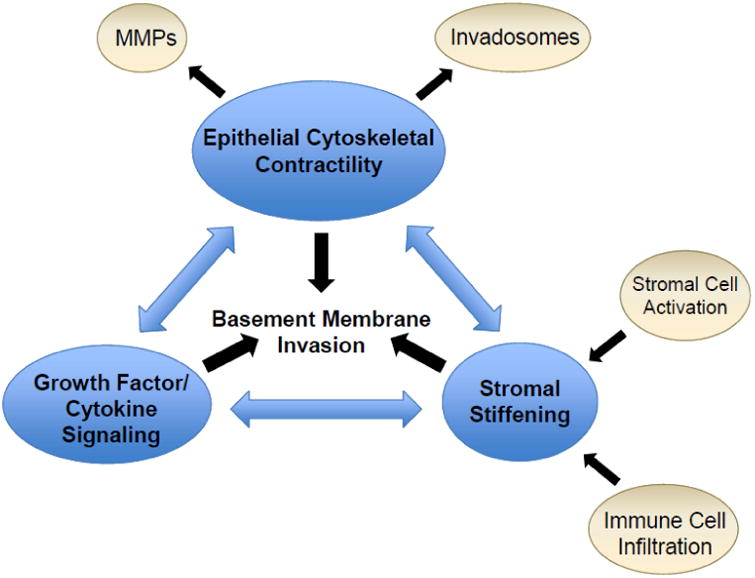
Inter-related and cross-regulating factors that promote basement membrane invasion in cancer progression. MMPs, matrix metalloproteinases.
2. Clinical significance of basement membrane invasion
Breast, pancreas, and liver cancer are three important malignancies of epithelial origin that occur with considerable incidence and affect distinct demographic populations. The three organs have distinct anatomy and microenvironments and the molecular compositions of their basement membranes are different (Figure 2). Clinically, each of these carcinomas pose a unique set of challenges to diagnosis and treatment. However, for all three tumors, breaching of the basement membrane remains the key step that differentiates a lesion that is easily treatable from one that is highly lethal. For each type of cancer, the need to understand the factors that regulate basement membrane invasion is critical for different reasons.
Figure 2.
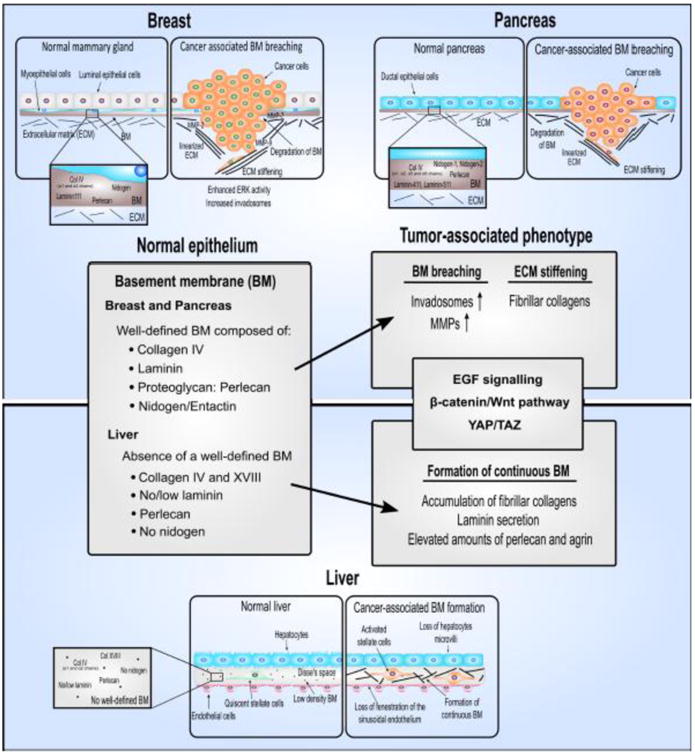
Primary components of the basement membrane in healthy breast, pancreas, and liver tissue and the changes that occur in the basement membrane during development of fibrosis and cancer.
Ductal carcinoma in situ (DCIS) is the earliest detectable form of breast cancer and the most common (80-90%) in situ carcinoma of the breast [1]. Histologically, it is characterized by proliferation of neoplastic cells within the lumen of mammary ducts. The surrounding layer of myoepithelial cells and the basement membrane essentially remain intact in these lesions [2]. This is in contrast to invasive breast cancer (IBC) in which malignant cells invade and break through the basement membrane. Since the advent of screening mammography, the incidence of DCIS diagnosis has increased dramatically and DCIS now accounts for 20% of all breast cancers [3]. DCIS is thought to be a non-obligate precursor to IBC because the vast majority of IBCs are accompanied by adjacent DCIS [4,5]. However, only 20-50% of patients with DCIS later develop IBC in the same quadrant of the same breast [6,7], indicating that a significant proportion of DCIS never progresses to IBC. Although DCIS is non-lethal, IBC is frequently a systemic disease in which patients succumb to metastatic spread of the cancer. Therefore, current recommendations for DCIS treatment include surgery, radiation, and hormone therapy upon diagnosis, which are essentially the same as for IBC, except for systemic chemotherapy. Because of the high rate of DCIS detection in asymptomatic individuals and its poorly quantified risk of progression to IBC, there is concern that prevailing standards of care over-diagnose and over-treat cancers that may never cause illness or death [8]. DCIS and IBC are remarkably similar at the genetic level [9–12] and there are currently no molecular markers to accurately predict progression of DCIS to IBC [3]. Therefore, while active surveillance has been proposed as an alternative management approach to DCIS [13], it remains unclear which patients may be safely watched without aggressive upfront treatment. Therefore, it is within this clinical context that it is critically important to identify the molecular factors that mediate basement membrane invasion in breast cancer: to prognosticate which DCIS tumors may progress to IBC and to identify drug targets that prevent progression to IBC.
Analogous to DCIS and IBC, pancreatic intraepithelial neoplasia (PanIN) is a non-lethal precursor to invasive pancreatic ductal adenocarcinoma (PDAC). PanINs are relatively common lesions found in 16% of pancreatic specimens resected for benign disease processes. The incidence of PanINs increases to 60% in chronic pancreatitis, considered to be a risk factor for PDAC, and PanINs are found in 80% of pancreata with PDAC [14]. There are three grades of PanINs that correspond to progressively greater architectural distortion of the duct, loss of polarity of the columnar epithelia, and nuclear pleomorphism, culminating in PDAC with invasion of the basement membrane. Importantly, acquisition of genetic mutations appear to occur in a stepwise fashion that begins with KRAS2 in PanIN-1, progressing to inactivation of p16/CDKN2A in PanIN-2, and inactivation of TP53 and MAD4/DPC4 (SMAD4) in PanIN-3 [15]. The frequency and rate of progression from PanIN to PDAC is unknown, but a 1% probability of a single PanIN progressing to PDAC has been estimated based on the prevalence of PanIN and PDAC [15].
PDAC is one of the deadliest malignancies. Most patients present with locally advanced or metastatic disease and only 15-20% are candidates for potentially curative surgical resection. The overall 5-year survival of PDAC is only 5%. Therefore, it is highly desirable to be able to identify PanINs that have a high risk of progressing to PDACs and intervene prior to invasive disease. However, in contrast to breast cancer, there are no low-cost non-invasive screening modalities for PanINs that have high sensitivity and specificity. Screening is currently considered investigational and limited to individuals with the highest risk, including those with hereditary pancreatitis, familial forms of cancer susceptibility, and strong family history of PDAC [16]. In addition, because pancreatic resection carries non-trivial risks of morbidity and mortality, the risk-benefit balance of managing non-lethal PanIN lesions is weighted toward a non-surgical approach. Thus, it is paramount to identify effective drug targets that prevent neoplastic cells from invading the basement membrane and the progression of PanIN toward PDAC.
Hepatocellular carcinoma (HCC) is an aggressive tumor with a poor prognosis and is one of the few cancers for which incidence is increasing worldwide [17]. HCC most commonly occurs in the setting of underlying chronic liver disease and continual cycles of necrosis, inflammation, and tissue repair are thought to lead to a characteristic progression of nodular lesions. The multistep process begins with a benign regenerative nodule, and progresses to a low-grade dysplastic nodule, then to a high-grade dysplastic nodule, and ultimately HCC [18]. Interestingly, the earliest mutations detected in this progression are telomerase promoter mutations that result in telomerase reactivation. Telomerase promoter mutations are found in 6% of low-grade dysplastic nodules, 19% of high-grade dysplastic nodules, and 61% of early HCC [19]. Subsequent oncogenic mutations that promote HCC progression are numerous and heterogeneous, but the molecular signatures of the tumors fall into two broad subtypes. One subtype is characterized by activation of cell proliferation signals, which may include the p53, Ras/ERK, or Akt/mTOR pathways, and is more aggressive. The other subtype is dominated by the activation of Wnt/β-catenin signaling [20]. Distinct from this tumor progression process, hepatocellular adenoma is a rare benign tumor that occurs most frequently in women taking oral contraception. Hepatocellular adenomas can be broadly classified into those that harbor hepatocyte nuclear factor 1α mutations or those that carry β-catenin mutations. Adenomas with β-catenin activation have a higher risk of malignant transformation [21].
Hepatocytes are specialized epithelial cells with unique apical-basolateral polarity and in the healthy state, do not have a classically defined basement membrane (Figure 2). During chronic liver disease, the low-density extracellular matrix (ECM) of the liver is progressively replaced by accumulation of fibrillar collagens as fibrosis develops. This process is accompanied by a loss of fenestration of the sinusoidal endothelium, called capillarization of the sinusoids, and physical changes in the hepatocytes in which they lose their microvilli [22,23]. In addition, collagen, laminin, perlecan, and agrin deposition is markedly increased until a continuous basement membrane is eventually formed, which is an important pathological feature of liver fibrosis [24–27]. Due to these unique histological characteristics of the liver, HCC is distinguished from high-grade dysplastic nodules by the invasion of neoplastic cells into the stroma of portal tracts [18]. Early HCC may be treated with curative intent by surgical resection or by liver transplantation if there is significant underlying liver dysfunction. However, most patients (85%) present with advanced disease and have median survival times of less than one year [28].
3. Stromal stiffness as a risk factor for cancer progression
Increased stromal stiffness may serve to promote cancer progression in two distinct ways. First, greater matrix rigidity of the underlying parenchyma may increase the risk of invasive cancer development. Second, the stiffened stroma of the tumor matrix may further promote the invasiveness and aggressiveness of malignant cells. The association between the stiffness of the underlying parenchymal substrate and cancer risk is suggested by epidemiological observations that identify clinical risk factors for various types of cancers. For example, women with high mammographic breast density, which may reflect higher levels of fibrillar collagen deposition and stiffer breast tissue, have a 4.6-fold increased risk of breast cancer as compared to women with low mammographic density [29–31]. Likewise, chronic pancreatitis, with resultant stiffened pancreatic parenchyma due to fibrosis, is a significant risk factor for developing PDAC [32,33]. Importantly, chronic liver disease due to any cause, albeit viral (e.g. hepatitis C), toxic (e.g. alcohol), metabolic (e.g. non-alcoholic steatohepatitis), or inflammatory (e.g. autoimmune hepatitis), eventually progresses to liver fibrosis and cirrhosis. Liver cirrhosis is the most important independent risk factor for HCC development [34]. Eighty to ninety percent of people with HCC have underlying cirrhosis and greater severity of cirrhosis correlates with greater risk of developing HCC. These observations suggest that the milieu of stiffer tissue rigidity may be generally permissive for the development of different types of primary tumors.
Experimental data provide the mechanism through which rigid tissue matrix may increase cancer risk. Matrix rigidity has profound effects on the behavior of non-malignant and malignant mammary epithelial cells. Mammary epithelial cells cultured on soft matrix gels (<400Pa) similar to the rigidity of normal breast tissue form growth-arrested polarized acini with adherens junctions and central lumens [35]. Increasing gel matrix rigidity to the stiffness found in breast tumors (>5kPa) disrupted acini formation and cell polarization, induced integrin clustering, activated focal adhesion kinase (FAK), enhanced Rho-dependent cytoskeletal contractility, and enhanced growth factor-dependent extracellular signal-regulated kinase (ERK) activation. Importantly, mammary epithelial cells transformed by over-expression of epidermal growth factor (EGF) receptor demonstrated disorganized growth and increased Rho-dependent cell spreading on soft gels. Inhibition of Rho/Rho-associated protein kinase (ROCK) signaling and ERK activation restored tissue polarity and reduced proliferation of these transformed epithelial cells. FAK-ROCK-ERK signaling leads to increased expression of a breast cancer-associated proliferative transcriptome and potentiation of cell growth and survival [35–38]. Inhibiting myosin contractility, ROCK, or ERK could reduce cell growth and survival. Reducing ERK activity decreased the expression of more than 95% of the tension-induced genes to that observed in mammary epithelial cells cultured on low-density gels. These findings suggest that ECM stiffness per se likely promotes ERK-dependent growth and survival signaling to modulate mammary tissue behavior [35–37]. Importantly, a direct link between ECM stiffness, focal adhesion assembly, and growth factor receptor signaling was illustrated by a series of studies in which focal adhesions were enhanced through expression of a mutant β1-integrin and ERK signaling in response to EGF stimulation in mammary epithelial cells on a soft ECM [35]. Moreover, increased matrix rigidity also modulates microRNAs (miRNAs) that regulate malignancy. miRNA-18a expression is upregulated through integrin-dependent stimulation of Myc and Wnt in response to a stiffer tissue ECM stroma and promotes mammary tumor progression by reducing the levels of the tumor suppressor phosphatase and tensin homolog (PTEN) in the mammary epithelium [39]. Interestingly, high miRNA-18a expression levels correlate with ECM rigidity and poor prognosis in patients with luminal breast cancers, providing a mechanistic link between increased matrix stiffness and risk of malignant progression [39]. These findings suggest that stromal rigidity has an important regulatory role both in pre-malignant and malignant epithelial cells along the spectrum of breast cancer development.
Recent findings suggest a mechanism through which increased tissue rigidity of fibrotic livers may create an environment permissive for HCC development. Liver fibrosis is associated with increased liver tissue stiffness, as determined by non-invasive imaging modalities such as ultrasound-based transient and shear-wave elastography [40] and magnetic resonance elastography [41]. Atomic force microscopy measurements demonstrate that normal liver matrix is around 150Pa and is increased to 1-6kPa in experimental models of liver fibrosis [42]. Increased matrix stiffness activates stellate cells and portal fibroblasts, the primary cell types responsible for abnormal ECM deposition during the development of fibrosis [43,44]. Importantly, increased matrix rigidity also has a direct inhibitory effect on hepatocyte function by activating mechano-signal transduction through FAK and decreasing expression of hepatocyte nuclear factor 4 alpha (HNF4α), a master transcriptional regulator of hepatic function. Moreover, expression of HNF4α in primary hepatocytes on stiff matrix can be rescued by inhibition of ROCK signaling, indicating a critical role of the FAK-ROCK signaling network in modulating hepatocyte function in response to tissue mechanics [42]. In addition to regulating function, HNF4α is also a tumor suppressor. HNF4α expression is reduced in fibrotic livers and forced re-expression of HNF4α can attenuate the progression of fibrosis [42,45,46]. Reduced expression of HNF4α expression promotes hepatocyte proliferation [47] and activation of mesenchymal genes [48]. Moreover, HNF4α expression is also reduced in HCC and forced re-expression of HNF4α inhibits tumor progression [49–54]. These findings suggest that increased matrix rigidity in fibrotic livers may create an environment permissive for HCC development by decreasing HNF4α expression in hepatocytes. Increased matrix stiffness increases the proliferation and chemotherapeutic resistance of HCC cell lines [55]. In addition, FAK signaling is required for the progression of a c-Met/β-catenin driven in vivo mouse model of HCC [56], suggesting that the stiffened matrix of a fibrotic liver may activate FAK signaling to promote HCC progression.
In addition to the rigidity of the parenchymal substrate, it has become increasingly evident that collagen deposition, remodeling, and cross-linking of the tumor stroma also play a key role in promoting tumor cell invasion (Figure 3). A biophysical analysis of human breast tumors revealed that there is an increase in total collagen that becomes progressively thickened and linearized as the nonmalignant normal breast transitions to DCIS, and thereafter to IBC. The invasive front of the tumors, in particular, was found to display a consistent and significant enhancement of aligned collagen fibers that atomic force microscopy indentation quantified as stiffer. The regions of tumor tissue that were stiffer also showed enhanced mechano-signaling, as demonstrated by activated β1 integrin, FAK, myosin light chain, and nuclear localization of Yes-associated protein (YAP) [57].
Figure 3.
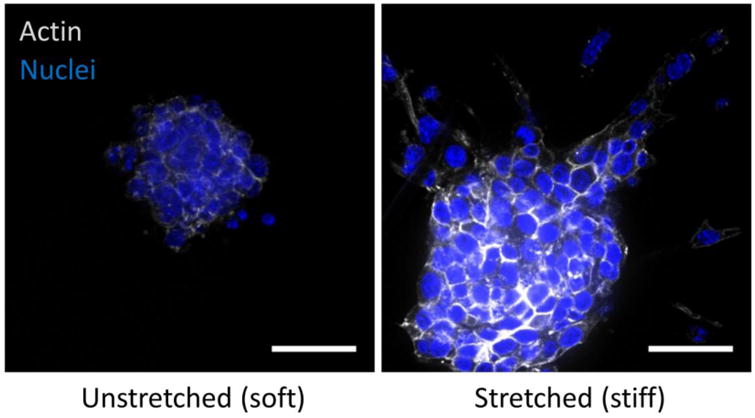
Malignant mammary epithelial cells cultured in reconstituted basement membrane as spherical multicellular organoids and then embedded in collagen hydrogels placed under either 0 (unstretched - soft) or 10% strain (stretched - stiff) in a tension bioreactor. After three days, cultures were fixed and stained for actin cytoskeleton (phalloidin) and nuclei (DAPI) to assess the extent of tumor cell invasion and migration (scale bar = 20 μm).
Moreover, PDAC is an example of a highly fibrotic tumor in which the stiffened tumor stroma may play an important role in promoting tumor aggressiveness and mediating therapy resistance. Survival of patients with PDAC does not correlate with total fibrillar collagen content within the tumor, but is significantly linked to increased thickness of collagen fibers adjacent to PDAC epithelium [58]. Importantly, tumor genotype significantly modulates surrounding tumor stromal fibrosis and tumor progression. Genotypic analysis of PDACs has led to the notion that there may be distinct genotypes/phenotypes in PDACs that could be broadly divided into two categories: classical and basal-like. Basal-like PDACs are associated with worse patient prognosis and are characterized by perturbed transforming growth factor β (TGFβ) signaling with mutant SMAD4 or significantly lower SMAD4 expression [59–61]. In genetically engineered mouse models, Kras mutation and reduced TGFβ signaling in the pancreatic epithelia lead to ROCK-dependent increased cellular contractility, activation of signal transducer and activator of transcription 3 (STAT3), increased YAP activity, induced expression of connective tissue growth factor, and ECM remodeling, resulting in deposition of thicker and stiffer collagen fibers [58]. A similar phenotype of increased fibrosis and PDAC development was recapitulated in mice with the Kras mutation and expression of a β1-integrin clustering mutant with elevated mechano-signal transduction, indicating that cytoskeletal contractility of epithelia could drive stiffening of the periductal ECM. These findings further illustrate the dynamic interplay between the tumor genotype and the phenotypic expression of the surrounding tumor stroma.
4. Force-mediated invasion of the basement membrane
Invasion through the basement membrane requires malignant cells to acquire a motile phenotype, which involves the manifestation of actin-rich protrusive structures at their leading migratory edge, termed invadosomes. In addition, to pass through the basement membrane, invading cells must secrete MMPs that break down the basement membrane ECM proteins. Formation of invadosomes and secretion of MMPs are interconnected processes and both are stimulated by stiffened matrix. Membrane type 1 (MT1)-MMP, MMP-2, and MMP-9 are concentrated within invadosomes and serve the critical function of ECM lysis at the invading edge of the cell [62–64]. MT1-MMP is a master switch proteinase because it activates MMP-2 and MMP-13 and directly degrades a host of ECM species including collagen types I, II, III, and IV, laminin 1 and 5, fibronectin, vitronectin, aggrecan, and fibrin [65]. MMP-2 and MMP-9 are both type IV collagenases shown to be important for the degradation of the basement membrane in tumor invasion [66]. Invadosomes are essential for MMP-dependent invasion of cells across ECM barriers [67]. Moreover, live cell imaging demonstrated that degradation of the ECM quickly followed the recruitment of MT1-MMP to the invadosome [68], linking invadosome formation and ECM degradation as key steps to basement membrane invasion.
Rho/ROCK-mediated actomyosin contractility is a major regulator of invadosome formation and turnover [69], which in turn is modulated by matrix rigidity. Increased ECM stiffness directly increases both the number and activity of invadosomes, thereby promoting invasion [70]. Activated FAK and p130Cas localize to invadosomes, suggesting that they may be transducing the mechanical cues of a stiffened matrix into downstream signals that promote further ECM degradation activity. Formation of invadosomes in breast carcinoma cells is regulated by matrix stiffness [70–72], and stiff ECM prolongs the lifespan of invadosomes [73]. Activation of the Wnt pathway leads to high expression of MMP-7, a transcriptional target of β-catenin, which is associated with the functional loss of the PTEN gene, the most common first event associated with the triple-negative breast cancer subtype [74]. Phosphoinositide 3-kinases has been shown to be involved in invadosome formation because its knockdown results in reduction in the number of invadosomes [75–77]. Many growth factors have been shown to affect invadopodia formation and activity [75]. Among them, EGF induces invadopodia formation [78,79], TGF-β [80,81], heparin binding (HB)-EGF [82,83], vascular endothelial growth factor [84], and hepatocyte growth factor [85] increase invadopodia numbers. Many of these growth factor pathways converge on common signaling hubs, especially Src kinase, phosphoinositide 3-kinases, and Rho family GTPases, which ultimately control invadopodia and podosomes [86,87]. These fundamental mechanisms are likely adopted for many types of invasive cancers since invadosome formation has been shown to be important in the progression of PDAC [88–91] and HCC [92–95] as well.
Importantly, increased matrix stiffness and epithelial contractility synergizes with growth factor and cytokine signaling to induce the invasive phenotype in malignant cells. Relevant for IBC, stiffened matrix gels enhanced growth factor-dependent ERK activation in mammary epithelial cells [35]. While culture of non-malignant mammary epithelial cells within stiff matrix increased colony size and disrupted acini formation, it was not sufficient to induce an invasive phenotype. On the other hand, activation of epidermal growth factor receptor 2 (HER2/neu) signaling alone induced cell proliferation and filling of the acinar lumen, but also failed to promote tumor cell invasion. It was when there was both stiffened matrix and HER2/neu activation that the mammary epithelial cells invaded into the gels along collagen fibrils [37]. The ability of tissue mechanics to potentiate growth factor or cytokine signaling was also demonstrated in PDAC in which loss of TGFβ signaling and elevated β1-integrin mechano-signal transduction enhanced STAT3 signaling to induce further matricellular fibrosis and tumor aggressiveness [58]. Similarly, in HCC, stiffened matrix enhanced hepatocyte growth factor-dependent ERK, AKT, STAT3, and cyclin D1 activation [55]. These findings suggest that matrix rigidity and cellular contractility potentiate growth factor- and cytokine-regulated oncogenic pathways implicated in invasion across distinct cancer types. Indeed, the cooperation between tissue mechanics and soluable factors may be a fundamental mechanism that extends beyond tumor biology since matrix stiffness also modifies the response of embryonic stem cells to morphogens [96].
5. Conclusion
Recent findings that demonstrate the pathogenic role of elevated mechano-signal transduction in the breast, pancreas, and liver illustrate the generalizability of these fundamental mechanisms as well as the unique pathophysiological features and clinical challenges of each disease process. In all three organs, greater interstitial matrix rigidity, either of the underlying parenchymal substrate or the remodeled tumor stroma, is a significant risk factor for tumor development and progression. These examples show that matrix rigidity can cooperate with and cross-regulate oncogenes in different ways. In IBC, increased EGF receptor signaling synergizes with FAK activation and Rho-dependent cytoskeletal contractility to promote tumor invasion. In PDAC, SMAD4 mutations leading to altered TGFβ signaling can induce increased tumor stromal stiffening and promote tumor aggressiveness. In the liver, increased matrix rigidity in fibrotic liver disease inhibits the expression of the HNF4α tumor suppressor and creates an environment permissive for HCC development. However, all three types of cancers likely use similar fundamental mechanisms of force-mediated invadosome formation and MMP-dependent ECM degradation to invade through the basement membrane. Importantly, tissue mechanics and epithelial contractility amplify growth factor and cytokine signaling pathways implicated in promoting the invasive phenotype of all three tumors. As illustrated by the three epithelial organs we highlighted in this review, the similarities and differences in how epithelial cells interact with their interstitial environment will provide important insights into the mechanisms that enable adenocarcinomas to breach the basement membrane and the development of potential targeted therapies to limit this progression.
Highlight.
Invasion of malignant cells through the basement membrane is mediated through three inter-related factors: stromal stiffening, epithelial cytoskeletal contractility, and growth factor/cytokine signaling. In this review, we highlight the similar as well as different ways these factors promote tumor progression in three distinct epithelial cancers.
Acknowledgments
We thank Laralynne Przybyla and Fuiboon Kai for the image of malignant mammary epithelial invasion. This work was supported by NIH/NCI grants U01-CA202241-01, R01-CA192914-01, and USMRAA (DOD) BC122990 to V.M.W. as well as NIH K08-DK093708 and P30-DK026743 to T.T.C.
Abbreviations
- DCIS
Ductal carcinoma in situ
- EGF
Epidermal growth factor
- ECM
Extracellular matrix
- ERK
Extracellular signal-regulated kinase
- FAK
Focal adhesion kinase
- HCC
Hepatocellular carcinoma
- HNF4α
Hepatocyte nuclear factor 4 alpha
- HER2/neu
Human epidermal growth factor receptor 2
- IBC
Invasive breast cancer
- MMPs
Matrix metalloproteinases
- PDAC
Pancreatic ductal adenocarcinoma
- PanIN
Pancreatic intraepithelial neoplasia
- PTEN
Phosphatase and tensin homolog
- ROCK
Rho/Rho-associated protein kinase
- STAT3
Signal transducer and activator of transcription 3
- TGFβ
Transforming growth factor β
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allred DC. Ductal carcinoma in situ: terminology, classification, and natural history. J Natl Cancer Inst Monogr. 2010;2010:134–138. doi: 10.1093/jncimonographs/lgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, Marchio C, Reis-Filho JS. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology. 2010;57:171–192. doi: 10.1111/j.1365-2559.2010.03568.x. [DOI] [PubMed] [Google Scholar]
- 3.Cowell CF, Weigelt B, Sakr RA, Ng CK, Hicks J, King TA, Reis-Filho JS. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol. 2013;7:859–869. doi: 10.1016/j.molonc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allred DC, Wu Y, Mao S, Nagtegaal ID, Lee S, Perou CM, Mohsin SK, O'Connell P, Tsimelzon A, Medina D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14:370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 5.Evans AJ, Pinder SE, Snead DR, Wilson AR, Ellis IO, Elston CW. The detection of ductal carcinoma in situ at mammographic screening enables the diagnosis of small, grade 3 invasive tumours. Br J Cancer. 1997;75:542–544. doi: 10.1038/bjc.1997.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Page DL, Dupont WD, Rogers LW, Jensen RA, Schuyler PA. Continued local recurrence of carcinoma 15-25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76:1197–1200. doi: 10.1002/1097-0142(19951001)76:7<1197::aid-cncr2820760715>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Sanders ME, Schuyler PA, Dupont WD, Page DL. The natural history of low-grade ductal carcinoma in situ of the breast in women treated by biopsy only revealed over 30 years of long-term follow-up. Cancer. 2005;103:2481–2484. doi: 10.1002/cncr.21069. [DOI] [PubMed] [Google Scholar]
- 8.Harding C, Pompei F, Burmistrov D, Welch HG, Abebe R, Wilson R. Breast Cancer Screening, Incidence, and Mortality Across US Counties. JAMA Intern Med. 2015;175:1483–1489. doi: 10.1001/jamainternmed.2015.3043. [DOI] [PubMed] [Google Scholar]
- 9.Hannemann J, Velds A, Halfwerk JB, Kreike B, Peterse JL, van de Vijver MJ. Classification of ductal carcinoma in situ by gene expression profiling. Breast Cancer Res. 2006;8:R61. doi: 10.1186/bcr1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter D, Lahti-Domenici J, Keshaviah A, Bae YK, Argani P, Marks J, Richardson A, Cooper A, Strausberg R, Riggins GJ, Schnitt S, Gabrielson E, Gelman R, Polyak K. Molecular markers in ductal carcinoma in situ of the breast. Mol Cancer Res. 2003;1:362–375. [PubMed] [Google Scholar]
- 12.Buerger H, Otterbach F, Simon R, Poremba C, Diallo R, Decker T, Riethdorf L, Brinkschmidt C, Dockhorn-Dworniczak B, Boecker W. Comparative genomic hybridization of ductal carcinoma in situ of the breast-evidence of multiple genetic pathways. J Pathol. 1999;187:396–402. doi: 10.1002/(SICI)1096-9896(199903)187:4<396∷AID-PATH286>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 13.Ryser MD, Worni M, Turner EL, Marks JR, Durrett R, Hwang ES. Outcomes of Active Surveillance for Ductal Carcinoma in Situ: A Computational Risk Analysis. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol. 2003;16:996–1006. doi: 10.1097/01.MP.0000087422.24733.62. [DOI] [PubMed] [Google Scholar]
- 15.Hruban RH, Maitra A, Goggins M. Update on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol. 2008;1:306–316. [PMC free article] [PubMed] [Google Scholar]
- 16.Canto MI, Hruban RH, Fishman EK, Kamel IR, Schulick R, Zhang Z, Topazian M, Takahashi N, Fletcher J, Petersen G, Klein AP, Axilbund J, Griffin C, Syngal S, Saltzman JR, Mortele KJ, Lee J, Tamm E, Vikram R, Bhosale P, Margolis D, Farrell J, Goggins M. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142:795–796. doi: 10.1053/j.gastro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrick JL, Kelly SP, Altekruse SF, McGlynn KA, Rosenberg PS. Future of Hepatocellular Carcinoma Incidence in the United States Forecast Through 2030. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.64.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roskams T, Kojiro M. Pathology of early hepatocellular carcinoma: Conventional and molecular diagnosis. Semin Liver Dis. 2010;30:17–25. doi: 10.1055/s-0030-1247129. [DOI] [PubMed] [Google Scholar]
- 19.Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, Zucman-Rossi J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–1992. doi: 10.1002/hep.27372. [DOI] [PubMed] [Google Scholar]
- 20.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–1239. doi: 10.1053/j.gastro.2015.05.061. [DOI] [PubMed] [Google Scholar]
- 21.Zucman-Rossi J, Jeannot E, Van Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, Bacq Y, Leteurtre E, Paradis V, Michalak S, Wendum D, Chiche L, Fabre M, Mellottee L, Laurent C, Partensky C, Castaing D, Zafrani ES, Laurent-Puig P, Balabaud C, Bioulac-Sage P. Genotype-phenotype correlation in hepatocellular adenoma: New classification and relationship with HCC. Hepatology. 2006;43:515–524. doi: 10.1002/hep.21068. [DOI] [PubMed] [Google Scholar]
- 22.Friedman SL. The Cellular basis of hepatic strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 23.Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim Biophys Acta - Mol Basis Dis. 2013;1832:876–883. doi: 10.1016/j.bbadis.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Clément B, Rescan PY, Baffet G, Loréal O, Lehry D, Campion JP, Guillouzo A, Diseases L. Hepatocytes may produce laminin in fibrotic liver and in primary culture. Hepatology. 1988;8:794–803. doi: 10.1002/hep.1840080417. [DOI] [PubMed] [Google Scholar]
- 25.Maher JJ, Friedman SL, Roll FJ, Bissell DM. Immunolocalization of laminin in normal rat liver and biosynthesis of laminin by hepatic lipocytes in primary culture. Gastroenterology. 1988;94:1053–1062. doi: 10.1016/0016-5085(88)90566-5. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi FB, Biagini G, Ballardini G, Cenacchi G, Faccani A, Pisi E, Laschi R, Liotta L, Garbisa S. Basement membrane production by hepatocytes in chronic liver disease. Hepatology. 1984;4:1167–1172. doi: 10.1002/hep.1840040612. [DOI] [PubMed] [Google Scholar]
- 27.Hahn E, Wick G, Pencev D, Timpl R. Distribution of basement membrane proteins in normal and fibrotic human liver: collagen type IV, laminin, and fibronectin. Gut. 1980;21:63–71. doi: 10.1136/gut.21.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crissien AM, Frenette C. Current management of hepatocellular carcinoma. Gastroenterol Hepatol (N Y) 2014;10:153–61. doi:2014 Mar;10(3):153-61. [PMC free article] [PubMed] [Google Scholar]
- 29.Boyd NF, Li Q, Melnichouk O, Huszti E, Martin LJ, Gunasekara A, Mawdsley G, Yaffe MJ, Minkin S. Evidence That Breast Tissue Stiffness Is Associated with Risk of Breast Cancer. PLoS One. 2014;9:1–8. doi: 10.1371/journal.pone.0100937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyd NF, Martin LJ, Bronskill M, Yaffe MJ, Duric N, Minkin S. Breast Tissue Composition and Susceptibility to Breast Cancer. J Natl Cancer Inst. 2010;102 doi: 10.1093/jnci/djq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyd NF, Martin LJ, Yaff MJ, Minkin S. Mammographic density and breast cancer risk : current understanding and future prospects. Breast Cancer Res. 2011;13:1–12. doi: 10.1186/bcr2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bang UC, Benfield T, Hyldstrup L, Bendtsen F, Beck Jensen JE. Mortality, cancer, and comorbidities associated with chronic pancreatitis: a Danish nationwide matched-cohort study. Gastroenterology. 2014;146:989–994. doi: 10.1053/j.gastro.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 33.Duell EJ, Lucenteforte E, Olson SH, Bracci PM, Li D, Risch HA, Silverman DT, Ji BT, Gallinger S, Holly EA, Fontham EH, Maisonneuve P, Bueno-de-Mesquita HB, Ghadirian P, Kurtz RC, Ludwig E, Yu H, Lowenfels AB, Seminara D, Petersen GM, La Vecchia C, Boffetta P. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4) Ann Oncol. 2012;23:2964–2970. doi: 10.1093/annonc/mds140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology. 2004 doi: 10.1053/j.gastro.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 35.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 36.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–4343. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubashkin MG, Cassereau L, Bainer R, Dufort CC, Yui Y, Ou G, Paszek MJ, Davidson MW, Chen Y, Weaver VM. Force Engages Vinculin and Promotes Tumor Progression by Triphosphate. Cancer Res. 2014;74:4597–4612. doi: 10.1158/0008-5472.CAN-13-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, Ou G, Wijekoon AC, Levental KR, Gilbert PM, Hwang ES, Chen YY, Weaver VM. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferraioli G, Tinelli C, Dal Bello B, Zicchetti M, Filice G, Filice C, Liver G. Fibrosis Study, Accuracy of real-time shear wave elastography for assessing liver fibrosis in chronic hepatitis C: a pilot study. Hepatology. 2012;56:2125–2133. doi: 10.1002/hep.25936. [DOI] [PubMed] [Google Scholar]
- 41.Loomba R, Wolfson T, Ang B, Hooker J, Behling C, Peterson M, Valasek M, Lin G, Brenner D, Gamst A, Ehman R, Sirlin C. Magnetic resonance elastography predicts advanced fibrosis in patients with nonalcoholic fatty liver disease: a prospective study. Hepatology. 2014;60:1920–1928. doi: 10.1002/hep.27362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, Espanol-Suner R, Willenbring H, Weaver VM, Chang TT. Physiological Ranges of Matrix Rigidity Modulate Primary Mouse Hepatocyte Function In Part Through Hepatocyte Nuclear Factor 4 Alpha. Hepatology. 2016 doi: 10.1002/hep.28450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- 44.Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B, Uemura M, Janmey PA, Wells RG. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–8. doi: 10.1152/ajpgi.00412.2010. doi:ajpgi.00412.2010[pii]10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W, Hu PF, Deng X, Tan YX, Zhang JP, Ning BF, Shi J, Zhang X, Wang HY, Lin Y, Xie WF. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut. 2010;59:236–46. doi: 10.1136/gut.2008.174904. [DOI] [PubMed] [Google Scholar]
- 46.Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, Fukumitsu K, Handa K, Tian J, Kaestner KH, Vodovotz Y, Locker J, Soto-Gutierrez A, Fox IJ. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest. 2015;125:1533–1544. doi: 10.1172/JCI73137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonzo Ja, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J Biol Chem. 2012;287:7345–56. doi: 10.1074/jbc.M111.334599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, Bonzo JA, Gonzalez FJ, Alonzi T, Amicone L, Tripodi M. The stable repression of mesenchymal program is required for hepatocyte identity: A novel role for hepatocyte nuclear factor 4?? Hepatology. 2011;53:2063–2074. doi: 10.1002/hep.24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lazarevich NL, Shavochkina Da, Fleishman DI, Kustova IF, Morozova OV, Chuchuev ES, Patyutko YI. Deregulation of hepatocyte nuclear factor 4 (HNF4)as a marker of epithelial tumors progression. Exp Oncol. 2010;32:167–171. doi:46/862[pii] [PubMed] [Google Scholar]
- 50.Fitamant J, Kottakis F, Benhamouche S, Tian HS, Chuvin N, Parachoniak CA, Nagle JM, Perera RM, Lapouge M, Deshpande V, Zhu AX, Lai A, Min B, Hoshida Y, Avruch J, Sia D, Campreciós G, Mc Clatchey AI, Llovet JM, Morrissey D, Raj L, Bardeesy N. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep. 2015;10:1692–1707. doi: 10.1016/j.celrep.2015.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ning BF, Ding J, Yin C, Zhong W, Wu K, Zeng X, Yang W, Chen YX, Zhang JP, Zhang X, Wang HY, Xie WF. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010;70:7640–7651. doi: 10.1158/0008-5472.CAN-10-0824. [DOI] [PubMed] [Google Scholar]
- 52.Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in Mice Is Associated with a Downregulation in the Expression of Hepatocyte Nuclear Factors. Hepatology. 2004;39:1038–1047. doi: 10.1002/hep.20155. [DOI] [PubMed] [Google Scholar]
- 53.Yin C, Lin Y, Zhang X, Chen YX, Zeng X, Yue HY, Hou JL, Deng X, Zhang JP, Han ZG, Xie WF. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology. 2008;48:1528–1539. doi: 10.1002/hep.22510. [DOI] [PubMed] [Google Scholar]
- 54.Späth GF, Weiss MC. Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J Cell Biol. 1998;140:935–946. doi: 10.1083/jcb.140.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, Benten D, Forbes SJ, Wells RG, Iredale JP. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shang N, Arteaga M, Zaidi A, Stauffer J, Cotler SJ, Zeleznik-Le NJ, Zhang J, Qiu W. FAK Is required for c-Met/β-catenin-driven hepatocarcinogenesis. Hepatology. 2015;61:214–226. doi: 10.1002/hep.27402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol. 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016 doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, Danenberg KL, Tempero MA, Spellman PT, Hanahan D, Gray JW. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–504. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailey P, Chang DK, Nones K, Johns AL, Patch A, Gingras M, Miller DK, Christ AN, Bruxner TJC, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Giry-laterriere M, Rooman I, Chou A, Pajic M, Scarlett CJ, Andreia V, Kench JG, Lovell JA, Merrett ND, Christopher W, Musgrove EA, Bailey UH, Hofmann O, Sutherland RL, Wheeler DA, Waddell N, Andrew V. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 61.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, Bentrem DJ, Talamonti MS, Iacobuzio-Donahue CA, Hollingsworth MA, Yeh JJ. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lagarrigue F, Dupuis-Coronas S, Ramel D, Delsol G, Tronchère H, Payrastre B, Gaits-Iacovoni F. Matrix metalloproteinase-9 is upregulated in nucleophosmin-anaplastic lymphoma kinase-positive anaplastic lymphomas and activated at the cell surface by the chaperone heat shock protein 90 to promote cell invasion. Cancer Res. 2010;70:6978–6987. doi: 10.1158/0008-5472.CAN-10-0861. [DOI] [PubMed] [Google Scholar]
- 63.Stetler-Stevenson WG, Kelly T, Lin CY, Yeh Y, Mueller SC, Chen WT. Binding and Localization of Mr 72,000 Matrix Metalloproteinase at Cell Surface Invadopodia. Cancer Res. 1993;53:3159–3164. [PubMed] [Google Scholar]
- 64.Takahara H, Howard L, Thompson EW, Sato H, Seiki M, Yeh Y, Chen WT. Transmembrane/cytoplasmic domain-mediated membrane type 1-matrix metalloprotease docking to invadopodia is required for cell invasion. Proc Natl Acad Sci U S A. 1997;95:7959–7964. doi: 10.1073/pnas.94.15.7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poincloux R, Lizárraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–24. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 66.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- 67.Hagedorn EJ, Ziel JW, Morrissey MA, Linden LM, Wang Z, Chi Q, Johnson SA, Sherwood DR. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J Cell Biol. 2013;201:903–913. doi: 10.1083/jcb.201301091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: Defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 69.Linder S, Wiesner C, Himmel M. Degrading Devices : Invadosomes in Proteolytic Cell Invasion. Annu Rev Cell Dev Biol. 2011;27:185–211. doi: 10.1146/annurev-cellbio-092910-154216. [DOI] [PubMed] [Google Scholar]
- 70.Alexander NR, Branch KM, Parekh A, Clark ES, Iwueke IC, Guelcher SA. Extracellular Matrix Rigidity Promotes Invadopodia Activity. Curr Biol. 2008;18:1295–1299. doi: 10.1016/j.cub.2008.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parekh A, Ruppender NS, Branch KM, Lin J, Boyer PD, Candiello JE, Merryman WD, Guelcher SA, Weaver AM. Sensing and Modulation of Invadopodia across a Wide Range of Rigidities. Biophys J. 2011;100:573–582. doi: 10.1016/j.bpj.2010.12.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, Charriere GM, Maridonneau-parini I. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun. 2014;5:5343. doi: 10.1038/ncomms6343. [DOI] [PubMed] [Google Scholar]
- 73.Collin O, Tracqui P, Stephanou A, Usson Y, Clément-lacroix J, Planus E. Spatiotemporal dynamics of actin-rich adhesion microdomains: influence of substrate flexibility. J Cell Sci. 2005;119:1914–1925. doi: 10.1242/jcs.02838. [DOI] [PubMed] [Google Scholar]
- 74.Dey N, Young B, Abramovitz M, Bouzyk M, Barwick B, De P. Differential Activation of Wnt-β-Catenin Pathway in Triple Negative Breast Cancer Increases MMP7 in a PTEN Dependent Manner. PLoS One. 2013;8:1–17. doi: 10.1371/journal.pone.0077425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoshino D, Branch KM, Weaver AM. Signaling inputs to invadopodia and podosomes. J Cell Biol. 2013;126:2979–2989. doi: 10.1242/jcs.079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoshino D, Jourquin J, Emmons SW, Miller T, Goldgof M, Costello K, Tyson DR, Brown B, Lu Y, Prasad NK, Zhang B, Mills GB, Yarbrough WG, Quaranta V, Seiki M, Weaver AM. Network Analysis of the Focal Adhesion to Invadopodia Transition Identifies a PI3K-PKC a Invasive Signaling Axis. Sci Signal. 2012;5:1–15. doi: 10.1126/scisignal.2002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamaguchi H, Yoshida S, Muroi E, Yoshida N, Kawamura M, Kouchi Z, Nakamura Y, Sakai R, Fukami K. Phosphoinositide 3-kinase signaling pathway mediated by p110a regulates invadopodia formation. J Cell Biol. 2011;193:1275–1288. doi: 10.1083/jcb.201009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mader CC, Oser M, Magalhaes MAO, Bravo-cordero JJ, Condeelis J, Koleske AJ, Gil-henn H. An EGFR – Src – Arg – Cortactin Pathway Mediates Functional Maturation of Invadopodia and Breast Cancer Cell Invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mandal S, Johnson KR, Wheelock MJ. TGF- β induces formation of F-actin cores and matrix degradation in human breast cancer cells via distinct signaling pathways. Exp Cell Res. 2008;314:3478–3493. doi: 10.1016/j.yexcr.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 81.Pignatelli J, Tumbarello DA, Schmidt RP, Turner CE. Hic-5 promotes invadopodia formation and invasion during TGF- b –induced epithelial– mesenchymal transition. J Cell Biol. 2012;197:421–437. doi: 10.1083/jcb.201108143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201:279–292. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hayes KE, Walk EL, Ammer AG, Kelley LC, Martin KH, Weed SA. Ableson kinases negatively regulate invadopodia function and invasion in head and neck squamous cell carcinoma by inhibiting an HB-EGF autocrine loop. Oncoge. 2013;32:4766–4777. doi: 10.1038/onc.2012.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lucas JT, Jr, Salimath BP, Slomiany MG, Rosenzweig SA. Regulation of invasive behavior by vascular endothelial growth factor is HEF1-dependent. Oncogene. 2010;29:4449–4459. doi: 10.1038/onc.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rajadurai CV, Havrylov S, Zaoui K, Vaillancourt R, Stuible M, Naujokas M, Zuo D, Tremblay ML, Park M. Met receptor tyrosine kinase signals through a cortactin – Gab1 scaffold complex, to mediate invadopodia. J Cell Sci. 2012;125:2940–2953. doi: 10.1242/jcs.100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Linder S, Aepfelbacher M. Podosomes : adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/S0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 87.Murphy DA, Courtneidge SA. The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haage A, Schneider IC. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014;28:3589–3599. doi: 10.1096/fj.13-245613. [DOI] [PubMed] [Google Scholar]
- 89.Neel NF, Rossman KL, Martin TD, Hayes TK, Yeh JJ, Der CJ. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol Cell Biol. 2012;32:1374–86. doi: 10.1128/MCB.06291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Welsch T, Keleg S, Bergmann F, Bauer S, Hinz U, Schmidt J. Actinin-4 expression in primary and metastasized pancreatic ductal adenocarcinoma. Pancreas. 2009;38:968–76. doi: 10.1097/MPA.0b013e3181b28d6f. [DOI] [PubMed] [Google Scholar]
- 91.Botta GP. Constitutive K-Ras(G12D power) Activation of ERK2 Specifically Regulates Oncogenesis of Pancreatic Cancer Cells in 3D. 2012 doi: 10.1158/1541-7786.MCR-11-0399. http://search.proquest.com/docview/1037812716?accountid=50433. [DOI] [PMC free article] [PubMed]
- 92.Chakraborty S, Lakshmanan M, Swa HLFF, Chen J, Zhang X, Ong YS, Loo LS, Akıncılar SC, Gunaratne J, Tergaonkar V, Hui KM, Hong W. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun. 2015;6:6184. doi: 10.1038/ncomms7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wong CCL, Wong CM, Tung EKK, Man K, Ng IOL. Rho-kinase 2 is frequently overexpressed in hepatocellular carcinoma and involved in tumor invasion. Hepatology. 2009;49:1583–1594. doi: 10.1002/hep.22836. [DOI] [PubMed] [Google Scholar]
- 94.Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH, Chen SP, Bian XW, Guan XY, Lin MC, Zeng YX, Kung HF, Xie D. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut. 2012;61:278–289. doi: 10.1136/gut.2011.239145. [DOI] [PubMed] [Google Scholar]
- 95.Chen P, Xi Q, Wang Q, Wei P. Downregulation of microRNA-100 correlates with tumor progression and poor prognosis in colorectal cancer. Med Oncol. 2014;31:235. doi: 10.1007/s12032-014-0235-x. [DOI] [PubMed] [Google Scholar]
- 96.Przybyla L, Lakins JN, Weaver VM. Tissue Mechanics Orchestrate Wnt-Dependent Human Embryonic Stem Cell Differentiation. Cell Stem Cell. 2016:1–14. doi: 10.1016/j.stem.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]