

Abstract
Objectives
To assess the accuracy of blood lactate and L:P molar as a screen for mitochondrial, respiratory chain, or fatty acid oxidation disorders in children with pediatric acute liver failure (PALF); to determine if serum lactate ≥ 2.5 mmol/L or L:P molar ratio ≥ 25 correlated with biochemical variables of clinical severity; and to determine if lactate or L:P molar is associated with clinical outcome at 21 days.
Study design
Retrospective review of demographic, clinical, laboratory, and outcome data for PALF study group participants who had lactate and pyruvate levels collected on the same day.
Results
Of 986 participants, 110 had lactate and pyruvate levels collected on the same day. Of the 110, the etiology of PALF was a mitochondrial disorder in eight (7%), indeterminate in 65 (59%), and an alternative diagnosis in 37 (34%). Lactate, pyruvate, and L:P molar ratio were similar among the three etiologic groups. There was no significant association between the initial lactate or L:P molar ratio and biochemical variables of clinical severity or clinical outcome at 21 days.
Conclusions
A serum lactate ≥ 2.5 mmol/L and/or elevated L:P molar ratio was common in all causes of PALF, not limited to those with a mitochondrial etiology, and did not predict 21 day clinical outcome.
Keywords: mitochondrial liver disease, biomarker, liver transplant
Pediatric acute liver failure (PALF) is a rare and potentially fatal illness caused by heterogeneous insults including infections, drugs and toxins, autoimmune liver disease, genetic/metabolic diseases, ischemia/reperfusion injury, and, in a relatively large proportion, a cause is not identified (1). PALF continues to have a high mortality rate and is the third leading indication in childhood for liver transplantation (2). Primary mitochondrial disorders, including mitochondrial DNA (mtDNA) depletion syndromes, occur in 1 in 5000 live births and are a known cause of PALF in children under 2 years of age (3–5). One of the biochemical features suggestive of a mitochondrial disorder is a serum lactate concentration ≥2.5mmol/L, especially in cases with respiratory chain alterations or mtDNA depletion syndrome(6). However, elevated blood lactate levels can be caused by factors other than primary mitochondrial function including poor collection technique, tissue hypoxia/ischemia, thiamine deficiency, or secondary mitochondrial dysfunction (7, 8). Conversely, patients with certain mitochondrial diseases, such as DNA polymerase gamma (POLG1)-associated disease, MPV17 deficiency, Leber Hereditary Optic Neuropathy (LHON), Leigh disease, Kearns-Sayre syndrome, and complex I deficiency, may have normal or minimally elevated lactate levels even in the setting of a metabolic crisis (9). The lactate to pyruvate (L:P) molar ratio is proposed to be a better screening test for mitochondrial disorders, as the L:P molar ratio reflects the equilibrium between the product and substrate of the reaction catalyzed by lactase dehydrogenase and indirectly reflects the NADH: NAD+ cytoplasmic redox state(10). When cellular respiration or mitochondrial oxidative metabolism is impaired, such as in inborn errors of components of the mitochondrial respiratory chain, there is an increase in reducing equivalents (excess NADH and absence of NAD+) which results in an elevated L:P molar ratio. In the past an L:P molar ratio ≥25 has been considered to be highly suggestive of respiratory chain dysfunction(11). However, an elevated lactate or an elevated L:P molar ratio could also represent secondary mitochondrial dysfunction occurring as a result of severe liver disease. Therefore, we sought to understand whether lactate and the L:P molar ratio could be used to accurately distinguish primary mitochondrial causes of PALF. Additionally, we sought to determine whether elevated lactate and an elevated L:P molar ratio represented more severe liver disease and thus predicted poor clinical outcomes.
The Mitochondrial Liver Diseases Working Group of the Childhood Liver Disease Research Network (ChiLDReN) recently recommended screening for mitochondrial disorders in infants and children with severe liver dysfunction, including those presenting with PALF, by examining for elevated blood lactate and the L:P molar ratio (12). However, the role and utility of these screening tests for mitochondrial disorders in patients with PALF have not been systematically determined. We hypothesized that an elevated serum lactate ≥ 2.5 mmol/L in combination with an elevated L:P molar ratio ≥25 would identify patients with primary mitochondrial causes of PALF and that these elevated laboratory values early during PALF onset would be predictive of clinical outcomes. To address these hypotheses, we utilized the PALF Study Group dataset to: (1) assess the accuracy of blood lactate and L:P molar ratio as a screen for mitochondrial, respiratory chain, or fatty acid oxidation disorders in children with PALF; (2) determine if serum lactate ≥2.5 mmol/L or L:P molar ratio ≥ 25 correlated with biochemical variables that reflect clinical severity in children with PALF; and (3) determine if lactate level or L:P molar ratio is associated with clinical outcomes at 21 days in PALF patients.
Methods
Data were obtained from the PALF Study Group registry, a National Institutes of Health-supported, multicenter, prospective study initiated in 1999 that collects data and specimens on children <18 years of age with PALF from 24 participating centers in the United States, Canada, and the United Kingdom (ClinicalTrials.gov: NCT00986648). Definitions used and study methodology have been previously reported (1, 13). The enrollment criteria for PALF required 1) the presence of severe hepatic dysfunction occurring within 8 weeks of onset of illness, 2) no known underlying chronic liver disease, and 3) a liver-based coagulopathy (not corrected with vitamin K) with an international normalized ratio (INR) ≥1.5 or prothrombin time (PT) ≥15 seconds in patients with encephalopathy or an INR ≥2.0 or PT ≥ 20 seconds in patients without encephalopathy. Enrollment occurred as soon as possible after hospital admission to the study site. The study protocol was approved by the individual centers’ Institutional Review Boards. Evaluation and management of each participant was based on local standard-of-care, however, the PALF study group had agreed-upon guidelines for optimal evaluation of PALF at different ages (1). Data collected by each site were transmitted to a central data-coordinating center for data editing and quality control procedures.
Participants in the PALF study dataset who had a serum lactate and pyruvate concentration obtained on the same day (within 7 days of study enrollment) were identified. Demographic information including sex, race, ethnicity, age at enrollment, was collected. Laboratory data used in this analysis included the following: the first serum lactate and pyruvate levels drawn on the same day, the resulting calculated L:P molar ratio, and aspartate aminotransferase (AST), alanine transaminase (ALT), and INR drawn on the same day as the lactate and pyruvate levels. Minimum glucose during the first 7 days of enrollment was also used in analysis. Participants were classified into three diagnosis groups based on the final determination of the underlying etiology of PALF: 1) primary mitochondrial disease diagnosis, 2) other confirmed cause (e.g., infectious, toxic or drug-induced [including acetaminophen], autoimmune, genetic, ischemia and others), and 3) indeterminate cause (no other etiology determined). For this analysis, the term primary mitochondrial disease includes subjects with a final diagnosis of mitochondrial, respiratory chain, or fatty acid oxidation disorders. Clinical outcome at 21 days (alive without liver transplant, death without transplant, or liver transplantation) was recorded for each participant.
Statistical Analyses
Descriptive statistics were used to characterize the participants by age at enrollment, sex, race, ethnicity and baseline lab values. Due to small sample size, the power to detect clinically meaningful differences between the three diagnosis groups is minimal; therefore, p-values are not shown for the comparison among the three diagnostic categories. Spearman correlations were used to estimate the association between lactate, the L:P molar ratio, and laboratory values within each diagnostic group. The exact Pearson Chi-square test was used to determine if the proportion of outcomes differed between the two lactate groups (<2.5 or ≥ 2.5) and the two L:P molar ratio groups (ratio <25 or ≥ 25). All statistical analyses were performed using SAS 9.3 software (SAS Institute, Cary, NC).
Results
For this study, data were analyzed for participants enrolled between December 27, 1999 and December 31, 2010. Of 986 participants in the PALF Study dataset, 537 had a serum lactate level recorded and, of these, 110 had both serum lactate and pyruvate drawn on the same day and were included in this analysis. The median time between hospital admission and enrollment in the PALF Study was 2 days (IQR: 1–4 days). Likewise, the median time between hospital admission and lactate and pyruvate measurement was 2 days (IQR: 1–4 days). Of these 110, 74 (67%) had a lactate level ≥ 2.5 mmol/L (Figure 1; available at www.jpeds.com). The median age at enrollment of these participants was 2.0 years (interquartile range 0.5–4.8 years) and 63 (57%) were male. Eight (7.3%) participants had a final diagnosis of a mitochondrial disorder, 37 (33.6%) had another confirmed diagnosis (other diagnosis group), and 65 (59.1%) had an indeterminate diagnosis. Of the eight participants with a final diagnosis of mitochondrial disorder, three had mitochondrial disease, three had a respiratory chain disorder, and two had drug-induced liver injury (non-acetaminophen) plus mitochondrial disease. Age, sex, race, and Hispanic ethnicity were similar among the three diagnosis groups (Table I). Baseline AST and ALT were lower in the mitochondrial diagnosis group.
Figure 1.

(online only): Flowchart of Lactate and Pyruvate Availability in all PALF Subject
Table 1.
Demographics and Clinical Characteristics of Subjects
| All participants with lactate and pyruvate values on same day | All (n = 110) | Mito Group (n= 8) | Indeterminate group (n = 65) | Other group (n = 37) |
|---|---|---|---|---|
|
| ||||
| Age at enrollment (yrs) | ||||
| Median | 2.0 | 0.8 | 2.0 | 2.3 |
| Q1 – Q3 | 0.5 – 4.8 | 0.2 – 2.5 | 0.7 – 6.2 | 0.1 – 7.6 |
|
| ||||
| Male | 63 (57%) | 7(88%) | 35(54%) | 21(57%) |
|
| ||||
| Race | ||||
| White | 87(81%) | 6(75%) | 54 (84%) | 27(77%) |
| Black | 14(13%) | 2(25%) | 8 (13%) | 4(11%) |
| Asian | 3(3%) | 0(0%) | 2 (3%) | 1(3%) |
| American Indian | 3 (3%) | 0(0%) | 0 (0%) | 3(9%) |
| Unknown | 3 | 0 | 1 | 2 |
|
| ||||
| Hispanic | 24(21%) | 2(25%) | 8 (22%) | 14(22%) |
|
| ||||
| Baseline Lab Measures | ||||
|
| ||||
| AST (IU/L) | ||||
| n | 109 | 8 | 64 | 37 |
| Median | 1668.0 | 251.5 | 1530.0 | 2359.0 |
| Q1–Q3 | 630.0 – 3038.0 | 125.0 – 831.0 | 651.5–2964.5 | 973.0–4880.0 |
|
| ||||
| ALT (IU/L) | ||||
| n | 109 | 8 | 64 | 37 |
| Median | 1293.0 | 96.0 | 1339.5 | 1513.0 |
| Q1–Q3 | 289.0 – 2690.0 | 25.5 – 213.0 | 472.4–3140.5 | 526.0–2273.0 |
|
| ||||
| AST/ALT | ||||
| n | 109 | 8 | 64 | 37 |
| Median | 1.40 | 2.73 | 1.23 | 1.86 |
| Q1–Q3 | 0.93 – 2.38 | 1.94–4.80 | 0.74 – 1.72 | 1.07 – 2.55 |
|
| ||||
| INR | ||||
| n | 100 | 6 | 60 | 34 |
| Median | 3.0 | 3.8 | 3.2 | 2.8 |
| Q1–Q3 | 2.1– 4.4 | 2.9– 5.3 | 2.1 – 5.1 | 2.2–3.9 |
|
| ||||
| Glucose (mg/dL) | ||||
| n | 110 | 8 | 65 | 37 |
| Median | 105.5 | 84.5 | 103.0 | 107.0 |
| Q1–Q3 | 78.0–130.0 | 57.0–134.0 | 77.0–119.0 | 91.0–136.0 |
|
| ||||
| Lactate:Pyruvate (L:P) Molar Ratio | ||||
| n | 110 | 8 | 65 | 37 |
| Median | 22.5 | 20.6 | 24.4 | 22.7 |
| Q1–Q3 | 15.0–33.3 | 12.8–36.2 | 15.9–31.5 | 15.0–48.0 |
| Min-Max | 2.8–170.0 | 12.0–125.7 | 2.8–120.0 | 4.5–170.0 |
Lactate, Pyruvate, and the L:P Molar Ratio Were Similar in All Diagnostic Groups
Examining the distribution of lactate and pyruvate levels by diagnosis group, lactate tended to be higher in the mitochondrial disease group but there was considerable overlap among the three groups (Figure 2). Pyruvate values were comparable among the three groups.(Figure 2). The L:P molar ratio was elevated in subjects in all three groups (Figure 2 and Table I). Surprisingly, only 25% (2/8) of the participants in the mitochondrial group had an elevated L:P molar ratio (≥25), whereas 46% (17/37) in the other diagnosis group, and 48% (31/65) in the indeterminate diagnosis group had elevated L:P molar ratio. We next examined the L:P molar ratio as a function of serum lactate or pyruvate for each diagnosis group (Figure 3). Lactate and L:P molar ratio were correlated in all 3 diagnostic groups with the strongest correlation being in the mitochondrial group (r=0.81 p=0.015), followed by the other diagnoses: (r= 0.62, p<0.001), and indeterminate groups (r=0.48, p<0.001). Pyruvate levels did not correlate with L:P molar ratio; thus the blood lactate was primarily responsible for the elevation of L:P molar ratio in individual subjects.
Figure 2.
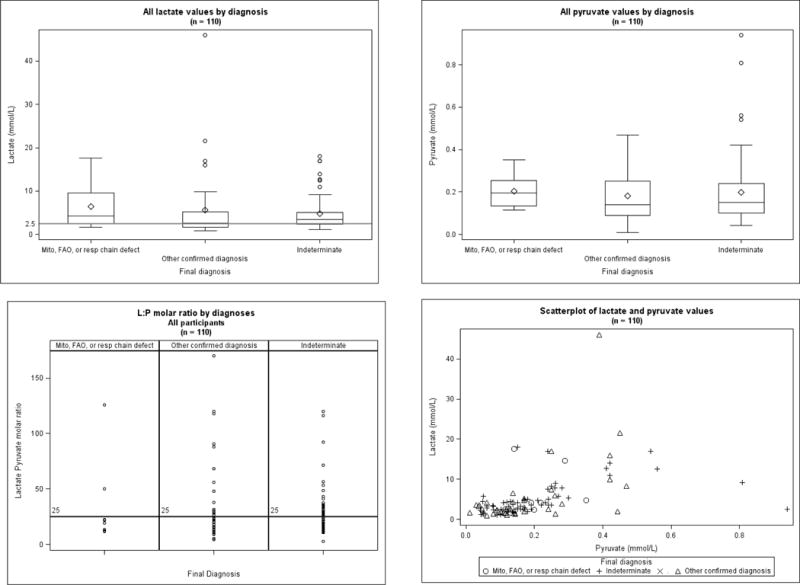
Distribution of blood lactate levels, pyruvate levels, and lactate: pyruvate molar ratios of PALF participants (n=110)
Figure 3.
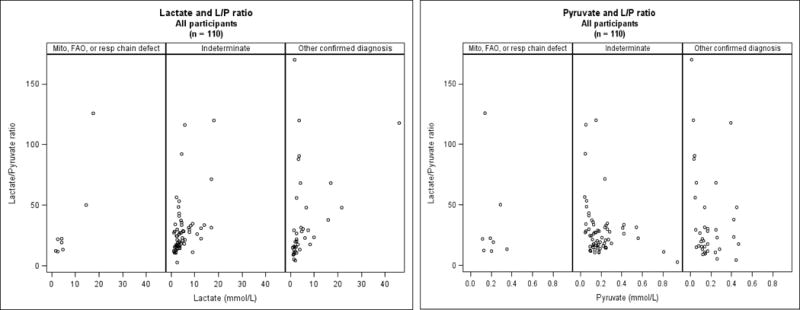
Lactate to Pyruvate (L:P) Molar Ratio Relationship to Serum Lactate or Pyruvate Levels by Diagnostic Group (n=110)
The Association Between Lactate and L:P Molar Ratio with Biochemical Variables that Reflect Clinical Severity in Children with PALF
We next examined the correlations between L:P molar ratio and lactate level with ALT, AST, the AST/ALT ratio (a proposed indicator of mitochondrial injury), INR and minimum glucose levels within each diagnostic group (Tables II and III; available at www.jpeds.com). No significant relationships of the L:P molar ratio with any of these clinical variables were observed in any of the diagnosis groups. Lactate level was associated with INR in the other confirmed diagnosis group (r = 0.64, p < 0.001). The correlation coefficient in the mitochondrial group was similar in magnitude (0.54 for INR), however the number of participants in the mitochondrial group was small and the correlations were not statistically different from zero.
Table 2.
(online only): Correlations between L:P molar ratio and biochemical values
| Lactate and pyruvate measured on same day (n = 110) |
Mitochondrial (n = 8) |
Confirmed Other (n=37) |
Indeterminate diagnosis (n=65) |
|
|---|---|---|---|---|
|
| ||||
| L:P molar ratio | ||||
| n | 110 | 8 | 37 | 65 |
| Median | 22.5 | 20.6 | 22.7 | 24.4 |
| Q1, Q3 | 15.0, 33.3 | 12.8, 36.2 | 15.0, 47.9 | 15.9, 31.5 |
| Min, Max | 2.8, 170.0 | 12.0, 125.7 | 4.5, 170.0 | 2.8, 120.0 |
|
| ||||
| Spearman correlations with L:P | ||||
|
| ||||
| AST/ALT ratio (on same day as L:P) | ||||
| n | 105 | 8 | 35 | 62 |
| Median | 1.3 | 2.8 | 1.7 | 1.1 |
| Q1, Q3 | 0.9, 2.4 | 2.1, 4.8 | 1.0, 2.6 | 0.6, 1.7 |
| Min, Max | 0.2, 16.3 | 0.9, 16.3 | 0.4, 5.4 | 0.2, 6.5 |
| correlation with L:P | 0.11 | 0.05 | 0.29 | 0.07 |
|
| ||||
| INR (on same day as L:P) | ||||
| n | 95 | 6 | 32 | 57 |
| Median | 2.9 | 4.1 | 2.7 | 3.1 |
| Q1, Q3 | 2.0, 4.4 | 2.5, 5.3 | 2.0, 3.7 | 2.1, 4.8 |
| Min, Max | 1.2, 25.9 | 1.6, 8.2 | 1.3, 5.5 | 1.2, 25.9 |
| correlation with L:P | 0.03 | 0.20 | 0.28 | −0.07 |
|
| ||||
| Minimum Glucose (mg/dl) | ||||
| n | 110 | 8 | 37 | 65 |
| Median | 90.5 | 76.0 | 93.0 | 86.0 |
| Q1, Q3 | 71, 119 | 57.0, 116.0 | 77.0, 134.0 | 69.0, 118.0 |
| Min, Max | 1.6, 681 | 47.0, 173.0 | 1.6, 681.0 | 27.0, 327.0 |
| correlation with L:P | 0.16 | −0.01 | 0.22 | 0.20 |
Table 3.
(online only): Correlations between Lactate and biochemical values
| Lactate (n = 110) | Mitochondrial (n = 8) |
Confirmed Other (n=37) |
Indeterminate diagnosis (n=65) |
|
|---|---|---|---|---|
|
| ||||
| Lactate (mmol/L) | ||||
| n | 110 | 8 | 37 | 65 |
| Median | 3.3 | 4.2 | 2.6 | 3.4 |
| Q1, Q3 | 2.2, 5.2 | 2.5, 9.6 | 1.7, 5.2 | 2.4, 5.1 |
| Min, Max | 0.9, 46.0 | 1.6, 17.6 | 0.9, 46.0 | 1.1,18.0 |
|
| ||||
| Spearman correlations with Lactate | ||||
|
| ||||
| AST/ALT ratio (on same day as Lactate) | ||||
| n | 105 | 8 | 35 | 62 |
| Median | 1.3 | 2.8 | 1.7 | 1.1 |
| Q1, Q3 | 0.9, 2.4 | 2.1, 4.8 | 1.0, 2.6 | 0.6, 1.7 |
| Min, Max | 0.2, 16.3 | 0.9, 16.3 | 0.4, 5.4 | 0.2, 6.5 |
| correlation with Lactate | 0.15 | 0.48 | 0.41 | 0.01 |
|
| ||||
| INR (on same day as Lactate) | ||||
| n | 95 | 6 | 32 | 57 |
| Median | 2.9 | 4.1 | 2.7 | 3.1 |
| Q1, Q3 | 2.0, 4.4 | 2.5, 5.3 | 2.0, 3.7 | 2.1, 4.8 |
| Min, Max | 1.2, 25.9 | 1.6, 8.2 | 1.3, 5.5 | 1.2, 25.9 |
| correlation with Lactate | 0.28*** | 0.54 | 0.64*** | 0.10 |
|
| ||||
| Minimum Glucose (mg/dl) | ||||
| n | 110 | 8 | 37 | 65 |
| Median | 90.5 | 76.0 | 93.0 | 86.0 |
| Q1, Q3 | 71.0, 119.0 | 57.0, 116.0 | 77.0, 134.0 | 69.0, 118.0 |
| Min, Max | 1.6, 681.0 | 47.0, 173.0 | 1.6, 681.0 | 27.0, 327.0 |
| correlation with | 0.02 | −0.23 | 0.08 | 0.09 |
p < 0.01
p < 0.01
p < 0.001
P-values within diagnoses are adjusted using Stepdown Bonferroni method.
Relationship Between Lactate, L:P Molar Ratio, and Clinical Outcome
Finally, we examined the relationship between initial lactate and L:P molar ratio with clinical outcomes (alive, death, or transplant) at 21 days after enrollment. Outcomes (Table IV) did not differ significantly based on either lactate level (<2.5 or ≥2.5) or L:P molar ratio (<25 or ≥25). We next examined clinical outcomes based on combination of lactate (<2.5 or ≥2.5 mmol/L) and L:P molar ratio (<25 or ≥25). Outcomes were not significantly different among these four combinations (Table IV).
Table 4.
21 Day Clinical Outcomes for participants with lactate and pyruvate measured on day 0 (n= 92)
| Alive | Transplant | Dead | p-value | |
|---|---|---|---|---|
| L:P ≥ 25 (n = 44) | 20 (45%) | 13 (30%) | 11 (25%) | 0.51 |
| L:P < 25 (n = 48) | 27 (56%) | 13 (27%) | 8 (17%) |
| Alive | Transplant | Dead | p-value | |
|---|---|---|---|---|
| Lactate ≥2.5 (n = 65) | 32(49%) | 19(29%) | 14(21%) | 0.87 |
| Lactate <2.5(n= 27) | 15(56%) | 7(26%) | 5(19%) |
| Alive | Transplant | Dead | p-value | |
|---|---|---|---|---|
| L ≥2.5 and L:P ≥25 (N = 37) | 17 (46%) | 10(27%) | 10(27%) | 0.80 |
| L <2.5 and L:P ≥25 (N= 7) | 3 (43%) | 3(43%) | 1 (14%) | |
| L≥2.5 and L:P <25 (n = 28) | 15(54%) | 9(32%) | 4 (14%) | |
| L <2.5 and L:P <25 (n = 20) | 12(60%) | 4(20%) | 4(20%) |
| Results from nominal logistic model of 21 day outcomes for participants with lactate and pyruvate measured on day 0 (n = 92) | |||
|---|---|---|---|
| Odds Ratio (reference is survival) | 95 % Confidence Interval | p-value | |
| Death | |||
| Lactate ≥ 2.5 | 1.09 | 0.31–3.80 | 0.89 |
| L:P ratio ≥ 25 | 1.82 | 0.59–5.59 | 0.30 |
| LTx | |||
| Lactate ≥ 2.5 | 1.18 | 0.39–3.55 | 0.77 |
| L:P ratio ≥ 25 | 1.30 | 0.48–3.53 | 0.61 |
Discussion
Establishing a diagnosis of a primary mitochondrial disorder in a pediatric patient who presents in ALF is of critical importance as it can impact prognosis and treatment choices, including the decision about whether to consider liver transplantation as a potential therapeutic option. However, establishing the diagnosis may be complicated and requires a high index of suspicion as children with mitochondrial disease can have heterogeneous clinical presentations with any combination of brain, muscle, heart, kidney and liver manifestations. This multicenter study evaluated the utility of the readily available serum lactate and the L:P molar ratios as biomarkers to suggest a mitochondrial cause of PALF. We found that elevated serum lactate level and an elevated L:P molar ratio were relatively common in all causes of PALF and were not specific for a mitochondrial etiology. In addition, there was no relationship between the L:P molar ratio with biochemical indicators of the severity of liver injury or with clinical outcomes (alive, death, or liver transplantation). Thus, L:P molar ratios appear to have limited clinical utility in children presenting with PALF.
There are several possible explanations for the poor performance of serum lactate or L:P molar ratio in distinguishing primary mitochondrial from other causes of PALF. First, it is possible that an underlying respiratory chain defect was present in a subset of participants who were ultimately diagnosed with another cause of PALF or were classified as indeterminate, perhaps increasing their susceptibility to or actually causing the liver failure. In this regard, Helbling et al (6) examined for mtDNA depletion in liver explants removed at the time of liver transplantation in 28 children with known non-mitochondrial chronic liver diseases and 45 with PALF (divided into those with viral, drug-induced and unknown causes) compared with healthy controls and patients with known genetically confirmed mtDNA depletion. Levels of mtDNA were in the definite range of mtDNA depletion in 34% of the PALF patients but in none of the chronic liver disease patients. Furthermore, 7 of 43 (16%) PALF patients vs. 7 of 240 (3%) controls who underwent gene sequencing had single variants in mtDNA depletion genes (DGUOK or POLG), suggesting that the heterozygote state for these gene variants may be a risk factor for PALF. Thus, elevated serum lactate level and L:P molar ratio observed in many of our PALF study participants from all diagnosis groups may indicate underlying non-diagnosed gene variants causing or predisposing to mitochondrial dysfunction.
A second possible explanation for our findings is that non-mitochondrial based liver injury may lead secondarily to reduced functioning of components of mitochondrial respiratory complexes causing altered redox status, culminating in elevated serum lactate and L:P molar ratio in PALF patients regardless of etiology. Supporting this secondary mitochondrial dysfunction hypothesis are recent data provided by Lane et al (9) who used sophisticated techniques to assess mitochondrial function in end-stage liver disease. These investigators performed blue native polyacrylamide gel electrophoresis (PAGE) with immunodetection of respiratory chain complexes I–V, measured biochemical activity of respiratory chain complexes II and IV and quantified mtDNA copy number in 45 explanted livers removed during transplantation. Abnormal mitochondrial function was frequently present in this cohort: ten of 40 patients (25 %) had a defect of one or more respiratory chain enzyme complexes on blue native PAGE, 20 patients (44 %) had low activity of complex II and/or IV, and ten (22 %) had a reduced mtDNA copy number. Importantly, combined respiratory chain deficiency and reduced amounts of mtDNA were detected in all three patients with ALF, none of whom had pathologic variants in DGUOK, POLG or MPV17, the three most common genetic causes of mtDNA depletion and liver failure. All six patients diagnosed with liver tumors showed variable alterations in mitochondrial function. These authors concluded that mitochondrial dysfunction may occur secondary to a wide range of liver diseases of non-mitochondrial etiology, supporting secondary mitochondrial dysfunction as a cause of elevated lactate and L:P molar ratio in our PALF participants.
A final possible explanation for our findings is that the elevated lactate level and L:P molar ratio were caused by altered fluid balance, hypotension, impaired tissue perfusion, hypoxia or other factors that commonly occur in critically ill PALF patients. Although classic teaching that an elevated L:P molar ratio is characteristic of primary mitochondrial respiratory chain dysfunction and could be used to differentiate between primary mitochondrial dysfunction and these other causes, in fact, secondary altered cellular respiration can similarly elevate the L:P molar ratio(10).
There are several limitations to this study. First, there was no uniform standard procedure mandated in the PALF study for obtaining specimens for lactate and pyruvate, thus there may be bias for which patients had these levels obtained. In addition, lactate levels can be artificially elevated if a patient is moving during phlebotomy or if there is prolonged use of a tourniquet when obtaining the blood sample. Likewise, pyruvate levels can be inaccurate if the blood sample is taken when the patient is post-prandial or if the sample is not immediately transferred into 8% perchlorate on ice within 30 seconds of being drawn(7, 8, 10). However, each site in the PALF study is a major pediatric center experienced in collecting specimens for clinical use of these tests. In addition, although spurious values of either lactate or pyruvate can affect the L:P molar ratio, we expect that artificial elevation in L:P molar ratios would have uniformly affected all three diagnosis groups. The second limitation is that based on the design of the PALF Study, follow-up information was only collected for 21 days after enrollment. Therefore, clinical outcome results beyond that time period were not available. The third limitation is that although this multicenter study is the largest study to date of L:P molar ratios in PALF, the number of participants was relatively small (110 patients total, only 8 of whom had mitochondrial disorders) because only 11% of participants had same day determinations of lactate and pyruvate, limiting our power to detect significant correlations. In addition, there may be bias in terms of which subjects had lactate and pyruvate levels obtained based on clinical suspicion. The data from this study suggest that PALF participants who were younger and white, and who had higher baseline AST/ALT ratios and INR, were more likely to have lactate and pyruvate measures drawn on the same day (Table V; available at www.jpeds.com). Finally, the data from this study is only applicable to children in acute liver failure and cannot be extrapolated to the diagnostic work-up for mitochondrial disorders in other settings.
Table 5.
(online only): Demographics and Clinical Characteristics of all participants in PALF
| All participants with lactate and pyruvate values on same day | All (n = 986) |
In Lactate/Pyruvate analysis (n=110) |
Not in Lactate/Pyruvate Analysis (n = 876) |
P value |
|---|---|---|---|---|
|
| ||||
| Age at enrollment (yrs) | <0.001 | |||
| Median | 4.6 | 2.0 | 5.5 | |
| Q1 – Q3 | 0.8–13.3 | 0.5 – 4.8 | 0.9–13.7 | |
|
| ||||
| Male | 495 (50%) | 63 (57%) | 432(49%) | 0.12 |
|
| ||||
| Race | 0.03 | |||
| White | 706(73%) | 87(81%) | 619 (72%) | |
| Black | 129(13%) | 14(13%) | 115(13%) | |
| Asian | 86(9%) | 3(3%) | 83 (10%) | |
| American Indian | 16(2%) | 3(3%) | 13 (2%) | |
| Mixed | 32(3%) | 0(0%) | 32 (4%) | |
| Unknown | 17 | 3 | 14 | |
|
| ||||
| Hispanic | 193(20%) | 24(21%) | 86(19%) | 0.53 |
|
| ||||
| Baseline Lab Measures | ||||
|
| ||||
| AST (IU/L) | 0.96 | |||
| n | 878 | 109 | 769 | |
| Median | 1664.5 | 1668.0 | 1664.0 | |
| Q1–Q3 | 436– 3584 | 630.0 – 3038.0 | 422.0–3599.0 | |
|
| ||||
| ALT (IU/L) | 0.20 | |||
| n | 878 | 109 | 769 | |
| Median | 1507.5 | 1293.0 | 1544.0 | |
| Q1–Q3 | 426 – 3318 | 289.0 – 2690.0 | 429.0–3349.0 | |
|
| ||||
| AST/ALT | 0.011 | |||
| n | 878 | 109 | 769 | |
| Median | 1.18 | 1.40 | 1.16 | |
| Q1–Q3 | 0.75–1.95 | 0.93 – 2.38 | 0.73 – 1.89 | |
|
| ||||
| INR | 0.011 | |||
| n | 890 | 100 | 790 | |
| Median | 2.6 | 3.0 | 2.5 | |
| Q1–Q3 | 2.0–3.8 | 2.1– 4.4 | 2.0 – 3.8 | |
|
| ||||
| Glucose (mg/dL) | 0.13 | |||
| n | 921 | 110 | 811 | |
| Median | 97.0 | 105.5 | 96.0 | |
| Q1–Q3 | 75.0–123.0 | 78.0–130.0 | 74.0–123.0 | |
In summary, the findings of this study suggest that mitochondrial dysfunction reflected by serum lactate and the L:P molar ratio is common in PALF. Although it is possible that a significant number of patients being diagnosed with indeterminate PALF truly have an undiagnosed primary mitochondrial disorder, it seems much more likely that secondary mitochondrial dysfunction occurs in a large number of PALF patients independent of cause of acute liver failure. The data in this study clearly suggest that the diagnosis of a mitochondrial disorder and subsequent clinical decisions (including consideration for liver transplant) should not be based solely on blood lactate or the L:P molar ratio. In the future, genetic testing using next generation sequencing techniques (including whole exome sequencing or targeted gene panels) with rapid turn-around may become available to quickly identify those children with a primary mitochondrial etiology of PALF(14).
Acknowledgments
We thank Edward Doo, MD, and Averell H. Sherker, MD (National Institutes of Health), as well as members of the Data Coordinating Center, University of Pittsburgh (directed by Steven H. Belle, PhD, MScHyg).
Funded by National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health (U01 DK072146 and T32 DK067009).
List of Abbreviations
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- INR
International normalized ratio
- L
P: Lactate: pyruvate molar ratio
- MtDNA
Mitochondrial DNA
- PALF
Pediatric acute liver failure
- PT
Prothrombin time
Appendix
Additional members of the PALF Study Group include:
Current Sites, Principal Investigators, and Coordinators –Kathryn Bukauskas, RN, CCRC, Madeline Schulte, RN, BSN, Clinical Research Coordinator (Children’s Hospital of Pittsburgh of UPMC, Pittsburgh, Pennsylvania); Michelle Hite, MA, CCRC (Children’s Hospital Colorado, Aurora, Colorado); Kathleen M. Loomes, MD, Elizabeth B. Rand, MD, David Piccoli, MD, Deborah Kawchak, MS, RD, Christa Seidman, Clinical Research Coordinator (Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania); Rene Romero, MD, Saul Karpen, MD, PhD, Liezl de la Cruz-Tracy, CCRC (Emory University, Atlanta, Georgia); Vicky Ng, MD, Kelsey Hunt, Clinical Research Coordinator (Hospital for Sick Children, Toronto, Ontario, Canada); Girish C. Subbarao, MD, Ann Klipsch, RN, Sarah Munson, Clinical Research Coordinator (Indiana University Riley Hospital, Indianapolis, Indiana); Lisa Sorenson, PhD, Susan Kelly, RN, BSN, Katie Neighbors, MPH, CCRC (Lurie Children’s Hospital of Chicago, Chicago, Illinois); Philip J. Rosenthal, MD, Shannon Fleck, Clinical Research Coordinator (University of California San Francisco, San Francisco, California); Mike A. Leonis, MD, PhD, John Bucuvalas, MD, Tracie Horning, Clinical Research Coordinator (University of Cincinnati, Cincinnati, Ohio); Norberto Rodriguez Baez, MD, Shirley Montanye, RN, Clinical Research Coordinator, Margaret Cowie, Clinical Research Coordinator (University of Texas Southwestern, Dallas, Texas); Simon P. Horslen, MD, Karen Murray, MD, Melissa Young, Clinical Research Coordinator, Heather Nielson, Clinical Research Coordinator, Jani Klein, Clinical Research Coordinator (University of Washington, Seattle, Washington); David A. Rudnick, MD, PhD, Ross W. Shepherd, MD, Kathy Harris, Clinical Research Coordinator (Washington University, St. Louis, Missouri).
Previous Sites, Principal Investigators, and Coordinators – Saul J. Karpen, MD, PhD, Alejandro De La Torre, Clinical Research Coordinator (Baylor College of Medicine, Houston, Texas); Dominic Dell Olio, MD, Deirdre Kelly, MD, Carla Lloyd, Clinical Research Coordinator (Birmingham Children’s Hospital, Birmingham, United Kingdom); Steven J. Lobritto, MD, Sumerah Bakhsh, MPH, Clinical Research Coordinator (Columbia University, New York, New York); Maureen Jonas, MD, Scott A. Elifoson, MD, Roshan Raza, MBBS (Harvard Medical School, Boston, Massachusetts); Kathleen B. Schwarz, MD, Wikrom W. Karnsakul, MD, Mary Kay Alford, RN, MSN, CPNP (Johns Hopkins University, Baltimore, Maryland); Anil Dhawan, MD, Emer Fitzpatrick, MD (King’s College Hospital, London, United Kingdom); Nanda N. Kerkar, MD, Brandy Haydel, CCRC, Sreevidya Narayanappa, Clinical Research Coordinator (Mt. Sinai School of Medicine, New York, New York); M. James Lopez, MD, PhD, Victoria Shieck, RN, BSN (University of Michigan, Ann Arbor, Michigan).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: None of the authors have any conflicts of interest to disclose
Trial registration ClinicalTrials.gov: NCT00986648
Contributor Information
Amy G. Feldman, Assistant Professor of Pediatrics, Digestive Health Institute, Children’s Hospital Colorado and Section of Pediatric Gastroenterology, Hepatology and Nutrition, Department of Pediatrics, University of Colorado School of Medicine, 13123 E. 16th Ave., Aurora, Colorado 80045, Phone: 720-777-5354, Fax: 720-777-7277.
Ronald J. Sokol, Professor and Vice Chair of Pediatrics, Chief of Section of Pediatric Gastroenterology, Hepatology and Nutrition and Digestive Health Institute, Children’s Hospital Colorado Department of Pediatrics, University of Colorado School of Medicine, 13123 E. 16th Ave., Box B290, Aurora, Colorado 80045, U.S.A., Phone: 720-777-6669, Fax: 720-777-7277.
Regina M. Hardison, Epidemiology Data Center Graduate School of Public Health, University of Pittsburgh, 127 Parran Hall 130 Desoto Street, Pittsburgh, PA 15261
Estella M. Alonso, Professor of Pediatrics, Northwestern University Feinberg School of Medicine Division of Gastroenterology, Hepatology and Nutrition, Ann and Robert H. Lurie Children’s Hospital of Chicago 225 E Chicago Ave, Chicago IL 60611, Phone 312-227-4606, Fax 312- 227-9645.
Robert H. Squires, Professor of Pediatrics, University of Pittsburgh School of Medicine, Department of Pediatrics, Division of Gastroenterology, Hepatology and Nutrition, Children’s Hospital of Pittsburgh of UPMC, 4401 Penn Ave, Pittsburgh, PA 15224, Phone: 412-692-8648; Fax: 412-692-8355.
Michael R. Narkewicz, Professor of Pediatrics, Digestive Health Institute, Children’s Hospital Colorado and The Section of Pediatric Gastroenterology, Hepatology and Nutrition, Department of Pediatrics, University of Colorado School of Medicine, 13123 E. 16th Ave., Aurora, Colorado 80045, Phone: 720-777-6669, Fax: 720-777-7277.
References
- 1.Squires RH, Jr, Shneider BL, Bucuvalas J, Alonso E, Sokol RJ, Narkewicz MR, et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. The Journal of pediatrics. 2006;148(5):652–8. doi: 10.1016/j.jpeds.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDiarmid SV, Anand R, Lindblad AS, Group SR Studies of Pediatric Liver Transplantation: 2002 update. An overview of demographics, indications, timing, and immunosuppressive practices in pediatric liver transplantation in the United States and Canada. Pediatric transplantation. 2004;8(3):284–94. doi: 10.1111/j.1399-3046.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 3.Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3(1):9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Hegarty R, Hadzic N, Gissen P, Dhawan A. Inherited metabolic disorders presenting as acute liver failure in newborns and young children: King’s College Hospital experience. Eur J Pediatr. 2015;174(10):1387–92. doi: 10.1007/s00431-015-2540-6. [DOI] [PubMed] [Google Scholar]
- 5.Alam S, Lal BB. Metabolic Liver Diseases Presenting as Acute Liver Failure in Children. Indian Pediatr. 2016 doi: 10.1007/s13312-016-0913-1. [DOI] [PubMed] [Google Scholar]
- 6.Helbling D, Buchaklian A, Wang J, Wong LJ, Dimmock D. Reduced mitochondrial DNA content and heterozygous nuclear gene mutations in patients with acute liver failure. Journal of pediatric gastroenterology and nutrition. 2013;57(4):438–43. doi: 10.1097/MPG.0b013e31829ef4b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chariot P, Ratiney R, Ammi-Said M, Herigault R, Adnot S, Gherardi R. Optimal handling of blood samples for routine measurement of lactate and pyruvate. Arch Pathol Lab Med. 1994;118(7):695–7. [PubMed] [Google Scholar]
- 8.Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, et al. The in-depth evaluation of suspected mitochondrial disease. Molecular genetics and metabolism. 2008;94(1):16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touati G, Rigal O, Lombes A, Frachon P, Giraud M, Ogier de Baulny H. In vivo functional investigations of lactic acid in patients with respiratory chain disorders. Archives of disease in childhood. 1997;76(1):16–21. doi: 10.1136/adc.76.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Debray FG, Mitchell GA, Allard P, Robinson BH, Hanley JA, Lambert M. Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin Chem. 2007;53(5):916–21. doi: 10.1373/clinchem.2006.081166. [DOI] [PubMed] [Google Scholar]
- 11.Lane M, Boczonadi V, Bachtari S, Gomez-Duran A, Langer T, Griffiths A, et al. Mitochondrial dysfunction in liver failure requiring transplantation. J Inherit Metab Dis. 2016;39(3):427–36. doi: 10.1007/s10545-016-9927-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molleston JP, Sokol RJ, Karnsakul W, Miethke A, Horslen S, Magee JC, et al. Evaluation of the child with suspected mitochondrial liver disease. Journal of pediatric gastroenterology and nutrition. 2013;57(3):269–76. doi: 10.1097/MPG.0b013e31829ef67a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Squires RH, Dhawan A, Alonso E, Narkewicz MR, Shneider BL, Rodriguez-Baez N, et al. Intravenous N-acetylcysteine in pediatric patients with nonacetaminophen acute liver failure: a placebo-controlled clinical trial. Hepatology. 2013;57(4):1542–9. doi: 10.1002/hep.26001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4(118):118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]