

Abstract
COL4A1 and COL4A2 are extracellular matrix proteins that form heterotrimers and are present in nearly all basement membranes in every organ. In the past decade, COL4A1 and COL4A2 mutations have been identified to cause a multi-system disorder for which penetrance and severity of constituent phenotypes can greatly vary. Here, we compare the outcomes of more than 100 mutations identified in patients and data from a murine allelic series to explore the presence of genotype-phenotype correlations – many of which are shared among other types of collagen. We find that there is a frequency bias for COL4A1 over COL4A2 mutations and that glycine (Gly) substitutions within the triple helical domain are the most common class of mutations. Glycine is most often replaced by a charged amino acid, however the position of the mutation, and not the properties of the substituting amino acid, appear to have a greater influence on disease severity. Moreover, the impact of position is not straightforward. Observations from a murine allelic series suggest that mutations in the NC1 domain may result in relatively mild phenotypes via a ‘quantitative’ mechanism similar to other types of collagens, however, this effect was not apparent in human reports. Importantly, other position-dependent effects had differential impacts depending on the phenotype of interest. For example, the severity of cerebrovascular disease correlated with an amino-to-carboxy severity gradient for triple-helical glycine substitutions whereas the penetrance and severity of myopathy and nephropathy appear to involve a functional sub-domain(s). Greater understanding of genotype-phenotype correlations and the interaction of consequences of different mutations will be important for patient prognosis and care and for developing mechanism-based therapeutics to treat individual components of this emerging syndrome.
1) Introduction
Type IV collagens are network-forming basement membrane collagens 1 and are encoded by six genes; collagen, type IV, alpha 1 (COL4A1) through 6 (COL4A6) in mammals 2, 3. COL4A1 and COL4A2 are present in almost all basement membranes, while the distributions of COL4A3, COL4A4, COL4A5 and COL4A6 are restricted to a smaller number of basement membranes present in the eye, ear and kidney 3, 4, 5. In humans, COL4A1 and COL4A2 share a common locus at 13q34 where they are arranged head to head with a bidirectional promoter 6–9. COL4A1 (52 exons) and COL4A2 (48 exons) encode proteins of 1669 and 1712 amino acids, respectively, which share 45% identity. COL4A1 and COL4A2 both contain the same three major domains: The non-collagenous (NC1) domain is a globular domain at the carboxy terminus that is required for the initiation of heterotrimer formation between one COL4A2 and two COL4A1 proteins (α1(IV)2 α2(IV)) 10, 11. The majority of each COL4A1 and COL4A2 protein comprises a collagenous triple helical domain characterized by repetition of Gly-Xaa-Yaa motifs (where Xaa and Yaa are variable amino acids). In contrast to fibrillar collagens, COL4A1 and COL4A2 have more than 20 interruptions of the Gly-Xaa-Yaa repeats that encompass protein interaction domains and confer local flexibility 12, 13. Following assembly in the endoplasmic reticulum, heterotrimers are transported to the Golgi where they are packaged for vesicular release from the cell 2, 3. Once they reach the extracellular space, heterotrimers participate in macromolecular network organizations whereby two heterotrimers associate via their NC1 domains, and four heterotrimers form anti-parallel lateral interactions via association of their 7S domains.
A targeted mutation of the Col4a1/Col4a2 locus on murine Chromosome 8 generated mice with null alleles for both Col4a1 and Col4a2 and provided the first insights into their biological roles 14. This work showed that formation of the collagen IV network is dispensable for initial basement membrane assembly, but critical for its structural integrity. Mice homozygous for the Col4a1;Col4a2 null allele died between embryonic days (E)10.5–11.5 with disrupted basement membranes including Reichert’s membrane 14. In contrast, heterozygous mice were described as having no overt phenotype. Soon after, heterozygous, semi-dominant pathogenic COL4A1 and COL4A2 mutations were identified in humans and in multiple mouse lines that model the pathology in patients 15–17. Consistent with the broad distribution of α1(IV)2α2(IV) heterotrimers in nearly all basement membranes, COL4A1 and COL4A2 mutations cause multisystem disorders with abnormalities in the vasculature, brain, eyes, kidneys and muscles being the most commonly reported to date 18–20. Cerebrovascular disease (CVD) is one of the most notable consequences of COL4A1 and COL4A2 mutations and comprises a growing constellation of clinical manifestations including porencephaly, small-vessel disease, leukoencephalopathy, intracranial aneurysms and recurrent intracerebral hemorrhages (ICH). Other cerebral defects include calcification and cortical malformations similar to those observed in Walker-Warburg syndrome and muscle-eye-brain disease. Patients also frequently present with ocular disease (microphthalmia, cataracts, anterior segment dysgenesis, Axenfeld Rieger syndrome, glaucoma, optic nerve hypoplasia and retinal vascular tortuosity) kidney disease (hematuria and renal cysts of variable severity) and muscular defects (cramps, elevated serum creatine kinase level and muscular dystrophy). An excellent review of clinical consequences of COL4A1 and COL4A2 was recently published 20. It is likely that the present observations are not exhaustive and that pathology in additional tissues and organs will be identified as studies advance.
As of April 2016, there were 93 and 12 mutations identified in human COL4A1 and COL4A2, respectively, and 13 and 3 in the corresponding murine genes (not including three targeted Col4a1 alleles). This sample size seemed appropriate to explore emerging genotype–phenotype correlations. Identifying genotype–phenotype correlations can be useful for patient management, genetic counseling, cohort stratifications for drug response or adverse events in clinical trials and for providing a better understanding of the biological function of distinct protein sub-domains. Equivalent analyses were performed with similar numbers of patients who have mutations in other types of collagens and these studies have provided valuable insight. The purpose of this review is to draw upon comparisons with other types of collagens and the current literature of human COL4A1 and COL4A2 mutations and mouse models to help identify potential genotype–phenotype correlations and understand their implications.
2) Genotype–phenotype correlations in patients with mutations in other types of collagens
Type IV collagens are part of the collagen superfamily comprising 28 members that have the defining feature of a triple-helical domain 1. Mutations in multiple types of collagens contribute to a diverse constellation of human diseases. There is great a depth of knowledge of disease mechanism for many types of collagens which can be instructive for understanding genotype–phenotype correlations for COL4A1 and COL4A2 mutations. However, despite commonalities, members of the collagen superfamily are structurally and functionally diverse and there is no universal disease mechanism. For example, type VI collagen mutations can cause muscular dystrophies that are notable for the breadth of variation in disease severity. The type VI collagens are non-fibrillar collagens and the α1(VI)α2(VI)α3(VI) heterotrimers form beaded microfibrils important for anchoring the basement membrane to other components of the extracellular matrix. Type VI collagen mutations can cause both Ulrich congenital muscular dystrophy (a severe neonatal or early childhood disease) and Bethlem myopathy (where ambulation is often retained into adulthood), which were considered to be distinct diseases until molecular characterization revealed a genotype–phenotype relationship that influences disease expressivity. Dominant missense mutations of Gly residues in the triple helical domain are a common class of type VI collagen mutations and the most severe cases cluster within a small region of ~15 amino acids near the amino terminus of the triple helical domain that is proposed to represent a functional subdomain important for muscle function 21.
The type II collagens represent a similar example of phenotypic dichotomy attributable to genotype. Type II collagens are fibril forming α1(II)3 homotrimers with expression in chondrocytes and in the ocular vitreous. COL2A1 mutations cause Stickler syndrome type I, characterized by high myopia, vitreoretinal degeneration, retinal detachment and cataracts. On the other end of the disease spectrum resulting from COL2A1 mutations are severe forms of skeletal dysplasia, chondrodysplasia and achondrogenesis. In a relatively clear example of genotype–phenotype correlations, the preponderance of mutations causing Stickler syndrome are highly disruptive mutations that likely result in functionally null alleles. In a published series of 188 Stickler syndrome type 1 probands, the predominant cause was loss-of-function mutations comprising splice site alterations, nonsense mutations, or insertion/deletions (indels). Only five of the 77 mutations that caused Stickler syndrome were Gly substitutions in the triple helical domain 22. There is further evidence to suggest that the Gly substitutions that cause Stickler syndrome type 1 are near the amino terminal quarter of the protein. In contrast, Gly substitutions nearer the carboxy terminus – especially those for which Gly is replaced by a bulkier amino acid – are more often associated with severe forms of skeletal dysplasia, achondrogenesis or severe short stature phenotypes 22–25.
In a departure from distinct clinical entities, vascular Ehlers–Danlos syndrome (vEDS) is caused by dominant mutations in COL3A1. Type III collagens are fibril forming α1(III)3 homotrimers and the triple helical domain consists of 343 strict repeats of the Gly-Xaa-Yaa motif. Typical consequences of vEDS are arterial dissections, bowel perforations and uterine ruptures 26, 27. A recent report 28 of genotype–phenotype associations in 146 vEDS index patients with molecular diagnosis found 126 distinct mutations that fit into five distinct categories: Gly substitutions in the triple helical domain (56.3%), splice-site and in-frame indels (28.6%), haploinsufficiency (5.5%), non-Gly substitutions in the triple helical domain (3.2%) and mutations outside of the triple-helical domain (6.3%). As a measure of relative severity the age at diagnosis was younger for patients with in-frame splice site mutations (25 years) than for those with Gly substitutions (34 years) and both were younger than for patients with other classes of mutations (45 years). Although there was a frequency bias for substitutions of Gly to more destabilizing amino acids (Aspartic acid, Glutamic acid, Valine), no correlation with disease severity was observed 28. Instead, the position of the glycine substitutions appears to play a role with those nearer the carboxy terminus being more severe 27. Moreover, the prognosis was better for patients who did not have Gly substitutions or in-frame coding mutations as they were generally spared from digestive complications. In a separate study of 630 index cases for which survival was the outcome measure, patients with triple helical glycine substitutions had significantly shorter survival than patients with null mutations, however, in this case the identity of the substituting amino acid did influence the outcome but the position of the mutation did not 29.
Perhaps the richest source of information for collagen-related genotype–phenotype correlations comes from the type I collagens. Unlike collagen II and III that form homotrimers, collagen I (like collagen IV) form α1(I)2α2 (I) heterotrimers. Mutations in COL1A1 and COL1A2 cause Osteogenesis imperfecta (OI), a genetically and clinically heterogeneous disorder characterized by skeletal deformities and fragile bones that are susceptible to fracture. An early influential analysis of approximately 70 COL1A1 and COL1A2 mutations identified genotype–phenotype correlations that underscored the distinction between the quantitative and qualitative effects of mutations 30. As larger sets of patients became available a clearer understanding emerged that appears to have broad applicability across many types of collagens 31. Mutations were associated with milder forms of OI if they caused quantitative defects (mutations in the carboxyl-terminal domain that preclude the mutant protein from integrating into heterotrimers and frameshifts and null alleles that trigger loss of mRNA leading to a reduction in protein levels). In contrast, qualitative defects (such as Gly substitutions in the triple helical domain, exon skipping and rearrangements) responsible for protein structural defects were associated with more severe forms of OI 30, 32. Because mutations within the triple helical domain are common and associated with severe forms of disease they have been the subjects of greatest interest. A definitive study of more than 800 COL1A1 and COL1A2 Gly mutations and altered splice sites in the triple helical domain allowed the discovery and confirmation of a number of genotype–phenotype correlations 31. Of the 832 independent mutations, 682 resulted in triple helical Gly substitutions (391 in COL1A1 and 291 in COL1A2) and 150 altered splice sites (102 in COL1A1 and 48 in COL1A2) 31. COL1A1 splice site mutations often shifted the open reading frame resulting in absence of the mutant chain and manifested as quantitative mutations that were rarely severe (lethal). In contrast, triple helical glycine substitutions produced mutant proteins that incorporate into heterotrimers leading to important qualitative differences. Large-scale analyses of the effects of triple helical Gly substitutions produce a number of interesting genotype–phenotype correlations. Gly is represented by four codons: GGU, GGC, GGA, GGG and transitions or transversions of either of the first two guanine nucleotides results in Gly substitutions to Alanine (Ala), Arginine (Arg), Aspartic acid (Asp), Cysteine (Cys), Glutamic acid (Glu), Serine (Ser), Tryptophan (Trp) or Valine (Val). One third of the Gly substitutions were lethal - especially when the substituting amino acid is charged (Asp and Glu), or has a branched side chain (Arg and Val). When these substitutions were non-lethal they associated with severe outcomes compared to substitutions for Cys, Glu, or Ala. Furthermore, the influence of amino acid substitutions on disease severity was superimposed on the specific location of the mutation. For instance, when comparing only substitutions of Cys for Gly there was a relatively smooth phenotypic gradient from mild to severe in an amino-to-carboxy direction, suggesting a position effect 30. While in a larger patient cohort no linear relationship emerged, with few exceptions, Gly substitutions within the first 200 amino acids of COL1A1 were non-lethal (on the mild end of the OI spectrum) whereas similar mutations closer to the carboxy-terminal end were often severe 31. Additionally, there were two specific sub-domains associated with lethal phenotypes that align with major ligand binding regions and gap regions without reported mutations that contain putative integrin-binding sites, suggesting critical functions 31. Mutations causing severe forms of OI were later described in a gap region, re-enforcing the hypothesis that these regions have important functional roles 33.
Notably, compared to COL1A1 mutations, COL1A2 mutations were identified much less frequently (291 vs 391 for Gly substitutions and 48 vs 102 for splice site mutations) and there were both similarities and differences in the outcomes 31. Among patients with COL1A2 glycine substitutions, those with Asp, Glu and Arg – but not Val – tended to have worse outcomes. COL1A2 Gly substitutions were predominantly non-lethal, with the exceptions of exon skipping mutations in the carboxyl half of the protein and glycine substitutions that occurred in eight regularly spaced clusters 31. The lethal clusters do not align with major ligand binding regions but were in regions suggested to be critical for interactions between proteoglycans and the collagen fibrils.
Overall, these studies revealed that mutations causing quantitative differences where the mutant allele is degraded or not incorporated into trimers are found less frequently and are associated with the mildest outcomes. Mutations that cause qualitative differences produce a wide range of phenotypes that depends on many factors including the gene involved and nature and location of the mutation. Analysis of COL1A1 demonstrated that, in addition to important functional subdomains, worse patient outcomes were associated with Gly substitutions for charged or branched amino acids and for Gly substitutions nearer the carboxy terminus. The position of mutations in COL1A2 appears to have less influence unless the occurred within one of the 8 regularly spaced clusters 31.
Mutations in the other type IV collagen isoforms also cause autosomal recessive (COL4A3 and COL4A4) or X-linked (COL4A5) Alport Syndrome and may offer insight into to genotype-phenotype correlations 34. The proteins encoded by these three genes form α3(IV)α4(IV)α5(IV) heterotrimers that are present in basement membranes of the glomerulus, cochlea and eye 10, 35. Alport Syndrome is characterized by progressive renal failure, sensorineural hearing loss, lenticonus, cataract and maculopathy 36, 37. Approximately 80–85% of Alport Syndrome cases are X-linked and caused by COL4A5 mutations 38, 39. Studies of large cohorts of males with X-linked Alport Syndrome reveal genotype-phenotype correlations that are often contradictory to those found in type I collagens. The consensus from studying over 1000 patients with COL4A5 mutations was that disease severity (age at onset for end stage renal failure) was greatest for truncating mutations, followed by splicing variants and finally missense mutations 40–44. Moreover, truncating or splicing mutations had two-fold greater odds of having ocular pathology compared to missense mutations 41. This is in contrast to the concept that emerged from type I collagens where outcomes for quantitative mutations are worse than for qualitative mutations. Heterotrimer stoichiometry can offer an explanation for this observation since severe loss of function mutations cannot be compensated for by the homologous chromosome, leaving cells with a virtual absence of COL4A5. Position dependent effects were also reported, however, these too appear to be opposite to those reported in type I collagens. Mutations in the amino terminal signal peptide, regardless of type, led to end stage renal failure at younger ages than mutations in the triple helical or NC1 domains 41. One study that only looked at triple helical glycine substitutions suggested that mutations in exons 1–20 (amino terminal) might have been less severe than those in exons 21–47 (carboxy terminal) 40 while a second, larger study found that mutations nearer the amino terminus were more severe 41. Finally, a recent study suggested that missense substitutions for bulkier amino acids were associated with more severe outcomes 43.
3) Genotype–phenotype correlations in patients with COL4A1 or COL4A2 mutations
This review includes 105 independent occurrences of COL4A1 or COL4A2 mutations of which, 93 were in COL4A1 (at 74 unique sites) and 12 in COL4A2 (at 10 unique sites) (Tables 1 and 2). All of the mutations described were dominant with equal representation in males (48%) and females (52%) and approximately equal numbers of inherited (47%) or sporadic (53%) incidences. Similar to other types of collagens, triple helical Gly substitutions represented the most prevalent class of mutations (68 out of 93) and the most common substitutions were for charged amino acids; Arg (30), Glu (11), and Asp (10). The remaining substitutions were Ser (7), Val (5), Ala (3), Leu (1), and a termination codon (1). No substitutions for tryptophan or cysteine, which are the only other substitutions possible with a single nucleotide change of glycine codons, were reported. Splice site mutations within the triple helical domain constitute the second most prevalent class of mutations (8 out of 93). Three of the splice site mutations caused in-frame deletion of an entire exon (exon 21, 25 or 31) and two mutations resulted in nonsense-mediated decay of the transcripts with reduced COL4A1 expression while the consequences of the other three were not reported. Other classes of mutations were much less frequent including Yaa position substitutions (3), substitutions within the repeat interruptions (3) and frame-shifting indels (3). Eight mutations outside of the triple helical domain have also been reported, seven of which were located in the NC1 domain and one substitution mutation of the start methionine.
Table 1.
| COL4A1 | COL4A2 | ||||
|---|---|---|---|---|---|
| Occurances | Locations | Occurances | Locations | ||
| Start codon | Met substitution | 1 | 1 | 0 | 0 |
| Triple helical domain | Gly of Gly-Xaa-Yaa motif | 68 | 50 | 6 | 6 |
| Arg (30), Glu (11), Asp (10), Ser (7), Val (5), Ala (3), Leu (1), Ter (1) | Arg (2), Glu (2), Asp (2) | ||||
| Yaa of Gly-Xaa-Yaa motif | 3 | 3 | 0 | 0 | |
| Pro to Leu, Met to Val, Gln to Glu | |||||
| Xaa of Gly-Xaa-Yaa motif | 0 | 0 | 4 | 2 | |
| Glu to Gly (3), Gln to Lys | |||||
| Repeat Interruptions | 3 | 3 | 0 | 0 | |
| Frameshift | 3 | 3 | 1 | 1 | |
| 2 deletions, 1 duplication | 1 deletion | ||||
| Splice-site | 8 | 8 | 0 | 0 | |
| NC1 domain | Missense | 4 | 3 | 1 | 1 |
| Nonsense | 1 | 1 | 0 | 0 | |
| Duplication | 1 | 1 | 0 | 0 | |
| Insertion | 1 | 1 | 0 | 0 | |
| Totals | 93 | 74 | 12 | 10 |
Table 2.

|
The low number of published COL4A2 mutations decreases the confidence for identifying broad trends, however, triple helical glycine substitutions (6) were also the most prevalent class of mutations where Gly was substituted for Arg (2), Glu (2) and Asp (2). One patient had a frame-shifting deletion and a second patient had a missense mutation, each occurring in the NC1 domain. There were also examples of triple helical Xaa-position substitutions (4), however, three of these were identified independently in the same amino acid and this variant was also identified in controls raising the possibility that this variant might be non-pathogenic. If so, the total number of COL4A2 mutations could be as low as nine with six being Gly substitutions in the triple helical domain. The failure so far to identify mutations in the repeat interruptions could represent differences between COL4A1 and COL4A2 or simply reflect the low number of COL4A2 mutations discovered to date.
Overall, COL4A2 mutations appear to associate with milder phenotypes (later age-at-onset of CVD, and fewer system abnormalities reported). However, because the limited number of COL4A2 mutations, we focus primarily on the 93 COL4A1 mutations for the investigation of genotype–phenotype correlations in patients. Fourteen COL4A1 mutations were presumed to reduce the amount of normal α1(IV)2 α2(IV) heterotrimers without producing abnormal heterotrimers and may therefore qualify as quantitative mutations: seven mutations in the NC1 domain, three frame-shifting indels, two frame-shifting splice mutations, one substitution of the start methionine, and one triple helical glycine substitution leading to a premature termination codon. Six out of the seven NC1 mutations and 12 out of the 14 total “quantitative” mutations were reported in patients with porencephaly and the remaining two patients had perinatal ICH. If one considers porencephaly and early age-at-onset as phenotypes on the severe end of the CVD spectrum, quantitative COL4A1 mutations appear to associate with relatively poor outcomes.
To compare the effects of qualitative measures, we focused on the 68 triple helical Gly substitutions in COL4A1. The frequencies of Arg, Glu, and Asp substitutions might suggest that the outcomes of these mutations are more severe, however this conclusion would need to be corrected for codon usage and integrate other potential factors. In four pairs of instances the same glycine residue was independently substituted by distinct amino acids (G498V, G498D; G655R, G655E; G708R, G708V; G990E, G990V). However, in all cases, the substituting amino acids are either branched or charged and so might all be expected to have severe consequences and there were no obvious differences that could be attributed to the identity of the substituting amino acid. To further investigate potential differential effects of substituting amino acids, we compared the consequences of the 30 Arg substitutions to the Ser (7) and Ala (3) substitutions. No obvious correlations were observed when comparing substitutions that were roughly position-matched. For example, G670R caused porencephaly, whereas G708R caused ocular defects but no CVD and G696S had adult onset ICH. Of four independent occurrences of G755R mutations, none lead to porencephaly and instead all result in childhood or adult onset CVD. In contrast, two independent occurrences for the nearby G749S mutation caused porencephaly. Although these analyses indicate that substitutions to branched amino acids occur more frequently, there is no clear evidence that they are associated with more severe outcomes.
Notably, the position of the mutation within the triple helical domain appeared to correlate with CVD severity. There are two Gly substitutions in the first quarter of the triple helical domain suggesting that Gly missense mutations near the amino-terminal end of the triple helical domain might cause milder phenotype. The 30 Arg substitutions occur at 20 unique sites allowing comparison of the effects of position for the same substituting amino acid. If one divides the triple helical domain roughly in half, 14 mutations are in the carboxy-terminal half (amino acid 805 and higher) and, of these, 12 caused porencephaly and two caused schizencephaly. In addition to early age-at-onset, these mutations were more often sporadic or de novo cases and were frequently associated with epileptic seizures and motor disorder and/or developmental delay. In contrast, of the 16 Gly to Arg mutations in the amino-terminal half of the protein, only five caused porencephaly, four mutations did not cause ICH/stroke, four mutations were identified in patients who were adults at the time of diagnosis and only four caused epileptic seizures.
In addition to modulating CVD severity, the location of the mutation may also influence the penetrance of other phenotypes. For instance, COL4A1 mutations cause ocular disease with high penetrance (72%), however the specific ocular defect depends on the position of the mutation. For example, mutations in the amino-terminal one-third of the protein caused retinal arteriolar tortuosity with complete penetrance, whereas mutations causing cataract, anterior segment dysgenesis, glaucoma and microphthalmia tended to occur closer to the carboxy terminus. Of interest, in the amino-terminal one-third of the protein there were mutations reported in six families with a clinical diagnosis of HANAC Syndrome (hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) 51–53, 112. The mutations cluster in exons 24 and 25 within a 31 amino acid region (G498–G528) of the triple helical domain that encompasses several integrin-binding domains. While nephropathy, myopathy and aneurysms are not restricted to patients with mutations in this region, these phenotypes may be disproportionately represented in patients from these families. These patients also had retinal arteriolar tortuosity and heart disease but other features including porencephaly, cerebral calcification, cataract, glaucoma, microcephaly or epileptic seizures were not described. Notably, while three Gly substitutions within exon 25 were reported in patients with HANAC syndrome, a splice site mutation causing an in-frame deletion of this exon was found in a patient with bilateral diffuse white matter abnormalities, multiple clinically asymptomatic hemorrhages and finally an acute ICH but without aneurysms, myopathy or renal involvement 56. Observations from patients with triple helical Gly substitutions support the existence of one or more functional subdomains within this domain that may associate with differential co-morbidities.
There are a number of limitations that demand approaching genotype–phenotype correlations in patients with caution. Most of the mutations occurred only once and to label the ‘severity’ of a mutation based on a sample size of one risks oversimplification of many complex genetic, environmental and stochastic interactions that are integrated in the phenotype. Even in cases of recurrent mutations, the conclusions are not simple. There were eight amino acids that had multiple hits and G755R and G773R were each reported in four independent studies. While the outcomes for G755R were comparable (all patients had ICH, leukoencephalopathy and cataracts with an age of onset between 10 and 21 years, and did not cause porencephaly or epilepsy) those for G773R were less so (three mutations caused porencephaly or severe stroke whereas one mutation cause only very mild white matter changes). Genetic context differences could explain some of the discordance in the expressivity resulting from a given mutation. However, phenotypic differences within families who are more closely matched genetically, including probands with porencephaly born to asymptomatic parents who carried mutations, were also observed. There are multiple possible explanations for these cases including genetic modifiers, mosaicism, biological variation or environmental influences.
4) Genotype–Phenotype correlations in Col4a1 and Col4a2 mutant mice
Studies of mouse models with Col4a1 and Col4a2 mutations are a powerful way to simplify the genetic and environmental influences and compliment patient studies. In addition to the Col4a1;Col4a2 null mutation 14, and a Col4a1 conditional mutation 101, 110, there are presently 17 mutations reported in Col4a1 and Col4a2 that cause pathology in multiple tissues and organs 16, 17, 100, 104 (Fig. 1). Because of procedural differences between research groups, notably the focus and depth of analysis, it is difficult to directly compare graded levels of relative severities for each mutation. Importantly, the genetic context in which the mutations are studied also has a significant effect on the penetrance and/or severity of pathology 18, 80, 101, 111. For example, Col4a1 mutant mice maintained on a pure C57BL/6J background have severe ocular dysgenesis, myopathy and ICH that are all suppressed in mutant mice that were crossed for a single generation to the CAST/EiJ inbred strain 15, 18, 80, 101, 111. Notably, the 129SvEvTac inbred strain also suppressed anterior segment dysgenesis, but not ICH, indicating that genetic context can have differential effects on distinct phenotypes and suggesting heterogeneity in the pathogenic mechanisms underlying different Col4a1-related phenotypes 101.
Figure 1. Summary of reported phenotypes for murine Col4a1 and Col4a2 mutations.

(A) Schematic diagram illustrating murine mutations reported in the literature. (B) List of Col4a1 and Col4a2 mutations and the phenotypes that have been reported for each.
To systematically investigate potential genotype–phenotype correlations, nine Col4a1 and one Col4a2 mutations 100 were uniformly backcrossed to the C57BL/6J genetic background 101, 102. The allelic series comprised seven missense mutations of glycine residues in the triple helical domain (six in COL4A1, one in COL4A2), one missense mutation in the NC1 domain of COL4A1, and a point mutation of splice acceptor site that results in the deletion of exon 41 (Δex41) but retains the open reading frame (Fig. 2A). By controlling environmental factors and the genetic context, studies using this allelic series demonstrated allelic heterogeneity between mutations. The severity of ICH was compared between strains of mutant mice aged for 7–9 months and different classes of mutations that implicated three potential genotype–phenotype correlations were identified 101, 102. First was a domain effect whereby the NC1 domain mutation (Col4a1S1582P), which presumably reflects a quantitative mutation (Fig. 2B), caused less severe ICH than the triple helical domain mutations (Fig. 2C). Second, for point mutations within the triple helical domain, there was a position effect whereby mutations nearer the carboxy termini (where heterotrimer winding is initiated) caused more severe ICH than mutations nearer the amino termini. Third, there appeared to be a ‘class effect’ whereby Col4a1Δex41 was more severe than missense mutations even when compared to a glycine point mutation within exon 41 (Col4a1G1180D). The pathogenicity resulting from COL4A1 and COL4A2 mutations is generally attributed to impaired secretion and concomitant intracellular accumulation and extracellular deficiency of mutant heterotrimers. Notably, ICH severity in this allelic series correlated with the relative levels of heterotrimer accumulation between mutations (Fig. 2B and C)101, 102.
Figure 2. Allelic heterogeneity for heterotrimer biosynthesis, intracerebral hemorrhage and myopathy.
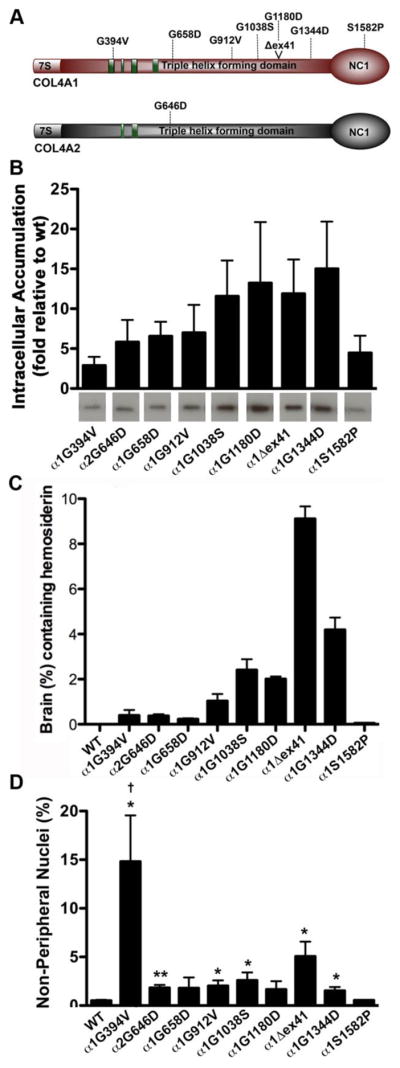
(A) Schematic diagram illustrating the mutations in the allelic series. All mutations are in the triple helical domain, except COL4A1S1582P, which is in the C-terminal NC1 domain. COL4A1Δex41 is a splice site mutation causing deletion of exon 41. (B) Western blot quantification demonstrating that the mutation in the NC1 domain (COL4A1S1582P) had relatively low levels intracellular heterotrimer accumulation and that there is a position-dependent graded severity of heterotrimer accumulation for mutations within the triple helical domain whereby mutations nearer the NC1 domain had higher levels of heterotrimer accumulation. (C) Quantification of intracerebral hemorrhage revealed that COL4A1Δex41 mutation leads to the most severe phenotype and that point mutations in the triple helical domain nearer the C-terminus tended to cause more (or more severe) hemorrhages. (D) Quantification of non-peripheral nuclei revealed that the Col4a1G394V mutation, which is in an integrin-binding domain, causes the most severe myopathy. For all groups, n>5. Comparisons between wild-type (Col4a1+/+) and mutant mice were performed using Student’s t-test; *, p<0.05. **, p<0.01. For comparison among the different strains, a one-way ANOVA followed by a Tukey’s post hoc test was performed; †, p<0.05. Panels A, B and D are modified from Kuo et al. (2014) 102 and Panel C is modified from Jeanne et al. (2015) 101.
An interesting contrast was observed when myopathy was quantified in this same allelic series 102. Similar to the previous trends, mice with a presumed qualitative mutation in the NC1 domain (Col4a1S1582P) had little or no phenotype and myopathy resulting from the Col4a1Δex41 mutation was more severe than that resulting from the ‘position-matched’ point mutation (Col4a1G1180D) (Fig. 2D). However, in contrast to the trends observed in ICH, there was no apparent position effect for mutations within the triple helical domain. Instead, Col4a1G394V, which was among the mutations with the least intracellular accumulation and mildest ICH, had the most severe myopathy demonstrating an interesting dichotomy for this mutation. Col4a1G394V occurs near a putative integrin-binding site adjacent to the cluster of HANAC mutations, in a region that may represent a functional sub-domain with an important role in muscle development and function 102.
Kidney abnormalities (albuminuria and thin or split glomerular basement membranes) were previously reported in Col4a1+/Δex41 mice 15 but had not been examined in other strains from the allelic series. Here we show that hematuria shares a profile that resembles ICH for the Col4a1 mutations whereby the Col4a1S1582P mutation had almost no phenotype, the Col4a1Δex41 mutation was worse than Col4a1G1180D and mutations nearer the carboxy termini were more severe than those nearer the amino termini (Fig. 3A). These data may suggest that hematuria has a vascular origin or that hematuria and ICH share pathogenic mechanisms. When we measured albuminuria the results were similar with the notable exception that the Col4a2 mutation (Col4a2G646D) was disproportionately severe compared to Col4a1 mutations (Fig. 3B). This observation might suggest that COL4A2 is more important for nephropathy or that this mutation occurs within a functional sub-domain important for renal pathology. In support of the latter alternative, the Col4a1G658Dmutation, which is nearby, lead to the most severe albuminuria amongst the Col4a1 point mutations.
Figure 3. Relative measures of hematuria and albuminuria in mice from an allelic series of Col4a1 and Col4a2 mutations.

We collected urine and measured levels of hematuria and albuminuria using colorimetric dipsticks and a clinical chemistry autoanalyzer, respectively (described in Gould et al. (2006) 15). A). The proportion of animals of each genotype with no, mild, moderate or severe hematuria are indicated as percentages. Sample sizes for both each genotype are indicated at the top. B) We measured albuminuria by calculating the ratio of albumin to creatinine (mg/g) and indicated the proportion of animals of each genotype with no, mild (<30), moderate (30–60) or severe (>60) albuminuria as a percentage. Sample sizes for both each genotype are indicated at the top.
Another potential genotype–phenotype correlation was reported in an allelic series of three mutations: Col4a1G627W, Col4a1K950E and Col4a1G1046D (also called Bru, Raw and Svc, respectively) 16. The authors reported that the Gly substitutions (Col4a1G627W and Col4a1G1046D) caused relatively more severe ocular and renal phenotypes than a Lys substitution affecting the Yaa-position amino acid on the Gly-Xaa-Yaa motif (Col4a1K950E), which only causes a retinal vascular phenotype. Col4a1G627W was identified in a different genetic background, nonetheless, Col4a1K950E and Col4a1G1046D were studied in similar genetic contexts and the position of the mutation within the Gly-Xaa-Yaa repeat is a plausible explanation for the mild phenotype(s) in Col4a1K950E mice. An initial report of these alleles suggested that the glomerular basement membrane was unremarkable but Bowman’s capsule had basement membrane defects and hypertrophic cells 16 which differed from the ultrastructural glomerular basement membrane abnormalities 15, albuminurea and hematuria described for the other alleles. More recently, a detailed characterization of renal pathology cause by Col4a1K950E and Col4a1G1046D mutations was described extensive pathology and potentially distinct involvement of glomerular and tubular defects 107. Notably, differences in focus, techniques and study depth underscore the challenges of comparing genotype–phenotype data between research groups. A targeted allele that is analogous to a human HANAC-causing mutation (Col4a1G498V) was also recently published 104 and while the relative severities of the phenotypes are difficult to compare in isolation, this report also includes a thorough characterization of renal pathology and supports the existence of one or more amino-terminal sub-domain(s) that disproportionately influence the severity of myopathy and nephropathy.
Despite a limited number of available mutations, findings from studies using murine models of Col4a1 mutations, including two allelic series, provide insights into disease mechanisms and support the conclusion that there are genotype–phenotype correlations. The data suggest that 1) mutations in the NC1 domain (and likely other ‘quantitative’ mutations) may associate with milder outcomes than those in the triple-helical domain; 2) Gly substitutions in the triple helical domain have position-dependent effects on biosynthesis that correlate with CVD, but not other phenotypes; 3) in frame, exon-skipping mutations within the triple helical domain may be disproportionately severe compared to Gly substitutions 4) Yaa-position substitutions may be less severe than Gly substitutions 5) one or more functional sub-domains near the amino-termini may be involved in myopathy and possibly nephropathy. The fact that genetic background and genotype–phenotype correlations had differential effects on different phenotypes suggest that there is mechanistic heterogeneity.
5) Conclusions and perspectives
Mutations in COL4A1 and COL4A2 have been studied for nearly a decade and, compared to type I collagen mutations underlying OI and COL4A5 mutations underlying Alport Syndrome, the number of patients from which to draw genotype-phenotype correlations is relatively limited. However, this retrospective analysis suggests the existence of genotype–phenotype correlations for COL4A1 and COL4A2 mutations that are consistent with observations for some other types of collagens. In both humans and mice, there is a frequency bias for mutations in COL4A1 over COL4A2. This inequity likely reflects a combination of ascertainment bias and true biological differences. Mutations in COL4A1 were reported several years before mutations in COL4A2 and routine COL4A2 screening in patients lagged. As mutation detection shifts from candidate gene sequencing to exome analyses this bias should disappear. However, ascertainment bias alone does not explain the discrepancies in the numbers of mutations in each gene. The genes are roughly equal in size and would be expected to acquire mutations at equivalent rates in patients and during random chemical mutagenesis. It is possible that milder outcomes for COL4A2 mutations may explain the frequency difference in mutations identified. It is interesting to note however that in the murine allelic series, the Col4a2 mutation essentially behaved like a position-matched Col4a1 mutation with the exception of one phenotype (albuminurea) for which this mutation was more severe.
As is the case for other types of collagens, Gly substitutions were the most common class of mutation in patients. Mutations in 50 out of 437 triple helical Gly residues in COL4A1 and six out of 428 triple helical Gly residues for COL4A2 have been identified. While substitutions for branched amino acids occurred more frequently, the identity of the substituting amino acid did not appear to have a strong influence on disease severity. Instead, there appear to be important positional influences; mutations located near the amino terminus of COL4A1 are associated with milder pathology, and the severity of CVD tends to be worse for Gly substitutions that are closer to the carboxyl end of the triple helical domain. Finally, mutations that clustered near the region containing integrin-binding domains appear to be associated with a greater likelihood for patients to have myopathy and nephropathy suggesting the existence of a functional subdomain influencing the penetrance of distinct phenotypes.
In findings from the murine allelic series the mutation with presumed quantitative effects (S1582P in the NC1 domain) had milder effects on heterotrimer biosynthesis, ICH and myopathy than most mutations with presumed qualitative effects (mutations in the triple helical domain). In contrast, in patients, qualitative COL4A1 mutations do not appear to be associated with more severe outcomes than quantitative mutations. As there are biases toward identification of mutations with severe outcomes in early studies, it is possible that these classes of mutations will diverge as more patients are reported with a broader spectrum of mutations. An additional caveat is that the molecular consequences of most mutations were inferred and not tested, which could lead to erroneous classification. Importantly, although porencephaly and early age-at-onset CVD were considered to be more severe than adult age-at-onset CVD determining the relative outcome severities is subjective and remains a pervasive challenge. There is the additional challenge in integrating multiple co-morbid phenotypes, some of which appear to be mechanistically distinct. In contrast, characterizations of OI severity relies on well-established criteria for bone fragility that are more quantitative and focused on a relatively defined outcome. Finally, the roles of different environmental factors (including anti-coagulants, surgical delivery, and exercise) 15, 101 must all be considered as they may contribute to misleading conclusions.
In contrast to patient studies, the allelic series of mutant mice comprises less saturation of mutations across the protein or for each class of mutation. For example, there is only one presumed quantitative mutation in the NC1 domain. Although the effect was consistent across phenotypes (and consistent with the absence of phenotypes in mice heterozygous for Col4a1;Col4a2 null alleles) these findings are based upon observations for a single allele and it would be overly simplistic to presume that mutations across the entire domain will behave equally. However, these resources are powered by standardized environments, genetic homogeneity and large sample sizes for an individual mutation which all improve reproducibility. Continued efforts to characterize pathology in Col4a1 and Col4a2 mutant mice will improve our understanding of the phenotypic spectrum of Col4a1– and Col4a2– related disease. Better definition of the genotype-phenotype correlations and patient outcomes will help to understand risks including co-morbidities or age-at-onset and will ultimately impact therapeutic approaches, genetic counseling and patient care. This information will ideally come from large, prospective, longitudinal studies to detail the natural histories of patients for whom there are biochemical data, and quantitative outcome measures for multiple phenotypes. Together, these and other advances may reveal pathways that can be targeted therapeutically to reduce, delay or prevent aspects of this syndrome.
Table 3.

|
Highlights.
We discuss genotype-phenotype correlations emerging from 105 COL4A1 and COL4A2 mutations in patients and 17 Col4a1 and Col4a2 mutations in mice. The results have similarities and differences when compared to genotype-phenotype correlations for other types of collagens. Triple helical glycine substitutions are the most frequent class of mutation. Glycine is most often replaced by a charged amino acid however the position of the mutation appears to be more important than the identity of the substituting amino acid. Data from mice, but not humans, suggest that NC1 domain mutations might be less damaging that triple helical domain mutations. Moreover, for cerebrovascular disease there appears to be a position-dependent effect whereby triple helical mutations nearer the carboxy terminus may be more damaging than those nearer the amino terminus. However, this association may not hold true for all phenotypes and there appears to be at least one functionalsubdomain near the amino terminal end of the triple helical domain in which mutations are have divergent outcome severities for different sub-phenotypes. The involvement of multiple organs and a lack of quantitative phenotypic measures make it difficult to draw firm conclusions and the different components of the multisystem disorder likely involve different pathogenic mechanisms, which must be considered.
Acknowledgments
We thank Dr. Cassandre Labelle-Dumais for comments on the manuscript and assistance with the figures. This study was funded in part by an American Heart Association post-doctoral fellowship to Dr. Jeanne, National Institute of Neurological Disease and Stroke research grant to Dr. Gould (NS083830) and support to the Department of Ophthalmology from the National Eye Institute (core grant EY02162) and Research to Prevent Blindness (unrestricted grant).
Abbreviations
- COL4A1
collagen type IV alpha 1
- COL4A2
collagen type IV alpha 2
- CVD
cerebrovascular disease
- HANAC
hereditary angiopathy with nephropathy, aneurysms and muscle cramps
- ICH
intracerebral hemorrhages
- OI
Osteogenesis imperfecta
- NC1
non collagenous 1
- vEDS
vascular Ehlers-Danlos syndrome
- Ala
Alanine
- Arg
Arginine
- Asp
Aspartic acid
- Cys
Cysteine
- Glu
Glutamic acid
- Gly
Glycine
- Ser
Serine
- Trp
Tryptophan
- Val
Valine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mao M, Alavi MV, Labelle-Dumais C, Gould DB. Type IV Collagens and Basement Membrane Diseases: Cell Biology and Pathogenic Mechanisms. Curr Top Membr. 2015;76:61–116. doi: 10.1016/bs.ctm.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. 2008;71:357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem. 1993;268:26033–26036. [PubMed] [Google Scholar]
- 5.Sado Y, Kagawa M, Kishiro Y, Sugihara K, Naito I, Seyer JM, Sugimoto M, Oohashi T, Ninomiya Y. Establishment by the rat lymph node method of epitope-defined monoclonal antibodies recognizing the six different alpha chains of human type IV collagen. Histochem Cell Biol. 1995;104:267–275. doi: 10.1007/BF01464322. [DOI] [PubMed] [Google Scholar]
- 6.Pollner R, Schmidt C, Fischer G, Kühn K, Poschl E. Cooperative and competitive interactions of regulatory elements are involved in the control of divergent transcription of human Col4A1 and Col4A2 genes. FEBS Lett. 1997;405:31–36. doi: 10.1016/s0014-5793(97)00152-x. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt C, Pollner R, Poschl E, Kühn K. Expression of human collagen type IV genes is regulated by transcriptional and post-transcriptional mechanisms. FEBS Lett. 1992;312:174–178. doi: 10.1016/0014-5793(92)80929-b. [DOI] [PubMed] [Google Scholar]
- 8.Pollner R, Fischer G, Poschl E, Kühn K. Regulation of divergent transcription of the genes coding for basement membrane type IV collagen. Annals of the New York Academy of Sciences. 1990;580:44–54. doi: 10.1111/j.1749-6632.1990.tb17916.x. [DOI] [PubMed] [Google Scholar]
- 9.Poschl E, Pollner R, Kühn K. The genes for the alpha 1(IV) and alpha 2(IV) chains of human basement membrane collagen type IV are arranged head-to-head and separated by a bidirectional promoter of unique structure. EMBO J. 1988;7:2687–2695. doi: 10.1002/j.1460-2075.1988.tb03122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boutaud A, Borza DB, Bondar O, Gunwar S, Netzer KO, Singh N, Ninomiya Y, Sado Y, Noelken ME, Hudson BG. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J Biol Chem. 2000;275:30716–30724. doi: 10.1074/jbc.M004569200. [DOI] [PubMed] [Google Scholar]
- 11.Trüeb B, Gröbli B, Spiess M, Odermatt BF, Winterhalter KH. Basement membrane (type IV) collagen is a heteropolymer. J Biol Chem. 1982;257:5239–5245. [PubMed] [Google Scholar]
- 12.Hofmann H, Voss T, Kühn K, Engel J. Localization of flexible sites in thread-like molecules from electron micrographs. Comparison of interstitial, basement membrane and intima collagens. Journal of Molecular Biology. 1984;172:325–343. doi: 10.1016/s0022-2836(84)80029-7. [DOI] [PubMed] [Google Scholar]
- 13.Bella J, Liu J, Kramer R, Brodsky B, Berman HM. Conformational effects of Gly-X-Gly interruptions in the collagen triple helix. Journal of Molecular Biology. 2006;362:298–311. doi: 10.1016/j.jmb.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 14.Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 15.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, John SWM. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 16.Van Agtmael T, Schlötzer-Schrehardt U, McKie L, Brownstein DG, Lee AW, Cross SH, Sado Y, Mullins JJ, Pöschl E, Jackson IJ. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet. 2005;14:3161–3168. doi: 10.1093/hmg/ddi348. [DOI] [PubMed] [Google Scholar]
- 17.Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SWM. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 18.Mao M, Smith RS, Alavi MV, Marchant JK, Cosma M, Libby RT, John SWM, Gould DB. Strain-Dependent Anterior Segment Dysgenesis and Progression to Glaucoma in Col4a1 Mutant Mice. Invest Ophthalmol Vis Sci. 2015;56:6823–6831. doi: 10.1167/iovs.15-17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet. 2012;21:R97–R110. doi: 10.1093/hmg/dds346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meuwissen MEC, Halley DJJ, Smit LS, Lequin MH, Cobben JM, de Coo R, van Harssel J, Sallevelt S, Woldringh G, van der Knaap MS, de Vries LS, Mancini GMS. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med. 2015;17:843–853. doi: 10.1038/gim.2014.210. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, Hu Y, Donkervoort S, Flanigan KM, Swoboda KJ, Winder TL, Weiss RB, Bönnemann CG. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 2013;34:1558–1567. doi: 10.1002/humu.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoornaert KP, Vereecke I, Dewinter C, Rosenberg T, Beemer FA, Leroy JG, Bendix L, Björck E, Bonduelle M, Boute O, Cormier-Daire V, De Die-Smulders C, Dieux-Coeslier A, Dollfus H, Elting M, Green A, Guerci VI, Hennekam RCM, Hilhorts-Hofstee Y, Holder M, Hoyng C, Jones KJ, Josifova D, Kaitila I, Kjaergaard S, Kroes YH, Lagerstedt K, Lees M, LeMerrer M, Magnani C, Marcelis C, Martorell L, Mathieu M, McEntagart M, Mendicino A, Morton J, Orazio G, Paquis V, Reish O, Simola KOJ, Smithson SF, Temple KI, Van Aken E, van Bever Y, van den Ende J, Van Hagen JM, Zelante L, Zordania R, De Paepe A, Leroy BP, De Buyzere M, Coucke PJ, Mortier GR. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010;18:872–880. doi: 10.1038/ejhg.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barat-Houari M, Dumont B, Fabre A, Them FT, Alembik Y, Alessandri J-L, Amiel J, Audebert S, Baumann-Morel C, Blanchet P, Bieth E, Brechard M, Busa T, Calvas P, Capri Y, Cartault F, Chassaing N, Ciorca V, Coubes C, David A, Delezoide A-L, Dupin-Deguine D, El Chehadeh S, Faivre L, Giuliano F, Goldenberg A, Isidor B, Jacquemont M-L, Julia S, Kaplan J, Lacombe D, Lebrun M, Marlin S, Martin-Coignard D, Martinovic J, Masurel A, Melki J, Mozelle-Nivoix M, Nguyen K, Odent S, Philip N, Pinson L, Plessis G, Quélin C, Shaeffer E, Sigaudy S, Thauvin C, Till M, Touraine R, Vigneron J, Baujat G, Cormier-Daire V, Le Merrer M, Geneviève D, Touitou I. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur J Hum Genet. 2016;24:992–1000. doi: 10.1038/ejhg.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortier GR, Weis M, Nuytinck L, King LM, Wilkin DJ, De Paepe A, Lachman RS, Rimoin DL, Eyre DR, Cohn DH. Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder. J Med Genet. 2000;37:263–271. doi: 10.1136/jmg.37.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura G, Haga N, Kitoh H, Tanaka Y, Sonoda T, Kitamura M, Shirahama S, Itoh T, Nakashima E, Ohashi H, Ikegawa S. The phenotypic spectrum of COL2A1 mutations. Hum Mutat. 2005;26:36–43. doi: 10.1002/humu.20179. [DOI] [PubMed] [Google Scholar]
- 26.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 27.Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996;135:163–181. [PubMed] [Google Scholar]
- 28.Frank M, Albuisson J, Ranque B, Golmard L, Mazzella J-M, Bal-Theoleyre L, Fauret A-L, Mirault T, ND, Mousseaux E, Boutouyrie P, Fiessinger J-Nel, Emmerich J, Messas E, Jeunemaitre X. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular. Ehlers–Danlos syndrome. 2015;23:1657–1664. doi: 10.1038/ejhg.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV) Genet Med. 2014;16:881–888. doi: 10.1038/gim.2014.72. [DOI] [PubMed] [Google Scholar]
- 30.Byers PH, Wallis GA, Willing MC. Osteogenesis imperfecta: translation of mutation to phenotype. J Med Genet. 1991;28:433–442. doi: 10.1136/jmg.28.7.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Körkkö J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila-Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgleish R, Byers PH. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forlino A, Marini JC. Osteogenesis imperfecta: prospects for molecular therapeutics. Mol Genet Metab. 2000;71:225–232. doi: 10.1006/mgme.2000.3039. [DOI] [PubMed] [Google Scholar]
- 33.Bodian DL, Chan T-F, Poon A, Schwarze U, Yang K, Byers PH, Kwok P-Y, Klein TE. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet. 2009;18:463–471. doi: 10.1093/hmg/ddn374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 35.Borza DB, Bondar O, Ninomiya Y, Sado Y, Naito I, Todd P, Hudson BG. The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J Biol Chem. 2001;276:28532–28540. doi: 10.1074/jbc.M103690200. [DOI] [PubMed] [Google Scholar]
- 36.Colville DJ, Savige J. Alport syndrome. A review of the ocular manifestations. NOPG. 1997;18:161–173. doi: 10.3109/13816819709041431. [DOI] [PubMed] [Google Scholar]
- 37.Colville D, Savige J, Morfis M, Ellis J, Kerr P, Agar J, Fasset R. Ocular manifestations of autosomal recessive Alport syndrome. NOPG. 1997;18:119–128. doi: 10.3109/13816819709057125. [DOI] [PubMed] [Google Scholar]
- 38.Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248:1224–1227. doi: 10.1126/science.2349482. [DOI] [PubMed] [Google Scholar]
- 39.Hostikka SL, Eddy RL, Byers MG, Höyhtyä M, Shows TB, Tryggvason K. Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome-linked Alport syndrome. Proc Natl Acad Sci USA. 1990;87:1606–1610. doi: 10.1073/pnas.87.4.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gross O, Netzer K-O, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. 2002;17:1218–1227. doi: 10.1093/ndt/17.7.1218. [DOI] [PubMed] [Google Scholar]
- 41.Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, Schrier RW. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010;21:876–883. doi: 10.1681/ASN.2009070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC. X-linked Alport syndrome: natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol. 2000;11:649–657. doi: 10.1681/ASN.V114649. [DOI] [PubMed] [Google Scholar]
- 43.Tsiakkis D, Pieri M, Koupepidou P, Demosthenous P, Panayidou K, Deltas C. Genotype-phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin Genet. 2012;82:297–299. doi: 10.1111/j.1399-0004.2012.01849.x. [DOI] [PubMed] [Google Scholar]
- 44.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer K-O, Flinter F, Pirson Y, Dahan K, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J Am Soc Nephrol. 2003;14:2603–2610. doi: 10.1097/01.asn.0000090034.71205.74. [DOI] [PubMed] [Google Scholar]
- 45.Breedveld G, de Coo IF, Lequin MH, Arts WFM, Heutink P, Gould DB, John SWM, Oostra B, Mancini GMS. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J Med Genet. 2006;43:490–495. doi: 10.1136/jmg.2005.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang X, Xiao X, Jia X, Li S, Li M, Guo X, Liu X, Zhang Q. Mutation analysis of the genes associated with anterior segment dysgenesis, microcornea and microphthalmia in 257 patients with glaucoma. Int J Mol Med. 2015 doi: 10.3892/ijmm.2015.2325. [DOI] [PubMed] [Google Scholar]
- 47.Garel C, Rosenblatt J, Moutard ML, Heron D, Gelot A, Gonzales M, Miné E, Jouannic JM. Fetal intracerebral hemorrhage and COL4A1 mutation: promise and uncertainty. Ultrasound in obstetrics & gynecology: the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2013;41:228–230. doi: 10.1002/uog.12268. [DOI] [PubMed] [Google Scholar]
- 48.Weng Y-C, Sonni A, Labelle-Dumais C, De Leau M, Kauffman WB, Jeanne M, Biffi A, Greenberg SM, Rosand J, Gould DB. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol. 2012;71:470–477. doi: 10.1002/ana.22682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoneda Y, Haginoya K, Kato M, Osaka H, Yokochi K, Arai H, Kakita A, Yamamoto T, Otsuki Y, Shimizu S-i, Wada T, Koyama N, Mino Y, Kondo N, Takahashi S, Hirabayashi S, Takanashi J-i, Okumura A, Kumagai T, Hirai S, Nabetani M, Saitoh S, Hattori A, Yamasaki M, Kumakura A, Sugo Y, Nishiyama K, Miyatake S, Tsurusaki Y, Doi H, Miyake N, Matsumoto N, Saitsu H. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann Neurol. 2013;73:48–57. doi: 10.1002/ana.23736. [DOI] [PubMed] [Google Scholar]
- 50.Giorgio E, Vaula G, Bosco G, Giacone S, Mancini C, Calcia A, Cavalieri S, Di Gregorio E, Rigault De Longrais R, Leombruni S, Pinessi L, Cerrato P, Brusco A, Brussino A. Two families with novel missense mutations in COL4A1: When diagnosis can be missed. J Neurol Sci. 2015;352:99–104. doi: 10.1016/j.jns.2015.03.042. [DOI] [PubMed] [Google Scholar]
- 51.Plaisier E, Chen Z, Gekeler F, Benhassine S, Dahan K, Marro B, Alamowitch S, Paques M, Ronco P. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am J Med Genet A. 2010;152A:2550–2555. doi: 10.1002/ajmg.a.33659. [DOI] [PubMed] [Google Scholar]
- 52.Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology. 2009;73:1873–1882. doi: 10.1212/WNL.0b013e3181c3fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, Dracon M, Fardeau M, Van Agtmael T, Kerjaschki D, Antignac C, Ronco P. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 54.Zenteno JC, Crespí J, Buentello-Volante B, Buil JA, Bassaganyas F, Vela-Segarra JI, Diaz-Cascajosa J, Marieges MT. Next generation sequencing uncovers a missense mutation in COL4A1 as the cause of familial retinal arteriolar tortuosity. Graefes Arch Clin Exp Ophthalmol. 2014;252:1789–1794. doi: 10.1007/s00417-014-2800-6. [DOI] [PubMed] [Google Scholar]
- 55.Magnin E, Ayrignac X, Berger E, Mine M, Tournier-Lasserve E, Labauge P. Late Diagnosis of COL4A1 Mutation and Problematic Vascular Risk Factor Management. Eur Neurol. 2014;72:150–152. doi: 10.1159/000360532. [DOI] [PubMed] [Google Scholar]
- 56.Coutts SB, Matysiak-Scholze U, Kohlhase J, Innes AM. Intracerebral hemorrhage in a young man. CMAJ. 2011;183:E61–4. doi: 10.1503/cmaj.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vahedi K, Kubis N, Boukobza M, Arnoult M, Massin P, Tournier-Lasserve E, Bousser MG. COL4A1 mutation in a patient with sporadic, recurrent intracerebral hemorrhage. Stroke. 2007;38:1461–1464. doi: 10.1161/STROKEAHA.106.475194. [DOI] [PubMed] [Google Scholar]
- 58.Livingston J, Doherty D, Orcesi S, Tonduti D, Piechiecchio A, La Piana R, Tournier-Lasserve E, Majumdar A, Tomkins S, Rice G, Kneen R, van der Knaap M, Crow Y. COL4A1 mutations associated with a characteristic pattern of intracranial calcification. Neuropediatrics. 2011;42:227–233. doi: 10.1055/s-0031-1295493. [DOI] [PubMed] [Google Scholar]
- 59.Tonduti D, Pichiecchio A, La Piana R, Livingston JH, Doherty DA, Majumdar A, Tomkins S, Mine M, Ceroni M, Ricca I, Balottin U, Orcesi S. COL4A1-related disease: raised creatine kinase and cerebral calcification as useful pointers. Neuropediatrics. 2012;43:283–288. doi: 10.1055/s-0032-1325116. [DOI] [PubMed] [Google Scholar]
- 60.Corlobe A, Tournier-Lasserve E, Mine M, Menjot de Champfleur N, Carra Dalliere C, Ayrignac X, Labauge P, Arquizan C. COL4A1 mutation revealed by an isolated brain hemorrhage. Cerebrovasc Dis. 2013;35:593–594. doi: 10.1159/000351520. [DOI] [PubMed] [Google Scholar]
- 61.John S, Jehi L, Manno EM, Conway DS, Uchino K. COL4A1 gene mutation--beyond a vascular syndrome. Seizure. 2015;31:19–21. doi: 10.1016/j.seizure.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 62.Lemmens R, Maugeri A, Niessen HWM, Goris A, Tousseyn T, Demaerel P, Corveleyn A, Robberecht W, van der Knaap MS, Thijs VN, Zwijnenburg PJG. Novel COL4A1 mutations cause cerebral small vessel disease by haploinsufficiency. Hum Mol Genet. 2013;22:391–397. doi: 10.1093/hmg/dds436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deml B, Reis LM, Maheshwari M, Griffis C, Bick D, Semina EV. Whole exome analysis identifies dominant COL4A1 mutations in patients with complex ocular phenotypes involving microphthalmia. Clin Genet. 2014;86:475–481. doi: 10.1111/cge.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smigiel R, Cabala M, Jakubiak A, Kodera H, Sasiadek MJ, Matsumoto N, Sasiadek MM, Saitsu H. Novel COL4A1 mutation in an infant with severe dysmorphic syndrome with schizencephaly, periventricular calcifications, and cataract resembling congenital infection. Birth Defects Res Part A Clin Mol Teratol. 2016;106:304–307. doi: 10.1002/bdra.23488. [DOI] [PubMed] [Google Scholar]
- 65.Coupry I, Sibon I, Mortemousque B, Rouanet F, Mine M, Goizet C. Ophthalmological features associated with COL4A1 mutations. Arch Ophthalmol. 2010;128:483–489. doi: 10.1001/archophthalmol.2010.42. [DOI] [PubMed] [Google Scholar]
- 66.Sibon I, Coupry I, Menegon P, Bouchet J-P, Gorry P, Burgelin I, Calvas P, Orignac I, Dousset V, Lacombe D, Orgogozo J-M, Arveiler B, Goizet C. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol. 2007;62:177–184. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 67.Aguglia U, Gambardella A, Breedveld GJ, Oliveri RL, Le Piane E, Messina D, Quattrone A, Heutink P. Suggestive evidence for linkage to chromosome 13qter for autosomal dominant type 1 porencephaly. Neurology. 2004;62:1613–1615. doi: 10.1212/01.wnl.0000123113.46672.68. [DOI] [PubMed] [Google Scholar]
- 68.Gasparini S, Qualtieri A, Ferlazzo E, Cianci V, Patitucci A, Spadafora P, Aguglia U. Normal immunofluorescence pattern of skin basement membranes in a family with porencephaly due to COL4A1 G749S mutation. Neurol Sci. 2016;37:459–463. doi: 10.1007/s10072-015-2435-3. [DOI] [PubMed] [Google Scholar]
- 69.Vermeulen RJ, Peeters-Scholte C, Van Vugt JJM, Van Vught JJMG, Barkhof F, Rizzu P, van der Schoor SRD, van der Knaap MS. Fetal origin of brain damage in 2 infants with a COL4A1 mutation: fetal and neonatal MRI. Neuropediatrics. 2011;42:1–3. doi: 10.1055/s-0031-1275343. [DOI] [PubMed] [Google Scholar]
- 70.Shah S, Kumar Y, McLean B, Churchill A, Stoodley N, Rankin J, Rizzu P, van der Knaap M, Jardine P. A dominantly inherited mutation in collagen IV A1 (COL4A1) causing childhood onset stroke without porencephaly. Eur J Paediatr Neurol. 2010;14:182–187. doi: 10.1016/j.ejpn.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 71.Rouaud T, Labauge P, Tournier-Lasserve E, Mine M, Coustans M, Deburghgraeve V, Edan G. Acute urinary retention due to a novel collagen COL4A1 mutation. Neurology. 2010;75:747–749. doi: 10.1212/WNL.0b013e3181eee440. [DOI] [PubMed] [Google Scholar]
- 72.Shah S, ELLARD S, KNEEN R, LIM M, OSBORNE N, Rankin J, Stoodley N, van der Knaap M, WHITNEY A, Jardine P. Childhood presentation of COL4A1 mutations. Dev Med Child Neurol. 2012;54:569–574. doi: 10.1111/j.1469-8749.2011.04198.x. [DOI] [PubMed] [Google Scholar]
- 73.Colin E, Sentilhes L, Sarfati A, Mine M, Guichet A, Ploton C, Boussion F, Delorme B, Tournier-Lasserve E, Bonneau D. Fetal intracerebral hemorrhage and cataract: think COL4A1. J Perinatol. 2014;34:75–77. doi: 10.1038/jp.2013.135. [DOI] [PubMed] [Google Scholar]
- 74.Slavotinek AM, Garcia ST, Chandratillake G, Bardakjian T, Ullah E, Wu D, Umeda K, Lao R, Tang PLF, Wan E, Madireddy L, Lyalina S, Mendelsohn BA, Dugan S, Tirch J, Tischler R, Harris J, Clark MJ, Chervitz S, Patwardhan A, West JM, Ursell P, de Alba Campomanes A, Schneider A, Kwok PY, Baranzini S, Chen RO. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects. Clin Genet. 2015;88:468–473. doi: 10.1111/cge.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xia X-Y, Li N, Cao X, Wu Q-Y, Li T-F, Zhang C, Li W-W, Cui Y-X, Li X-J, Xue C-Y. A novel COL4A1 gene mutation results in autosomal dominant non-syndromic congenital cataract in a Chinese family. BMC Med Genet. 2014;15:97. doi: 10.1186/s12881-014-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vahedi K, Boukobza M, Massin P, Gould DB, Tournier-Lasserve E, Bousser M-G. Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology. 2007;69:1564–1568. doi: 10.1212/01.wnl.0000295994.46586.e7. [DOI] [PubMed] [Google Scholar]
- 77.Meuwissen MEC, de Vries LS, Verbeek HA, Lequin MH, Govaert PP, Schot R, Cowan FM, Hennekam R, Rizzu P, Verheijen FW, Wessels MW, Mancini GMS. Sporadic COL4A1 mutations with extensive prenatal porencephaly resembling hydranencephaly. Neurology. 2011;76:844–846. doi: 10.1212/WNL.0b013e31820e7751. [DOI] [PubMed] [Google Scholar]
- 78.Decio A, Tonduti D, Pichiecchio A, Vetro A, Ciccone R, Limongelli I, Giorda R, Caffi L, Balottin U, Zuffardi O, Orcesi S. A novel mutation in COL4A1 gene: a possible cause of early postnatal cerebrovascular events. Am J Med Genet A. 2015;167A:810–815. doi: 10.1002/ajmg.a.36907. [DOI] [PubMed] [Google Scholar]
- 79.Plancher JM, Hufnagel RB, Vagal A, Peariso K, Saal HM, Broderick JP. Case of Small Vessel Disease Associated with COL4A1 Mutations following Trauma. Case Rep Neurol. 2015;7:142–147. doi: 10.1159/000431309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Labelle-Dumais C, Dilworth DJ, Harrington EP, De Leau M, Lyons D, Kabaeva Z, Manzini MC, Dobyns WB, Walsh CA, Michele DE, Gould DB. COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 2011;7:e1002062. doi: 10.1371/journal.pgen.1002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tomotaki S, Mizumoto H, Hamabata T, Kumakura A, Shiota M, Arai H, Haginoya K, Hata D. Severe Hemolytic Jaundice in a Neonate with a Novel COL4A1 Mutation. Pediatr Neonatol. 2014 doi: 10.1016/j.pedneo.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 82.Lichtenbelt KD, Pistorius LR, de Tollenaer SM, Mancini GM, de Vries LS. Prenatal genetic confirmation of a COL4A1 mutation presenting with sonographic fetal intracranial hemorrhage. Ultrasound in obstetrics & gynecology: the official journal of the International Society of Ultrasound in Obstetrics and Gynecology. 2012;39:726–727. doi: 10.1002/uog.11070. [DOI] [PubMed] [Google Scholar]
- 83.van der Knaap MS, Smit LME, Barkhof F, Pijnenburg YAL, Zweegman S, Niessen HWM, Imhof S, Heutink P. Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol. 2006;59:504–511. doi: 10.1002/ana.20715. [DOI] [PubMed] [Google Scholar]
- 84.Smit LM, Barth PG, Valk J, Njiokiktjien C. Familial porencephalic white matter disease in two generations. Brain Dev. 1984;6:54–58. doi: 10.1016/s0387-7604(84)80010-8. [DOI] [PubMed] [Google Scholar]
- 85.Takenouchi T, Ohyagi M, Torii C, Kosaki R, Takahashi T, Kosaki K. Porencephaly in a fetus and HANAC in her father: variable expression of COL4A1 mutation. Am J Med Genet A. 2015;167A:156–158. doi: 10.1002/ajmg.a.36823. [DOI] [PubMed] [Google Scholar]
- 86.NIWA T, AIDA N, Osaka H, Wada T, Saitsu H, IMAI Y. Intracranial Hemorrhage and Tortuosity of Veins Detected on Susceptibility-weighted Imaging of a Child with a Type IV Collagen α1 Mutation and Schizencephaly. Magn Reson Med Sci. 2015;14:223–226. doi: 10.2463/mrms.2014-0060. [DOI] [PubMed] [Google Scholar]
- 87.Leung M, Lewis E, Humphreys P, Miller E, Geraghty M, Lines M, Sell E. COL4A1 mutation in a pediatric patient presenting with post-ictal hemiparesis. Can J Neurol Sci. 2012;39:654–657. doi: 10.1017/s0317167100015420. [DOI] [PubMed] [Google Scholar]
- 88.Bilguvar K, Diluna ML, Bizzarro MJ, Bayri Y, Schneider KC, Lifton RP, Günel M, Ment LR Group PaBT. COL4A1 mutation in preterm intraventricular hemorrhage. J Pediatr. 2009;155:743–745. doi: 10.1016/j.jpeds.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.HARTEMAN JC, Groenendaal F, van HAASTERT IC, LIEM KD, STROINK H, BIERINGS MB, HUISMAN A, de Vries LS. Atypical timing and presentation of periventricular haemorrhagic infarction in preterm infants: the role of thrombophilia. Dev Med Child Neurol. 2012;54:140–147. doi: 10.1111/j.1469-8749.2011.04135.x. [DOI] [PubMed] [Google Scholar]
- 90.de Vries LS, Koopman C, Groenendaal F, Van Schooneveld M, Verheijen FW, Verbeek E, Witkamp TD, van der Worp HB, Mancini G. COL4A1 mutation in two preterm siblings with antenatal onset of parenchymal hemorrhage. Ann Neurol. 2009;65:12–18. doi: 10.1002/ana.21525. [DOI] [PubMed] [Google Scholar]
- 91.Değerliyurt A, Ceylaner G, Koçak H, Bilginer Gürbüz B, Cihan BS, Rizzu P, Ceylaner S. A new family with autosomal dominant porencephaly with a novel Col4A1 mutation. Are arachnoid cysts related to Col4A1 mutations? Genet Couns. 2012;23:185–193. [PubMed] [Google Scholar]
- 92.Rødahl E, Knappskog PM, Majewski J, Johansson S, Telstad W, Kråkenes J, Boman H. Variants of anterior segment dysgenesis and cerebral involvement in a large family with a novel COL4A1 mutation. Am J Ophthalmol. 2013;155:946–953. doi: 10.1016/j.ajo.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 93.Murray LS, Lu Y, Taggart A, Van Regemorter N, Vilain C, Abramowicz M, Kadler KE, Van Agtmael T. Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet. 2014;23:283–292. doi: 10.1093/hmg/ddt418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vilain C, Van Regemorter N, Verloes A, David P, Van Bogaert P. Neuroimaging fails to identify asymptomatic carriers of familial porencephaly. Am J Med Genet. 2002;112:198–202. doi: 10.1002/ajmg.10452. [DOI] [PubMed] [Google Scholar]
- 95.Ha TT, Sadleir LG, Mandelstam SA, Paterson SJ, Scheffer IE, Gecz J, Corbett MA. A mutation in COL4A2 causes autosomal dominant porencephaly with cataracts. Am J Med Genet A. 2016;170A:1059–1063. doi: 10.1002/ajmg.a.37527. [DOI] [PubMed] [Google Scholar]
- 96.Gunda B, Mine M, Kovács T, Hornyák C, Bereczki D, Várallyay G, Rudas G, Audrezet M-P, Tournier-Lasserve E. COL4A2 mutation causing adult onset recurrent intracerebral hemorrhage and leukoencephalopathy. J Neurol. 2014;261:500–503. doi: 10.1007/s00415-013-7224-4. [DOI] [PubMed] [Google Scholar]
- 97.Yoneda Y, Haginoya K, Arai H, Yamaoka S, Tsurusaki Y, Doi H, Miyake N, Yokochi K, Osaka H, Kato M, Matsumoto N, Saitsu H. De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly. Am J Hum Genet. 2012;90:86–90. doi: 10.1016/j.ajhg.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Verbeek E, Meuwissen MEC, Verheijen FW, Govaert PP, Licht DJ, Kuo DS, Poulton CJ, Schot R, Lequin MH, Dudink J, Halley DJ, de Coo RIF, den Hollander JC, Oegema R, Gould DB, Mancini GMS. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur J Hum Genet. 2012;20:844–851. doi: 10.1038/ejhg.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, Valant V, Greenberg SM, Rosand J, Gould DB. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am J Hum Genet. 2012;90:91–101. doi: 10.1016/j.ajhg.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Favor J, Gloeckner CJ, Janik D, Klempt M, Neuhäuser-Klaus A, Pretsch W, Schmahl W, Quintanilla-Fend L. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics. 2007;175:725–736. doi: 10.1534/genetics.106.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jeanne M, Jorgensen J, Gould DB. Molecular and Genetic Analyses of Collagen Type IV Mutant Mouse Models of Spontaneous Intracerebral Hemorrhage Identify Mechanisms for Stroke Prevention. Circulation. 2015;131:1555–1565. doi: 10.1161/CIRCULATIONAHA.114.013395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuo DS, Labelle-Dumais C, Mao M, Jeanne M, Kauffman WB, Allen J, Favor J, Gould DB. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum Mol Genet. 2014;23:1709–1722. doi: 10.1093/hmg/ddt560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Loscertales M, Nicolaou F, Jeanne M, Longoni M, Gould DB, Sun Y, Maalouf FI, Nagy N, Donahoe PK. Type IV collagen drives alveolar epithelial-endothelial association and the morphogenetic movements of septation. BMC Biol. 2016;14:59. doi: 10.1186/s12915-016-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen Z, Migeon T, Verpont MC, Zaidan M, Sado Y, Kerjaschki D, Ronco P, Plaisier E. HANAC Syndrome Col4a1 Mutation Causes Neonate Glomerular Hyperpermeability and Adult Glomerulocystic Kidney Disease. J Am Soc Nephrol. 2016;27:1042–1054. doi: 10.1681/ASN.2014121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lyon: Bruised (Bru) Mouse News Lett. 1984. Google Scholar [computer program] [Google Scholar]
- 106.Van Agtmael T, Bailey MA, Schlötzer-Schrehardt U, Craigie E, Jackson IJ, Brownstein DG, Megson IL, Mullins JJ. Col4a1 mutation in mice causes defects in vascular function and low blood pressure associated with reduced red blood cell volume. Hum Mol Genet. 2010;19:1119–1128. doi: 10.1093/hmg/ddp584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jones FE, Bailey MA, Murray LS, Lu Y, McNeilly S, Schlötzer-Schrehardt U, Lennon R, Sado Y, Brownstein DG, Mullins JJ, Kadler KE, Van Agtmael T. ER stress and basement membrane defects combine to cause glomerular and tubular renal disease resulting from Col4a1 mutations in mice. Disease Models and Mechanisms. 2016;9:165–176. doi: 10.1242/dmm.021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thaung C, West K, Clark BJ, McKie L, Morgan JE, Arnold K, Nolan PM, Peters J, Hunter AJ, Brown SDM, Jackson IJ, Cross SH. Novel ENU-induced eye mutations in the mouse: models for human eye disease. Hum Mol Genet. 2002;11:755–767. doi: 10.1093/hmg/11.7.755. [DOI] [PubMed] [Google Scholar]
- 109.Taylor SH, Al-Youha S, Van Agtmael T, Lu Y, Wong J, McGrouther DA, Kadler KE. Tendon is covered by a basement membrane epithelium that is required for cell retention and the prevention of adhesion formation. PLoS ONE. 2011;6:e16337. doi: 10.1371/journal.pone.0016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alavi MV, Mao M, Pawlikowski BT, Kvezereli M, Duncan JL, Libby RT, John SWM, Gould DB. Col4a1 mutations cause progressive retinal neovascular defects and retinopathy. Sci Rep. 2016;6:18602. doi: 10.1038/srep18602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gould DB, Marchant JK, Savinova OV, Smith RS, John SWM. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet. 2007;16:798–807. doi: 10.1093/hmg/ddm024. [DOI] [PubMed] [Google Scholar]
- 112.Plaisier E, Alamowitch S, Gribouval O, Mougenot B, Gaudric A, Antignac C, Roullet E, Ronco P. Autosomal-dominant familial hematuria with retinal arteriolar tortuosity and contractures: a novel syndrome. Kidney Int. 2005;67:2354–2360. doi: 10.1111/j.1523-1755.2005.00341.x. [DOI] [PubMed] [Google Scholar]