

Abstract
Many diseases accompanied by chronic inflammation are connected with dysregulated activation of macrophage subpopulations. Recently, we reported that nitro-fatty acids (NO2-FAs), products of metabolic and inflammatory reactions of nitric oxide and nitrite, modulate macrophage and other immune cell functions. Bone marrow cell suspensions were isolated from mice and supplemented with macrophage colony-stimulating factor (M-CSF) or granulocyte-macrophage colony-stimulating factor (GM-CSF) in combination with NO2-OA for different times. RAW 264.7 macrophages were used for short-term (1–5 min) experiments. We discovered that NO2-OA reduces cell numbers, cell colony formation, and proliferation of macrophages differentiated with colony-stimulating factors (CSFs), all in the absence of toxicity. In a case of GM-CSF-induced bone marrow-derived macrophages (BMMs), NO2-OA acts via downregulation of signal transducer and activator of transcription 5 (STAT5) and extracellular signal-regulated kinase (ERK) activation. In the case of M-CSF-induced BMMs, NO2-OA decreases activation of M-CSFR and activation of related PI3K and ERK. Additionally, NO2-OA also attenuates activation of BMMs. In aggregate, we demonstrate that NO2-OA regulates the process of macrophage differentiation and that NO2-FAs represent a promising therapeutic tool in the treatment of inflammatory pathologies linked with increased accumulation of macrophages in inflamed tissues.
Keywords: nitro-oleic acid, nitro-fatty acids, differentiation, inflammation, macrophages, growth factors, signaling pathways
Graphical Abstract
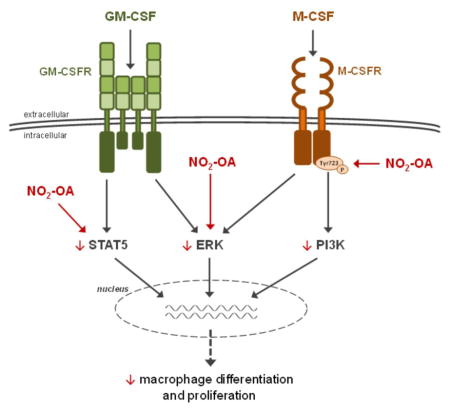
Introduction
Macrophages play a role in almost every aspect of the biology of an organism; including development, homeostasis and repair and immune responses to pathogens [1]. Differentiation of macrophages from bone marrow precursors is a complex process that involves several developmental stages that are induced and controlled by different growth factors such as macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) [2]. Both colony-stimulating factors (CSFs) are multifunctional cytokines that regulate differentiation, proliferation, and survival of monocyte/macrophage precursors. M-CSF is constitutively and ubiquitously produced by many tissues, where it maintains macrophage populations important for tissue homeostasis. GM-CSF, in contrast, has low basal circulating levels and is often elevated during immune reactions [3–6]. Therefore, both CSFs were used as model inducers of differentiation of bone marrow-derived macrophages (BMMs) from bone marrow cells and precursors (BMCs).
The process of CSF-induced macrophage differentiation is regulated via several signaling pathways (e.g. signal transducer and activator of transcription 5, STAT5; PU.1; extracellular signal-regulated kinase, ERK; phosphatidylinositol 3-kinase, PI3K; and receptor for M-CSF, M-CSFR). These signaling pathways are triggered in the early phase of BMM differentiation, followed by additional upregulation of M-CSFR and PU.1 expression as well as macrophage proliferation during the middle and late phase of this process [4, 7, 8]. Importantly, macrophages reach final maturation after activation by numerous signals from their microenvironment and dysregulation of their functions may result in the development and progression of cardiovascular, chronic inflammatory, and autoimmune diseases [9–12]. Recent studies have demonstrated that proliferative activity of BMCs can be affected by relatively mild systemic inflammatory stimuli, which are associated with elevated levels of M-CSF and GM-CSF. This leads to increased differentiation and proliferation of monocytes/macrophages in the blood and different tissues [13–18]. Moreover, it is suggested that CSFs are important components of the microenvironment, responsible for BMM functional specialization to M1/M2 phenotype [19, 20]. While M1 macrophages possess characteristic microbicidal and tumoricidal activities, M2 macrophages are involved in the control of wound healing, tissue repair, and remodeling [21]. Thus, M-CSF- and GM-CSF-induced differentiation of macrophages represents a viable pharmacological target for the future treatment of chronic and inflammatory diseases.
Inflammatory and metabolic reactions both promote associated with increased levels of reactive nitric oxide, nitrite and oxygen-derived species. The exposure of endogenous unsaturated fatty acids (FAs) to these reactive molecules results in generation of several nitrative and oxidative derivatives, including electrophilic nitro-fatty acid derivatives (NO2-FAs). Mechanism of NO2-FA signaling will depends on their relative distribution in hydrophobic and hydrophilic microenvironments, esterification and relative concentrations of nucleophilic targets for Michael addition. The predominant signaling reactions of NO2-FAs are a consequence of electrophilic nitroalkylation of nucleophilic amino acids, leading to post-translational protein modifications [22, 23]. This nitroalkylation of predominantly hyperreactive protein cysteine residues leads to alterations in structure, function, and subcellular distribution of target proteins, such as receptors, transcription factors, enzymes, and ion channels (e.g. STATs; nuclear factor kappa B, NF-κB; Keap1/Nrf2 system, and peroxisome proliferator-activated receptor gamma, PPAR-γ) [22, 24–38]. NO2-FAs are endogenously present in tissues, body fluids, and membranes with total levels (free plus esterified form) in the high nM range once dissociated from nucleophilic binding partners, with these concentrations affected by dietary, metabolic, and inflammatory status [37, 39, 40]. For example, intramitochondrial NO2-FA concentrations after a brief priod of ischemia-reoxygenation was ~1 μM. Importantly, NO2-FAs were shown to exhibit significant anti-inflammatory actions and their production is considered an adaptive reaction that contributes to regulating and mediating the resolution of inflammation [22, 23, 32]. Therefore, NO2-FAs may represent a viable class of mediators for the treatment of metabolic disorders and different inflammatory related diseases.
Recently, we have reported that nitro-oleic acid (NO2-OA) modulates the activation of both M1 and M2 macrophages [33] as well as macrophage adhesion on endothelial cells and their chemotaxis [38]. Moreover, NO2-OA reduces the total numbers of macrophages accumulated in different types of an inflamed tissues – including atherosclerotic plaques [36], lung tissue [34], and heart atrium [35], affirming the potential of NO2-FA as mediators that can regulate diverse macrophage functions. Herein, we reveal that NO2-OA also influences also the process of M-CSF- and GM-CSF-induced macrophage differentiation, which could represent another crucial finding for successful realization of clinical trials. In this study, BMCs were isolated from mice and supplemented with M-CSF or GM-CSF. Differentiated BMMs were then activated with LPS/IFN-γ or IL-4 for evaluating of their activation phenotype. We analyzed the total numbers of BMMs and their colonies as well as their activation status. Finally our analysis was focused on the molecular mechanisms of NO2-OA-dependent regulation of different signaling pathways (STAT5, ERK, and PI3K), associated with early phase of macrophage differentiation and activation of M-CSFR.
Materials and methods
Reagents and Solutions
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). NO2-OA was synthesized as previously [37–40] and consisted of an equimolar mixture of regioisomers of (E)-9- and 10-nitro-octadec-9-enoic acid (1:1; for structures see Supplement Figure 1), which displays signaling activity equivalent to that of each pure regioisomers. The NO2-OA was diluted to 100 mM solution in methanol and stored at −80°C. For experimental applications, a 10 mM solution of NO2-OA in methanol was prepared and diluted in Dulbecco’s Modified Eagle’s Medium (DMEM; PAN-Biotech, Aidenbach, Germany) to obtain 100 μM NO2-OA (stored in +4°C no more than 2 weeks). All stocks were prepared and stored in sterile, low-binding tubes [33]. In our experiments we used final NO2-OA concentration 1 or 3 μM. The concentrations of NO2-OA were selected based on previous studies, a lack of toxicity [33–36, 38], and responses that have been induced by physiologically and pharmacologically relevant concentrations of NO2-FAs in vivo [37, 39, 40].
Primary Cell Culture and Treatment
Native BMCs were isolated from healthy C57BL/6J mice (males, 25–30 g, 12–14 weeks old; Masaryk University, Brno, Czech Republic) by flushing femora and tibiae [41]. The experiments were approved by the Animal Care Committee and were in accordance with the EU and NIH Guide for Care and Use of Laboratory Animals. Complete cell suspension was seeded on 6-well plates in 1 ml of DMEM with 2.5% low endotoxin fetal bovine serum (FBS; PAA, Pasching, Austria) and 1% penicillin-streptomycin. The cells were immediately supplemented with 20 ng/ml M-CSF or GM-CSF (BioLegend, San Diego, CA, USA) and with 3 μM NO2-OA, and cultured in a humidified incubator at 37°C and 5% CO2. Schematic representation of BMC experimental setup is available in Figure 1A. The CSF concentrations were selected according to literature [15, 42, 43]. The cells used as controls were not supplemented with CSFs and/or NO2-OA. To study the late phase of macrophage differentiation and maturation, the second and the fourth day after the start of the experiment, cultivated BMMs were re-supplemented with 0.5 ml of fresh DMEM with 2.5% FBS, 1% penicillin-streptomycin, and with proportional amount of NO2-OA and M-CSF or GM-CSF. By the seventh day, media with nonadherent cells were removed and replaced with 1 ml of fresh DMEM with 2.5% FBS, 1% penicillin-streptomycin, and with 3 μM NO2-OA (without M-CSF and GM-CSF). Immediately after that, adherent BMMs were activated by combination of 500 ng/ml LPS (E. coli serotype 026:B6) and 50 ng/ml IFN-γ or by 20 ng/ml IL-4 for 24 h. To study the middle phase of macrophage differentiation, adherent BMMs were harvested after 2 days from the start of experiment. To study the early phase of macrophage differentiation, adherent and nonadherent BMCs were harvested after 30 min from the start of the experiment.
Figure 1. Schematic representation of experimental setup.

Upper parts briefly describe cell preparation, treatment, and cultivation time. Lower parts summarize days and types of measurements as well as cell type used. (A) BMC experiments. (B) RAW 264.7 experiments.
Cell Line and Treatment
Murine peritoneal macrophages RAW 264.7 (ATCC, Manassas, VA, USA) were grown in DMEM with 10% FBS and 1% gentamycin in a humidified incubator at 37°C and 5% CO2 [33]. The RAW 264.7 cells were used in short-termed experiments, where the isolation procedure was not compatible using BMCs. The cells were routinely checked for mycoplasma contamination. Before the experiment, the RAW 264.7 macrophages were seeded on 6-well plates in 1 ml of FBS free DMEM with 1% penicillin-streptomycin. After 6 hours, the RAW 264.7 cells were supplemented with 50 ng/ml M-CSF or GM-CSF and with 1 μM NO2-OA. For schematic representation of RAW 264.7 experimental setup see Figure 1B. The RAW 264.7 cells were harvested after 5-min treatment.
Cell Counting
Quantification of BMM numbers was performed by cell counting. Pictures of culture dish wells (magnification 40x) used for counting were taken through Eclipse TS100 Inverted Microscope (Nikon, Japan) after 7 days of BMM differentiation in DMEM supplemented with 20 ng/ml M-CSF or GM-CSF (BioLegend, San Diego, CA, USA) and with 3 μM NO2-OA (for details see part Primary cell culture and Treatment as well as Figure 1A).
Methylcellulose Assay
To study the ability of BMCs/BMMs to differentiate, proliferate, and form colonies in a semi-solid media in response to M-CSF or GM-CSF induction, The Colony Forming Cell Assay (R&D systems, Minneapolis, MN, USA) was performed according to supplier’s instructions. Native BMCs were isolated and cultivated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in solution (ratio 1:3) of methylcellulose and DMEM (with 2.5% FBS and 1% penicillin-streptomycin) for 15 days. The third, sixth, ninth, and twelfth day after the start of experiment, cultivated cells were re-supplemented with fresh methylcellulose/DMEM solution with proportional amount of NO2-OA and M-CSF or GM-CSF. Quantification of nonadherent BMM colonies was performed by colony counting. Pictures of culture dish wells (magnification 10x) used for counting were taken through Eclipse TS100 Inverted Microscope (Nikon, Japan) after 15 days of BMM differentiation in methylcellulose semi-solid media.
Viability and proliferation
The measurement of cell viability was based on the total cellular mass of adherent cells using the detergent-compatible BCA protein assay reagent (Bio-Rad, Hercules, CA, USA) with bovine serum albumin as a standard [44]. None of the tested compounds decreased total protein levels below the nontreated BMCs/BMMs (data not shown). The proliferation of adherent BMMs was detected by ATP Cell Viability Test (BioThema, Sweden) and results are displayed as percentage of nontreated BMCs/BMMs.
Cytotoxicity
Potential cytotoxic effects of NO2-OA were determined by measurement of lactate dehydrogenase (LDH) in cell media using the Cytotoxicity Detection KitPLUS (Roche, Pleasanton, CA, USA). Samples (80 μl) were mixed with reaction mixture in a ratio of 1:1 and incubated at room temperature for 30 min. The absorbance was measured at 490 nm using a SPECTRA Rainbow microplate reader (Tecan, Mannedorf, Switzerland). Murine macrophages RAW 264.7 lysed by supplier-provided lysis buffer were used as positive control.
Determination of NO Production
Changes in NO production were measured indirectly via the accumulation of nitrites, the end product of NO metabolism, in media samples using spectrophotometrical Griess assay [33]. Samples (150 μl) were mixed with Griess reagent in a ratio of 1:1 and incubated at room temperature for 15 min. The absorbance was measured at 542 nm using a SPECTRA Rainbow microplate reader (Tecan, Mannedorf, Switzerland).
Western blot technique
Expression and/or activation of proteins was detected after 5 min-treatment in lysates of adherent RAW 264.7 macrophages, after 30 min-treatment in mixture of lysates from both adherent and nonadherent BMCs or after 2 days and 8 days of differentiation in lysates from adherent BMMs. Total cellular protein lysates were harvested by disrupting the cells using SDS lysis buffer [45]. In 2 day-experiments samples were not diluted to the same protein concentration, rather normalization was performed using β-actin expression. Protein samples were subjected to SDS-polyacrylamide gel electrophoresis separation on a 12.5% acrylamide gel and transferred onto polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore, Billerica, MA, USA). After incubation in blocking buffer (5% nonfat dried milk in solution of Tris-buffered saline and Tween 20, TBS-T), membranes were probed with primary antibodies overnight at 4°C; primary antibodies against β-actin (cat. no. sc-47778; Santa Cruz Biotechnology, Dallas, TX, USA), iNOS (cat. no. 610431; BD Transduction Transduction Laboratories, Lexington, KY, USA), Arginase I (cat. no. 9819; Cell Signaling Technology, Danvers, MA, USA), M-CSFR (cat. no. 3152; Cell Signaling Technology), phospho-M-CSFR (Tyr723; cat. no. 3155; Cell Signaling Technology), PI3K p85 (cat. no. 4257; Cell Signaling Technology), phospho-PI3K p85/p55 (Tyr458/Tyr199; cat. no. 4228; Cell Signaling Technology), STAT5 (cat. no. 9358; Cell Signaling Technology), phospho-STAT5 (Tyr694; cat. no. 9359; Cell Signaling Technology), p44/42 MAPK (ERK1/2) (cat. no. 4695; Cell Signaling Technology), phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204; cat. no. 4370; Cell Signaling Technology), PU.1 (cat. no. 2266; Cell Signaling Technology), CCAAT/enhancer binding protein beta (C/EBPβ; cat. no. 3087; Cell Signaling Technology), and Avian myelocytomatosis virus oncogene cellular homolog (c-Myc; cat. no. 9402; Cell Signaling Technology) were used. Following incubation, membranes were washed three times in TBS-T and then incubated with secondary anti-mouse or anti-rabbit antibodies conjugated with horseradish peroxidase (Cell Signaling Technology) for an hour at room temperature. The blots were visualized by SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA) and exposed to CP-B X-ray films (Agfa, Brno, Czech Republic). The relative levels of proteins were quantified by scanning densitometry using the ImageJ program (National Institutes of Health, Bethesda, MD, USA) and the individual band density value was expressed in arbitrary units (optical density, OD) [45]. The data in graphs represents the ratio between OD values of bands detecting protein of interest and OD values of bands detecting β-actin (Figure 3B, 4B, and 4C), or the ratio between OD values of bands detecting phosphorylated (p-) and the total protein form (Figure 5B, C, and E–H).
Figure 3. The effect of NO2-OA on M-CSFR, PU.1, C/EBPβ, and c-Myc expression in CSF-induced BMMs differentiated for 7 days.

Native BMCs were isolated and cultivated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in DMEM for 7 days. Adherent BMMs were then activated with LPS (500 ng/ml) and IFN-γ (50 ng/ml) or with IL-4 (20 ng/ml) for 24 h. For schematic representation of BMC experimental setup see Figure 1A. (A) Expression of M-CSFR and β-actin in adherent BMMs; Western blot pictures represent one of three individual experiments. (B) Assessment of M-CSFR expression in adherent BMMs; n=3. A p value of less than 0.05 was considered significant and statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared to the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively. (C) Expression of PU.1, C/EBPβ, c-Myc, and β-actin in adherent BMMs; representative Western blot pictures.
Figure 4. The effect of NO2-OA on M-CSFR, PI3K, STAT5, ERK 1/2, PU.1, and β-actin expression in BMMs differentiated for 2 days.

Native BMCs were isolated and cultivated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) for 2 days. For schematic representation of BMC experimental setup see Figure 1A. (A) Expression of M-CSFR, PI3K, STAT5, ERK 1/2, PU.1, and β-actin in adherent BMMs; Western blot pictures represent one of three or four individual experiments. (B, C) Assessment of M-CSFR and PU.1 expression in adherent BMMs; n=3–4. A p value of less than 0.05 was considered significant and statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared to the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively. (D, E) Pearson correlation between M-CSFR or PU.1 expression and β-actin expression in adherent BMMs; n=3. A p value of less than 0.05 was considered significant. (F) Assessment of BMM proliferation; n=3. A p value of less than 0.05 was considered significant and statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared to the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively.
Figure 5. The effect of NO2-OA on STAT5, ERK, M-CSFR, and PI3K activation during early phase of differentiation.

Native BMCs were isolated and treated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) for 30 min (A–C). RAW 264.7 macrophages were treated with M-CSF (50 ng/ml) or GM-CSF (50 ng/ml) and NO2-OA (1 μM) for 5 min (D–H). For schematic representation of experimental setups see Figure 1. (A) Activation of STAT5 (Tyr694) and ERK 1/2 (Thr202/Tyr204) in adherent and nonadherent BMCs; Western blot pictures represent one of four individual experiments. (B, C) Assessment of STAT5 and ERK 1/2 phosphorylation in adherent and nonadherent BMCs; n=4. (D) Activation of STAT5 (Tyr694), ERK 1/2 (Thr202/Tyr204), M-CSFR (Tyr723), and PI3K (Tyr458) in RAW 264.7; Western blot pictures represent one of four individual experiments. (E–H) Assessment of STAT5, ERK 1/2, M-CSFR, and PI3K phosphorylation in RAW 264.7; n=4. A p value of less than 0.05 was considered significant and statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared to the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively.
Statistics
Data was statistically analyzed using one-way analysis of variance (ANOVA), which was followed by Bonferonni’s multiple comparison test (GraphPad Prism 5.01), or Pearson correlation (GrafPad Prism 5.01). All data is reported as means ± SEM. A p < 0.05 was considered significant. The statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared with the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively. The statistical significance for the Pearson correlation was labeled as *p.
Results
NO2-OA reduces numbers, proliferation, and colony formation of CSF-differentiated BMMs
Native BMCs were isolated and cultivated with i) M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in DMEM for 7 days (Figure 2A, 2B, and 2D) or ii) M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in methylcellulose solution for 15 days (Figure 2C). A schematic representation of the BMC experimental design is in Figure 1A. Importantly, NO2-OA significantly reduced numbers of CSF-induced BMMs (Figure 2A) and their proliferation activity (Figure 2B) as well as numbers of CSF-induced BMM colonies (Figure 2C), in the absence of an impact on cell morphology (Figure 2D). GM-CSF emerged as a more potent stimulator of BMM differentiation, proliferation, and colony formation compared with M-CSF (Figure 2A–D). BMCs cultivated without supplementation of CSFs displayed only modest ability to differentiate or proliferate and did not form colonies (Figure 2A–D). Compared to positive control, neither NO2-OA, nor M-CSF/GM-CSF increased LDH level in BMM supernatants following 24 hours of treatment (data not shown).
Figure 2. The effect of NO2-OA on BMM numbers, proliferation, and colony formation.

Native BMCs were isolated and cultivated i) with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in DMEM for 7 days (A, B, and D) or ii) with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in methylcellulose solution for 15 days (C). For schematic representation of BMC experimental setup see Figure 1A. (A) Quantification of adherent BMM numbers; n=4. (B) Assessment of BMM proliferation; n=5. (C) Quantification of nonadherent BMM colonies; n=3. A p value of less than 0.05 was considered significant and statistical comparison is presented as follows: a, b, c, d, and e represents the group of data statistically significant when compared to the data in the first (a), second (b), third (c), fourth (d), and fifth (e) bar, respectively. (D) Representative images of adherent BMMs; magnification 40x.
NO2-OA supresses expression of M-CSFR and activation of BMMs
The presence and maturation of differentiated BMMs was confirmed by expression of M-CSFR, PU.1, C/EBPβ, and c-Myc, as well as BMM activation potential. All these parameters were sensitive to NO2-OA treatment, which inhibited M-CSFR expression (Figure 3) and the activation phenotype of BMMs (Supplement Figure 2). M-CSFR expression (cell surface form; 170 kDa) was detected in lysates from adherent BMMs after 7 days of differentiation (Figure 3A and 3B). Both M-CSF and GM-CSF upregulated M-CSFR protein expression (Figure 3A and 3B). The M1 activation of BMMs was triggered by LPS (500 ng/ml) in combination with IFN-γ (50 ng/ml) and M2 activation was stimulated by IL-4 (20 ng/ml) for the next 24 hours. All three groups of BMMs were sensitive to LPS/IFN-γ, which induced downregulation of M-CSFR expression (Figure 3A and 3B) and upregulated iNOS expression (Supplement Figure 2A) and NO production (Supplement Figure 2B). In comparison with LPS/IFN-γ, IL-4 caused significant but rather mild reduction of M-CSFR levels in GM-CSF-induced BMMs (Figure 3A and 3B). The expression of Arginase I was provoked only in GM-CSF-induced BMMs treated with IL-4 (Supplement Figure 2A). Other selected markers of macrophage maturation, namely PU.1, C/EBPβ, and c-Myc, were also detected in lysates from adherent BMMs after 7 days of differentiation. While PU.1 and C/EBPβ were upregulated by LPS/IFN-γ, c-Myc was elevated only after IL-4 (Figure 3C).
NO2-OA suppresses expression of M-CSFR and PU.1 in GM-CSF-elicited BMMs
Native BMCs were isolated and cultivated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in DMEM for 2 days (Figure 1A and 4). Expression of the total protein forms of M-CSFR, PI3K, STAT5, ERK, PU.1, and β-actin was detected in lysates from adherent BMMs (Figure 4A). Importantly, NO2-OA significantly downregulated expression of M-CSFR (Figure 4B) and PU.1 (Figure 4C) in GM-CSF-induced BMMs. Both CSFs provoked changes in β-actin expression (Figure 4A) reflecting elevation in overall cellular metabolism and proliferative capacity (Figure 4F). GM-CSF was confirmed as a strong stimulator of macrophage differentiation, since GM-CSF-induced BMMs showed more extensive expression of all proteins compared with M-CSF-induced or control BMMs (Figure 4A). With the exception of PU.1 (Figure 4E), the changes in expression of M-CSFR (Figure 4D) and other proteins (data not shown) correlated with changes in β-actin expression. The NO2-OA-induced reduction of M-CSFR (Figure 4B) and PU.1 (Figure 4C) was significant even when normalized to β-actin expression. Expression of other proteins (PI3K, STAT5, and ERK) was unchanged when normalized to β-actin expression (data not shown). Additionally, the effect of NO2-OA on macrophage proliferation and expression of CD11b was studied by flow cytometry (Figure 1A and Supplement Figure 3). NO2-OA attenuated the elevation of both control and CSF-induced BMM populations after 2 day-differentiation (Supplement Figure 3A and 3B). The expression of CD11b in control and CSF-induced BMMs was not affected by NO2-OA treatment in equal protein concentrations of cells (Supplement Figure 3C).
NO2-OA rapidly regulates STAT5, ERK, M-CSFR, and PI3K activation in BMMs during initial phases of CSF-induced differentiation
To evaluate activation of signaling pathways, i) native BMCs were isolated and treated with M-CSF (20 ng/ml) or GM-CSF (20 ng/ml) and NO2-OA (3 μM) in DMEM for 30 min (Figure 1A); ii) RAW 264.7 macrophages were treated with M-CSF (50 ng/ml) or GM-CSF (50 ng/ml) and NO2-OA (1 μM) for 5 min (Figure 1B). Lysates from both adherent and nonadherent BMCs were analyzed for phosphorylation/expression of different proteins and signaling molecules (STAT5, ERK, M-CSFR, and PI3K). Activation of STAT5 (Tyr694 phosphorylation) was triggered only by GM-CSF supplementation and was profoundly inhibited by NO2-OA treatment after 30 min of cell incubation (Figure 5A and 5B). The phosphorylation of ERK 1/2 (Thr202/Tyr204) was increased in both M-CSF- and GM-CSF-treated BMCs and was significantly inhibited by NO2-OA (Figure 5A and 5C). GM-CSF more extensively induced ERK activation, compared with M-CSF (Figure 5A and 5C). The phosphorylation of M-CSFR (Tyr723) and PI3K (Tyr458) was unaffected by CSFs and NO2-OA after 30 min of BMC treatment (data not shown). Because the isolation procedure of BMCs did not permit shorter experimental time frames, RAW 264.7 macrophages were employed [33, 45]. RAW 264.7 cells were supplemented with M-CSF or GM-CSF and treated with NO2-OA for 5 min. Both STAT5 and ERK activation in RAW 264.7 macrophages (Figure 5D–F) were reflective of BMC responses after treatment for 30 min (Figure 5A–C). Importantly, M-CSF induced significant elevation of M-CSFR (Tyr723) and PI3K (Tyr458) phosphorylation in RAW 264.7 macrophages, which was completely inhibited by NO2-OA treatment (Figure 5D, 5G, and 5H).
Discussion
The pleiotropic anti-inflammatory signaling actions of NO2-FAs remain to be fully characterized, especially in the context of the regulation of inflammatory cell function and signaling pathway activities [32]. We recently reported that and exemplary fatty acid nitroalkene derivative, NO2-OA, when administered in vivo leads to a reduction in macrophage accumulation inflamed tissues following metabolic and inflammatory stress (such as lungs, atrial fibrotic lesions, and atherosclerotic plaques) [34–36], suggesting a potential role for NO2-FAs in macrophage differentiation and maturation. Notably, the mechanisms accounting for NO2-OA regulation of these processes remains to be elucidated. Herein, we discovered that NO2-OA inhibits M-CSF and GM-CSF-induced macrophage differentiation and regulates the activation phenotype of BMMs. In concert with these actions, NO2-OA modulates diverse signaling pathways involved in M-CSF-induced differentiation, reinforcing the significant potential for NO2-FAs in influencing various macrophage functions.
First, NO2-OA markedly reduces CSF-induced BMM numbers, proliferation, and BMM colony formation at 2, 7, and 15 days. Importantly, NO2-OA did not change cell morphology or induce cytotoxic responses. The presence of differentiated BMMs was confirmed by detection of M-CSFR (also known as CSF-1R or CD115), a key marker of macrophage phenotype [4, 8, 19, 42]. M-CSFR levels were upregulated in both CSF-differentiated BMMs and reduced by NO2-OA treatment. These results are consistent with our previous findings that NO2-OA (i) reduces the macrophage numbers detected in lung tissue of mice with hypoxia-induced pulmonary hypertension [34], (ii) partially prevents the M1 macrophage accumulation in fibrotic atrium of angiotensin II-treated mice [35], and (iii) downregulates monocyte/macrophage accumulation in atherosclerotic plaques in a murine model of atherosclerosis [36]. Since these pathologies are also linked with increased levels of M-CSF and GM-CSF [13–18], we suggest that NO2-FA-mediated reduction of macrophage infiltration is a consequence of the inhibition of macrophage differentiation and proliferation.
Considering the processes of cell activation and functional specialization as an integral and final part of cell maturation, CSF-differentiated BMMs were treated by M1 (LPS/IFN-γ) or M2 (IL-4) macrophage activators [21]. These experiments were motivated by the previous observation of inhibitory effects of NO2-FAs towards both M1 and M2 macrophage subpopulations, where NO2-OA was shown to significantly reduce the LPS-stimulated production of both pro-inflammatory and immunoregulatory mediators (including NO, superoxide and tumor growth factor-β, TGF-β) as well as iNOS expression [33]. NO2-OA also decreased IL-4-induced macrophage responses by inhibiting Arginase-I expression and TGF-β production. These effects were mediated via downregulation of STAT, MAPK and NF-κB signaling responses [33]. The current study also confirmed that NO2-OA downregulates LPS/IFN-γ-induced production of NO and expression of iNOS (markers of M1 activation) as well as IL-4-activated expression of Arginase I (a marker of M2 activation) also in CSF-differentiated BMMs.
In addition to general markers of macrophage polarization, we now report the expression of additional critical markers of macrophage maturation, namely transcription factors PU.1, C/EBPβ, and c-Myc, which regulate cell differentiation and survival, as well as proliferation. Importantly, PU.1 and C/EBPβ control the expression of several myeloid-specific genes, including M-CSFR and receptor for GM-CSF (GM-CSFR) [8, 46]. While PU.1 and C/EBPβ are upregulated during macrophage maturation and are involved in development of both M1 and M2 phenotypes [8, 47, 48], c-Myc is repressed in the late phase of macrophage differentiation and is considered as a specific marker of M2 activation [8, 49, 50]. In the present study, NO2-OA modulated the expression of transcription factors selectively induced by CSFs, LPS/IFN-γ and IL-4. PU.1 was elevated in M-CSF-induced BMMs treated with either LPS/IFN-γ or IL-4 and in GM-CSF-induced BMMs stimulated by LPS/IFN-γ. C/EBPβ was effectively increased in CSF-induced BMMs treated with LPS/IFN-γ acting as a marker of M1 activation. As expected, c-Myc was upregulated only in IL-4-polarized BMMs elicited by both CSFs used. Furthermore, NO2-OA reduces both LPS/IFN-γ and IL-4-induced changes in C/EBPβ, PU.1, and c-Myc expression, supporting the concept that NO2-FA regulate macrophage maturation and functional specialization to the M1/M2 phenotype. These observations lend additional perspective to reports of the impact of NO2-FAs on macrophages with M1 phenotype, such as the inhibition of LPS-induced iNOS expression, iNOS activity, and NO production by nitro-arachidonic acid, nitro-linoleic acid (NO2-LA), and cholesteryl-nitrolinoleate in the macrophage cell line J774.1 [26, 51]. Moreover, NO2-OA and NO2-LA decreased LPS-stimulated secretion of inflammatory cytokines in RAW 264.7 cells [25]. Also, the prophylactic administration of NO2-OA limits local and systemic inflammatory responses (e.g. production of inflammatory cytokines, expression of chemokines, and expression of adhesion molecules) and the severity of multiorgan dysfunction (kidney, liver, and heart) in mice exposed to LPS [52].
Interestingly, we also found that NO2-OA and macrophage activators reduce M-CSFR expression in CSF-induced BMMs. Significant degradation of the cell surface form of M-CSFR was already described in mouse macrophage cell line BAC-1.2F5 stimulated either by LPS/IFN-γ or by IL-4 [53, 54]. In general, it is assumed that the process of receptor internalization probably serves as a negative feedback loop that prevents ongoing signaling and thus attenuates proliferative effect of M-CSF [53, 55]. These events can also account for the ability of NO2-OA to reduce macrophage cell numbers during the process of differentiation.
In order to reveal the effect of NO2-OA on the second day of BMM treatment, we determined the numbers of BMMs and the total expression of M-CSFR, PI3K, STAT5, ERK, and PU.1, all factors involved in macrophage differentiation [3, 56–58]. Both CSFs provoked changes in β-actin expression reflecting elevation in overall cellular metabolism, including differentiation and proliferative capacity. Except for PU.1, the comparison of β-actin expression with the expression of each individual protein above-mentioned showed highly significant correlation. We found a substantial increase in M-CSFR expression initiated by both CSFs, whereas the expression of PU.1 was elevated only by GM-CSF. Importantly, the effect of GM-CSF on M-CSFR and PU.1 expression was inhibited by NO2-OA. This reduction in expression was significant even if normalized to β-actin expression. These results confirmed that M-CSFR and PU.1 help regulate the middle and the late phase of CSF-induced macrophage differentiation [4, 7, 8]. As expected, the expression of other proteins (PI3K, STAT5, and ERK), known to be activated within early phase of differentiation [3, 56–58], was not changed when normalized to β-actin. The CSF-induced changes in β-actin expression were in accordance with the increase in BMM populations after 2 days of differentiation. These findings affirm that NO2-OA regulates macrophage proliferation. The presence of CD11b, a marker of leukocytes (including monocytes/macrophages, neutrophils, and natural killer cells) [42], was not affected by NO2-OA when the cells were diluted to equal concentration. We hypothesize that CD11b expression might be regulated by signaling pathways other than those influenced by NO2-OA (PI3K, STAT5, and ERK). Similar results were observed during the monocyte and neutrophil maturation process, where CD11b was partially induced independently of differentiating agents [59].
BMCs and RAW 264.7 macrophages were employed in subsequent analyses focused on the effect of NO2-OA on activation of molecules involved in CSF-induced macrophage differentiation, including M-CSFR, PI3K, STAT5, and ERK [3]. M-CSFR activation triggers auto-phosphorylation of several intracellular receptor tyrosine residues, leading to initiation of different signaling cascades, e.g. PI3K or MAPKs. Although the regulation of cellular processes by activated GM-CSFR is mediated by STAT5 and to a lesser extent MAPK proteins [3, 56–58]. RAW 264.7 cells were used in short time frame experiments evaluating the facile M-CSFR and PI3K phosphorylation that peaks between 1 to 5 minutes [16, 55, 60]. NO2-OA inhibited M-CSF-induced phosphorylation of M-CSFR, an effect associated with the reduced activation of PI3K. Additionally, NO2-OA diminishes GM-CSF-mediated phosphorylation of STAT5. The activation of ERK was common for both CSFs used and was sensitive to NO2-OA inhibition. Due to the lack of a suitable antibody for determination of GM-CSFR phosphorylation, this issue must be elucidated in the future studies.
The effect of NO2-OA on STAT5 and ERK is consistent with the inhibitory effect of NO2-OA on STAT1, 3, and 6 as well as MAPK phosphorylation in macrophages [33, 36]. For example, a murine model of atherosclerosis, Apolipoprotein E-deficient mice, revealed that chronic administration of NO2-OA decreases macrophage infiltration and, due to inhibition of STAT1 phosphorylation, also reduces foam cell formation [36]. Also, NO2-OA downregulates LPS-induced activation of STAT1, STAT3, and MAPKs (ERK, p38 MAPK, and c-Jun N-terminal kinase) as well as IL-4-elicited activation of STAT6 and STAT3 in RAW 264.7 macrophages [33]. Additionally, there is a suppressive effect of NO2-OA and NO2-LA on LPS-stimulated STAT1 phosphorylation in RAW 264.7 cells [31]. We speculate that NO2-OA-mediated reduction of ERK activation might also be linked with reduced numbers of BMMs observed after 7 days of macrophage differentiation. This mechanism may also account for the reduction of smooth muscle cell proliferation upon attenuation of ERK activation after NO2-OA treatment in a murine model of pulmonary hypertension [34].
The signaling actions of NO2-FA are based on the electrophilic nature of the β-carbon adjacent to the nitro-bonded carbon. This promotes the alkylation of nucleophilic amino acid residues (e.g. cysteine thiolate and to a lesser extent the imidazole moiety of histidine and the ε-amino group of lysine residues), yielding new carbon-carbon or carbon-heteroatom bond frameworks that reversibly modify receptors, transcription factors and enzymes [24–31, 37]. For example, NO2-OA alkylates nucleophilic residues of angiotensin II receptor type 1 (AT1R) and impacts the propagation of downstream signaling events (e.g. intracellular inositol-1,4,5-triphosphate and calcium mobilization) via this covalent interaction between NO2-OA and AT1R [29]. We hypothesize that NO2-OA could also interact with other types of receptors (e.g. M-CSFR and GM-CSFR), promoting alterations in receptor phosphorylation and the binding of protein kinases or other binding partners. In human and mouse M-CSFR, the extracellular domain consists of five Ig-like domains containing highly conserved cysteine residues. These domains are involved in the intrachain disulfide bonding and can undergo covalent modification [61], thus representing potential target for NO2-FA action. The high-affinity, functional GM-CSFR is composed of a specific ligand-binding α subunit and a common β subunit, which are members of the class 1 subgroup of the cytokine receptor superfamily and contain a number of conserved motifs, including four spatially, conserved cysteine residues that are essential for activation of receptor [62]. These moieties could be modified by NO2-FAs.
Although it is traditionally recognized that the regulation of kinases occurs via phosphorylation, MAPKs are also sensitive to electrophile-dependent regulation. For example the electrophiles 4-hydroxy-2-trans-nonenal covalently reactis with histidine residues within the ERK2 kinase phosphorylation lip to inhibit ERK enzymatic activity and signaling [63]. Thus, NO2-OA could similarly impact M-CSF- and GM-CSF-induced ERK phosphorylation and activation, and lead to an overall reduction of M-CSFR or GM-CSFR activation. STAT proteins are also likely to be regulated by oxidative and electrophilic alkylation reactions, in addition to tyrosine phosphorylation. STATs contain highly reactive cysteine residues, which are crucially involved in regulation of their activity (e.g. nucleocytoplasmic shuttling) [64]. Based on these precepts, we propose a similar mechanism for NO2-OA action in downregulation of GM-CSF-triggered STAT5 phosphorylation.
There are some limitations in clinically translating the observations of this study. First, the in vitro experiments with isolated cells do not model all of the conditions operative in the bone marrow during macrophage differentiation. Importantly, bone marrow is a complex organ containing many different hematopoietic and non-hematopoietic cell types, surrounded by niche characterized by compartments of extracellular matrix, specific pH, oxygen levels, and concentrations of nutrients and growth factors [65]. All of these biochemical and microenvironmental factors can also contribute to the regulation of monocytes/macrophage differentiation, which is necessarily simplified in in vitro models. Second, although this study mainly focuses on the anti-proliferative and anti-inflammatory effects of NO2-FAs, these species may be affecting other metabolic and signaling pathways in bone marrow due to an ability to post-translationally modify diverse proteins. Thus, one cannot exclude the possibility that NO2-FAs influence the process of macrophage differentiation and maturation via additional mechanisms.
Conclusions
The impact of NO2-OA on M-CSFR and GM-CSFR dependent regulation of macrophage signaling via STAT5, ERK, and PI3K-mediated mechanisms can account for downstream NO2-FA regulation of macrophage differentiation. While M-CSF and GM-CSF share some mutual pathways (e.g. ERK), they could differ in several signaling molecules (e.g. STAT5). Nevertheless, we cannot exclude the involvement of other signaling cascades (e.g. NF-κB, PPAR-γ, and Keap1/Nrf2) being potentially operative in the regulation of CSF-induced signaling by the pleiotropic actions of fatty acid nitroalkenes. In aggregate, this study reinforced that there are anti-inflammatory regulatory actions of NO2-OA in mature macrophages, supporting that NO2-FAs may represent new drug candidates suitable for deployment against chronic and inflammatory diseases having a complex pathogenesis.
Supplementary Material
Highlights.
NO2-OA reduces numbers of differentiated macrophages.
NO2-OA affects early phase of growth factor-induced macrophage differentiation.
NO2-OA acts via downregulation of M-CSFR, STAT5, ERK, and PI3K activation.
NO2-OA decreases activation of differentiated macrophages.
Acknowledgments
We thank Lenka Vystrcilova for excellent technical assistance and Sylva Novotna for the expert grammar analysis. This work was supported by the Czech Science Foundation (no. 13-40824P and 17-08066Y) and by the Ministry of Education, Youth and Sports (projects no. LD15069 and no. LQ1605 from the National Program of Sustainability II). BAF was supported by NIH grants R01-HL-058115, R01-HL-64937, PO1-HL-103455.
This article is based upon work from the COST Action BM1404 Mye-EUNITER (www.mye-euniter.eu), supported by COST (European Cooperation in Science and Technology). COST is part of the EU Framework Program Horizon 2020.
Abbreviations
- AT1R
angiotensin II receptor type 1
- BMC
bone marrow cell and precursor
- BMM
bone marrow-derived macrophage
- C/EBPβ
CCAAT/enhancer binding protein beta
- c-Myc
Avian myelocytomatosis virus oncogene cellular homolog
- CSF
colony-stimulating factor
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ERK
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GM-CSFR
granulocyte-macrophage colony-stimulating factor receptor
- IFN-γ
interferon gamma
- IL-4
interleukin 4
- iNOS
inducible nitric oxide synthase
- NO2-LA
nitro-linoleic acid
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- M-CSF
macrophage colony-stimulating factor
- M-CSFR
macrophage colony-stimulating factor receptor
- NF-κB
nuclear factor kappa B
- NO2-FA
nitro-fatty acid
- NO
nitric oxide
- NO2-OA
nitro-oleic acid
- OD
optical density
- PBS
phosphate buffered saline
- PI3K
phosphatidylinositol 3-kinase
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- STAT
signal transducer and activator of transcription
- TBS-T
Tris-buffered saline and Tween 20
- TGF-β
Tumor growth factor-β
Footnotes
Conflict of Interest
Bruce A. Freeman and Steven R. Woodcock acknowledge an interest in Complexa, Inc., as scientific founder/shareholder (BAF) and consultant (SRW). All other authors confirm that there are no conflicts of interest in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–55. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakajima H. Role of transcription factors in differentiation and reprogramming of hematopoietic cells. Keio J Med. 2011;60(2):47–55. doi: 10.2302/kjm.60.47. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34(2):81–9. doi: 10.1016/j.it.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14(11):628–38. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18(1):39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol. 2005;25(5):405–28. doi: 10.1615/critrevimmunol.v25.i5.50. [DOI] [PubMed] [Google Scholar]
- 7.Zhu J, Emerson SG. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene. 2002;21(21):3295–313. doi: 10.1038/sj.onc.1205318. [DOI] [PubMed] [Google Scholar]
- 8.Valledor AF, et al. Transcription factors that regulate monocyte/macrophage differentiation. J Leukoc Biol. 1998;63(4):405–17. doi: 10.1002/jlb.63.4.405. [DOI] [PubMed] [Google Scholar]
- 9.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31(7):1506–16. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Price LC, et al. Inflammation in pulmonary arterial hypertension. Chest. 2012;141(1):210–21. doi: 10.1378/chest.11-0793. [DOI] [PubMed] [Google Scholar]
- 11.Nahrendorf M, Swirski FK. Monocyte and macrophage heterogeneity in the heart. Circ Res. 2013;112(12):1624–33. doi: 10.1161/CIRCRESAHA.113.300890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wermuth PJ, Jimenez SA. The significance of macrophage polarization subtypes for animal models of tissue fibrosis and human fibrotic diseases. Clin Transl Med. 2015;4:2. doi: 10.1186/s40169-015-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiba Y, et al. M-CSF accelerates neointimal formation in the early phase after vascular injury in mice: the critical role of the SDF-1-CXCR4 system. Arterioscler Thromb Vasc Biol. 2007;27(2):283–9. doi: 10.1161/01.ATV.0000250606.70669.14. [DOI] [PubMed] [Google Scholar]
- 14.Devaraj S, et al. C-reactive protein induces M-CSF release and macrophage proliferation. J Leukoc Biol. 2009;85(2):262–7. doi: 10.1189/jlb.0808458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anzinger JJ, et al. Murine bone marrow-derived macrophages differentiated with GM-CSF become foam cells by PI3Kgamma-dependent fluid-phase pinocytosis of native LDL. J Lipid Res. 2012;53(1):34–42. doi: 10.1194/jlr.M018887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuhrman B, et al. Ox-LDL induces monocyte-to-macrophage differentiation in vivo: Possible role for the macrophage colony stimulating factor receptor (M-CSF-R) Atherosclerosis. 2008;196(2):598–607. doi: 10.1016/j.atherosclerosis.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 17.Le Meur Y, et al. Macrophage accumulation at a site of renal inflammation is dependent on the M-CSF/c-fms pathway. J Leukoc Biol. 2002;72(3):530–7. [PubMed] [Google Scholar]
- 18.Davies LC, et al. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat Commun. 2013;4:1886. doi: 10.1038/ncomms2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleetwood AJ, et al. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178(8):5245–52. doi: 10.4049/jimmunol.178.8.5245. [DOI] [PubMed] [Google Scholar]
- 20.Fleetwood AJ, et al. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. 2009;86(2):411–21. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- 21.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freeman BA, et al. Nitro-fatty acid formation and signaling. J Biol Chem. 2008;283(23):15515–9. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trostchansky A, Rubbo H. Nitrated fatty acids: mechanisms of formation, chemical characterization, and biological properties. Free Radic Biol Med. 2008;44(11):1887–96. doi: 10.1016/j.freeradbiomed.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Schopfer FJ, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A. 2005;102(7):2340–5. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui T, et al. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281(47):35686–98. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferreira AM, et al. Macrophage activation induces formation of the anti-inflammatory lipid cholesteryl-nitrolinoleate. Biochem J. 2009;417(1):223–34. doi: 10.1042/BJ20080701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villacorta L, et al. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am J Physiol Heart Circ Physiol. 2007;293(1):H770–6. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iles KE, et al. Fatty acid transduction of nitric oxide signaling: nitrolinoleic acid mediates protective effects through regulation of the ERK pathway. Free Radic Biol Med. 2009;46(7):866–75. doi: 10.1016/j.freeradbiomed.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, et al. Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ Res. 2010;107(4):540–8. doi: 10.1161/CIRCRESAHA.110.218404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonacci G, et al. Electrophilic fatty acids regulate matrix metalloproteinase activity and expression. J Biol Chem. 2011;286(18):16074–81. doi: 10.1074/jbc.M111.225029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichikawa T, et al. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology. 2008;149(8):4086–94. doi: 10.1210/en.2007-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delmastro-Greenwood M, Freeman BA, Wendell SG. Redox-dependent anti-inflammatory signaling actions of unsaturated fatty acids. Annu Rev Physiol. 2014;76:79–105. doi: 10.1146/annurev-physiol-021113-170341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ambrozova G, et al. Nitro-oleic acid modulates classical and regulatory activation of macrophages and their involvement in pro-fibrotic responses. Free Radic Biol Med. 2016;90:252–60. doi: 10.1016/j.freeradbiomed.2015.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klinke A, et al. Protective effects of 10-nitro-oleic acid in a hypoxia-induced murine model of pulmonary hypertension. Am J Respir Cell Mol Biol. 2014;51(1):155–62. doi: 10.1165/rcmb.2013-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudolph TK, et al. Nitrated fatty acids suppress angiotensin II-mediated fibrotic remodelling and atrial fibrillation. Cardiovasc Res. 2016;109(1):174–84. doi: 10.1093/cvr/cvv254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudolph TK, et al. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30(5):938–45. doi: 10.1161/ATVBAHA.109.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batthyany C, et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281(29):20450–63. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ambrozova G, et al. Nitro-Oleic Acid Inhibits Vascular Endothelial Inflammatory Responses and The Endothelial-Mesenchymal Transition. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbagen.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsikas D, et al. Nitro-fatty acids occur in human plasma in the picomolar range: a targeted nitro-lipidomics GC-MS/MS study. Lipids. 2009;44(9):855–65. doi: 10.1007/s11745-009-3332-4. [DOI] [PubMed] [Google Scholar]
- 40.Baker PR, et al. Red cell membrane and plasma linoleic acid nitration products: Synthesis, clinical identification, and quantitation. PNAS. 2004;101(32):11577–82. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maceckova M, et al. Bone marrow-derived macrophages exclusively expressed caveolin-2: The role of inflammatory activators and hypoxia. Immunobiology. 2015;220(11):1266–74. doi: 10.1016/j.imbio.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 42.Francke A, et al. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. J Histochem Cytochem. 2011;59(9):813–25. doi: 10.1369/0022155411416007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lacey DC, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol. 2012;188(11):5752–65. doi: 10.4049/jimmunol.1103426. [DOI] [PubMed] [Google Scholar]
- 44.Krejcova D, et al. The effect of different molecular weight hyaluronan on macrophage physiology. Neuro Endocrinol Lett. 2009;30(Suppl 1):106–11. [PubMed] [Google Scholar]
- 45.Pekarova M, et al. Asymmetric dimethylarginine regulates the lipopolysaccharide-induced nitric oxide production in macrophages by suppressing the activation of NF-kappaB and iNOS expression. Eur J Pharmacol. 2013;713(1–3):68–77. doi: 10.1016/j.ejphar.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Mak KS, et al. PU.1 and Haematopoietic Cell Fate: Dosage Matters. Int J Cell Biol. 2011;2011:808524. doi: 10.1155/2011/808524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pham TH, et al. CCAAT enhancer-binding protein beta regulates constitutive gene expression during late stages of monocyte to macrophage differentiation. J Biol Chem. 2007;282(30):21924–33. doi: 10.1074/jbc.M611618200. [DOI] [PubMed] [Google Scholar]
- 48.Shibata Y, et al. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15(4):557–67. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 49.Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27(50):6462–72. doi: 10.1038/onc.2008.312. [DOI] [PubMed] [Google Scholar]
- 50.Pello OM, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119(2):411–21. doi: 10.1182/blood-2011-02-339911. [DOI] [PubMed] [Google Scholar]
- 51.Trostchansky A, et al. Synthesis, isomer characterization, and anti-inflammatory properties of nitroarachidonate. Biochemistry. 2007;46(15):4645–53. doi: 10.1021/bi602652j. [DOI] [PubMed] [Google Scholar]
- 52.Wang H, et al. Nitro-oleic acid protects against endotoxin-induced endotoxemia and multiorgan injury in mice. Am J Physiol Renal Physiol. 2010;298(3):F754–62. doi: 10.1152/ajprenal.00439.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dello Sbarba P, et al. Interleukin-4 rapidly down-modulates the macrophage colony-stimulating factor receptor in murine macrophages. J Leukoc Biol. 1996;60(5):644–50. doi: 10.1002/jlb.60.5.644. [DOI] [PubMed] [Google Scholar]
- 54.Baccarini M, et al. IFN-gamma/lipopolysaccharide activation of macrophages is associated with protein kinase C-dependent down-modulation of the colony-stimulating factor-1 receptor. J Immunol. 1992;149(8):2656–61. [PubMed] [Google Scholar]
- 55.Huynh J, et al. CSF-1 receptor signalling from endosomes mediates the sustained activation of Erk1/2 and Akt in macrophages. Cell Signal. 2012;24(9):1753–61. doi: 10.1016/j.cellsig.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 56.Mossadegh-Keller N, et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature. 2013;497(7448):239–43. doi: 10.1038/nature12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119(15):3383–93. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- 58.Lehtonen A, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. 2002;71(3):511–9. [PubMed] [Google Scholar]
- 59.Drayson MT, et al. Cell proliferation and CD11b expression are controlled independently during HL60 cell differentiation initiated by 1,25 alpha-dihydroxyvitamin D(3) or all-trans-retinoic acid. Exp Cell Res. 2001;266(1):126–34. doi: 10.1006/excr.2001.5200. [DOI] [PubMed] [Google Scholar]
- 60.Yu W, et al. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J Biol Chem. 2012;287(17):13694–704. doi: 10.1074/jbc.M112.355610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu W, et al. CSF-1 receptor structure/function in MacCsf1r−/− macrophages: regulation of proliferation, differentiation, and morphology. J Leukoc Biol. 2008;84(3):852–63. doi: 10.1189/jlb.0308171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bagley CJ, et al. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood. 1997;89(5):1471–82. [PubMed] [Google Scholar]
- 63.Sampey BP, et al. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Mol Pharmacol. 2007;71(3):871–83. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 64.Marg A, et al. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol. 2004;165(6):823–33. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.