

Abstract
Vascular endothelial (VE)‐cadherin, a major endothelial adhesion molecule, regulates vascular permeability, and increased vascular permeability has been observed in several cancers. The aim of this study was to elucidate the role of the NEDD8‐Cullin E3 ligase, in maintaining barrier permeability. To this end, we investigated the effects of the inhibition of Cullin E3 ligases, by using inhibitors and knockdown techniques in HUVECs. Furthermore, we analyzed the mRNA and protein levels of the ligases by quantitative RT‐PCR and Western blotting, respectively. The results revealed that NEDD8‐conjugated Cullin 3 is required for VE‐cadherin‐mediated endothelial barrier functions. Treatment of HUVECs with MLN4924, a chemical inhibitor of the NEDD8‐activating enzyme, led to high vascular permeability due to impaired cell–cell contact. Similar results were obtained when HUVECs were treated with siRNA directed against Cullin 3, one of the target substrates of NEDD8. Immunocytochemical staining showed that both treatments equally depleted VE‐cadherin protein localized at the cell–cell borders. However, quantitative RT‐PCR showed that there was no significant difference in the VE‐cadherin mRNA levels between the treatment and control groups. In addition, cycloheximide chase assay revealed that the half‐life of VE‐cadherin protein was dramatically reduced by Cullin 3 depletion. Together, these findings suggest that neddylated Cullin 3 plays a crucial role in endothelial cell barrier function by regulating VE‐cadherin.
Keywords: Cullin 3, endothelial barrier function, MLN4924, neddylation, vascular endothelial‐cadherin
Vascular endothelial barrier function is essential for organ homeostasis. In many pathological conditions such as cancer, diabetes, and acute inflammation, barrier dysfunction is observed.1 For example, tumor‐derived inflammatory mediators lead to increased paracellular leakage of plasma fluid and protein, and leukocyte infiltration in vital tissues. Therefore, it is important to understand the molecular mechanism of barrier function in order to develop a novel therapeutic strategy for diseases involving vascular leakage.
The homeostasis of barrier function is maintained by a balance between positive and negative signaling to VE cells. For example, in inflammatory tissues, thrombin and histamine stimulate ECs and induce the formation of intercellular gaps, leading to an increase in endothelial permeability.2, 3 Conversely, angiopoietin 1 and sphingosine‐1‐phosphate stabilize endothelial barrier integrity.4 These two signals strengthen vascular VE‐cadherin (also known as cadherin‐5 and CD144)‐dependent EC adhesion.5 Vascular endothelial‐cadherin is the major driving force for endothelial barrier functions. Vascular endothelial‐cadherin associates to form homophilic dimers and joins neighboring ECs, and it is a component of endothelial cell‐to‐cell AJs. The cytoplasmic tail domain of VE‐cadherin forms an AJ complex by associating with the armadillo family of proteins, β‐ and p120‐catenins.6 Endothelial cell permeability is controlled by the modulation of the VE‐cadherin signal pathway through phosphorylation and complex formation. For example, inflammatory mediators such as histamine and thrombin promote the phosphorylation of VE‐cadherin and lead to loss of association with β‐catenin and p120‐catenin, which leads to weakened endothelial cell–cell adhesion, resulting in increased EC permeability. Although several studies have reported the post‐translational modification of VE‐cadherin through phosphorylation, little is known about the regulation of VE‐cadherin through the ubiquitin‐mediated degradation pathway.
Neddylation,7 a conjugation of the ubiquitin‐like protein Nedd8 to its target protein, is a crucial post‐translational modification. MLN4924, an inhibitor of NAE, inhibits CUL‐RING UbE3 ligase activity, which requires CUL protein activation by NEDD8. MLN4924 also suppresses the survival, as well as growth and metastasis, of several tumors. Furthermore, MLN4924 has been shown to block angiogenesis in various models in vitro and in vivo.8 Therefore, this inhibitor is currently in phase I clinical trials for the treatment of cancer.9, 10, 11, 12, 13
The aim of this study was to understand the mechanisms underlying barrier dysfunction, and to elucidate the role of neddylation activity in maintaining barrier permeability. We found that MLN4924 suppressed VE‐cadherin protein production in ECs. Furthermore, we showed that NEDD8‐conjugated CUL3 is essential for the homeostasis of VE barrier functions through VE‐cadherin protein turnover.
Materials and Methods
Reagents and antibodies
Anti‐VE‐cadherin antibody was purchased from R&D Systems (Minneapolis, MN, USA). MLN4924 was purchased from Funakoshi Chemical Co. (Tokyo, Japan). Anti‐CUL3 (clone CUL3‐9, catalog# SAB4200180), anti‐Flag mAb, M2, and mouse monoclonal anti‐β‐actin antibody (clone AC‐15) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Anti‐β‐catenin antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐p120catenin (catalog# 610133) antibody was purchased from BD Transduction Laboratories (San Diego, CA, USA).
Cell culture
The HUVECs were purchased from Cell Systems (Kirkland, WA, USA) and were maintained in endothelial growth medium EGM‐2 (Lonza, Walkersville, MD, USA). All experiments were carried out with HUVECs at passages 2–4.
Lentiviral vector construction
Lentiviral vectors were constructed by inserting cDNAs encoding a Flag‐tagged RNAi‐resistant‐CUL3 construct into the lentiviral expression vector CSII‐CMV‐MCS‐IRES2‐Bsd. Lentiviral vectors were generated in 293T cells. Lentiviral expression and packaging vectors were kindly provided by Dr. Miyoshi (RIKEN BioResource Center, Tsukuba, Japan).
RNA interference
The siRNAs were purchased from Sigma‐Aldrich (Table S1). MISSION siRNA Universal Negative Control SIC‐001 (Sigma‐Aldrich) was used as the control siRNA. siRNA was transfected with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) at a concentration of 20 nM, according to the manufacturer's protocol.
Construction of RNAi‐resistant Flag‐tagged CUL3 plasmid
The siRNA‐resistant lentivirus‐Flag‐CUL3 was constructed by mutating 5′‐gagtgtatgagttcctatt‐3′ to 5′‐ gaAtgCatgTCTAGCtaCC‐3′ using PCR‐based mutagenesis without changing the amino acid sequence.
Western blot analysis
Whole protein extracts from HUVECs (20 μg) were subjected to SDS‐PAGE. The electrophoresed proteins were then transferred to PVDF membranes, which were then blocked with 5% skim milk in 0.05% Tween‐20/PBS for 30 min, followed by incubation with a primary antibody (1:1000 [v/v]). After washing with 0.05% Tween‐20/PBS, the membrane was treated with the appropriate HRP‐conjugated IgG antibodies (1:4000 [v/v]; Promega, Madison, WI, USA). Proteins were detected using enhanced chemiluminescence and were imaged on an LAS‐4000 luminescent image analyzer (Fujifilm, Tokyo, Japan).
Isolation of RNA, cDNA library synthesis, and quantitative RT‐PCR
Total RNAs were isolated from HUVECs using ISOGEN II (Nippon Gene, Tokyo, Japan) according to the manufacturer's protocol. One microgram of RNA was used for first‐strand synthesis using High Capacity RNA‐to‐cDNA Master Mix (Applied Biosystems, Foster City, CA, USA). Real‐time PCR was carried out (FastStart Universal SYBR Green Master ROX; Roche Diagnostics, Basel, Switzerland) using the ABI 7300/7500 Real‐Time PCR system (Applied Biosystems). Primers used for amplification were as follows: VE‐cadherin forward, 5′‐TGACGTGAACGACAACTGGG‐3′ and reverse, 5′‐GACGCATTGAACAACCGATG‐3′; and GAPDH forward, 5′‐TGCACCACCAACTGCTTAGC‐3′ and reverse, 5′‐GGCATGGACTGTGGTCATGAG‐3′.
Fluorescence immunostaining
The HUVECs were seeded on gelatin‐coated coverslips and then incubated with EGM‐2 for 24 h. MLN4924 or siRNA targeting human CUL3 or control siRNA were transfected into the HUVECs, followed by culturing for 72 h. The cells were fixed with 4% paraformaldehyde solution and washed three times with PBS. Triton X‐100‐permeated cells were blocked with PBS containing 4% BSA (4% BSA–PBS), and the cells were reacted with anti‐VE‐cadherin antibody (dilution, 1:1000) in 4% BSA–PBS overnight at room temperature. The cells were then washed several times with PBS followed by incubation with Alexa Fluor 488‐conjugated goat anti‐mouse IgG antibody (dilution, 1:1000; Invitrogen) in 4% BSA–PBS for 1 h at room temperature. After washing three times with PBS, fluorescent 3‐D images of the cells were obtained using a confocal laser microscope A1R (Nikon, Tokyo, Japan).
Endothelial cell permeability assay
The HUVECs were seeded onto Costar Transwell inserts (0.4‐μm pore size; Corning, New York, NY, USA). The next day, control siRNA, CUL3 siRNA, DMSO, or MLN4924 was transfected into the cells and cultured for an additional 2 days; FITC‐conjugated dextran (400 μg/mL) was then added to the upper wells. After 2 h of additional incubation at 37°C, the medium in the lower wells was collected, and the fluorescence intensity was measured, with 485 and 538 nm as the excitation and emission wavelengths, respectively, using a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cycloheximide chase assay
The HUVECs were treated with CONT or CUL3 siRNA and then incubated for 72 h. After HUVECs were treated with 25 μg/mL cycloheximide (Wako, Osaka, Japan), the cells were lysed at various time points (0, 2, 4, 6, 9, and 12 h). Western blot analyses for VE‐cadherin were carried out as described, and the protein levels were expressed as a ratio of the level at the 0‐min time point.
Statistical analysis
Data were acquired from a minimum of three independent experiments. The results are represented as means ± SE. Comparisons between two groups were carried out using Student's t‐test. Differences were considered significant if P‐values were <0.05. All statistical analyses were carried out with GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Results
MLN4924 led to loss of endothelial cell–cell contact, resulting in vascular leakage
In order to clarify the effect of NAE activity on the barrier functions of ECs, confluent HUVECs were treated with MLN4924 (100 nM) or DMSO for 72 h. With MLN4924 treatment, the ECs were extended, and cell–cell contacts were largely disrupted (Fig. 1a), whereas the ECs were tightly attached to each other in the absence of MLN4924 treatment. Furthermore, to investigate the effects of MLN4924 on EC permeability, we undertook a fluorescein leakage assay using HUVECs. After the cells were treated with MLN4924 at different concentrations (0, 20, 40, 60, 80, and 100 nM) or DMSO for 72 h in the upper compartment of the Transwell chambers, endothelial leakage was investigated using FITC‐labeled dextran (Fig. 1b). MLN4924‐treatment dramatically increased vascular permeability in a dose dependent‐manner (Fig. 1c). The level of endothelial leakage after treatment with 100 nM MLN4924 was approximately equal to that after thrombin treatment (data not shown). These results suggested that neddylation activity is essential for the formation of endothelial cell–cell contact and that it positively regulates endothelial permeability.
Figure 1.
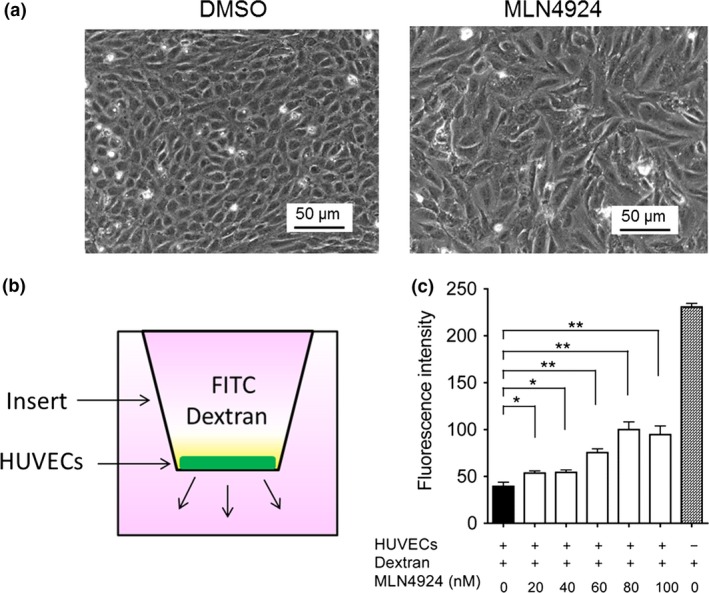
NEDD8‐activating enzyme activity is required for endothelial barrier function. (a) MLN4924 abrogated endothelial cell–cell contacts. After HUVECs were pretreated with MLN4924 (100 nM) or DMSO for 72 h, the cells were analyzed by phase‐contrast microscopy. Bar = 50 μm. (b) Schematic representation of a Transwell chamber for assaying transport across an endothelial monolayer. (c) MLN4924 increased endothelial cell permeability in a dose‐dependent manner. After HUVECs were treated with 20, 40, 60, 80, or 100 nM MLN4924 for 72 h, FITC–dextran was added in the apical chamber. After incubation for 1 h, the amount of FITC–dextran in the bottom chamber was measured. *P < 0.05; **P < 0.01.
Neddylation activity is required for VE‐cadherin protein production
To reveal the molecular mechanisms underlying this phenomenon, we first analyzed the effects of MLN4924 on the VE‐cadherin complex by examining the protein levels of essential signal components, including VE‐cadherin, p120 catenin, and β‐catenin in cultured HUVECs. Cells were treated with MLN4924 (100 nM) or DMSO for 72 h. MLN4924 treatment significantly reduced VE‐cadherin protein levels (Fig. 2a), while the protein levels of p120 catenin and β‐catenin remained constant (Fig. 2a). We also undertook immunofluorescence staining with an antibody against VE‐cadherin following MLN4924 treatment. As shown in Figure 2(b), compared with control cells, MLN4924 treatment resulted in a marked decrease in the immunofluorescence intensity of VE‐cadherin on endothelial cell–cell junctions.
Figure 2.
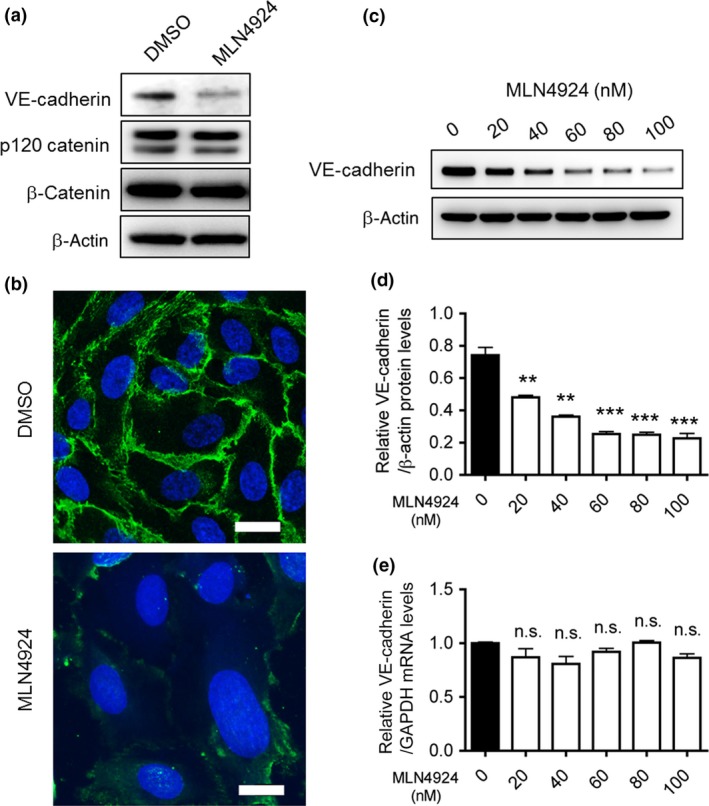
NEDD8‐activating enzyme activity essential for vascular endothelial (VE)‐cadherin protein production. (a) HUVECs were transfected with MLN4924 (80 nM) or DMSO for 72 h. The proteins were extracted and separated using 10% SDS–polyacrylamide gels. VE‐cadherin, p120 catenin, and β‐catenin proteins were then detected by Western blotting. β‐Actin was used as a loading control. (b) HUVECs cultured on gelatin‐coated cover slips were treated with MLN4924 (100 nM) or DMSO. The cells were fixed and stained with anti‐VE‐cadherin antibody followed by Alexa 488‐conjugated goat anti‐mouse IgG. Scale bar = 20 μm. (c) After HUVECs were treated with 20, 40, 60, 80, or 100 nM MLN4924 for 72 h, VE‐cadherin proteins were detected by Western blotting. Anti‐β‐actin antibody was used as an internal control. (d) Endogenous protein levels of VE‐cadherin were quantified using ImageJ software (National Institutes of Health, Bethesda, MD). **P < 0.01; ***P < 0.001. (e) VE‐cadherin mRNA levels were quantified by quantitative RT‐PCR in MLN4924 (20, 40, 60, 80, or 100 nM)‐treated HUVECs, and normalized to GAPDH mRNA levels. n.s., not significant.
In order to clarify the effect of MLN4924 on VE‐cadherin itself more precisely, different concentrations of MLN4924 were tested for their effect on VE‐cadherin protein and mRNA levels in HUVECs. After HUVECs were cultured with MLN4924 (0, 20, 40, 60, 80, or 100 nM) for 72 h, Western blot and qRT‐PCR analyses were carried out. MLN4924 treatment decreased the levels of VE‐cadherin protein in a dose‐dependent manner (Fig. 2c,d), while the mRNA levels remained constant (Fig. 2e). The suppression largely occurred with MLN4924 concentrations >60 nM (up to 70% inhibition; Fig. 2c,d).
Cullin 3 is required for VE‐cadherin protein production
Cullin UbE3 ligases require modification by NEDD8 for their activation,7 and the effect of MLN4924 on VE‐cadherin production in HUVECs appears to be caused by CUL inactivation. Therefore, we investigated the involvement of each member of the CUL family (CUL1, 2, 3, 4A, and 5) expressed in HUVECs in MLN4924‐induced abrogation of VE‐cadherin production. MISSION siRNAs (Sigma‐Aldrich) targeting CUL1, 2, 3, 4A, or 5 were used to treat HUVECs prior to the detection of VE‐cadherin protein and mRNA by Western blot analysis and qRT‐PCR, respectively. When HUVECs were transfected with CUL siRNA targeting each of the five genes at a concentration of 20 nM, the production of each CUL mRNA decreased significantly, as evidenced by the results of semiquantitative RT‐PCR analyses (data not shown). Interestingly, only CUL3 siRNA, but not the other CUL siRNAs, significantly suppressed VE‐cadherin protein production (Fig. 3a,b), while the mRNA expression levels remained constant (Fig. 3c). We further analyzed the protein levels of the essential components of the VE‐cadherin complex, including VE‐cadherin, p120 catenin, and β‐catenin, in CUL3‐knockdown HUVECs. As shown in Figure 4(a), the protein level of VE‐cadherin, but not those of p120 catenin and β‐catenin, decreased dramatically when CUL3 was knocked down in HUVECs. We also carried out immunofluorescence staining with an antibody against VE‐cadherin under CUL3 knockdown. As shown in Figure 4(b), compared with control cells, CUL3 knockdown resulted in a marked decrease of the immunofluorescence intensity of VE‐cadherin on CUL3‐depleted endothelial cell–cell junctions. We further confirmed the specificity of CUL3 knockdown on the reduction (by approximately 50%) of VE‐cadherin protein levels by using two kinds of siRNAs against CUL3 (Fig. 4c–e). In addition, we generated the corresponding RNAi‐resistant construct for CUL3 siRNA#1 and undertook rescue experiments. As shown in Figure 4(f), the decrease in VE‐cadherin protein levels recovered completely following overexpression of the RNAi‐resistant CUL3 cDNA construct (Fig. 4f).
Figure 3.
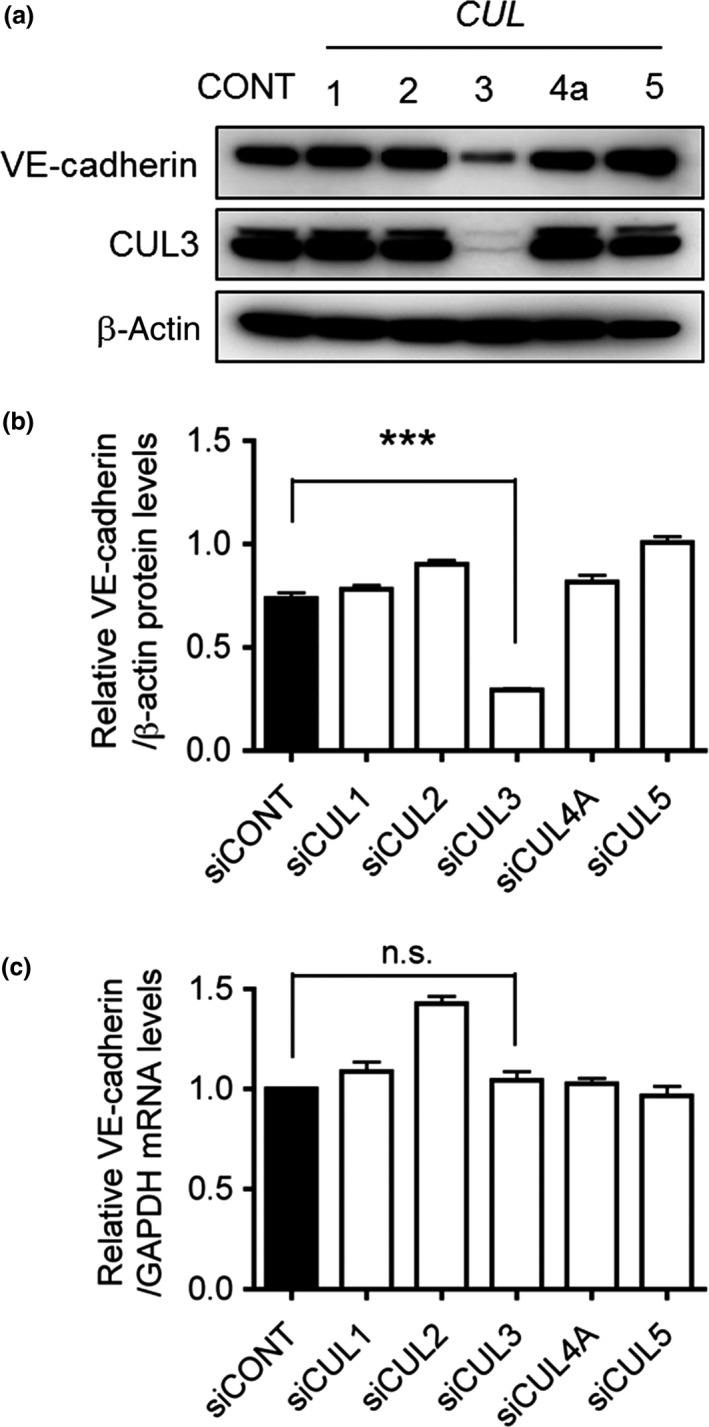
Small interfering RNA‐based screening of Cullins (CULs) responsible for regulating vascular endothelial (VE)‐cadherin. (a) HUVECs were transfected with MISSION siRNAs (Sigma‐Aldrich) against control (CONT), CUL1,CUL2,CUL3,CUL4A, and CUL5, followed by culturing for 72 h. The proteins were extracted and separated using 10% SDS–polyacrylamide gels. VE‐cadherin proteins were then detected by Western blot analysis. β‐Actin was used as a loading control. (b) Quantification of VE‐cadherin protein levels in CONT,CUL1,CUL2,CUL3,CUL4A, and CUL5 knockdown cells was carried out using ImageJ software. ***P < 0.001. (c) VE‐cadherin mRNA levels were quantified by quantitative RT‐PCR in CONT,CUL1,CUL2,CUL3,CUL4A, or CUL5 siRNA‐transfected HUVECs, and normalized to GAPDH mRNA levels.
Figure 4.
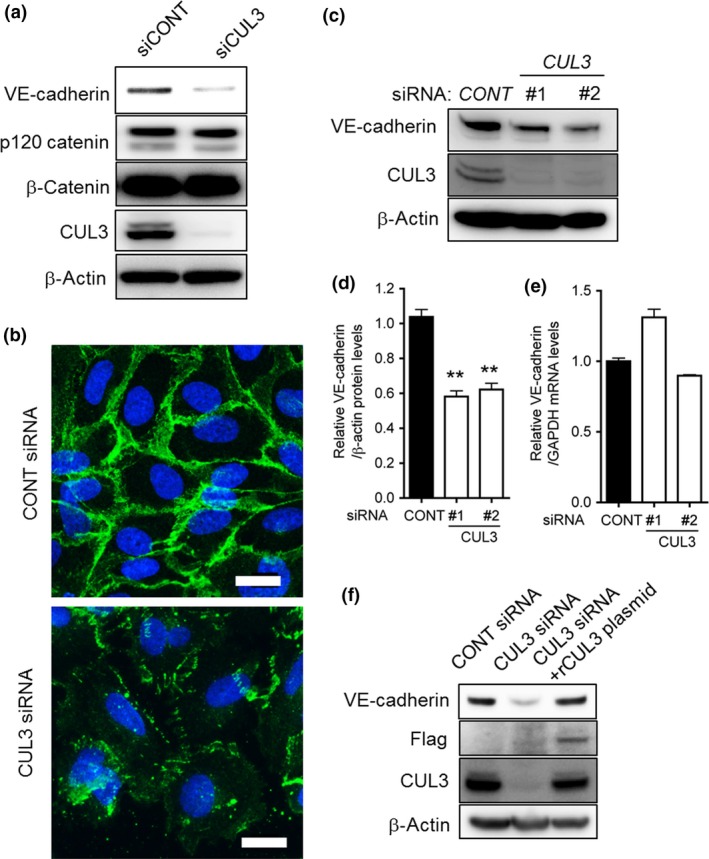
Cullin 3 (CUL3) is essential for vascular endothelial (VE)‐cadherin protein production. (a) Control or CUL3 siRNA‐transfected HUVECs were incubated for 72 h. The levels of VE‐cadherin, p120 catenin, β‐catenin, and CUL3 were determined by Western blot analysis. β‐Actin was used as the internal control. (b) HUVECs cultured on gelatin‐coated cover slips were treated with control (CONT) siRNA or CUL3 siRNA. The cells were fixed and stained with anti‐VE‐cadherin antibody followed by Alexa 488‐conjugated goat anti‐mouse IgG. Scale bar = 20 μm. (c) HUVECs were transiently transfected with control siRNA (left lane), CUL3 siRNA#1 (middle lane), and CUL3 siRNA#2 (right lane). Cells were harvested 72 h after transfection, and proteins were analyzed by Western blotting with anti‐VE‐cadherin and anti‐CUL3 antibodies. Anti‐β‐actin antibody was used as an internal control. (d) Quantification of Western blot analysis for VE‐cadherin protein levels in CONT siRNA,CUL3 siRNA#1, and CUL3 siRNA#2‐transfected HUVECs. **P < 0.01. (e) Quantitative RT‐PCR analyses of VE‐cadherin mRNA levels in control siRNA‐, CUL3 siRNA#1‐, and CUL3 siRNA#2‐transfected HUVECs. (F) An RNAi‐resistant CUL3 construct corresponding to siRNA#1 (CUL3) was generated. CUL3 siRNA#1‐resistant CUL3 constructs were expressed in CONT siRNA‐ or CUL3 siRNA‐transfected HUVECs using the Lentivirus gene expression system. HUVECs were incubated for 72 h, and VE‐cadherin, Flag, and CUL3 levels were detected by Western blotting.
Cullin 3 essential for maintenance of endothelial cell–cell contacts
To elucidate the role of CUL3 in endothelial cell–cell contacts, we carried out a cell permeability assay using CUL3‐depleted HUVECs. CUL3 or control siRNA were transfected into the HUVECs, followed by culturing for 72 h. As shown in Figure 5, cell–cell contacts were largely disrupted by CUL3 depletion, as assessed by phase‐contrast imaging (Fig. 5a). This effect was similar to that observed in the case of MLN4924‐treated ECs. Furthermore, EC permeability was dramatically increased by CUL3 depletion as well as MLN4924 treatment (Fig. 5b). These results indicated that the increase of vascular leakage by CUL3 knockdown was due to the depletion of VE‐cadherin protein production.
Figure 5.
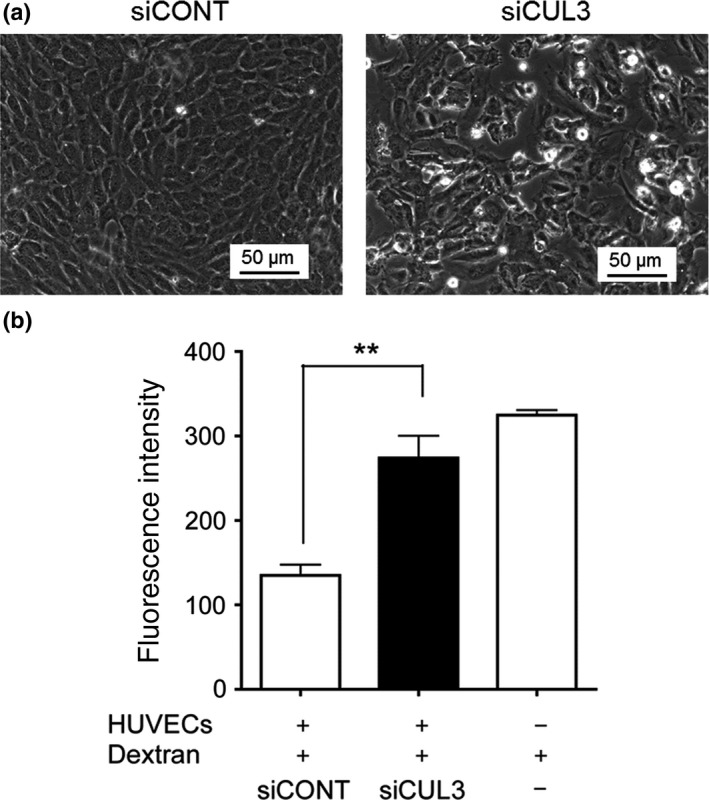
Cullin 3 (CUL3) required for endothelial barrier function. (a) Depletion of CUL3 abrogated endothelial cell–cell contacts. After HUVECs were pretreated with control (CONT) siRNA or CUL3 siRNA for 72 h, cells were analyzed by phase‐contrast microscopy. Bar = 50 μm. (b) CUL3 knockdown increased endothelial cell permeability. After HUVECs were treated with CONT siRNA or CUL3 siRNA for 72 h, FITC–dextran was added in the apical chamber. After incubation for 1 h, the amount of FITC–dextran in the bottom chamber was estimated. **P < 0.01.
Cullin 3 regulates VE‐cadherin protein turnover
Vascular endothelial‐cadherin protein levels in HUVECs were suppressed by CUL3 knockdown or MLN4924 treatment, while their mRNA levels remained unchanged. Therefore, we hypothesized that CUL3 regulates the protein metabolism of VE‐cadherin. To test this hypothesis, cycloheximide chase assay was carried out. After CONT siRNA‐ or CUL3 siRNA‐transfected HUVECs were cultured for 72 h, cycloheximide was added immediately, and the cells were incubated for 0, 2, 4, 6, 9, and 12 h. As shown in Figure 6, the half‐life of VE‐cadherin was shortened dramatically to <2 h by CUL3 depletion. These results suggested that neddylated CUL3 regulates vascular leakage by regulating VE‐cadherin protein turnover in ECs (Fig. 7).
Figure 6.
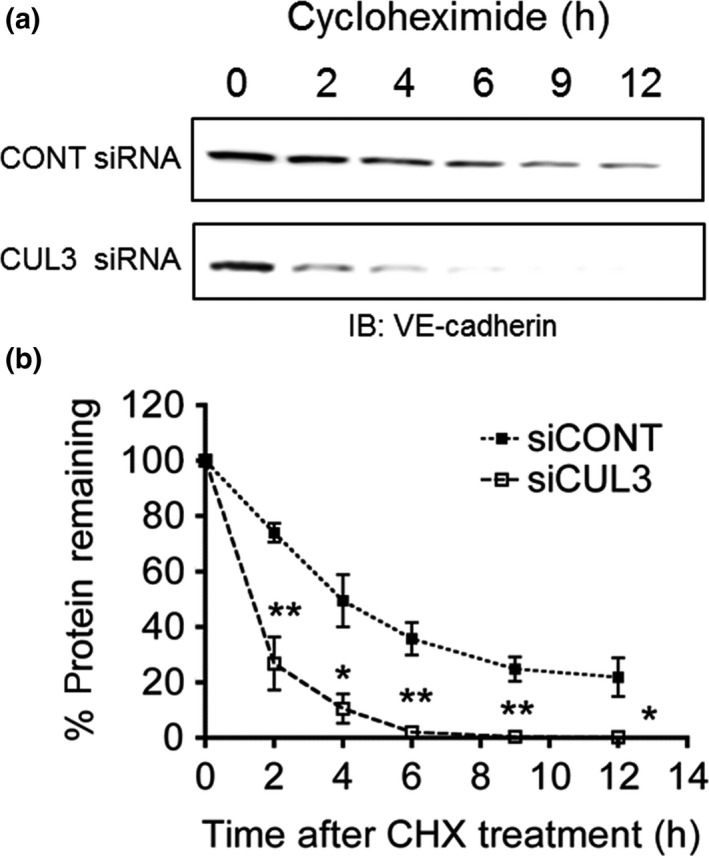
Cullin 3 (CUL3) regulates vascular endothelial (VE)‐cadherin turnover in HUVECs. (a) HUVECs were transfected with control (CONT) siRNA or CUL3 siRNA and cultured for 72 h, followed by cycloheximide (CHX) treatment for 0, 2, 4, 6, 9, and 12 h. After harvesting the HUVECs, proteins were subjected to SDS‐PAGE, followed by Western blotting with antibodies to VE‐cadherin and β‐actin. IB, immunoblot. (b) Quantification of CHX chase assay. Data represent the percentages of VE‐cadherin band intensity relative to that at time point 0. Relative band density values were measured using ImageJ software. *P < 0.05; **P < 0.01.
Figure 7.
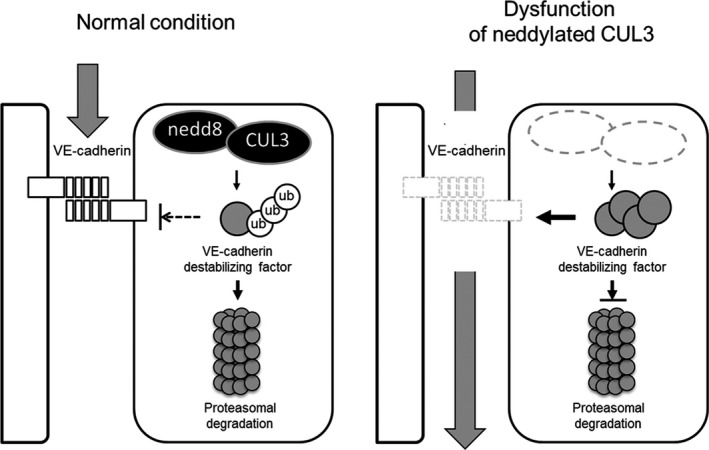
Scheme of the proposed molecular mechanism. Neddylated Cullin 3 (CUL3) regulates vascular endothelial (VE)‐cadherin turnover in endothelial cells. Under normal conditions, neddylated CUL3 complex constitutively degrades unidentified VE‐cadherin destabilizing factor(s) through a ubiquitin (ub)–proteasome system, thereby stabilizing endothelial barrier integrity by maintaining a high level of VE‐cadherin protein. Following treatment with a NEDD8‐activating enzyme inhibitor, MLN4924 or CUL3 siRNA, VE‐cadherin is depleted in the cells because of the accumulation of VE‐cadherin destabilizing factor(s). Consequently, endothelial vascular permeability is increased. Gray arrows denote vascular leakage.
Discussion
In this study, we showed, for the first time, that NAE activity is essential for regulating EC permeability (Fig. 1). Endothelial cells mainly form two types of homophilic AJs, which can be distinguished by the transmembrane adhesion molecules and their intracellular partners.14 Tight junctions are mediated by members of the claudin family (claudins 1, 5, and 12),9, 15, 16 occludin,17 and junctional adhesion molecules.18, 19 These molecules associate with zonula occludens 1/2 and protease activated receptor 2/3 at the intracellular region. However, AJs are mediated by VE‐cadherin, which interacts with the catenin family of proteins.20 Vascular endothelial‐cadherin is abundantly expressed throughout the vascular tree. Vascular endothelial‐cadherin null mice showed early embryo lethality at day 9.5 due to vascular defects.21 In the adult, blocking of VE‐cadherin by neutralizing antibodies induced a marked increase in vascular permeability.22, 23 Therefore, VE‐cadherin expression and organization at the AJs is a critical determinant for vascular functions. Hence, it is essential to elucidate the regulatory mechanisms underlying the expression and activation of VE‐cadherin in order to establish promising therapies for vascular barrier dysfunction.
Our data showed that the VE‐cadherin protein levels were dramatically decreased by MLN4924 treatment (Fig. 2). We also found that knockdown of CUL3, one of the substrate molecules of neddylation, suppressed VE‐cadherin protein production, resulting in increased EC permeability (Figs 1, 2, 3, 4). However, neither treatment inhibited the protein levels of other EC adhesion molecules such as p120 catenin, β‐catenin (Figs 2, 4), zonula occludens 1, claudin 5, or claudin 1 (data not shown). Our findings, together with the results of previous studies, suggested that neddylated CUL3 selectively and specifically targets VE‐cadherin‐mediated vascular permeability.
The cycloheximide chase assay showed that the half‐life of VE‐cadherin protein was decreased by CUL3 depletion (Fig. 5). However, VE‐cadherin gene expression levels were not different between control and CUL3‐knockdown HUVECs (Fig. 3). These data implied that CUL3 might be involved in the post‐translational regulation of VE‐cadherin. Recent studies have reported that VE‐cadherin protein levels are regulated at the post‐translational level through ubiquitination‐driven processing and lysosomal degradation. Orsenigo et al.24 showed that phosphorylation and K63‐linked ubiquitination of VE‐cadherin by Bradykinin stimulation is necessary for its internalization. Xiao et al.25 reported that the proteasome inhibitor prevents the internalization and fragmentation of cell‐surface VE‐cadherin. On the basis of our data and these previous reports, we hypothesized that the downregulation of VE‐cadherin by CUL3 depletion was due to increased degradation in the proteasome or lysosome. To test this hypothesis, we undertook rescue experiments in which chemical inhibitors of these processes, such as MG132 and chloroquine, were added to CUL3‐depleted HUVECs. However, treatment with the lysosomal inhibitor did not lead to the recovery of VE‐cadherin levels (Fig. S1). Treatment with the proteasome inhibitor dramatically reduced VE‐cadherin protein levels (Fig. S2). These data suggested that neddylated CUL3 could indirectly target VE‐cadherin by proteasomal degradation of certain unidentified destabilizing factor(s) for VE‐cadherin (Fig. 7).
Cullin 3 interacts with BTBP at the N‐terminal region to form UbE3‐ligase complexes. In these complexes, the BTBPs play a crucial role in the determination of substrate specificity.26 One hundred and eighty‐three genes have been reported to encode BTBPs in humans.27 Several CUL3–BTBP‐based E3 ligase complexes are known to be involved in various physiological events. For example, CUL3‐Keap1‐Nrf2 is essential for oxidant regulation,28 CUL3‐BAZF‐CBF1 is important for angiogenic switching,29 and CUL3‐SPOP‐Gli1/Gli2 participates in morphological determination during development.30, 31 These findings indicate that one of the CUL3–BTBP complexes could target one or more of the destabilizing factor(s) of VE‐cadherin by ubiquitination and subsequent degradation. Indeed, treatment with proteasome inhibitors such as MG132 and epoxomicin showed dramatically impaired production of VE‐cadherin in HUVECs (Fig. S2). These phenotypes were similar to those elicited following treatment with MLN4924 or CUL3 siRNA. Thus, the downregulation of VE‐cadherin protein levels by these treatments might be due to intracellular accumulation of the substrates of CUL3–BTBP‐based E3 ligase complexes, which function as VE‐cadherin regulators.
Several substrates of CUL3–BTBP‐based E3 ligase complexes have been identified to date.32 However, there is no evidence that the complex regulates the post‐translational modification of VE‐cadherin. Therefore, in the future, the BTBP and substrates responsible for this effect need to be identified in order to clarify the entire molecular mechanism of CUL3‐mediated VE‐cadherin protein turnover.
In conclusion, our results showed that neddylated CUL3 is crucial for endothelial barrier function and that this effect is mediated by VE‐cadherin. We believe that our data contribute to the understanding of the precise mechanism by which neddylated CUL3 regulates vascular EC permeability in humans and provide insights that may help establish a new strategy for the treatment of vascular barrier dysfunction‐associated diseases.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AJ
adherens junction
- BTBP
Bric‐a‐brac/Tramtrack/Broad complex (BTB) domain‐containing protein
- CONT
control
- CUL
Cullin
- EC
endothelial cell
- NAE
NEDD8‐activating enzyme
- NEDD8
neural precursor cell expressed, developmentally downregulated 8
- qRT‐PCR
quantitative RT‐PCR
- VE
vascular endothelium
Supporting information
Fig. S1. Detection of vascular endothelial (VE)‐cadherin in chloroquine‐treated Cullin 3 (CUL3)‐depleted HUVECs. HUVECs were treated with control siRNA (left lane), CUL3 siRNA (middle lane), and CUL3 siRNA + chloroquine (5 μg/mL; right lane). Cells were harvested 72 h after transfection, and proteins were analyzed by Western blotting with anti‐VE‐cadherin and anti‐CUL3 antibodies. Anti‐β‐actin antibody was used as an internal control.
Fig. S2. Detection of vascular endothelial (VE)‐cadherin in epoxomicin‐ or MG132‐treated HUVECs. HUVECs were treated with 1 μM epoxomicin or MG132 for 8 h. Cells were harvested and proteins were analyzed by Western blotting with anti‐VE‐cadherin and anti‐Cullin 3 antibodies. Anti‐β‐actin antibody was used as an internal control.
Table S1. List of siRNA identifiers used in this study.
Acknowledgments
We thank Yusuke Fujioka (Department of Biochemistry and Molecular Genetics, Ehime University) for professional assistance. We gratefully appreciate Drs. Hirofumi Inoue, Shinji Fukuda, Daisuke Nanba, Hidetaka Ohnuki, and Hisayo Fukuda‐Nishida of Ehime University for helpful discussions. This work was supported by Grants‐in‐Aid for Scientific Research (16H04698 to S.H. and 16K18421 to T.S.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Cancer Sci 108 (2017) 208–215
Funding Information
Ministry of Education, Culture, Sports, Science and Technology, Japan (16H04698, 16K18421).
References
- 1. Weis SM, Cheresh DA. Pathophysiological consequences of VEGF‐induced vascular permeability. Nature 2005; 437: 497–504. [DOI] [PubMed] [Google Scholar]
- 2. Amerongen GPV, Draijer R, Vermeer MA, van Hinsbergh VWM. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin – role of protein kinases, calcium, and RhoA. Circ Res 1998; 83: 1115–23. [DOI] [PubMed] [Google Scholar]
- 3. Gamble JR, Drew J, Trezise L et al Angiopoietin‐1 is an antipermeability and anti‐inflammatory agent in vitro and targets cell junctions. Circ Res 2000; 87: 603–7. [DOI] [PubMed] [Google Scholar]
- 4. Garcia JGN, Liu F, Verin AD et al Sphingosine 1‐phosphate promotes endothelial cell barrier integrity by Edg‐dependent cytoskeletal rearrangement. J Clin Invest 2001; 108: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell‐to‐cell adherens junctions. Arterioscler Thromb Vasc Biol 1999; 19: 2286–97. [DOI] [PubMed] [Google Scholar]
- 6. Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE‐cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 1998; 111: 1853–65. [DOI] [PubMed] [Google Scholar]
- 7. Hori T, Osaka F, Chiba T et al Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999; 18: 6829–34. [DOI] [PubMed] [Google Scholar]
- 8. Yao WT, Wu JF, Yu GY et al Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis 2014; 5: e1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soucy TA, Smith PG, Milhollen MA et al An inhibitor of NEDD8‐activating enzyme as a new approach to treat cancer. Nature 2009; 458: 732–6. [DOI] [PubMed] [Google Scholar]
- 10. Soucy TA, Smith PG, Rolfe M. Targeting NEDD8‐activated cullin‐RING ligases for the treatment of cancer. Clin Cancer Res 2009; 15: 3912–6. [DOI] [PubMed] [Google Scholar]
- 11. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8‐targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res 2010; 70: 10310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8‐activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res 2011; 71: 3042–51. [DOI] [PubMed] [Google Scholar]
- 13. Luo ZG, Yu GY, Lee HW et al The Nedd8‐activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 2012; 72: 3360–71. [DOI] [PubMed] [Google Scholar]
- 14. Dejana E. Endothelial cell‐cell junctions: happy together. Nat Rev Mol Cell Biol 2004; 5: 261–70. [DOI] [PubMed] [Google Scholar]
- 15. Liebner S, Fischmann A, Rascher G et al Claudin‐1 and claudin‐5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol 2000; 100: 323–31. [DOI] [PubMed] [Google Scholar]
- 16. Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin‐5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 1999; 147: 185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hirase T, Staddon JM, Saitou M et al Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci 1997; 110: 1603–13. [DOI] [PubMed] [Google Scholar]
- 18. Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol 2007; 7: 467–77. [DOI] [PubMed] [Google Scholar]
- 19. Gavard J. Endothelial permeability and VE‐cadherin A wacky comradeship. Cell Adh Migr 2014; 8: 158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giannotta M, Trani M, Dejana E. VE‐cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013; 26: 441–54. [DOI] [PubMed] [Google Scholar]
- 21. Vittet D, Buchou T, Schweitzer A, Dejana E, Huber P. Targeted null‐mutation in the vascular endothelial‐cadherin gene impairs the organization of vascular‐like structures in embryoid bodies. Proc Natl Acad Sci U S A 1997; 94: 6273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crosby CV, Fleming PA, Argraves WS et al VE‐cadherin is not required for the fort‐nation of nascent blood vessels but acts to prevent their disassembly. Blood 2005; 105: 2771–6. [DOI] [PubMed] [Google Scholar]
- 23. Corada M, Mariotti M, Thurston G et al Vascular endothelial‐cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A 1999; 96: 9815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orsenigo F, Giampietro C, Ferrari A et al Phosphorylation of VE‐cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun 2012; 3: doi:10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao KY, Allison DF, Kottke MD et al Mechanisms of VE‐cadherin processing and degradation in microvascular endothelial cells. J Biol Chem 2003; 278: 19199–208. [DOI] [PubMed] [Google Scholar]
- 26. Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin‐RING E3 ubiquitin ligases – ‘Ubiquitylation: mechanism and functions’ review series. EMBO Rep 2013; 14: 1050–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stogios PJ, Downs GS, Jauhal JJS, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol 2005; 6: R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobayashi A, Kang MI, Okawa H et al Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate for proteasomal degradation of Nrf2. Mol Cell Biol 2004; 24: 7130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohnuki H, Inoue H, Takemori N et al BAZF, a novel component of cullin3‐based E3 ligase complex, mediates VEGFR and Notch cross‐signaling in angiogenesis. Blood 2012; 119: 2688–98. [DOI] [PubMed] [Google Scholar]
- 30. Chen MH, Wilson CW, Li YJ et al Cilium‐independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 2009; 23: 1910–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang CB, Pan Y, Wang BL. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full‐length activators but not their repressors. Development 2010; 137: 2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3‐RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J 2013; 32: 2307–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Detection of vascular endothelial (VE)‐cadherin in chloroquine‐treated Cullin 3 (CUL3)‐depleted HUVECs. HUVECs were treated with control siRNA (left lane), CUL3 siRNA (middle lane), and CUL3 siRNA + chloroquine (5 μg/mL; right lane). Cells were harvested 72 h after transfection, and proteins were analyzed by Western blotting with anti‐VE‐cadherin and anti‐CUL3 antibodies. Anti‐β‐actin antibody was used as an internal control.
Fig. S2. Detection of vascular endothelial (VE)‐cadherin in epoxomicin‐ or MG132‐treated HUVECs. HUVECs were treated with 1 μM epoxomicin or MG132 for 8 h. Cells were harvested and proteins were analyzed by Western blotting with anti‐VE‐cadherin and anti‐Cullin 3 antibodies. Anti‐β‐actin antibody was used as an internal control.
Table S1. List of siRNA identifiers used in this study.