

Abstract
This study aimed to clarify the genomic factors associated with the diagnosis and prognosis of oral squamous cell carcinoma via next‐generation sequencing. We evaluated data from 220 cases of oral squamous cell carcinoma. Genomic DNA was eluted using formalin‐fixed, paraffin‐embedded samples, and targeted resequencing of 50 cancer‐related genes was performed. In total, 311 somatic mutations were detected in 220 patients, consisting of 68 synonymous mutations and 243 non‐synonymous mutations. Genes carrying mutations included TP53, CDKN2A, and PIK3CA in 79 (35.9%), 35 (15.9%), and 19 patients (8.6%), respectively. Copy number analysis detected amplification of PIK3CA and AKT1 in 38 (17.3%) and 11 patients (5.0%), respectively. Amplification of receptor tyrosine kinases was found in 37 patients (16.8%). Distant metastasis was noted in nine of 37 patients (24%) with receptor tyrosine kinase amplification, accounting for 43% of the 21 cases of distant metastasis. The cumulative 5‐year survival rate was 64.6% in the receptor tyrosine kinase amplification group vs 85.2% in the no receptor tyrosine kinase amplification group. Moreover, we identified significantly poorer prognosis in the TP53 mutation/receptor tyrosine kinase amplification group, for which the cumulative 5‐year survival rate was 41.6%. In conclusion, the results of this study demonstrated that receptor tyrosine kinase amplification is a prognostic factor for distant metastasis of oral squamous cell carcinoma, indicating the necessity of using next‐generation sequencing in clinical sequencing.
Keywords: Distant metastasis, gene amplification, next‐generation sequencing, oral squamous cell carcinoma, receptor tyrosine kinase
It has become possible in recent cancer genome research to analyze a large volume of genomic data from a human sample due to remarkable technological innovations including next‐generation sequencing (NGS). Large‐scale cancer genome projects, such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium, provided an overview of genomic alterations in many cancers. Such data accumulation in cancer genome analyses and development of molecular‐targeted drugs are fueling a shift in cancer treatment from conventional therapies selected on the basis of the organ of origin, histologic type, and stage of cancers to therapies selected according to the presence of gene mutations.
Annually, approximately 300 000 people worldwide develop oral cancer.1 The majority of oral malignancies arise from epithelial tissue, and squamous cell carcinoma is the predominant tumor type.2 Despite advances in diagnostic technology and therapeutic techniques, the survival rate of oral squamous cell carcinoma (OSCC) has improved by only 5% in the past 20 years, and the 5‐year survival rate of OSCC is 60%.3 Comprehensive analyses of gene mutations in head and neck squamous cell carcinoma (HNSCC) including OSCC using NGS have also been conducted, revealing substantial information about genomic alterations in HNSCC.4, 5, 6, 7, 8 However, there is little evidence for selecting stratified therapies based on genomic alterations. In addition, repositioning of drugs and agents used in other organs should be examined, as only one adaptive molecular‐targeted drug, cetuximab, an anti‐EGFR monoclonal antibody drug, is used in the treatment of HNSCC.
This study aimed to clarify the genomic factors associated with the diagnosis and prognosis of OSCC by integrating analyses of genetic alterations in OSCC and clinicopathological information via target resequencing using NGS.
Materials and Methods
Patients and genomic DNA samples
We investigated 220 patients who received a histological diagnosis of OSCC between 2001 and 2015 at the Department of Oral and Maxillofacial Surgery, Tokyo Medical and Dental University. Genomic DNA was extracted from formalin‐fixed, paraffin‐embedded (FFPE) tumor tissue from patients diagnosed with OSCC. Clinicopathological information was obtained from medical charts. The median follow‐up period was 44 months (range 0.5–165 months). This study was conducted in accordance with the Declaration of Helsinki, and approved by the ethics committee of Tokyo Medical and Dental University, Faculty of Dentistry (No. 1087).
DNA extraction
The tumor areas in FFPE tissues were marked and hand‐dissected using macrodissection methods to ensure tumor tissue inclusion, and genomic DNA was extracted from FFPE tissues using a QIAamp DNA FFPE Tissue Kit (Qiagen, Venlo, The Netherlands). The purified DNA was quantified using a Qubit DNA high‐sensitivity assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
Library preparation and sequencing
Library preparation was performed using an Ion AmpliSeq Library Kit 2.0 and Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific). The panel target's hotspot regions included more than 2800 COSMIC mutations of 50 cancer‐related genes (Table [Link], [Link]a and b). After library preparation, each amplicon library was quantified using an Agilent 2100 Bioanalyzer and Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced using an Ion Proton platform and Ion PI Chip (Thermo Fisher Scientific). The average read depths were approximately 1700.
Sequencing analysis
Data were analyzed using Torrent Suite Software v4.2.191 (Thermo Fisher Scientific) and Ion Reporter Software v4.6 (Thermo Fisher Scientific). The read alignments were performed using the human reference genome hg19. Detected variants with quality scores of <20 and allele frequencies of <4.0%9 were eliminated. Further, we filtered out possible germline mutations using the databases of the 1000 genomes project (http://www.internationalgenome.org/) and 5000 exomes project (http://evs.gs.washington.edu/EVS/), as matched normal tissue samples were not analyzed in this study. Copy number analyses were performed using Biomedical Genomics Workbench 2.5 (Qiagen). The algorithm implemented is based on a CNA detection tool called COpy Number Targeted Resequencing Analysis (CONTRA), which includes a module for efficiently creating a pseudo‐control from multiple samples.10 Non‐cancer samples derived from FFPE specimens of nine patients with oral leukoplakia were used to create pseudo‐control data. Each target region copy number was calculated using a log2 ratio. We used |2| ≤ adjusted fold‐change (log2) as the criterion for CNAs. More details about this method are given in the Supporting information (Doc. S1, Fig. [Link], [Link]a–b).
Statistical analysis
The associations between clinicopathological variables and gene aberrations were evaluated using Fisher's exact test or the chi‐squared test. Overall survival (OS) was measured as the time interval between the first date of visiting our department and that of the last follow‐up or death. Cox proportional hazard models were used to assess the univariate and multivariate prognostic significance of clinicopathological variables and tumor gene aberrations regarding OS. Survival curves were estimated according to the Kaplan–Meier method, and these differences were examined using the log‐rank test. All analyses were performed using PASW Statistics, version 18 (SPSS Inc., Chicago, IL, USA). Statistical analyses related to somatic mutations were performed for non‐synonymous mutations, excluding synonymous mutations.
Results
SMs and CNAs in patients with OSCC
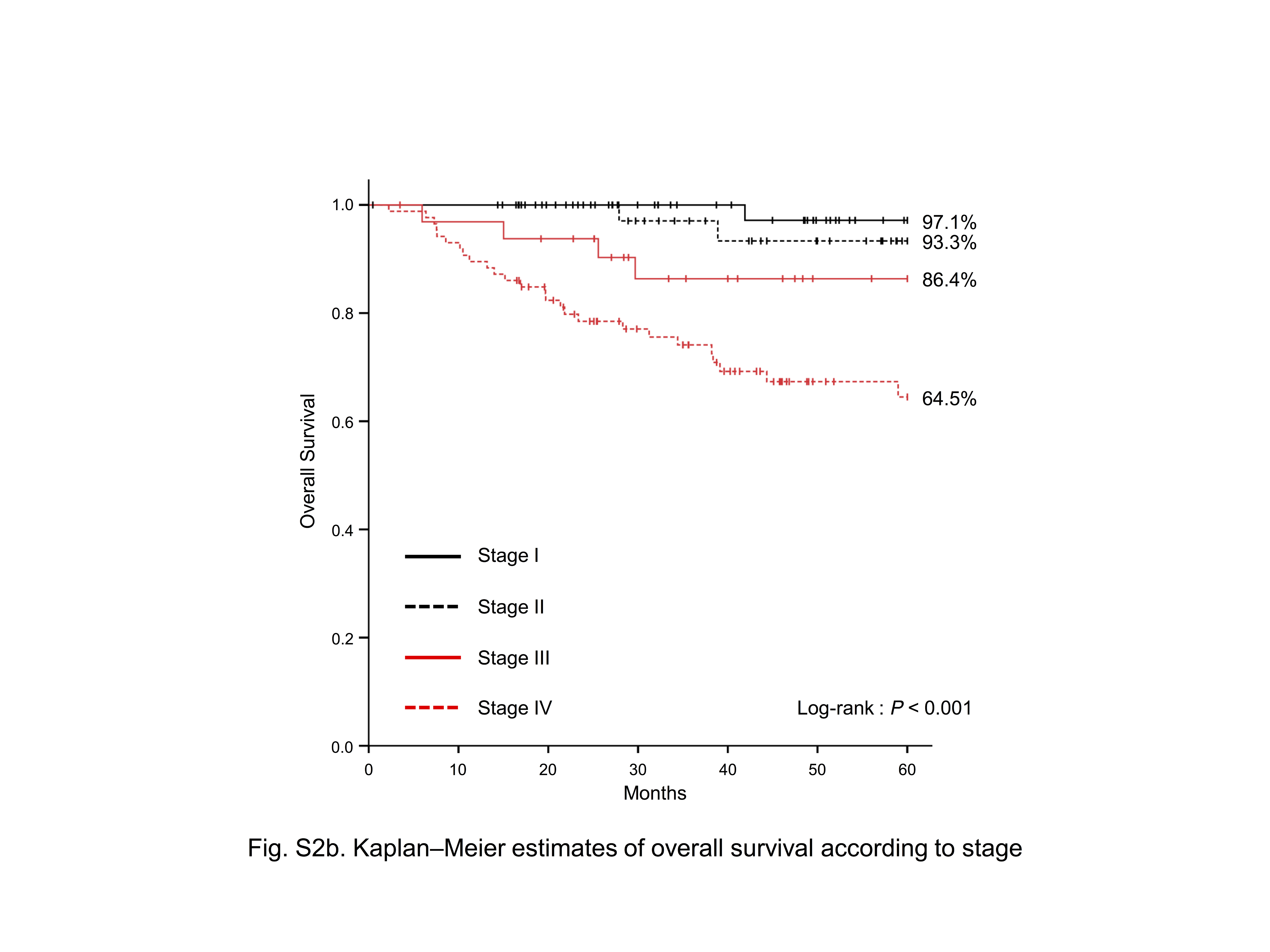
The 220 subjects consisted of 135 males and 85 females with a mean age of 58.7 years. Their oral subsites of tumor development were the tongue in 123 patients, gum in 64 patients, buccal mucosa in 21 patients, floor of the mouth in 10 patients, and hard palate in two patients (Table S2). The cumulative 5‐year survival rates of subsites were 81.8% in the tongue, 87.3% in the gum, and 68.8% in others (buccal mucosa, floor of the mouth, and hard palate) (Fig. S2a). Their tumor stages according to the UICC stage classification were stage I in 53 patients, stage II in 48 patients, stage III in 33 patients, and stage IV in 86 patients, with cumulative 5‐year survival rates of 97.1, 93.3, 86.4, and 64.5%, respectively (Table S2, Fig. S2b).
Figure 1 presents the results for the detection of somatic mutations (SMs) and copy number alterations (CNAs). Regarding SMs, 311 mutations were detected in 220 patients, producing an average of 1.41 mutations (0–23 mutations) per patient. The 311 mutations consisted of 68 synonymous mutations and 243 non‐synonymous mutations. The non‐synonymous mutations consisted of 161 missense mutations (66.3%), 49 nonsense mutations (20.2%), nine frame shift deletions (3.7%), 16 frame shift insertions (6.6%), one in‐frame shift deletion (0.4%), one in‐frame shift insertion (0.4%), and six splice site mutations (2.5%). Genes detected to have a mutation included TP53 in 79 patients (35.9%), CDKN2A in 35 patients (15.9%), PIK3CA in 19 patients (8.6%), NOTCH1 in nine patients (4.1%), HRAS in four patients (1.8%), RB1 in three patients (1.4%), FBXW7 in one patient (0.5%), and PTEN in onde patient (0.5%). No SM was detected in CSF1R, CTNNB1, EZH2, FGFR2, IDH1, JAK3, MPL, NPM1, and PTPN11.
Figure 1.

Mutational landscape of oral squamous cell carcinoma in 220 patients. Patients were stratified into four subgroups according to stage. The figure shows mutation burdens, clinicopathological features, somatic mutations, and copy number alterations in order from the top panel. Concerning the percentage of copy number alterations, the figure shows the deletion rates for tumor suppressor genes and amplification rates for oncogenes. Amp., amplification; Del., deletion.
Copy number analysis uncovered deletions of tumor suppressor genes, namely CDKN2A in 25 patients (11.4%), NOTCH1 in eight patients (3.6%), and SMAD4 in three patients (1.4%). Amplification was observed for PIK3CA in 38 patients (17.3%), AKT1 in 11 patients (5.0%), HRAS in eight patients (3.6%), and BRAF in six patients (2.7%). Amplification of receptor tyrosine kinases (RTKs) was found in 37 patients (16.8%). The amplified RTK genes included EGFR in 17 patients (7.7%), ERBB2 in seven patients (3.2%), FGFR1 in seven patients (3.2%), FGFR3 in six patients (2.7%), ERBB4 in three patients (1.4%), MET in three patients (1.4%), FLT3 in two patients (0.9%), KIT in one patient (0.5%), and PDGFRA in one patient (0.5%). Amplification of KIT and PDGFRA occurred in the same patient, in whom amplification of EGFR, ERBB2, FGFR3, and MET was also detected. No amplification of FGFR2 was detected.
The frequency of PIK3CA SMs was significantly higher in stages III/IV than in stages I/II (P = 0.023). Meanwhile, the frequency of PIK3CA amplification was significantly higher in stages I/II than in stages III/IV (P = 0.007). Examination of these frequencies separately for T‐ and N‐factors used for stage classification revealed that the frequency of PIK3CA SMs was significantly higher for patients with T3/4 lesions (P = 0.015), whereas the frequency of PIK3CA amplification was significantly higher in patients with N0 lesions (P = 0.028) (Table 1). Regarding mutations of TP53 and CDKN2A, no significant difference was detected between stages. Concerning HRAS, SMs were detected in four patients, and their stage was IV in all cases (Tables S3–S4). There was no relationship between the presence or absence of genetic alterations and age, gender, smoking history, alcohol history, subsite, or histological differentiation (Fig. S3 and Tables S3 and S4).
Table 1.
Association between stage and PIK3CA aberrations
| Variable | PIK3CA soamtic mutation | PIK3CA copy number alteration | ||||||
|---|---|---|---|---|---|---|---|---|
| Wild type | Mutation | P‐value | Direction | No amp. | Amp. | P‐value | Direction | |
| Stage | ||||||||
| I/II | 97 | 4 | 0.023 | Mutation worse | 75 | 25 | 0.007 | Amplification better |
| III/IV | 104 | 15 | 107 | 13 | ||||
| T status | ||||||||
| 1/2 | 148 | 9 | 0.015 | Mutation worse | 126 | 31 | 0.126 | |
| 3/4 | 53 | 10 | 56 | 7 | ||||
| N status | ||||||||
| 0 | 123 | 10 | 0.466 | 104 | 29 | 0.028 | Amplification better | |
| 1–3 | 78 | 9 | 78 | 9 | ||||
Amp., amplification.
Somatic mutations and CNAs with higher frequencies among previously reported genetic alterations in OSCC were examined via gene classification (e.g., RTKs, PI3K pathway genes, tumor suppressor genes, and Ras/Raf pathway genes) (Fig. 2). Among RTKs, CDKN2A, and PIK3CA, SMs and CNAs exhibited a mutually exclusive trend. In addition, comparisons of each gene revealed that deletion of CDKN2A was exclusive with SMs of TP53, whereas amplification of PIK3CA was cooperative with SMs of TP53.
Figure 2.
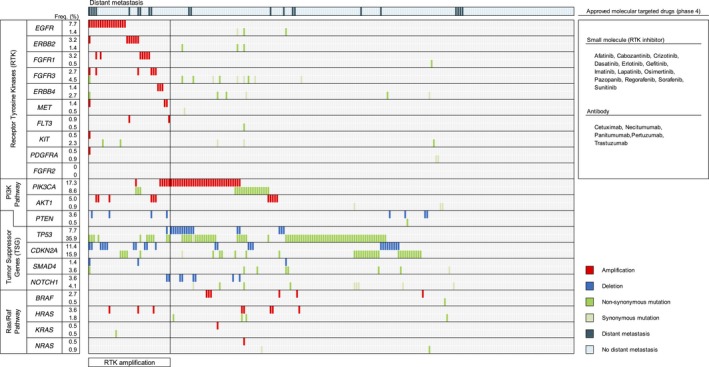
Key genes and pathways in oral squamous cell carcinoma. Somatic mutations and copy number alterations indicated mutual exclusivity for receptor tyrosine kinases (RTKs), CDKN2A, and PIK3CA. CDKN2A deletions were exclusive with TP53 somatic mutations, whereas PIK3CA amplifications were cooperative with TP53 somatic mutations. The number of patients with RTK amplification and distant metastasis was nine. Meanwhile, 24 (37 patients) and 43% (21 patients) had RTK amplification and distant metastasis, respectively. The right panel of the figure shows approved molecular‐targeted drugs against RTKs in malignant tumors. In head and neck squamous cell carcinoma (HNSCC), the only approved drug is cetuximab.
RTK amplification is predictive of distant metastasis in patients with OSCC
Distant metastasis was found in nine of 37 patients (24%) with RTK amplification. This accounted for 43% of the 21 cases of distant metastasis. Among the 220 patients, RTK amplification was detected in all three patients who were free of cervical lymph node metastasis (N0) but developed distant metastasis after therapy (Table 2). Moreover, in 21 patients who developed distant metastasis, the 10 patients who were clinically diagnosed with early‐stage cancer and developed distant metastasis after primary therapy included four patients with RTK amplification and four patients with poorly differentiated histology (data not shown). In addition, no RTK amplification was detected in any of the five patients who developed distant metastasis and had poorly differentiated histology (Table 2).
Table 2.
Summary of patients with receptor tyrosine kinase amplification and distant metastasis
| Patient No. | Age | Gender | Smoking history | Alcohol history | Subsite | FFPE sample | Histological differentiation | T status | N status | Stage | Distant metastasis | Outcome | RTK amplification | Somatic mutation | Copy number alteration | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amplification | Deletion | |||||||||||||||
| 1 | 56 | M | Never | Yes | BM | B | M/D | 1 | 0 | I | ‐ | NED | EGFR | CDKN2A, KIT | – | – |
| 2 | 29 | F | UNK | UNK | TON | B | W/D | 1 | 0 | I | – | LTF | EGFR | – | GNA11, JAK3 | CDKN2A, RB1 |
| 3 | 67 | M | Former | Yes | GUM | B | W/D | 1 | 0 | I | – | NED | MET | ATM | PIK3CA | – |
| 4 | 41 | F | Never | Yes | TON | B | W/D | 1 | 0 | I | – | NED | FLT3 | – | PIK3CA | HNF1A, NOTCH1, STK11 |
| 5 | 76 | F | Never | Yes | GUM | B | W/D | 1 | 0 | I | – | NED | MET | TP53 | PIK3CA | HNF1A, NOTCH1, PTEN, STK11, TP53 |
| 6 | 36 | M | UNK | UNK | TON | B | W/D | 2 | 0 | II | – | NED | EGFR | – | – | – |
| 7 | 21 | F | Never | No | TON | B | M/D | 2 | 0 | II | – | NED | EGFR | – | – | – |
| 8 | 78 | M | Former | Yes | GUM | P | M/D | 2 | 0 | II | – | NED | EGFR | KIT | – | CDKN2A, RB1 |
| 9 | 53 | M | Current | Yes | TON | B | W/D | 2 | 0 | II | – | NED | EGFR, FGFR1 | SMARCB1 | – | CDKN2A, RB1 |
| 10 | 30 | M | Current | No | TON | B | W/D | 2 | 0 | II | – | NED | FGFR1 | TP53 | – | CDKN2A |
| 11 | 35 | F | Never | No | TON | B | P/D | 2 | 0 | II | – | NED | EGFR | – | AKT1, HRAS | PTEN, RB1 |
| 12 | 35 | F | Current | Yes | TON | B | M/D | 2 | 0 | II | – | NED | EGFR | CDKN2A | GNA11, JAK3 | – |
| 13 | 52 | M | Former | Yes | GUM | B | M/D | 2 | 0 | II | – | NED | ERBB4 | – | PIK3CA | – |
| 14 | 30 | M | Current | Yes | TON | B | M/D | 2 | 0 | II | – | NED | EGFR | JAK2, RET | – | – |
| 15 | 65 | M | UNK | UNK | BM | B | P/D | 2 | 0 | II | – | NED | FGFR3 | – | AKT1, ALK | ATM, CDKN2A, RB1 |
| 16 | 35 | F | Never | No | TON | B | W/D | 2 | 0 | II | – | NED | ERBB4 | – | PIK3CA | – |
| 17 | 65 | F | Current | Yes | TON | B | P/D | 2 | 0 | II | – | NED | ERBB2 | CDKN2A | – | – |
| 18 | 74 | F | Never | No | GUM | B | M/D | 2 | 1 | III | – | NED | FGFR1 | – | – | – |
| 19 | 36 | M | Current | Yes | TON | B | M/D | 2 | 1 | III | – | LTF | EGFR | CDKN2A | – | – |
| 20 | 62 | F | Current | No | GUM | B | M/D | 3 | 0 | IVA | – | NED | EGFR | KRAS | ABL1 | – |
| 21 | 75 | F | Never | UNK | GUM | B | M/D | 4a | 0 | IVA | – | DID | EGFR | – | – | CDKN2A |
| 22 | 54 | F | Never | No | TON | P | M/D | 2 | 2b | IVA | – | NED | FGFR3 | TP53 | ABL1, AKT1, CSF1R, GNA11, HRAS, JAK3, RET, SMO | RB1 |
| 23 | 63 | M | Current | Yes | TON | B | W/D | 3 | 2b | IVA | – | NED | ERBB2 | – | – | – |
| 24 | 50 | M | Current | Yes | FOM | B | M/D | 3 | 2b | IVA | – | NED | FGFR1 | – | ABL1, GNA11, GNAQ, JAK3 | CDKN2A |
| 25 | 73 | M | Current | Yes | FOM | B | M/D | 4a | 2c | IVA | – | NED | ERBB2 | PIK3CA | PIK3CA | CDKN2A |
| 26 | 82 | F | Former | No | GUM | B | W/D | 4a | 2c | IVA | – | NED | ERBB4 | – | – | – |
| 27 | 62 | M | UNK | UNK | HP | B | M/D | 4a | 2c | IVA | – | DOD(P,N) | ERBB2 | – | – | CDKN2A |
| 28 | 45 | M | Never | Yes | GUM | B | W/D | 4b | 1 | IVB | – | NED | EGFR | TP53 | AKT1 | – |
| 29 | 58 | M | UNK | UNK | FOM | B | M/D | 2 | 0 | II | Lung | DOD(M) | EGFR | TP53 | – | CDKN2A, PTEN |
| 30 | 62 | M | Current | Yes | BM | B | W/D | 4a | 0 | IVA | Lung | DOD(M) | FGFR1 | CDKN2A, GNA11, PIK3CA, TP53 | – | – |
| 31 | 57 | F | Former | Yes | GUM | P | W/D | 4a | 0 | IVA | Lung, bone | DOD(M) | ERBB2, FGFR3 | PIK3CA | ABL1, GNA11, GNAQ, HRAS, JAK2, JAK3, RET, SMO | APC, SMAD4 |
| 32 | 24 | M | Never | No | TON | B | M/D | 1 | 2b | VIA | Lung | DOD(M) | EGFR, ERBB2, FGFR3, KIT, MET, PDGFRA | ATM, ERBB4, FGFR3, IDH2, KDR, RB1, SMAD4, STK11, TP53 | ABL1, GNA11, GNAQ, JAK3, RET | CDKN2A, RB1, SMAD4 |
| 33 | 55 | M | Never | No | BM | LN | W/D | 2 | 2b | IVA | Lung | DOD(N,M) | FGFR3 | CDH1, TP53 | ABL1, AKT1, RET | ATM, PTEN, RB1 |
| 34 | 58 | F | Never | Yes | TON | B | W/D | 2 | 2c | IVA | Lung | DOD(P,N,M) | ERBB2, FLT3 | – | – | – |
| 35 | 63 | F | Former | Yes | TON | B | M/D | 3 | 2c | IVA | Lung | DOD(N,M) | EGFR | TP53 | – | – |
| 36 | 53 | M | Never | Yes | TON | B | M/D | 4a | 2c | IVA | Lung, bone | DOD(M) | EGFR, FGFR1, FGFR3 | – | AKT1, GNA11, JAK3 | RB1 |
| 37 | 62 | F | Current | Yes | TON | B | M/D | 4a | 2c | IVA | Lung, bone, liver | DOD(M) | FGFR1 | APC,TP53 | – | – |
| 38 | 71 | M | Current | Yes | BM | LN | P/D | 2 | 1 | III | Lung | DOD(M) | – | CDKN2A, TP53 | – | – |
| 39 | 74 | F | Former | UNK | BM | P | M/D | 2 | 1 | III | Lung | DOD(M) | – | – | CSF1R | – |
| 40 | 66 | M | Current | Yes | GUM | B | W/D | 4a | 1 | IVA | Lung, bone | AWD | – | TP53 | – | CDKN2A |
| 41 | 73 | M | Never | Yes | TON | P | P/D | 1 | 2b | IVA | Lung | DOD(M) | – | SMO | – | – |
| 42 | 69 | M | Current | Yes | TON | P | W/D | 2 | 2b | IVA | Lung | DOD(M) | – | – | AKT, CSF1R, HRAS | RB1 |
| 43 | 65 | M | UNK | Yes | TON | B | P/D | 2 | 2b | IVA | Lung | DOD(M) | – | TP53 | – | – |
| 44 | 62 | M | Never | Yes | BM | B | P/D | 2 | 2b | IVA | Lung | DOD(M) | – | TP53 | PIK3CA | TP53 |
| 45 | 74 | M | Current | No | BM | B | P/D | 2 | 2b | IVA | Lung | DOD(M) | – | – | – | TP53 |
| 46 | 32 | M | Never | Yes | TON | B | M/D | 2 | 2b | IVA | Lung, bone | DOD(M) | – | SMARKB1 | – | – |
| 47 | 65 | M | Current | Yes | TON | B | M/D | 3 | 2b | IVA | Lung, bone, liver | DOD(M) | – | TP53 | – | – |
| 48 | 67 | M | Current | Yes | TON | B | W/D | 2 | 2c | IVA | Lung | DOD(M) | – | TP53 | PIK3CA | STK11, TP53 |
| 49 | 71 | F | Never | No | GUM | B | M/D | 4a | 2c | IVA | Lung | DOD(N,M) | – | – | – | – |
The upper part of the table shows patients with RTK amplification (Patients 1–28), the middle shows patients with both RTK amplification and distant metastasis (29–37), and the lower part shows patients with distant metastasis (38–49). AWD, alive with disease; B, biopsy sample; BM, buccal mucossa; DID, died of intercurrent disease; DOD, died of disease; F, female; FOM, floor of mouth; HP, hard palate; LN, lymph node sample; LTF, lost to follow up; M, male; M/D, moderately differentiated; NED, no evidence of disease; P, primary sample of surgical specimen; P/D, poorly differentiated; TON, tongue; UNK, unknown; W/D, well differentiated.
Univariate and multivariate analyses according to clinicopathological factors and genes were conducted using the Cox proportional hazard model (Table 3). In univariate analysis for OS, a statistically significant difference was detected for RTK amplification (hazard ratio [HR] = 2.662, 95% confidence interval [CI] = 1.290–5.491, P = 0.008) and CDKN2A deletion (HR = 2.442, 95% CI = 1.059–5.634, P = 0.036). The cumulative 5‐year survival rates were 64.6% (95% CI = 47.4–81.8) in the RTK amplification group (Fig. 3a) and 63.7% (95% CI = 41.6–85.8) in the CDNK2A deletion group (Fig. S4a). Regarding SMs of TP53 with the highest frequency, no statistically significant difference was detected (Fig. S4b). In addition, no statistically significant difference was detected in CNAs and SMs of genes in the PI3K and/or Ras/Raf pathways. In multivariate analysis, we considered RTK and CDKN2A CNAs, and clinicopathological poor prognostic factors for OS, namely, age, subsite, histological differentiation, and clinical stage, which could be predictive before treatment. This analysis revealed that RTK amplification was an independent prognostic factor (HR = 2.410, 95% CI = 1.056–5.498, P = 0.037) (Table 3).
Table 3.
Results of univariate and multivariate analysis for overall survival
| Variable | Category | Hazard ratio for death (95.0% CI) | P‐value |
|---|---|---|---|
| Univariate analysis | |||
| Age (years) | < 60 vs ≥ 60 | 1.402 (0.690–2.850) | 0.351 |
| Gender | Male vs Female | 0.754 (0.365–1.555) | 0.444 |
| Smoking | Non–smoker vs Smoker | 0.992 (0.472–2.086) | 0.984 |
| Alcohol | No alcohol use vs Alcohol use | 1.058 (0.463–2.418) | 0.893 |
| Subsite | Tongue/Gum vs Others (BM/FOM/HP) | 2.711 (1.289–5.700) | 0.009 |
| Histological differentiation | Well/Moderately vs Poorly | 2.014 (0.874–4.643) | 0.100 |
| cStage | cStage I–III vs cStage IV | 1.901 (0.953–3.792) | 0.068 |
| Stage | Stage I–III vs Stage IV | 6.505 (2.822–14.99) | <0.001 |
| RTK | No amplification vs Amplification | 2.662 (1.290–5.491) | 0.008 |
| PIK3CA | No amplification vs Amplification | 0.543 (0.166–1.781) | 0.314 |
| Wild type vs Mutation | 1.197 (0.365–3.925) | 0.767 | |
| RAS/RAF pathway | No amplification vs Amplification | 0.731 (0.175–3.057) | 0.668 |
| Wild type vs Mutation | 0.901 (0.123–6.599) | 0.918 | |
| CDKN2A | No deletion vs Deletion | 2.442 (1.059–5.634) | 0.036 |
| Wild type vs Mutation | 0.874 (0.337–2.264) | 0.781 | |
| TP53 | No deletion vs Deletion | 1.375 (0.419–4.510) | 0.599 |
| Wild type vs Mutation | 1.192 (0.593–2.397) | 0.622 | |
| Multivariate analysis | |||
| Age (years) | < 60 vs ≥ 60 | 1.256 (0.595–2.653) | 0.550 |
| Subsite | Tongue/Gum vs Others (BM/FOM/HP) | 2.170 (0.958–4.918) | 0.063 |
| Histological differentiation | Well/Moderately vs Poorly | 2.176 (0.917–5.162) | 0.078 |
| cStage | cStage I–III vs cStage IV | 1.824 (0.879–3.788) | 0.107 |
| RTK | No amplification vs Amplification | 2.410 (1.056–5.498) | 0.037 |
| CDKN2A | No deletion vs Deletion | 1.104 (0.398–3.059) | 0.849 |
RTK (EGFR, ERBB2, ERBB4, FGFR1, FGFR2, FGFR3, FLT3, KIT, MET, PDGFRA). RAS‐RAF pathway (BRAF, HRAS, KRAS, NRAS). BM, buccal mucosa; cStage, clinical stage; FOM, floor of mouth; HP, hard palate.
Figure 3.
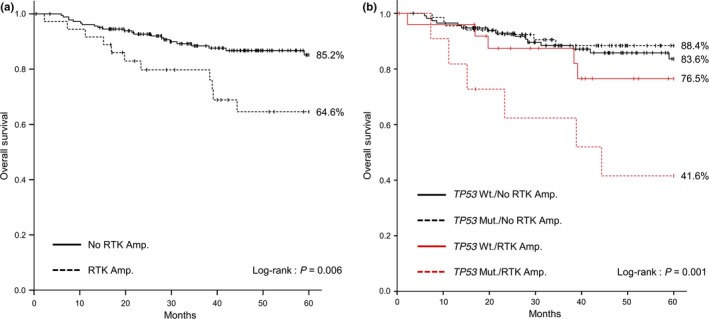
Kaplan–Meier estimates of overall survival (OS) among patients according to genomic variables. (a) Patients were stratified into two subgroups according to receptor tyrosine kinase (RTK) amplification status. The cumulative 5‐year survival rate was 85.2% (95% confidence interval [CI] = 79.1–91.3) in the no RTK Amp. group, vs 64.6% (95% CI = 47.4–81.8) in the RTK Amp. group. (b) Patients were stratified into four subgroups according to TP53 mutation and RTK amplification status. The cumulative 5‐year survival rates were 83.6% (95% CI = 75.6–91.7) in the TP53 Wt./No RTK Amp. group, 88.4% (95% CI = 80.2–96.6) in the TP53 Mut./No RTK Amp. group, 76.5% (95% CI = 58.2–94.9) in the TP53 Wt./RTK Amp. Group, and 41.6% (95% CI = 10.9–72.2) in the TP53 Mut./RTK Amp. group. Amp., amplification; Wt., wild type; Mut., mutation.
Although no statistically significant difference was detected in OS concerning the presence or absence of any SMs of TP53, comparing four groups according to the presence or absence of any TP53 mutation or RTK amplification revealed significantly poorer prognosis in the TP53 mutation/RTK amplification group (HR = 4.820, 95% CI = 1.869–12.43, P = 0.001) (Table 4 ). The cumulative 5‐year survival rate of this group was 41.6% (95% CI = 10.9–72.2) (Fig. 3b). Additionally, similar comparisons among the four groups detected significantly poorer prognosis associated with the presence of RTK amplifications irrespective of CDKN2A deletion (no CDKN2A deletion/RTK amplification: HR = 2.626, 95% CI = 1.103–6.248, P = 0.029; CDKN2A deletion/RTK amplification: HR = 3.517, 95% CI = 1.196–10.34, P = 0.022) (Table 5). The cumulative 5‐year survival rates of these two groups were 68.6 (95% CI = 48.6–88.6) and 56.3% (95% CI = 24.0–88.6), respectively (Fig. S4c). Examination of the cause of death in 25 cases in which CDKN2A deletion was detected revealed that two, one, two, and two patients died because of the primary lesion, cervical metastasis, distant metastasis, and another disease, respectively (data not shown).
Table 4.
Four‐groups analysis according to the presence or absence of TP53 and receptor tyrosine kinase genetic alterations
| Variable | Category | No. patients | No. deaths | Hazard ratio for death (95.0% CI) | P‐value |
|---|---|---|---|---|---|
| TP53/RTK | Wild type/No amplification | 116 | 15 | Reference | |
| Mutation/No amplification | 67 | 7 | 0.805 (0.328–1.976) | 0.636 | |
| Wild type/Amplification | 26 | 5 | 1.560 (0.567–4.294) | 0.389 | |
| Mutation/Amplification | 11 | 6 | 4.820 (1.869–12.43) | 0.001 |
Table 5.
Four‐groups analysis according to the presence or absence of CDKN2A and receptor tyrosine kinase genetic alterations
| Variable | Category | No. patients | No. deaths | Hazard ratio for death (95.0% CI) | P‐value |
|---|---|---|---|---|---|
| CDKN2A/RTK | No deletion/No amplification | 169 | 19 | Reference | |
| Deletion/No amplification | 14 | 3 | 2.398 (0.708–8.120) | 0.160 | |
| No deletion/Amplification | 26 | 7 | 2.626 (1.103–6.248) | 0.029 | |
| Deletion/Amplification | 11 | 4 | 3.517 (1.196–10.34) | 0.022 |
Discussion
The clinical application of NGS, such as in the clinical trial NCI Molecular Analysis for Therapy Choice (NCI‐MATCH), may provide insights into the genomic landscape of human cancers and identify therapeutic targets for molecular targeted agents.11 In this trial, only the malignant tissue will be screened.12 In our hospital, collecting sufficient matched normal oral tissue from patients with OSCC was challenging. This is a very common issue encountered in clinical contexts; therefore, we filtered out possible germline mutations using the databases of the 1000 Genomes Project and 5000 Exomes Project to identify SMs in the absence of matched normal controls. Recently, studies using similar approaches have been reported.8, 13 We expect that with increasing numbers of normal samples deposited in common databases, these methods can be further refined. Furthermore, the availability of these databases and methods allows future studies to decrease the cost of germline sequencing. This approach also obviates the issue of dealing with incidental or secondary findings that this generates.
The most prevalent genetic alteration detected in our cohort comprised a broad spectrum of TP53 mutations (35.9%). Other frequently mutated genes include CDKN2A (15.9%) and PIK3CA (8.6%), and this pattern of Japanese OSCC genetic mutations was compared with other studies. The genetic mutations of oral tongue carcinoma in Singapore exhibited similar frequencies of mutations in TP53 (38.3%) and PIK3CA (8.3%), whereas mutations in CDKN2A were less frequent.8 In the HNSCC data of TCGA, mutation frequencies in TP53, CDKN2A, and PIK3CA were 72%, 22%, and 21%, respectively.4 The TP53 mutation frequency in gingivo‐buccal OSCC in India was 62%.7 On the other hand, the mutation frequencies of TP53 in HNSCC and OSCC in COSMIC data (v78, released 05‐SEP‐16) were 32% and 42%, respectively (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). Similarly, patterns attributable to etiology and ethnicity have been observed for other cancer types.14
The results of this study demonstrated that RTK amplification is a predictive factor for distant metastasis of OSCC. Regulation failure induced by RTK alteration is known to cause intercellular/intracellular signaling perturbation, and RTK alterations have been reported in many cancers.15, 16 In HNSCC, genetic alterations of not only EGFR but also ERBB2, FGFR1, FGFR3, and MET have been reported.17, 18, 19, 20, 21 RTK gene alterations induce epithelial–mesenchymal transition,22 allowing tumor cells to acquire migration capabilities. Overexpression of EGFR was demonstrated to be an adverse prognostic factor in HNSCC.23 In addition, HER2 overexpression in breast and gastric cancers and FGFR1 overexpression in lung cancer have been identified as adverse prognostic factors.24, 25, 26 EGFR amplification was detected most frequently in this study, similar to previous reports.4, 19 Meanwhile, amplification of other RTKs such as ERBB2, FRFR1, and FGFR3 was also detected, albeit in a small number of patients, demonstrating that RTKs are adverse prognostic factors. Moreover, it was demonstrated that the group with both RTK amplification and TP53 mutations had significantly poorer prognosis than the other groups, although no significant difference was detected in OS regarding the presence or absence of TP53 mutations alone. This is likely because mutation‐caused loss of function of TP53 promoted tumor activation by RTK amplification, resulting in significant induction of distant metastasis. Meanwhile, CDKN2A, another tumor suppressor gene, has also been reported to undergo deletion in HNSCC.27 However, it was not demonstrated that a synergistic effect of CDKN2A deletion and RTK amplification on OS.
Our examination of stages detected a statistically significant difference in the frequency of genetic alterations in PIK3CA between stages as well as their factors, namely local tumor progression (T status) and cervical lymph node metastasis (N status), separately. Regarding SMs of PIK3CA and stages, Kozaki et al.28 reported that the frequency of SMs of PIK3CA was significantly higher in stage IV compared to stages I–III in OSCC, which is in line with our results. It has been found that precursor lesions of invasive cancers also exhibit PIK3CA amplification.29, 30 Given our result that the frequency of PIK3CA amplification was significantly higher in patients free of cervical lymph node metastasis, PIK3CA amplification is likely to reflect a tumor's early characteristics.
Since molecular‐targeted drugs became available, it has become common in cancer treatment to choose drugs based on the presence or absence of a gene mutation. RTKs can be the target molecules of various molecular‐targeted drugs. Cetuximab, an anti‐EGFR monoclonal antibody, has been demonstrated to provide clinical improvement in the treatment of locally advanced and recurrent/metastatic HNSCC.31, 32 Although clinical trials of various other RTK inhibitors and anti‐RTK monoclonal antibody drugs have been conducted,33, 34, 35, 36 no efficient therapy equivalent to that of cetuximab has been identified. The reason may be that in most of these trials, the subjects had recurrent or metastatic lesions. Therefore, examination of the administration, methods, and timing of treatment may facilitate good outcomes. The use of a molecular‐targeted drug as postoperative adjuvant therapy for the high‐risk group as identified by RTK amplification through the analysis of genomic alterations using biopsy tissues is an example.
Cigarette smoking is the most cited risk factor for oral cancer. It raises the risk of developing oral cancer by threefold, and concomitant alcohol consumption, acting synergistically, increases the risk 10–15‐fold.37 The risk of cancer is higher in tissues which are in close contact with ingested alcohol, such as the oral cavity, pharynx, and esophagus.38 However, it is not clear why alcohol use preferentially exerts a local carcinogenic effect. López‐Lázaro discussed that the cytotoxic effect of ethanol activates the division of the stem cells that maintain the deeper layers of the mucosa in homeostasis.38 Meanwhile, Hashibe et al.37 reported that a substantial proportion of head and neck cancers cannot be attributed to tobacco or alcohol use, particularly for oral cavity cancer.37 Therefore, we investigated the smoking status and alcohol consumption in our cohort; however, there were no significant correlations between the smoking status and/or alcohol consumption and SMs, CNAs, and clinicopathological features in our study. At the same time, smoking is known to affect the genome by causing certain types of mutation. The mutational events linked to smoking are traditionally reported as an increase in C>A mutations and a decrease in C>T mutations.39 However, Pickering et al.40 reported that smoking has only a minor impact on the types of mutations observed in oral tongue SCC and TCGA data also demonstrate that the genomic effects of smoking are tumor‐site specific. Although data are sparse because of limited sequencing lesions, we have also investigated the events of somatic mutations detected in our study; no significant differentiations were revealed (data not shown) between 106 mutations in the non‐smoking group (58 patients) and 159 in the smoking group (72 patients). These findings indicate that smoking and/or alcohol consumption only have a minor impact on carcinogenesis in oral cancer.
The panel that we used in this study does not include some important genes that have been detected in OSCC, such as CASP8 and FAT1 4, 5 (Table S1a). Moreover, we need additional consideration concerning the target region of each gene even though the panel is designed to target many hotspot regions (Table S1b). Oncogenes are recurrently mutated at the same amino acid positions, whereas tumor suppressor genes are mutated through truncating mutations throughout their length.41 Regarding NOTCH1, most of the mutations in hematopoietic tumors have been identified in the heterodimerization and C‐terminal polypeptide‐enriched proline, glutamate, serine, and threonine domain,42, 43 and the panel we used only targeted these regions. Conversely, it has been demonstrated that in HNSCC, mutations, including truncating mutations, have also been detected in the N‐terminal epidermal growth factor‐like ligand‐binding domain, and these mutations appeared to be loss‐of‐function mutations.5, 6 Therefore, a specific custom panel for OSCC with better target genes and regions may enable us to obtain more information and discover new treatments.
In conclusion, the results of this study using FFPE samples of cancer tissues and NGS demonstrate that RTK amplification is a prognostic prediction factor for distant metastasis of OSCC, indicating the necessity for using NGS in clinical sequencing. To achieve stratified therapies of OSCC based on genomic alterations, evidence must be accumulated.
Disclosure Statement
The authors have no conflicts of interest.
Abbreviations
- CI
confidence interval
- CNAs
copy number alterations
- FFPE
formalin‐fixed, paraffin‐embedded
- HNSCC
head and neck squamous cell carcinoma
- HR
hazard ratio
- NGS
next generation sequencing
- OSCC
oral squamous cell carcinoma
- OS
overall survival
- RTK
receptor tyrosine kinase
- SMs
somatic mutations
- TCGA
The Cancer Genome Atlas
Supporting information
Fig. S1. Receiver operating characteristic curve to determine the cut‐off point for (a) deletion and (b) amplification.
{kind=link}
{kind=link}
Fig. S2. Kaplan–Meier estimates of overall survival according to (a) subsite and (b) stage.
{kind=link}
{kind=link}
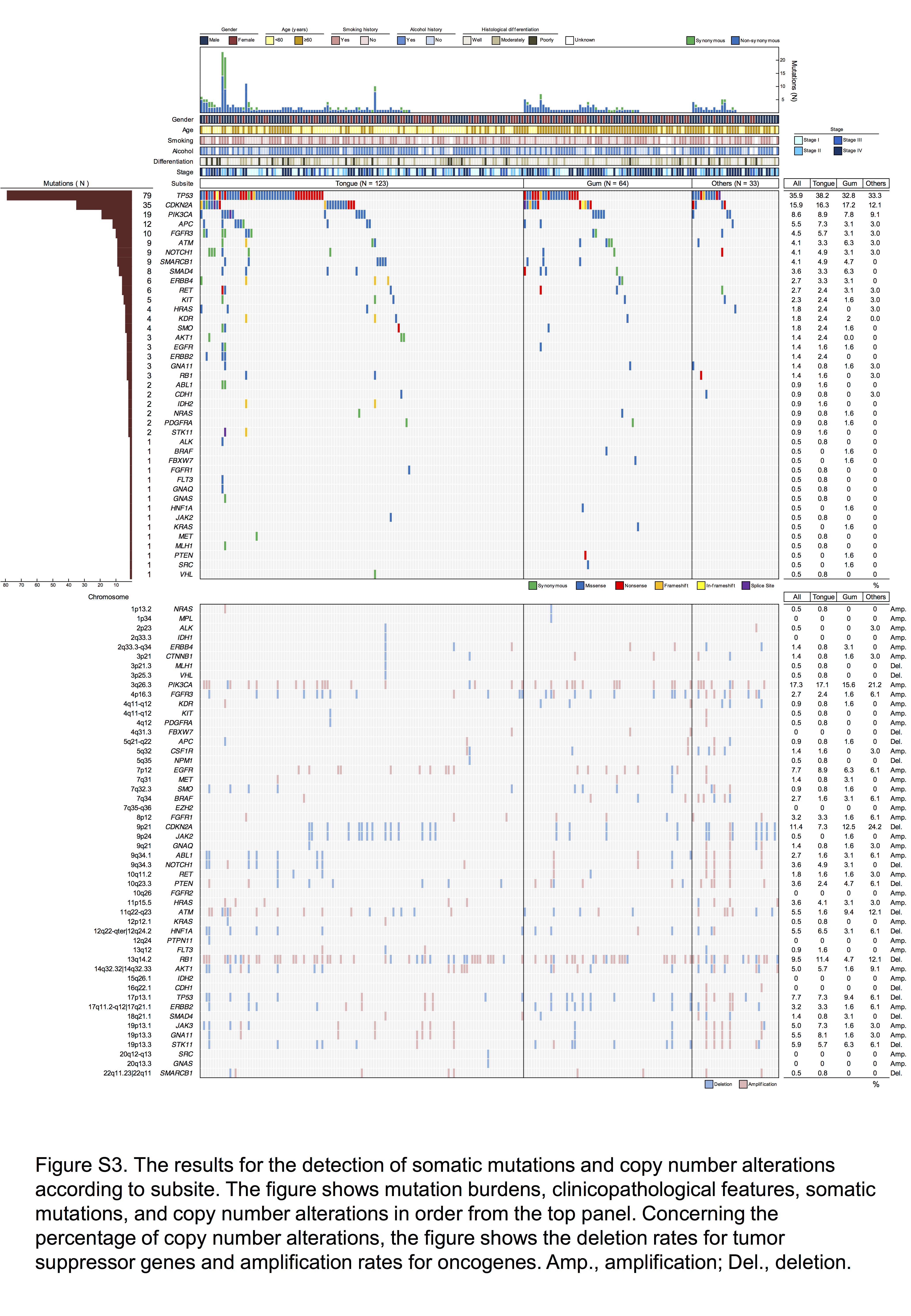
Fig. S3. The results for the detection of somatic mutations and copy number alterations according to subsite.
{kind=link}
Fig. S4. Kaplan–Meier estimates of overall survival according to (a) CDKN2A deletion status, (b) TP53 mutation status, and (c) CDKN2A deletion/receptor tyrosine kinase amplification status.
{kind=link}
{kind=link}
{kind=link}
Table S1. Information for (a) target genes and (b) target regions.
Table S2. Summary of clinicopathological data.
Table S3. Associations of clinicopathological variables with somatic mutations and copy number alterations in 50 genes.
Table S4. Somatic mutations and copy number alterations according to stage and subsite.
Doc. S1. Supplementary Materials and Methods concerned with copy number analyses.
Acknowledgments
This work was supported by JPSP KAKENHI Grant Number JP16H05510 (K.M.), Tailor‐made Medical Treatment Program, The Japan Agency for Medical Research and Development (AMED) (S.I. and H.K.), JPSP KAKENHI Grant Number JP16H02481 (S.I.), and partially supported by P‐DIRECT and P‐CREATE from AMED (J.I.). We thank the late Professor Ken‐ichi Kozaki for useful discussions. His intellect, his vistas, his human warmth, and his own experiences in the treatment for his oral cancer was truly an important contribution to all cancer research such that his reach will truly exceed his grasp. We thank Makiko Matsuda and Miwako Hamagaki for technical support.
Cancer Sci 108 (2017) 256–266
Funding Information
Japan Society for the Promotion of Science, (Grant/Award Number: ‘16H02481’, ‘16H05510’) Japan Agency for Medical Research and Development, Ministry of Education, Culture, Sports, Science, and Technology.
References
- 1. Ferlay J, Soerjomataram I, Dikshit R et al Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015; 136: E359–86. [DOI] [PubMed] [Google Scholar]
- 2. Barnes L, International Academy of Pathology, World Health Organization, International Agency for Research on Cancer . Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press, 2005; 430 p. [Google Scholar]
- 3. Chinn SB, Myers JN. Oral cavity carcinoma: current management, controversies, and future directions. J Clin Oncol 2015; 33: 3269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Network CGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015; 517: 576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stransky N, Egloff AM, Tward AD et al The mutational landscape of head and neck squamous cell carcinoma. Science 2011; 333: 1157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Agrawal N, Frederick MJ, Pickering CR et al Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011; 333: 1154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Consortium IPTotICG . Mutational landscape of gingivo‐buccal oral squamous cell carcinoma reveals new recurrently‐mutated genes and molecular subgroups. Nat Commun 2013; 4: 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vettore AL, Ramnarayanan K, Poore G et al Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med 2015; 7: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hadd AG, Houghton J, Choudhary A et al Targeted, high‐depth, next‐generation sequencing of cancer genes in formalin‐fixed, paraffin‐embedded and fine‐needle aspiration tumor specimens. J Mol Diagn 2013; 15: 234–47. [DOI] [PubMed] [Google Scholar]
- 10. Li J, Lupat R, Amarasinghe KC et al CONTRA: copy number analysis for targeted resequencing. Bioinformatics 2012; 28: 1307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abrams J, Conley B, Mooney M et al National Cancer Institute's precision medicine initiatives for the new national clinical trials network. Am Soc Clin Oncol Educ Book 2014: 71–6. [DOI] [PubMed] [Google Scholar]
- 12. Do K, O'Sullivan Coyne G, Chen AP. An overview of the NCI precision medicine trials‐NCI MATCH and MPACT. Chin Clin Oncol 2015; 4: 31. [DOI] [PubMed] [Google Scholar]
- 13. Suzuki A, Mimaki S, Yamane Y et al Identification and characterization of cancer mutations in Japanese lung adenocarcinoma without sequencing of normal tissue counterparts. PLoS ONE 2013; 8: e73484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan‐On W, Nairismägi ML, Ong CK et al Exome sequencing identifies distinct mutational patterns in liver fluke‐related and non‐infection‐related bile duct cancers. Nat Genet 2013; 45: 1474–8. [DOI] [PubMed] [Google Scholar]
- 15. Robertson SC, Tynan JA, Donoghue DJ. RTK mutations and human syndromeswhen good receptors turn bad. Trends Genet 2000; 16: 265–71. [DOI] [PubMed] [Google Scholar]
- 16. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010; 141: 1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chung CH, Parker JS, Karaca G et al Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004; 5: 489–500. [DOI] [PubMed] [Google Scholar]
- 18. Hama T, Yuza Y, Saito Y et al Prognostic significance of epidermal growth factor receptor phosphorylation and mutation in head and neck squamous cell carcinoma. Oncologist 2009; 14: 900–8. [DOI] [PubMed] [Google Scholar]
- 19. Sheu JJ, Hua CH, Wan L et al Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res 2009; 69: 2568–76. [DOI] [PubMed] [Google Scholar]
- 20. Tillman BN, Yanik M, Birkeland AC et al Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low‐risk patient as defined by targeted sequencing. Head Neck 2016; 38(Suppl. 1): E1646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chau NG, Perez‐Ordonez B, Zhang K et al The association between EGFR variant III, HPV, p16, c‐MET, EGFR gene copy number and response to EGFR inhibitors in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck Oncol 2011; 3: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grünert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4: 657–65. [DOI] [PubMed] [Google Scholar]
- 23. Bentzen SM, Atasoy BM, Daley FM et al Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. J Clin Oncol 2005; 23: 5560–7. [DOI] [PubMed] [Google Scholar]
- 24. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 25. Kim HR, Kim DJ, Kang DR et al Fibroblast growth factor receptor 1 gene amplification is associated with poor survival and cigarette smoking dosage in patients with resected squamous cell lung cancer. J Clin Oncol 2013; 31: 731–7. [DOI] [PubMed] [Google Scholar]
- 26. He C, Bian XY, Ni XZ et al Correlation of human epidermal growth factor receptor 2 expression with clinicopathological characteristics and prognosis in gastric cancer. World J Gastroenterol 2013; 19: 2171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reed AL, Califano J, Cairns P et al High frequency of p16 (CDKN2/MTS‐1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 1996; 56: 3630–3. [PubMed] [Google Scholar]
- 28. Kozaki K, Imoto I, Pimkhaokham A et al PIK3CA mutation is an oncogenic aberration at advanced stages of oral squamous cell carcinoma. Cancer Sci 2006; 97: 1351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Redon R, Muller D, Caulee K, Wanherdrick K, Abecassis J, du Manoir S. A simple specific pattern of chromosomal aberrations at early stages of head and neck squamous cell carcinomas: PIK3CA but not p63 gene as a likely target of 3q26‐qter gains. Cancer Res 2001; 61: 4122–9. [PubMed] [Google Scholar]
- 30. Woenckhaus J, Steger K, Werner E et al Genomic gain of PIK3CA and increased expression of p110alpha are associated with progression of dysplasia into invasive squamous cell carcinoma. J Pathol 2002; 198: 335–42. [DOI] [PubMed] [Google Scholar]
- 31. Bonner JA, Harari PM, Giralt J et al Radiotherapy plus cetuximab for squamous‐cell carcinoma of the head and neck. N Engl J Med 2006; 354: 567–78. [DOI] [PubMed] [Google Scholar]
- 32. Vermorken JB, Mesia R, Rivera F et al Platinum‐based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359: 1116–27. [DOI] [PubMed] [Google Scholar]
- 33. Massarelli E, Lin H, Ginsberg LE et al Phase II trial of everolimus and erlotinib in patients with platinum‐resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol 2015; 26: 1476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Argiris A, Ghebremichael M, Gilbert J et al Phase III randomized, placebo‐controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: an eastern cooperative oncology group trial. J Clin Oncol 2013; 31: 1405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsao AS, Liu S, Fujimoto J et al Phase II trials of imatinib mesylate and docetaxel in patients with metastatic non‐small cell lung cancer and head and neck squamous cell carcinoma. J Thorac Oncol 2011; 6: 2104–11. [DOI] [PubMed] [Google Scholar]
- 36. Harrington K, Temam S, Mehanna H et al Postoperative adjuvant lapatinib and concurrent chemoradiotherapy followed by maintenance lapatinib monotherapy in high‐risk patients with resected squamous cell carcinoma of the head and neck: a phase III, randomized, double‐blind, placebo‐controlled study. J Clin Oncol 2015; 33: 4202–9. [DOI] [PubMed] [Google Scholar]
- 37. Hashibe M, Brennan P, Chuang SC et al Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev 2009; 18: 541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. López‐Lázaro M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol 2016; 62: 149–52. [DOI] [PubMed] [Google Scholar]
- 39. Alexandrov LB, Nik‐Zainal S, Wedge DC et al Signatures of mutational processes in human cancer. Nature 2013; 500: 415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pickering CR, Zhang J, Neskey DM et al Squamous cell carcinoma of the oral tongue in young non‐smokers is genomically similar to tumors in older smokers. Clin Cancer Res 2014; 20: 3842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science 2013; 339: 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ellisen LW, Bird J, West DC et al TAN‐1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991; 66: 649–61. [DOI] [PubMed] [Google Scholar]
- 43. Baldus CD, Thibaut J, Goekbuget N et al Prognostic implications of NOTCH1 and FBXW7 mutations in adult acute T‐lymphoblastic leukemia. Haematologica 2009; 94: 1383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Receiver operating characteristic curve to determine the cut‐off point for (a) deletion and (b) amplification.
Fig. S2. Kaplan–Meier estimates of overall survival according to (a) subsite and (b) stage.
Fig. S3. The results for the detection of somatic mutations and copy number alterations according to subsite.
Fig. S4. Kaplan–Meier estimates of overall survival according to (a) CDKN2A deletion status, (b) TP53 mutation status, and (c) CDKN2A deletion/receptor tyrosine kinase amplification status.
Table S1. Information for (a) target genes and (b) target regions.
Table S2. Summary of clinicopathological data.
Table S3. Associations of clinicopathological variables with somatic mutations and copy number alterations in 50 genes.
Table S4. Somatic mutations and copy number alterations according to stage and subsite.
Doc. S1. Supplementary Materials and Methods concerned with copy number analyses.