

Abstract
FGF/FGFR gene aberrations such as amplification, mutation and fusion are associated with many types of human cancers including urothelial cancer. FGFR kinase inhibitors are expected to be a targeted therapy for urothelial cancer harboring FGFR3 gene alternations. ASP5878, a selective inhibitor of FGFR1, 2, 3 and 4 under clinical investigation, selectively inhibited cell proliferation of urothelial cancer cell lines harboring FGFR3 point mutation or fusion (UM‐UC‐14, RT‐112, RT4 and SW 780) among 23 urothelial cancer cell lines. Furthermore, ASP5878 inhibited cell proliferation of adriamycin‐resistant UM‐UC‐14 cell line harboring MDR1 overexpression and gemcitabine‐resistant RT‐112 cell line. The protein expression of c‐MYC, an oncoprotein, in gemcitabine‐resistant RT‐112 cell line was higher than that in RT‐112 parental cell line and ASP5878 decreased the c‐MYC expression in both RT‐112 parental and gemcitabine‐resistant RT‐112 cell lines. Once‐daily oral administration of ASP5878 exerted potent antitumor activities in UM‐UC‐14, RT‐112 and gemcitabine‐resistant RT‐112 xenograft models without affecting body weight. These findings suggest that ASP5878 has the potential to be an oral targeted therapy against urothelial cancer harboring FGFR3 fusion or FGFR3 point mutation after the acquisition of gemcitabine‐ or adriamycin‐resistance.
Keywords: Chemotherapy, FGFR3‐fusion or ‐mutation, molecular targeted drug therapy, oral dosing, urothelial cancer
Urothelial cancer can arise anywhere along the epithelial lining of urinary tract, including the bladder, renal pelvis and ureter. Although urothelial cancers arising in these various locations have similar morphology and gene expression profile,1 urothelial cancer occurs most frequently in the bladder. Bladder cancer is the most common malignancy involving the urinary system and the ninth most common malignancy worldwide.2 Bladder cancer is mainly divided into two groups by stage. The stage classification differentiates between non‐muscle invasive (Tis, Ta, and T1) and muscle‐invasive tumors (T2, T3, and T4) according to the depth of invasion. The standard therapy of muscle‐invasive bladder cancer is the combination of chemotherapeutic agents (GC and MVAC). However, despite reasonable response rates to chemotherapy in patients with locally advanced or metastatic bladder cancer, long‐term progression‐free survival rates remain insufficient,3 which is thought to be caused by the induction of MDR1 overexpression or the alterations in the apoptotic machinery including overexpression of c‐MYC, an oncoprotein.4, 5 Therefore, effective drugs against chemotherapy‐resistant bladder cancer are eagerly needed.
The mammalian FGF/FGFR family comprises 18 ligands and four main receptors (FGFR1–4). FGFs induce FGFR dimerization, followed by FGFR autophosphorylation and activation of downstream signaling pathways. In a variety of human cancers, aberrant activation of FGF/FGFR signaling promotes cellular proliferation, migration/invasion and angiogenesis.6 Five different FGFR3 point mutations such as R248C, S249C, G372C, Y375C, and K652E account for more than 90% of the point mutations of FGFR3, and S249C is the most common (48%) in bladder cancer.7 The frequency of FGFR3 point mutation in muscle‐invasive bladder cancer is lower than that in non‐muscle invasive bladder cancer [15% (7/47): invasive, 58% (58/100): non‐invasive].7 Another report shows that the frequencies of FGFR3 point mutations in primary muscle invasive urothelial tumors and metastases are 2% (2/161) and 9% (3/33), respectively.8 Recently, it has been also reported that FGFR3‐TACC3 and FGFR3‐BAIAP2L1, fusion genes were identified in some urothelial cancer cell lines and cancer tissue samples.9, 10 FGFR3 fusion genes are observed in 3% (3/114) of muscle‐invasive urothelial cancer.11 Therefore, clinical trials of FGFR inhibitors in urothelial cancer harboring FGFR3 fusion genes or point mutations are ongoing.12 The clinical relevance of FGFR3‐TACC3 has been suggested by the clinical report of JNJ‐42756493, a pan‐FGFR inhibitor, which exerts three out of four partial responses among patients with tumors harboring FGFR3‐TACC3 fusion genes.13 In a subset of urothelial cancer patients harboring FGFR3 gene alternation (FGFR3 fusion gene and point mutation) treated with BGJ398, the overall response rate in 25 evaluable patients was 36% and included one unconfirmed complete response and eight partial responses.14 In light of these reports, FGFR3 has been considered as an attractive target for novel therapy in urothelial bladder cancer.
In this report, we describe the preclinical profile of ASP5878, which is a selective FGFR inhibitor under clinical investigation (NCT 02038673), targeting FGFR3‐fusion or ‐mutation positive urothelial bladder cancer. Interestingly, ASP5878 suppressed the growth of FGFR3‐fusion or ‐mutation positive urothelial cancer cell lines even after the acquisition of chemoresistance. Our data indicate that ASP5878 is a potentially effective therapeutic agent for urothelial cancer patients whose tumors express FGFR3 mutation or ‐fusion after the acquisition of gemcitabine‐ or adriamycin‐ resistance.
Materials and Methods
Reagents
2‐[4‐({5‐[(2,6‐difluoro‐3,5‐dimethoxyphenyl)methoxy]pyrimidin‐2‐yl}amino)‐1H‐pyrazol‐1‐yl]ethan‐1‐ol [ASP5878, Fig. 1, 15] was synthesized at Astellas Pharma Inc. (Tokyo, Japan). ASP5878 was dissolved in DMSO or suspended in 0.5% methyl cellulose for in vitro and in vivo experiments, respectively. Gemcitabine was purchased from Eli Lilly Inc. (Indianapolis, IN, USA), and was dissolved in water or saline for in vitro and in vivo experiments, respectively. Adriamycin was purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan), and was dissolved in water.
Figure 1.
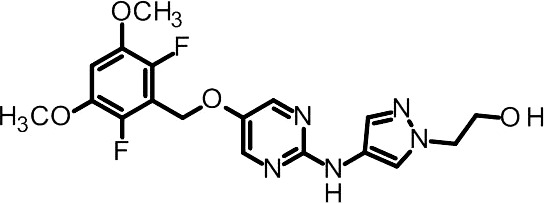
Chemical structure of ASP5878.
Cell lines
HT‐1197, HT‐1376, J82, RT4, SW 780, TCCSUP, and UM‐UC‐3 were purchased from ATCC (Manassas, VA, USA). 647‐V, BC‐3C, BFTC‐905, CAL‐29, KU‐19‐19, RT‐112, SW‐1710 and VM‐CUB1 were purchased from DSMZ (Braunschweig, Germany). EJ138, U‐BLC1, UM‐UC‐9 and UM‐UC‐14 were purchased from ECACC (Salisbury, UK). KMBC‐2 and T24 were purchased from JCRB Cell Bank (Osaka, Japan). BOY‐12E, and JMSU‐1 were provided by the RIKEN BRC (Tsukuba, Japan). These cell lines were cultured according to the guidelines from the suppliers.
To generate chemotherapy‐resistant cell lines, UM‐UC‐14 and RT‐112 cell lines were exposed to adriamycin and gemcitabine, respectively, whose concentrations were gradually increased up to 100 and 1000 ng/mL, respectively. Adriamycin‐resistant UM‐UC‐14 and gemcitabine‐resistant RT‐112 cell lines were maintained in the culture medium containing 50 ng/mL adriamycin and 1000 ng/mL gemcitabine, respectively.
In vitro cell growth assay
The cells were seeded in 96‐well plates at 2000 cells per well and incubated overnight. On the following day, the cells were exposed to ASP5878 for 4 days (JMSU‐1) or 5 days (other cell lines). The cell viability was measured with CellTiter‐Glo (Promega, Madison, WI, USA). Data are presented as means from a single experiment performed in duplicate.
MDR1 expression
Immunoblotting was performed using mouse anti‐MDR1 (D‐11) monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti‐β‐actin (13E5) monoclonal antibody (Cell Signaling Technology, Danvers, MA, USA).
Inhibition of in vitro FGFR3 phosphorylation
Cells were seeded in 100 mm dishes at 2 × 106 cells/10 mL/dish and cultured overnight. Media were replaced with ASP5878 containing media at the final concentrations of 0, 1, 10, 100 and 1000 nmol/L, respectively. The final concentration of DMSO in each dish was 0.1%. Following 2‐h incubation with ASP5878, cells were rinsed with PBS and collected. Cell pellet was obtained and lysed with cell lysis buffer containing phosphatase inhibitor (Thermo Fisher Scientific, Rockford, IL, USA) and protease inhibitor (Roche, Basel, Switzerland). Cell lysate was centrifuged and then supernatant was obtained as the sample for ELISA assay. Phosphorylated and total FGFR3 were measured by sandwich ELISA assay (DYC2719 and DYC766; R&D Systems, Minneapolis, MN, USA). The ratio of phosphorylated FGFR3 to total FGFR3 is calculated according to the formula: (phospho FGFR3 concentration [pg/mL])/(total FGFR3 concentration [pg/mL]). FGFR3 phosphorylation rate to the DMSO‐treated sample was calculated according to the formula: (phosphorylation ratio of ASP5878‐treated sample)/(phosphorylation ratio of DMSO‐treated sample) × 100 (%).
Immunoblotting for the downstream signaling of FGFR3 and c‐MYC
Cells were seeded in 100 mm dishes at 2 × 106 cells/10 mL per dish and cultured overnight. Media were replaced with ASP5878 containing media at the final concentrations of 0, 1, 10, 100 and 1000 nmol/L respectively. The final concentration of DMSO in each dish was 0.01%. Following 2‐h (for ERK and phospho‐ERK) or 48‐h (for c‐MYC) incubation with ASP5878, cells were rinsed with PBS and collected. The cells were lysed with cell lysis buffer (Cell Signaling Technology) containing phosphatase inhibitor (Thermo Scientific) and protease inhibitor (Roche), and protein levels of ERK, c‐MYC and actin, and phosphorylation levels of ERK were determined by immunoblotting. Antibodies were obtained from following sources: ERK (#9102; Cell Signaling Technology) and phospho‐ERK (Thr202/Tyr204) (#9101; Cell Signaling Technology), actin (A5441; Sigma‐Aldrich, St Louis, MO, USA), c‐MYC (#5605; Cell Signaling Technology).
In vivo tumor studies
Five‐week‐old male nude mice (BALB/c nu/nu) were purchased from Charles River Japan, Inc (Kanagawa, Japan). All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Astellas Pharma Inc. Furthermore, Astellas Pharma Inc., Tsukuba Research Center was accredited by AAALAC International. UM‐UC‐14, RT‐112 and gemcitabine‐resistant RT‐112 cell lines were subcutaneously inoculated into the flank of mice at 3 × 106, 1 × 106 and 1 × 106 cells/0.1 mL (matrigel: PBS = 1:1)/mouse, respectively and allowed to grow. The mice with tumor were divided into 4 or 5 groups (n = 5 or 10) so that the mean tumor volume of the groups were similar on Day 0. ASP5878 (0.3–10 mg/kg) was administered orally once daily to these xenografted mice. Intravenous gemcitabine (100 mg/kg) was given to them twice a week. Tumor volume was determined by length × width2 × 0.5. Matrigel were purchased from Corning Life Sciences (Tewksbury, MA, USA).
In vivo FGFR3 phosphorylation
Tumor samples were collected from UM‐UC‐14 tumor‐bearing mice at 0.5, 1, 2, 4, 6, 12, 18 and 24 h after single dose of ASP5878 and vehicle. Frozen tumor samples were lysed with cell lysis buffer containing phosphatase inhibitor (Thermo Fisher Scientific) and protease inhibitor (Roche). Phosphorylated and total FGFR3 were measured by sandwich ELISA assay.
Statistical analysis
Values are expressed as the mean ± SE. Differences between groups were analyzed using Dunnett's multiple comparison test. All data analysis was performed using the SAS statistical software (SAS Institute, Cary, NC, USA), with P‐values <0.05 considered significant.
Results
Kinase inhibition profile of ASP5878
Materials and Methods of kinase assay were described in the Supporting Information (Appendix S1). ASP5878 potently inhibited the tyrosine kinase activities of recombinant FGFR 1, 2, 3 and 4 with IC50 values of, 0.47, 0.60 0.74 and 3.5 nmol/L, respectively (Table S1). The selectivity of ASP5878 was profiled against a kinase panel of 128 human kinases. FGFRs, VEGFR2 and FMS were inhibited by more than 50% by ASP5878 (200 nmol/L) (Tables S1,S2).
Anti‐proliferative profile of ASP5878 in urothelial cancer and other FGFR‐dependent cell lines
ASP5878 inhibited cell growth of UM‐UC‐14 [FGFR3_S249C;16], RT‐112 [FGFR3‐TACC3;9], RT4 [FGFR3‐TACC3;9], SW 780 [FGFR3‐BAIAP2L1;10] and JMSU‐1 [FGFR1 overexpression;17] with IC50 values of <100 nmol/L (Fig. 2). ASP5878, however, was inactive (IC50 values ≥ 300 nmol/L) against other urotherial cancer cell lines without FGFR genetic alterations (Fig. 2). Additionally, ASP5878 also inhibited cell proliferation of NCI‐H1581 [FGFR1 amplification, lung;18], HSC‐39 [FGFR2 amplification, stomach;19], and Hep3B2.1‐7 [FGF19 amplification, liver;20] which is known as a FGF19/FGFR4‐dependent cell line (Table S3, Appendix S1). Thus, ASP5878 has potent anti‐proliferative effects in human cancer cell lines harboring gene alterations in FGF or FGFR.
Figure 2.
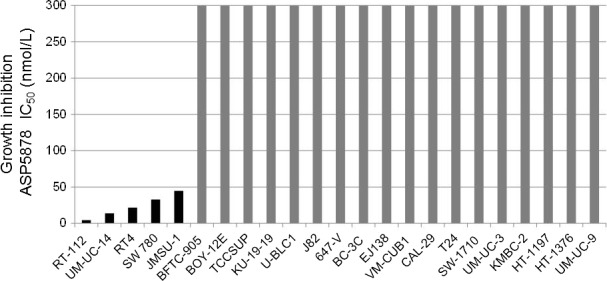
Cell panel assay for the identification of ASP5878‐sensitive bladder cancer cell lines. The 23 bladder cancer cell lines were treated with ASP5878 or 0.1% DMSO (control) for 4 (JMSU‐1) or 5 days (other cell lines). The cell viability on day 4 or day 5 was measured by quantitating the amount of ATP in cell lysate. The IC 50 value of ASP5878 on the cell proliferation of each cell line was indicated with each bar graph. Data are presented as means from a single experiment performed in duplicate.
Inhibitory effect of ASP5878 on FGFR3 and ERK phosphorylation in UM‐UC‐14 and RT‐112 cell lines
ASP5878 (1, 10, 100 and 1000 nmol/L) decreased phosphorylated FGFR3 in UM‐UC‐14 and RT‐112 cell lines after 2 h of treatment (Fig. 3a). ERK phosphorylation, a downstream signaling molecule in the cell lines, was inhibited by ASP5878 in a concentration‐dependent manner (Fig. 3b). Thus, ASP5878 inhibits FGFR3 phosphorylation and ERK phosphorylation in urothelial cancer cell lines harboring FGFR3 gene alternations.
Figure 3.
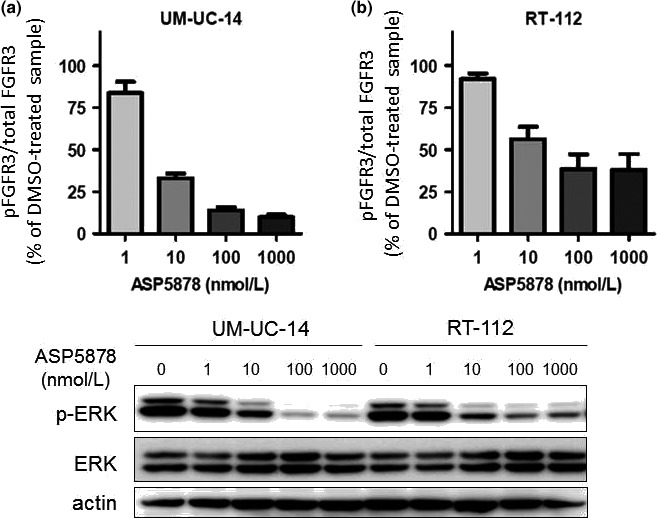
Inhibitory effects of ASP5878 on FGFR3 and ERK phosphorylation in UM‐UC‐14 and RT‐112 cell lines. (a) UM‐UC‐14 and RT‐112 cell lines are incubated for 2 h with each concentration of ASP5878 or 0.1% DMSO. Cells are then lysed and assessed FGFR3 phosphorylation rate by sandwich ELISA assay. (b) UM‐UC‐14 and RT‐112 cell lines are incubated for 2 h with each concentration of ASP5878 or 0.01% DMSO. Phosphorylated ERK (p‐ERK), ERK and actin were detected by immunoblotting.
Anti‐proliferative effects of ASP5878 in urothelial cancer cell lines with acquired resistance to adriamycin or gemcitabine
It has been reported that MDR1 mRNA levels in bladder cancer tissues are correlated with the resistance to adriamycin in bladder cancer patients.4 We therefore established adriamycin‐resistant UM‐UC‐14 cell line as described in “Materials and Methods” and compared MDR1 expression levels in the parental and the adriamycin‐resistant UM‐UC‐14 cell lines. The expression levels of MDR1 protein (Fig. 4a) and mRNA (Fig. S1, Appendix S1) in the adriamycin‐resistant UM‐UC‐14 cell line were much higher than those in the parental cell line. Adriamycin exhibited 8.7‐folds weaker anti‐proliferative effect in the adriamycin‐resistant UM‐UC‐14 cell line (IC50 = 58 ng/mL, 95% CI: 17–203, n = 3) than that in the parental cell line (IC50 = 6.7 ng/mL, 95% CI: 2.3–20, n = 3) (Fig. 4b). While ASP5878 exerted anti‐proliferative effects in both the parental and adriamycin‐resistant UM‐UC‐14 cell lines with similar IC50 values of 8.7 (95% CI: 2.3–32, n = 3) and 11 nmol/L (95% CI: 3.9–34, n = 3), respectively (Fig. 4c).
Figure 4.
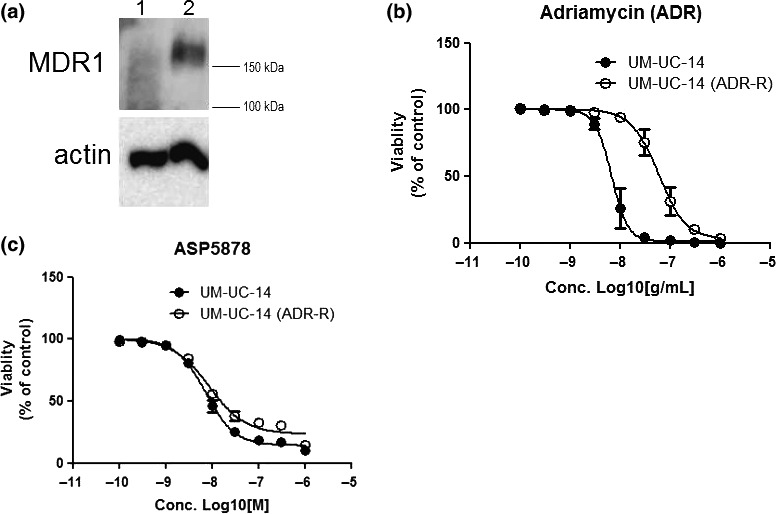
Anti‐proliferative effect of ASP5878 in adriamycin‐resistant UM‐UC‐14 cell line. (a) MDR1 protein expression in adriamycin‐resistant UM‐UC‐14 (lane 2) and parental UM‐UC‐14 (lane 1) cell lines was detected by immunoblotting. (b, c) Anti‐proliferative effect of ASP5878 (b) and adriamycin (c) in adriamycin‐resistant UM‐UC‐14 (ADR‐R) and parental UM‐UC‐14 cell lines. These cell lines were treated with ASP5878 or adriamycin for 5 days [control: 0.1% DMSO (ASP5878), water (adriamycin)]. Values are expressed as the mean ± SE from three separate experiments.
Gemcitabine is also one of the chemotherapeutic agents for invasive/metastatic bladder cancer. However, despite reasonable response rates to initial chemotherapy including gemcitabine in patients with locally advanced or metastatic bladder cancer, long‐term progression‐free survival rates remain insufficient.4 Therefore, we established gemcitabine‐resistant RT‐112 cell line as described in “Materials and Methods” and examined effects of ASP5878 on the proliferation and downstream signaling of FGFR3 in the gemcitabine‐resistant RT‐112 cell line. Gemcitabine inhibited the proliferation of the parental RT‐112 cell line with IC50 value of 0.95 ng/mL (95% CI: 0.18–5.0, n = 3) but not inhibited that of the gemcitabine‐resistant RT‐112 cell line up to 1000 ng/mL (Fig. 5a). ASP5878 inhibited the proliferation of the parental and the gemcitabine‐resistant RT‐112 cell lines with similar IC50 values of 8.7 (95% CI: 3.9–20, n = 3) and 10 nmol/L (95% CI: 3.7–27, n = 3), respectively (Fig. 5b) and decreased the level of ERK phosphorylation in the gemcitabine‐resistant RT‐112 cell line as with that in the parental cell line (Figs 3b,5c). It has been reported that gemcitabine‐resistant cell growth in urothelial cancer cells is related to upregulation of c‐MYC expression which is involved in cell proliferation.5 Upregulation of c‐MYC protein in the gemcitabine‐resistant RT‐112 cell line was also observed (Fig. 5d). Interestingly, ASP5878 decreased the expression of c‐MYC in both the gemcitabine‐resistant RT‐112 and the parental cell lines (Fig. 5d). From these findings, it is possible that ASP5878 can inhibit cell proliferation and c‐MYC expression independent on gemcitabine‐resistant status of urothelial cancer cell line harboring FGFR3 gene alternation. Thus, ASP5878 has growth inhibitory activities against urothelial cancer harboring FGFR3‐TACC3 fusion or FGFR3_S249C even after the acquisition of adriamycin or gemcitabine resistance.
Figure 5.
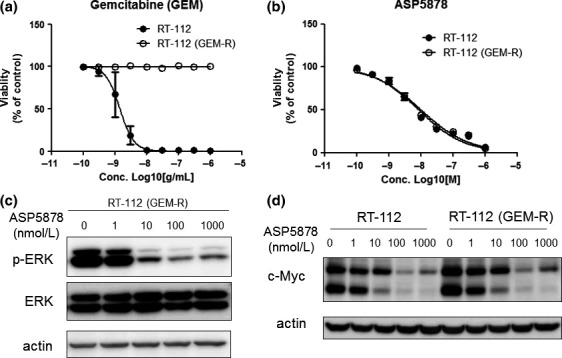
Anti‐proliferative effect of ASP5878 in gemcitabine‐resistant RT‐112 cell line. (a, b) Anti‐proliferative effect of gemcitabine (a) and ASP5878 (b) in gemcitabine‐resistant RT‐112 (GEM‐R) and parental RT‐112 cell lines. These cell lines were treated with gemcitabine and ASP5878 for 5 days [control: 0.1% DMSO (ASP5878), water (gemcitabine)]. Values are expressed as the mean ± SE from three separate experiments. (c) Gemcitabine‐resistant RT‐112 cell line was incubated for 2 h with each concentration of ASP5878 or 0.01% DMSO. Phosphorylated ERK (p‐ERK), ERK and actin were detected by immunoblotting. (d) RT‐112 and gemcitabine‐resistant RT‐112 cell lines were incubated for 48 h with each concentration of ASP5878 or 0.01% DMSO. c‐MYC and actin were detected by immunoblotting.
Antitumor activities of ASP5878 in urothelial cancer xenograft models
Once‐daily oral administration of ASP5878 dose‐dependently inhibited tumor growth and induced tumor regression at more than 1 mg/kg in UM‐UC‐14 subcutaneous xenograft mouse model (Fig. 6a). Single administration of ASP5878 (1, 3, and 10 mg/kg) inhibited FGFR3 phosphorylation in UM‐UC‐14 subcutaneous tumor and the duration of inhibition was dose‐dependent (Fig. 6b), which indicates a reasonable antitumor activity of ASP5878 in UM‐UC‐14 subcutaneous xenograft mouse model. ASP5878 also dose‐dependently inhibited tumor growth in RT‐112 (Fig. 6c) and gemcitabine‐resistant RT‐112 (Fig. 6d) subcutaneous xenograft mouse models. Body weight was not affected at any dose of ASP5878 examined in these experiments (data not shown). Thus, ASP5878 has the antitumor activities in urothelial cancer models harboring FGFR3_S249C or FGFR3‐TACC3 fusion after the acquisition of gemcitabine resistance.
Figure 6.
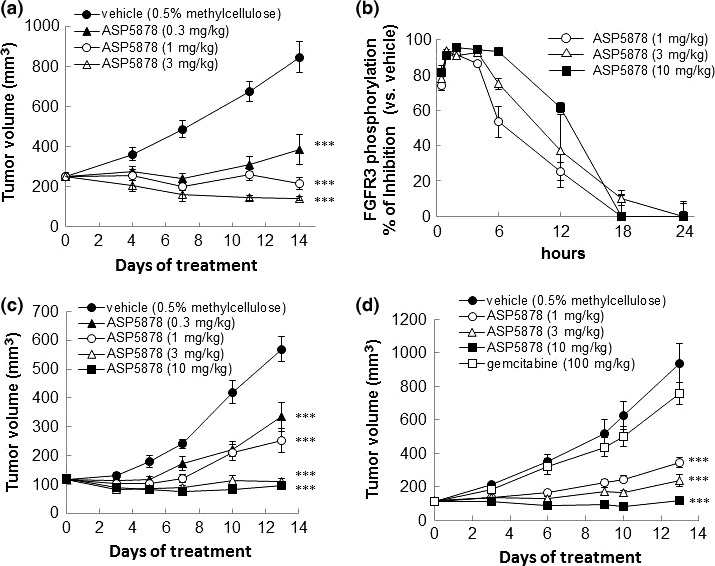
ASP5878 treatment leads to tumor regression in UM‐UC‐14, RT‐112 and gemcitabine‐resistant RT‐112 subcutaneous xenograft models. (a) ASP5878 was administered by oral gavage once daily to nude mice bearing UM‐UC‐14 tumors (n = 10). (b) Tumor samples were collected from UM‐UC‐14 tumor‐bearing mice at various time points (0.5, 1, 2, 4, 6, 12, 18 and 24 h) after single dose of ASP5878 and vehicle. Phosphorylated FGFR3 and total FGFR3 were measured by sandwich ELISA assay (n = 3). (c) ASP5878 was administered by oral gavage once daily to nude mice bearing RT‐112 tumors (n = 5). (d) ASP5878 or gemcitabine was administered by oral gavage once‐daily or intravenous injection twice‐weekly to nude mice bearing gemcitabine‐resistant RT‐112 tumors (n = 5). Each point represents the mean ± SE. Statistical analysis for antitumor tests was performed the values on the final day of each experiment (***P < 0.001, Dunnett's multiple comparison test).
Discussion
FGFR tyrosine kinases are frequently activated by diverse genetic alterations in cancer, and therefore, FGFR inhibitors may be effective in patients with FGFR genetic alterations. In this study, we examined the therapeutic potential of ASP5878 in urothelial cancer cell lines and xenografts harboring FGFR3 gene alternations. ASP5878, an FGFR tyrosine kinase inhibitor with a high selectivity against a number of other kinases (Tables S1,S2), has potent anti‐proliferative effects on FGFR1, 2, 3 and 4‐depencent cell lines (Table S3). In 23 urothelial cancer cell lines, ASP5878 inhibited the proliferation of RT‐112 and RT4 harboring FGFR3‐TACC3, SW 780 harboring FGFR3‐BAIAP2L1, UM‐UC‐14 harboring FGFR3_S249C and JMSU‐1 harboring FGFR1 overexpression (Fig. 2). FGFR3‐TACC3 displayed ligand‐independent constitutive activation of FGFR3 kinase activity and dimerization through a coiled‐coil domain in TACC3.9, 15 BAIAP2L1 has Bin‐Amphiphysin‐Rvs (BAR) domain which contributes to dimerization and constitutive activity in FGFR3‐BAIAP2L1 fusion protein.10 FGFR3_S249C mutation induces disulfide bond formation by introducing an additional cysteine in the extracellular domain of FGFR3, thereby causing constitutive dimerization and activation of the receptor.21 Aside from FGFR3 gene alternations, JMSU‐1 cell line, an urothelial cancer cell line harboring FGFR1 overexpression, has been demonstrated to have FGFR1‐dependent cell growth activity by using FGFR1 siRNA.17 These findings suggest that FGFR3‐TACC3, FGFR3‐BAIAP2L1, FGFR3_S249C mutations and FGFR1 overexpression may be predictors of the sensitivity to ASP5878 in urothelial cancer.
Currently, combination chemotherapy such as MVAC and GC are the first‐line therapy for metastatic bladder cancer patients. Unfortunately, the treatment success of bladder cancer is limited resulting in a median survival of 12–16 months.4 Treatment failure can be commonly caused by development of resistance to chemotherapy.3, 22
MDR1 is a cell membrane efflux pump involved in drug resistance. Expression of MDR1 was detected in both pre‐ and post‐chemotherapy tumor tissue samples from patients with bladder cancer and a higher expression in post‐chemotherapy patients was reported.23, 24 We also obtained adriamycin‐resistant UM‐UC‐14 cell line harboring MDR1 overexpression by stepwise increasing concentrations of adriamycin (Fig. 4). In addition, some studies have highlighted the important role of c‐MYC in the development of drug‐resistant phenotypes in cancer.25, 26 It has been reported that KU19‐19/GEM, gemcitabine‐resistant urothelial cancer cells, upregulated c‐MYC expression in the presence of gemcitabine and the growth of KU19‐19/GEM cells was suppressed by KSI‐3716, a c‐MYC inhibitor.5 As is the case of KU19‐19/GEM cells, we also successfully established gemcitabine‐resistant RT‐112 cell line harboring c‐MYC upregulation by stepwise exposure to gemcitabine (Fig. 5). Furthermore, c‐MYC overexpression has been observed in urothelial cancer tissues.27, 28, 29 Thus, c‐MYC is thought to be relevant to drug‐resistance in cancer. On the other hand, it has been reported that c‐MYC expression was decreased by PD173074, an FGFR inhibitor, in lung cancer cell lines harboring FGFR1 overexpression.30 Activated ERK, a downstream molecule of FGFR, stabilizes c‐MYC in melanoma cells.31 In the present study, ASP5878 also inhibited ERK phosphorylation and induced c‐MYC down‐regulation in urothelial cancer cell line harboring FGFR3 gene alternation independent on gemcitabine resistant status (Fig. 5d). These findings suggest that c‐MYC expression may be regulated by the FGFR/ERK signaling pathway.
Despite a lot of studies related to the mechanisms of chemoresistance, an effective therapy for chemoresistant urothelial cancer is still unestablished. Recently, FGFR inhibitors such as BGJ398 and CH5183284/Debio1347 have been reported to have an antitumor effect in FGFR3‐dependent urothelial cancer models.32, 33 And also several FGFR inhibitors including BGJ398, CH5183284/Debio1347, JNJ‐42756493 and AZD4547 are being developed for treatment of urothelial cancer. In a subset of urothelial cancer patients harboring FGFR3 gene alternation, JNJ‐42756493 and BGJ398 exerted partial responses. However, these FGFR inhibitors haven't been shown to have therapeutic potential against chemoresistant urotherial cancer in the preclinical models. Therefore, we evaluated ASP5878 for the treatment of chemoresistant urothelial cancer by using chemoresistant urothelial cancer cell lines. In adriamycin‐resistant UM‐UC‐14 cell lines, MDR1 expression was increased (Fig. 4a) and ASP5878 inhibited the proliferation in common with the parent UM‐UC‐14 cell line (Fig. 4b). Furthermore, ASP5878 inhibited the cell growth in gemcitabine‐resistant RT‐112 cells in vitro and in vivo studies (Figs 5b,6d). In addition to gemcitabine and adriamycin, cisplatin is also a key chemotherapeutic agent. However, we have not obtained the data that ASP5878 inhibits cell proliferation in cisplatin‐resistant cells, because we currently do not succeed in the establishment of cisplatin‐resistant cells. In the present study, we demonstrated antitumor activities of ASP5878 using mouse models xenografted with urothelial cancer cell lines (Fig. 6). Patient‐derived xenograft models are thought to be useful to make sure the efficacy of ASP5878 in urothelial cancer harboring FGFR3 gene alternation. These are future tasks to be confirmed. From these findings, ASP5878 may exert antitumor activity against adriamycin‐resistant and gemicitabne‐resistant urotherial cancer harboring FGFR3 gene alternations.
Hyperphosphatemia has been commonly observed with other FGFR inhibitors (e.g. JNJ‐42756493 and BGJ398).13, 14 In line with the findings, ASP5878 also induced serum phosphate increase in rodents (unpublished data). The safety, pharmacokinetics, and pharmacodynamics of ASP5878 are currently evaluated in clinical phase I study. In addition to the safety information, it has been reported that T 1/2 values of JNJ‐42756493 and BGJ398 are quite large according to clinical information of these compounds.13, 34 In the present study, we showed the duration of FGFR3 inhibitory activity after single administration of ASP5878 was relatively short in mice (Fig. 6b), which might be a benefit for the management of plasma phosphate levels.
In conclusion, ASP5878, a selective FGFR inhibitor, showed potent anti‐proliferative and antitumor activity in urothelial cancer cell line harboring FGFR3‐TACC3, FGFR3‐BAIAP2L1 or FGFR3 point mutation and their tumor xenografted models. ASP5878 also inhibited the proliferation of adriamycin‐resistant UM‐UC‐14 and gemcitabine‐resistant RT‐112 cell lines. These findings suggest that ASP5878, which is currently being evaluated in phase I clinical trials, has therapeutic potential against urothelial bladder cancers harboring FGFR3‐TACC3, FGFR3‐BAIAP2L1 or FGFR3 point mutation after the acquisition of gemcitabine‐ or adriamycin‐ resistance.
Disclosure Statement
All authors are employees of Astellas Pharma Inc.
Abbreviations
- 95% CI
95% confidential interval
- BAIAP2L1
BAI1‐associated protein 2‐like 1
- DMSO
dimethyl sulfoxide
- ERK
extracellular signal‐regulated kinase
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- GC
gemcitabine/cisplatin
- IC50
50% inhibitory concentration
- MDR
multidrug‐resistant transporter
- MVAC
methotrexate/vinblastine/adriamycin/cisplatin;
- PBS
phosphate‐buffered saline
- TACC3
transforming acid coiled coil 3
Supporting information
Appendix S1. Supporting Materials and Methods.
Figure S1. MDR1 mRNA expression in adriamycin‐resistant UM‐UC‐14 cell line.
Table S1. Kinase inhibitory profile of ASP5878 against 128 kinases.
Table S2. Inhibitory activity against 128 kinases.
Table S3. Anti‐proliferative effect of ASP5878 in FGFR‐dependent cell lines.
Cancer Sci 108 (2017) 236–242
References
- 1. Zhongfa Z, Furge KA, Yang XJ, The BT, Hansel DR. Comparative gene expression profiling analysis of urothelial carcinoma of the renal pelvis and bladder. BMC Med Genomics 2010; 3: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ploeg M, Aben KK, Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World J Urol 2009; 27: 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von der Maase H, Sengelov L, Roberts JT et al Long‐term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 2005; 23: 4602–8. [DOI] [PubMed] [Google Scholar]
- 4. Tada Y, Wada M, Migita T et al Increased expression of multidrug resistance‐associated proteins in bladder cancer during clinical course and drug resistance to doxorubicin. Int J Cancer 2002; 98: 630–5. [DOI] [PubMed] [Google Scholar]
- 5. Seo HK, Ahn KO, Jung NR et al Antitumor activity of the c‐Myc inhibitor KSI‐3716 in gemcitabine‐resistant bladder cancer. Oncotarget 2014; 5(2): 326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010; 10: 116–29. [DOI] [PubMed] [Google Scholar]
- 7. Tomlinson DC, Baldo O, Harnden P, Knowles MA. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol 2007; 213(1): 91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guancial EA, Werner L, Bellmunt J et al FGFR3 expression in primary and metastatic urothelial carcinoma of the bladder. Cancer Med 2014; 3: 835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet 2013; 22: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakanishi Y, Akiyama N, Tsukaguchi T et al Mechanism of oncogenic signal activation by the novel fusion kinase FGFR3‐BAIAP2L1. Mol Cancer Ther 2015; 14: 704–12. [DOI] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014; 507: 315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Costa R, Carneiro BA, Taxter T et al FGFR3‐TACC3 fusion in solid tumors: mini review. Oncotarget 2016; 7: 55924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tabernero J, Bahleda R, Dienstmann R et al Phase I dose‐escalation study of JNJ‐42756493, an oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015; 33: 3401–8. [DOI] [PubMed] [Google Scholar]
- 14. Pal SK, Rosenberg JE, Keam B et al Efficacy of BGJ398, a fibroblast growth factor receptor (FGFR) 1‐3 inhibitor, in patients (pts) with previously treated advanced/metastatic urothelial carcinoma (mUC) with FGFR3 alterations. ASCO Annual Meeting 2016 Abstract Number 4517.
- 15. Asaumi M, Futami T, Hisamichi H et al inventors; Astellas Pharma Inc., Kotobuki Pharm Co., Ltd., assignees. Nitrogen‐containing Aromatic Heterocyclic Compound. WO 2013/129369 A1 2013 Sep 6.
- 16. Miyake M, Ishii M, Koyama N et al 1‐tert‐butyl‐3‐[6‐(3,5‐dimethoxy‐phenyl)‐2‐(4‐diethylamino‐butylamino)‐pyrido[2,3‐d]pyrimidin‐7‐yl]‐urea (PD173074), a selective tyrosine kinase inhibitor of fibroblast growth factor receptor‐3 (FGFR3), inhibits cell proliferation of bladder cancer carrying the FGFR3 gene mutation along with up‐regulation of p27/Kip1 and G1/G0 arrest. J Pharmacol Exp Ther 2010; 332(3): 795–802. [DOI] [PubMed] [Google Scholar]
- 17. Tomlinson DC, Lamont FR, Shnyder SD, Knowles MA. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen‐activated protein kinase pathway in bladder cancer. Cancer Res 2009; 69: 4613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiss J, Sos ML, Seidel D et al Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010; 2: 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ueda T, Sasaki H, Kuwahara Y et al Deletion of the carboxyl‐terminal exons of K‐sam/FGFR2 by short homology‐mediated recombination, generating preferential expression of specific messenger RNAs. Cancer Res 1999; 59: 6080–6. [PubMed] [Google Scholar]
- 20. Sawey ET, Chanrion M, Cai C et al Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011; 19: 347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. di Martino E, L'Hôte CG, Kennedy W, Tomlinson DC, Knowles MA. Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type‐ and mutation‐specific manner. Oncogene 2009; 28: 4306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pectasides D, Pectasides M, Economopoulos T. Systemic chemotherapy in locally advanced and/or metastatic bladder cancer. Cancer Treat Rev 2006; 32: 456–70. [DOI] [PubMed] [Google Scholar]
- 23. Petrylak DP, Scher HI, Reuter V, O'Brien JP, Cordon‐Cardo C. P‐glycoprotein expression in primary and metastatic transitional cell carcinoma of the bladder. Ann Oncol 1994; 5: 835–40. [DOI] [PubMed] [Google Scholar]
- 24. Chen Z, Zhang Y, Zhang X et al Expression of multidrug‐associated protein, P‐glycoprotein, P53 and Bcl‐2 proteins in bladder cancer and clinical implication. J Tongji Med Univ 2001; 21: 56–8. [DOI] [PubMed] [Google Scholar]
- 25. Mizutani Y, Fukumoto M, Bonavida B, Yoshida O. Enhancement of sensitivity of urinary bladder tumor cells to cisplatin by c‐myc antisense oligonucleotide. Cancer 1994; 74: 2546–54. [DOI] [PubMed] [Google Scholar]
- 26. Knapp DC, Mata JE, Reddy MT, Devi GR, Iversen PL. Resistance to chemotherapeutic drugs overcome by c‐Myc inhibition in a Lewis lung carcinoma murine model. Anticancer Drugs 2003; 14(1): 39–47. [DOI] [PubMed] [Google Scholar]
- 27. Christoph F, Schmidt B, Schmitz‐Dräger BJ, Schulz WA. Over‐expression and amplification of the c‐myc gene in human urothelial carcinoma. Int J Cancer 1999; 84(2): 169–73. [DOI] [PubMed] [Google Scholar]
- 28. Schmitz‐Dräger BJ, Schulz WA, Jürgens B et al c‐myc in bladder cancer. Clinical findings and analysis of mechanism. Urol Res 1997; 25(Suppl 1): S45–9. [DOI] [PubMed] [Google Scholar]
- 29. Lipponen PK. Expression of c‐myc protein is related to cell proliferation and expression of growth factor receptors in transitional cell bladder cancer. J Pathol 1995; 175(2): 203–10. [DOI] [PubMed] [Google Scholar]
- 30. Malchers F, Dietlein F, Schöttle J et al Cell‐autonomous and non‐cell‐autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov 2014; 4(2): 246–57. [DOI] [PubMed] [Google Scholar]
- 31. Tsai WB, Aiba I, Long Y et al Activation of Ras/PI3K/ERK pathway induces c‐Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deaminase resistance in melanoma cells. Cancer Res 2012; 72: 2622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guagnano V, Furet P, Spanka C et al Discovery of 3‐(2,6‐dichloro‐3,5‐dimethoxy‐phenyl)‐1‐{6‐[4‐(4‐ethyl‐piperazin‐1‐yl)‐phenylamino]‐pyrimidin‐4‐yl}‐1‐methyl‐urea (NVP‐BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011; 54: 7066–83. [DOI] [PubMed] [Google Scholar]
- 33. Nakanishi Y, Akiyama N, Tsukaguchi T et al The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther 2014; 13: 2547–58. [DOI] [PubMed] [Google Scholar]
- 34. Sequist LV, Cassier C, Varga A et al Phase I study of BGJ398, a selective pan‐FGFR inhibitor in genetically preselected advanced solid tumors. AACR 105nd Annual Meeting 2014 Abstract CT326.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Materials and Methods.
Figure S1. MDR1 mRNA expression in adriamycin‐resistant UM‐UC‐14 cell line.
Table S1. Kinase inhibitory profile of ASP5878 against 128 kinases.
Table S2. Inhibitory activity against 128 kinases.
Table S3. Anti‐proliferative effect of ASP5878 in FGFR‐dependent cell lines.