

Abstract
Coxiella burnetii is an obligate intracellular pathogen and the causative agent of human Q fever. Replication of the bacterium within a large parasitophorous vacuole (PV) resembling a host phagolysosome is required for pathogenesis. PV biogenesis is a pathogen driven process that requires engagement of several host cell vesicular trafficking pathways to acquire vacuole components. The goal of this study was to determine if infection by C. burnetii modulates endolysosomal flux to potentially benefit PV formation. HeLa cells, infected with C. burnetii or left uninfected, were incubated with fluorescent transferrin (Tf) for 0–30 min, and the amount of Tf internalized by cells quantitated by high-content imaging. At 3 and 5 days, but not 1 day post-infection, the maximal amounts of fluorescent Tf internalized by infected cells were significantly greater than uninfected cells. The rates of Tf uptake and recycling were the same for infected and uninfected cells; however, residual Tf persisted in EEA.1 positive compartments adjacent to large PV after 30 min of recycling in the absence of labeled Tf. On average, C. burnetii-infected cells contained significantly more CD63-positive endosomes than uninfected cells. In contrast, cells containing large vacuoles generated by Chlamydia trachomatis exhibited increased rates of Tf internalization without increased CD63 expression. Our results suggest that C. burnetii infection expands the endosomal system to increase capacity for endocytic material. Furthermore, this study demonstrates the power of high-content imaging for measurement of cellular responses to infection by intracellular pathogens.
Keywords: endosome, lysosome, intracellular pathogen, endocytic trafficking, fluorescence microscopy, Chlamydia trachomatis, Coxiella burnetii
Introduction
Coxiella burnetii is an intracellular bacterial pathogen capable of infecting a broad range of host organisms. Symptomatic infection of humans, called Q fever, normally manifests as an acute flu-like disease with fever and body aches that can last several weeks. Less frequently, chronic endocarditis or hepatitis occurs (Eldin et al., 2017). Aerosolically-transmitted C. burnetii has a tropism for mononuclear phagoctyes with the pathogen initially targeting alveolar macrophages (Stein et al., 2005).
Following internalization of C. burnetii by host cells, the nascent pathogen-occupied vacuole matures through the endolysosomal pathway to acquire characteristics of a phagolysosome (Romano et al., 2007; Howe et al., 2010). The limiting membrane of the mature parasitophorous vacuole (PV) decorates with lysosome-associated membrane proteins (LAMPs), and the PV lumen acidifies and acquires active cathepsins (Howe et al., 2010). Concurrent with vacuole acidification is C. burnetii metabolic activation and translocation of effector proteins into the host cell via a Dot/Icm type 4B secretion system (T4BSS) (Newton et al., 2013). T4BSS effector proteins modify the PV into a replication-permissive niche by subverting several host cell functions including those involved in vesicular trafficking (Beare et al., 2011; Carey et al., 2011; Larson et al., 2016). The C. burnetii PV can occupy nearly the entire host cell cytoplasm. Accordingly, expansion of the nascent vacuole into a large replication-permissive niche is predicted to require substantial manipulation of the endosomal system.
Plasma membrane endocytosis is a continual process in mammalian cells (Huotari and Helenius, 2011). Degradation of internalized cargo within the endosomal pathway begins with vesicles generated by endocytosis and ends with lysosomal fusion (Saftig and Klumperman, 2009; Huotari and Helenius, 2011). Endocytic vesicles fuse with peripherally localized early endosomes (EEs) where the endocytosed material is sorted and either recycled out of the cell or transported further within the endosomal system. Fusion between EEs and new endocytic vesicles ceases after ~10 min as EEs undergo default maturation and become increasingly acidified (Maxfield and McGraw, 2004; Huotari and Helenius, 2011). Maturing endosomes also translocate toward the center of the cell, transitioning into perinuclear recycling endosomes where additional recycling away of lipids and proteins occurs prior to delivery of remaining cargo to late endosomes (LEs) or multivesicular bodies. In the final step of maturation, the lumenal contents of late endosomal vesicles are degraded by acid hydrolases delivered by lysosomal fusion (Huotari and Helenius, 2011).
Several membrane trafficking pathways participate in PV formation and pathogen growth. Defects in C. burnetii replication and PV expansion occur when endosomal trafficking is suppressed by inhibition of Rab5 or Rab7 (Romano et al., 2007; McDonough et al., 2013), or disruption of endolysosomal fusion proteins syntaxin-17 (McDonough et al., 2013) or VAMP7 (Campoy et al., 2013). Dysregulation of Rab1 (Campoy et al., 2011) and Rab24 (Gutierrez et al., 2005), key regulators of secretory and autophagic systems, respectively, also impairs C. burnetii growth in host cells. Furthermore, impaired homotypic fusion of the PV has been linked to subversion of the autophagic system by the C. burnetii effector CvpB/Cig2 via a mechanism involving phosphoinositide manipulation (Newton et al., 2014; Kohler et al., 2016; Martinez et al., 2016). Indeed, Coxiella vacuolar proteins (Cvp) are a group of Dot/Icm effectors that, when ectopically expressed in C. burnetii-infected cells, localize to the limiting membrane of the PV. C. burnetii mutants in cvpA, -B, -C, -D, and -E all exhibit defects in replication and PV formation (Larson et al., 2013, 2015; Martinez et al., 2014, 2016; Newton et al., 2014). When ectopically expressed, CvpA localizes to vesicles that label with the early/recycling endosome marker transferrin (Tf) receptor (Larson et al., 2013, 2015). Tf co-localization coincides with CvpA interactions with clathrin and the tetrameric clathrin adaptor protein complex AP2. The formation of clathrin-AP2 coats on the luminal surface of the plasma membrane triggers vesicle budding and endocytosis of many proteins implicated in PV biogenesis, including LAMP1, Tf receptor, and mannose-6-phosphate receptor (Braulke and Bonifacino, 2009; Saftig and Klumperman, 2009). The effector protein Cig57 also co-opts clathrin-mediated trafficking by interacting with the clathrin-coated pit accessory protein FCHO2 (Latomanski et al., 2016).
The C. burnetii PV phenotypically resembles a large, temporally stable phagolysosome. Endocytosis and endocytic recycling play key roles in endosomal compartment structure and function. Consequently, manipulation of endocytic trafficking is predicted to be an important strategy employed by C. burnetii to promote PV biogenesis. The objective of this study was to determine whether C. burnetii infection of host cells alters normal endocytic trafficking. Quantitative light microscopy coupled with high-content image analysis was conducted to measure endocytosis and recycling of fluorescently labeled Tf, a marker commonly used to track endocytic trafficking (Hirschmann et al., 2015). Cells infected with C. burnetii exhibited greater amounts of internalized Tf, with the largest increases occurring in cells with mature PV. This behavior was associated with a general expansion of the endocytic compartment. These data provide new details on manipulation of vesicular trafficking by C. burnetii to promote infection.
Materials and methods
Cultivation of the bacteria
C. burnetii Nine Mile phase II (clone 4, RSA439) was propagated in Vero (African green monkey kidney) cells (CCL-81; American Type Culture Collection [ATCC]) and Chlamydia trachomatis LGV-434, serotype L2, was propagated in HeLa 229 cells (CCL-2; ATCC). Bacteria were purified from host cells as previously described (Caldwell et al., 1981; Shannon and Heinzen, 2007). C. burnetii stocks were enumerated by measuring genome equivalents (GE) by quantitative PCR using primer/probe sets specific to the dotA gene (Howe et al., 2010). C. trachomatis stocks were enumerated by measuring inclusion-forming units as previously described (Moore et al., 2008).
Cell infection
Infected and uninfected HeLa cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, Rockville, MD) containing 10% fetal bovine serum (FBS) and incubated at 37°C in 5% CO2. Semi-confluent cell monolayers were trypsinized and 2.5 × 105 cells were seeded in T25 cell culture flasks (Corning, Corning, NY). Cells were infected with C. burnetii at a multiplicity of infection (MOI) of 50 based on GE, or left uninfected. The day before conducting Tf assays, matched flasks of infected or uninfected HeLa cells were trypsinized and 6 × 103 cells per well seeded in 96-well plastic bottom plates (Ibidi, Martinsried, Germany). For infections with C. trachomatis, 6 × 103 HeLa cells per well seeded in 96-well plastic bottom plates were infected at a MOI of 100 and incubated for 26 h. Infection conditions resulted in approximately 80% of cells infected with C. burnetii or C. trachomatis.
Tf internalization and recycling assays
Measurement of cellular Tf internalization and recycling was assessed using methods previously validated by Hirschmann et al. (2015) with several modifications. Cells in 96-well plates were washed once with phosphate buffered saline (PBS; 12.8 mM KH2PO4, 72.6 mM NaCl, 53.9 mM Na2HPO4, pH 7.4) and then serum starved for 30 min in 150 μl assay medium (DMEM, 1% BSA, 20 mM HEPES, pH 7.2) at 37°C and 5% CO2. Alexa Fluor 488-labeled transferrin (Tf488) (Thermo Fisher) was prepared according to the manufacturer's instructions and diluted in assay medium to 125 μg/ml. For Tf internalization, 96-well plates were placed in a 37°C water bath, and Tf internalization was initiated by aspirating wells and adding 100 μl of pre-warmed 37°C Tf488 suspension. All samples were assayed in triplicate wells and separate sample wells were left untreated (no Tf488) to control for background signal from cell autofluorescence. To halt uptake of Tf488, plates were placed on ice and immediately washed once with ice-cold PBS, once with ice-cold acid wash buffer (0.5% acetate, 0.5 M NaCl, pH 3.0) to remove remaining surface-bound probe, and then three times with PBS. Cell cultures used to measure Tf internalization were fixed with 4% paraformaldehyde in PBS for 30 min whereas cultures used to measure Tf recycling were placed back in the 37°C water bath and incubated with 100 μl of pre-warmed assay buffer containing 50 μM deferoxamine mesylate (Sigma) to prevent re-uptake of recycled Tf. Recycling was stopped with the addition of 4% paraformaldehyde. To prepare samples for imaging, wells were washed 3 times with PBS, blocked for 10 min with PBS containing 5% BSA, and permeabilized with PBS containing 0.1% Triton X-100 and 5% FBS for 5 min. The bacteria were immunostained with rabbit anti-C. burnetii or rabbit anti-C. trachomatis serum, and Alexa Fluor 594-conjugated donkey anti-rabbit secondary antibody (Thermo Fisher). Nucleic acids were stained with 2 μM Hoescht 33342 (Thermo Fisher) and cells were stained with HCS CellMask™ Deep Red (Thermo Fisher) whole cell stain (WCS) in PBS according to the manufacturer's specifications.
Image acquisition and analysis
Automated image acquisition with Zeiss Zen imaging software was used to collect 2 × 2 tiled images (2048 × 2048 pixels) of triplicate sample wells using a Carl Zeiss LSM 710 laser scanning microscope equipped with a Plan-APOCHROMAT 20 × /0.8 objective. Imaged fields were selected at random and acquired using autofocus mode with the pinhole completely open to simulate wide-field acquisition. A CellProfiler image analysis pipeline was designed for image segmentation, sorting of infected and uninfected cells, and measurement of signal intensities in each cell. The Hoescht (blue), Tf488 (green), C. burnetii (red), and WCS (far red) channels captured for each image were imported as gray-scale Tiffs into CellProfiler for analysis.
Data analysis
CellProfiler data were exported as spreadsheets for analysis. Tf488 and C. burnetii per cell measurements were extracted with Microsoft Excel 2013 and imported into GraphPad Prism 7.01 for removal of outliers (ROUT, Q = 1%) and to perform statistical calculations. The mean Tf488 integrated intensity per cell was used to compare amounts of internalized Tf488 in each sample after subtracting background signal due to autofluorescence in the green channel. Background fluorescence signal in the absence of Tf488 was calculated using matched cells and cell cultures (i.e., uninfected, infected, or cells containing large PV or inclusions) at the designated time points. Statistical significance was determined by two-way ANOVA using Tukey's test for multiple comparisons.
Immunofluorescence microscopy
For immunostaining of cellular markers, HeLa cells were fixed and permeabilized as described above, then incubated with primary antibodies against CD63 (BD Biosciences 556019), Tf receptor (ThermoFisher 136800), or EEA.1 (BD Biosciences 610456). Primary antibodies were detected with Alexa Fluor 568 conjugated donkey anti-mouse antibody (Thermo Fisher). A Carl Zeiss LSM 880 laser scanning microscope equipped with a Plan-APOCHROMAT 63 × /1.4 objective and Airyscan detector was used to capture super-resolution images of infected cells. For quantification of surface-localized Tf receptor, samples were fixed with 4% paraformaldehyde for 30 min, blocked 15 min with PBS plus 1% BSA, and incubated 45 min with 1:250 dilution of antibody against Tf receptor (ThermoFisher A11130) in PBS with 1% BSA. Image acquisition and quantitation of Tf receptor and CD63 were conducted as described for measurement of Tf488. Background signal was normalized using cells stained with anti-mouse Alexa Fluor 568 in the absence of primary antibody.
Results
Tf488 traffics to puncta adjacent to the C. burnetii PV.
Clathrin accessory proteins recognize Fe3+-loaded Tf bound to Tf receptor on the plasma membrane and sequester the complex into clathrin-coated pits that bud and form endocytic vesicles (Maxfield and McGraw, 2004; Braulke and Bonifacino, 2009). Internalized vesicles harboring the Tf complex fuse with early endosomes where Fe3+ is released and Tf and Tf receptor are recycled to the cell surface (Maxfield and McGraw, 2004; Hirschmann et al., 2015). To examine whether C. burnetii infection alters Tf subcellular localization, infected or uninfected HeLa cells loaded with Tf488 for 30 min were imaged by fluorescence microscopy. As expected, both infected and uninfected cells exhibited Tf488-positive vesicles dispersed throughout the cytoplasm (Figure 1). Additionally, in cells harboring large PV, prominent Tf488-positive puncta were juxtaposed with the vacuolar membrane, suggesting that C. burnetii may alter endocytic trafficking to recruit vesicles to the PV.
Figure 1.
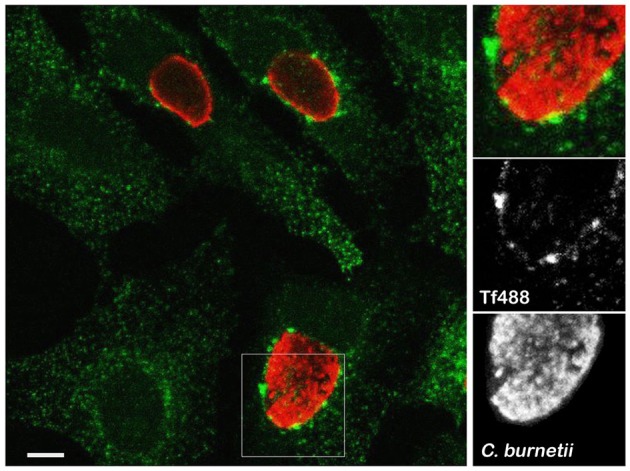
Large Tf puncta localize adjacent to the C. burnetii PV. Representative confocal fluorescence micrograph depicting Tf488 localization in HeLa cells infected for 3 days with C. burnetii. Cells incubated with Tf488 (green) for 30 min were fixed and stained with a rabbit anti-C. burnetii serum and Alexa Fluor 594 conjugated anti-rabbit secondary antibody (red). Scale bar, 10 μm.
Cells infected with C. burnetii exhibit more internalized Tf488
Measurement of internalized Tf over time is commonly used to determine the rate of cellular endocytosis (Hirschmann et al., 2015). To determine if C. burnetii infection alters endocytic trafficking, the amount of internalized Tf488 in cell cultures infected with C. burnetii for 1, 3, or 5 days was compared to internalized Tf488 in uninfected cell cultures using quantitative fluorescence microscopy. Cells were incubated with Tf488 for 0–30 min, then fixed and stained for imaging. CellProfiler was used to identify cells within each image and to quantitate multiple phenotypic parameters for each cell, including the intensity of light emitted by fluorescently labeled C. burnetii in the Alexa Fluor 594 (AF594) channel (Figure 2A). The level of C. burnetii infection varied among individual cells in infected cell cultures, ranging from cells with large PV that emitted strongly in the AF594 channel, to cells lacking C. burnetii that emitted little AF594 signal. Due to non-specific staining by the primary antibody used to label C. burnetii for imaging, even uninfected cell cultures emitted some AF594 signal. To examine the amounts of Tf488 internalized by only cells harboring large PV, CellProfiler was used to select a subset of cells from infected cell cultures that emitted strongly in the AF594 channel. A threshold for AF594 signal was set in CellProfiler above the background signal emitted by uninfected cell cultures. Cells selected based on these criteria contained obvious PV (Figure 2B) and comprised ~40% of all the cells in infected cell cultures at 3 or 5 days post-infection. Further validating these selection criteria, no cells with large PV were detected in infected cell cultures at 1 day post-infection, a time point prior to exponential growth of C. burnetii (Coleman et al., 2004).
Figure 2.

Summary of CellProfiler image segmentation and analysis pipeline. (A) Hoescht, Tf488, C. burnetii, and whole cell stain (WCS) channels were imported to CellProfiler as gray-scale Tiffs. In step 1, the IdentifyPrimaryObjects module was used to define C. burnetii. In step 2, the Hoescht channel was masked with C. burnetii objects to remove signal from bacterial nucleic acids, then the IdentifyPrimaryObjects module was applied to define host cell nuclei. In step 3, cell boundaries on the WCS channel were defined using the IdentifySecondaryObjects module to propagate from the host cell nuclei identified in step 2. In step 4, CellProfiler modules MeasureObjectIntensity and MeasureObjectShapeSize were used to quantitate multiple size, shape, and intensity parameters for each of the cell objects. (B) Representative fluorescence micrograph depicting segmentation and sorting of infected cells with large PV. Infected HeLa cells loaded with Tf488 (green) were stained with rabbit anti-C. burnetii serum and Alexa Fluor 594 conjugated anti-rabbit secondary antibody (red) and Hoescht (blue). (The green channel intensity was increased for better visibility of Tf488.) CellProfiler was used to sort infected cells (cyan outlines) by C. burnetii intensity per cell and to select those cells with large PV (yellow outlines). Scale bar, 20 μm.
The mean Tf488 intensities per cell of infected and uninfected cell cultures, as well as for the subset of cells in infected cell cultures harboring large PV, revealed a rapid increase in the amount of internalized Tf488 over the first 10 min of the endocytosis assay, with intracellular levels of Tf488 reaching steady-state at 20–30 min (Figure 3A). Compared to uninfected cell cultures, infected cell cultures at 3 or 5 days post-infection, but not 1 day post-infection, contained significantly more intracellular Tf488 at the 20 to 30 min time points. The mean Tf488 signal emitted by infected cell cultures at 3 and 5 days post-infection was 10 and 16% greater, respectively, than for uninfected cell cultures at 30 min, with the largest increases consistently occurring with cells infected 5 days. Increases in intracellular Tf488 in response to C. burnetii infection were more pronounced in cells harboring large PV. Relative to uninfected cell cultures at the 30 min time point, the mean Tf488 signal emitted by cells with large PV increased 21 and 36% at 3 and 5 days post-infection, respectively. Moreover, at the 10 to 30 min time points, the Tf488 signal emitted by the subset of cells harboring large PV was statistically greater than the total emission by infected cell cultures. To determine if C. burnetii infection alters the rate of Tf488 internalization, fluorescence signals were measured every minute for 5 min at 3 days post-infection. The amounts of intracellular Tf488 increased linearly within this time frame with the slope of the line equaling the apparent rate of internalization. Linear regression analysis of these data indicated similar apparent rates of internalization for uninfected cells, infected cells, and infected cells containing large PV (Figure 3B). The rate of internalization is dependent on the amount of cell surface receptor (Hirschmann et al., 2015). Consistent with uptake data, no increase in surface Tf receptor was detected between infected and uninfected cells at 3 days post-infection (Figure 3C).
Figure 3.

Cells infected with C. burnetii exhibit more internalized Tf. (A) HeLa cells infected with C. burnetii or left uninfected were incubated with Tf488 for the indicated times. CellProfiler was used to select only cells that emitted strong C. burnetii signal (Infected—large PV) from the total population of cells in infected cell cultures (Infected). The amount of intracellular Tf was measured by quantitative fluorescence microscopy. Plots depict percent Tf488 intensity calculated relative to uninfected cell cultures (Uninfected) at 0 min. A black asterisk indicates the mean Tf intensity of infected cell cultures is significantly greater than for uninfected cell cultures. A green asterisk indicates the mean Tf intensities of the subset of cells containing large PV is significantly greater than for infected or uninfected cell cultures. Statistical significance (P < 0.01) was determined by two-way ANOVA using Tukey's test for multiple comparisons with the mean number of cells counted per sample equal to 1038 ± 411. (B) Similar slopes were detected in the linear first 5 min of the Tf endocytosis assay, indicating increased internalized Tf by infected cells is not due to an increase in the rate of uptake (3 days post-infection). The plot depicts mean Tf488 signal intensity (integrated intensity per cell) with the mean number of cells counted at each time point equal to 1553 ± 178. (C) Increased internalized Tf by infected cells (total population) is not associated with increased surface expression of Tf receptor. Uninfected and infected cells (3 days post-infection) were fixed and stained for surface and total cellular Tf receptor. The mean number of cells counted per sample is equal to 2437 ± 582. Results are representative of 3 independent experiments and error bars indicate the standard errors from the means.
Cells harboring large C. trachomatis inclusions have more intracellular Tf and display an increased rate of Tf internalization
We measured Tf internalization by HeLa cells infected with C. trachomatis, an intracellular bacterial pathogen that also generates a large vacuole (inclusion) in which to replicate (Figure 4A). Cells infected with C. trachomatis for 26 h were loaded with Tf488 and analyzed with CellProfiler as above for C. burnetii. During the 30 min assay, there was no statistical difference between the mean Tf488 intensities of uninfected and infected cell cultures (Figure 4B). However, infected cells containing large inclusions (~45% of all cells) exhibited significantly greater internalized Tf488 that correlated with an increase in the apparent rate of internalization relative to the uninfected cells (Figure 4C). Thus, unlike C. burnetii infection, altered Tf trafficking was only associated with HeLa cells containing large chlamydial inclusions and associated with an elevated rate of Tf internalization.
Figure 4.

Cells harboring large C. trachomatis inclusions have more intracellular Tf and display an increased rate of Tf internalization. (A) Fluorescence micrograph of an infected HeLa cell showing a representative large inclusion harboring C. trachomatis (26 h post-infection). Cells incubated with Tf488 (green) for 30 min were fixed and stained with rabbit anti-C. trachomatis serum and Alexa Fluor 594 conjugated anti-rabbit secondary antibody (red). DNA (blue) was stained with Hoescht. Scale bar, 10 μm. (B,C) HeLa cells (26 h post-infection) infected with C. trachomatis or left uninfected were incubated with Tf488 for the indicated times. CellProfiler was also used to select only cells that emitted strong C. trachomatis signal (Infected—Large Inclusion) from the total population of cells in infected cell cultures (Infected). The amount of intracellular Tf was measured by quantitative fluorescence microscopy. (B) Graph depicting percent Tf488 intensity calculated relative to uninfected cell cultures (Uninfected) at 0 min. A green asterisk indicates the mean Tf intensities of the subset of cells containing large inclusions is significantly greater than for infected or uninfected cell cultures. Statistical significance (P < 0.005) was determined by two-way ANOVA using Tukey's test for multiple comparisons with the mean number of cells counted per sample is equal to 1062 ± 305. (C) Graph showing cells harboring large inclusions exhibit a greater apparent rate of Tf488 internalization as indicated by the steeper slope of the line within the linear first 6 min of the assay. The mean number of cells counted per sample is equal to 1338 ± 175. Results are representative of 3 independent experiments and error bars indicate the standard errors from the means. Statistical significance (asterisk, P < 0.0001) was determined by one-way ANOVA using Tukey's test for multiple comparisons.
Elevated intracellular Tf in C. burnetii-infected cells is associated with increased cellular CD63
To understand how C. burnetii infection could result in higher levels of intracellular Tf, we examined whether PV biogenesis expands the endosomal compartment. CD63 (LAMP3) is a membrane protein primarily located in late endosomes and lysosomes (Pols and Klumperman, 2009). As such, cellular CD63 content can be used as a general measure of the size of the endolysosomal system (Xinhan et al., 2011; Martina et al., 2014; Awad et al., 2015). Uninfected HeLa cells and cells infected for 3 days with C. burnetii, or 26 h with C. trachomatis, were stained for CD63 by immunofluorescence and CellProfiler used to measure the CD63 signal per cell. C. burnetii-infected cells contained significantly more cellular CD63 than uninfected cells (Figures 5A,B). No difference was observed between uninfected cells and cells infected with C. trachomatis. Thus, the increase in internalized Tf during C. burnetii infection correlates with increased endolysosomal content.
Figure 5.
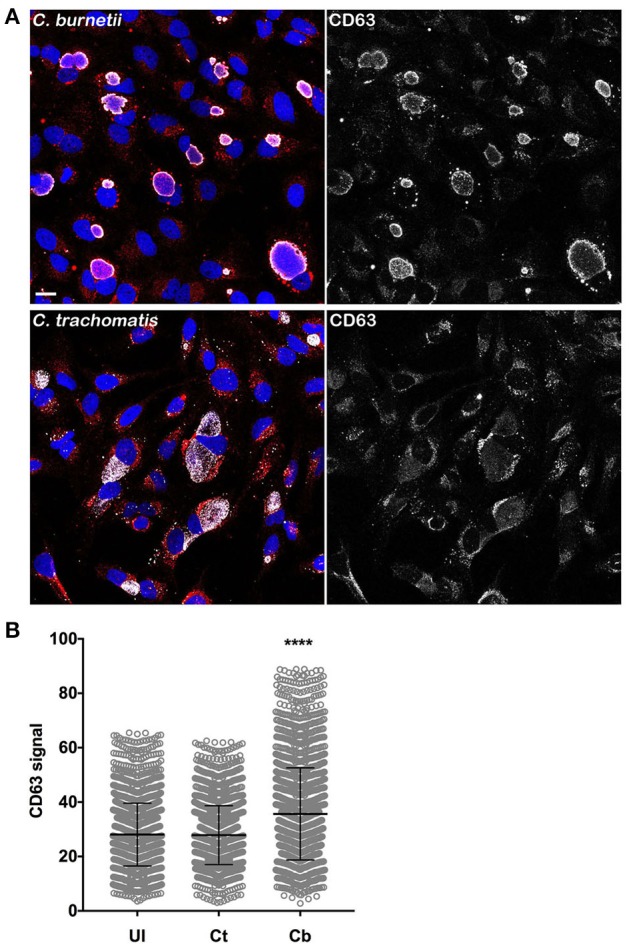
Elevated intracellular Tf in C. burnetii-infected cells is associated with increased cellular CD63. (A) HeLa cells infected for 3 days with C. burnetii, or 26 h with C. trachomatis, were stained for CD63 by immunofluorescence using anti-CD63 monoclonal antibody and Alexa Fluor 568 conjugated anti-mouse secondary antibody. Scale bar, 20 μm. (B) CellProfiler was used to measure the CD63 signal per cell. C. burnetii-infected cells contained significantly more cellular CD63 than uninfected cells. No difference was observed between uninfected and C. trachomatis-infected cells. Statistical significance (asterisk, P < 0.0001) was determined by two-way ANOVA using Tukey's test for multiple comparisons with the mean number of cells counted per sample is equal to 1894 ± 207.
C. burnetii infection does not alter the rate of Tf recycling and PV-associated Tf488 puncta persist after extensive recycling
After Fe3+ is released into early endosomes and transported to the cytosol, Tf remains bound to Tf receptor and the complex is recycled to the cell surface (Maxfield and McGraw, 2004; Hirschmann et al., 2015). To examine whether Tf recycling is altered in response to C. burnetii infection, HeLa cells infected for 3 days were loaded with Tf488 for 45 min and the decrease in internalized Tf488 was measured over time. Consistent with results shown in Figure 3, the mean amount of Tf488 internalized by infected cell cultures and infected cells harboring large PV, was significantly greater than for uninfected cell cultures at the start of the recycling assay (0 min) (Figure 6A). However, regression analysis of the linear first 5 min of the assay indicated the apparent rates of recycling for uninfected cells, infected cells, and infected cells containing large PV were similar. Thus, C. burnetii infection increases the amount of intracellular Tf without altering the rate of endocytic recycling.
Figure 6.

C. burnetii infection does not alter the rate of Tf recycling and PV-associated Tf puncta persist after extensive recycling. (A) HeLa cells infected with C. burnetii for 3 days or left uninfected were loaded with Tf488 for 45 min and the decrease in intracellular Tf488 was measured by quantitative fluorescence microscopy. Plots depict percent Tf488 intensity calculated relative to uninfected cell cultures (Uninfected) at 0 min. CellProfiler was used to select only cells that emitted high C. burnetii signal (Infected—Large PV) from infected cell cultures (Infected). Statistical significance (P < 0.01) was determined by two-way ANOVA using Tukey's test for multiple comparisons with the mean number of cells counted per sample equal to 945 ± 301. A green asterisk indicates the mean Tf intensity of the subset of cells containing large PV is significantly greater than for uninfected cells, while a black asterisk indicates the mean Tf intensities of the subset of cells containing large PV and infected cell cultures are significantly greater than for uninfected cells. However, regression analysis of the linear first 10 min of the assay indicate the apparent rates of recycling for uninfected cells, infected cells, and infected cells containing large PV are similar. Results are representative of 3 independent experiments and error bars indicate the standard errors from the means. (B) Representative confocal fluorescence micrographs depicting the intracellular localization of Tf488 (green) in HeLa cells infected with C. burnetii (red) for 3 days, and loaded with Tf488 for 45 min, then incubated in medium lacking Tf488. After a 0, 10, or 30 min incubation, samples were fixed and immunostained with rabbit anti-C. burnetii serum and Alexa Fluor 594 conjugated anti-rabbit secondary antibody. PV-associated Tf puncta persist after the 30 min chase. Scale bar, 10 μm. (C) Tf puncta remaining in HeLa cells (5 days post-infection) after Tf488 (green) labeling and a 30 min chase were immunostained for CD63 or EEA.1 using monoclonal antibodies and Alexa Fluor 568 conjugated anti-mouse secondary antibody (red). Bacterial and host DNA (blue) were stained with Hoescht. Tf puncta co-localize with EEA.1, and to a lesser extent, CD63. Scale bar, 10 μm.
Intriguingly, at the 30 min time point, when nearly all Tf488 is recycled out of uninfected cell cultures, a small amount was detected in cell cultures infected with C. burnetii. To examine this phenomenon further, high magnification confocal z-stack images of infected cells were acquired throughout the Tf488 recycling assay. A time-dependent decease in internalized Tf488 was evident when comparing images of cells at the start of the assay (0 min) to cells after 10 or 30 min of recycling (Figure 6B). However, in cells with large PV, Tf488 puncta remained visible adjacent to the PV membrane when little cytoplasmic Tf488 remained (Figure 6C). Persistent Tf puncta partially co-localized with EEA.1 in vesicles adjacent to CD63-positive endosomal compartments, suggesting Tf488 follows its canonical trafficking itinerary through the endosomal compartment during C. burnetii infection.
Discussion
Large size, pronounced fusogenecity, and temporal stability are characteristics of the C. burnetii PV that differentiate this specialized replication niche from typical phagolysosomes. Although knowledge of membrane trafficking pathways required for PV biogenesis has increased in recent years, it remains unclear how C. burnetii acquires materials for PV formation. Here, measurement of endocytic trafficking using fluorescently labeled Tf revealed that C. burnetii-infected cells have significantly more internalized Tf than uninfected cells. The greatest increases in intracellular Tf occur in cells infected with C. burnetii for longer periods of time and in cells harboring more bacteria, and consequently, larger PV. Increased intracellular Tf occurs without altering the apparent rates of Tf internalization or recycling. Furthermore, higher surface Tf receptor densities are not detected in infected cells. Perturbation of endocytic transport appears to occur throughout the course of C. burnetii PV maturation due to expansion of the endosomal compartment, as evidenced by increased cellular CD63. Further supporting this hypothesis is a recent report showing upregulation of the endocytic coat protein clathrin during the C. burnetii infectious process (Latomanski et al., 2016). Collectively, our data suggest establishment of a large, mature PV coincides with increased capacity for endocytic material within the endosomal system.
In the recycling assay, Tf-positive puncta persist next to the PV after extensive recycling in the absence of Tf488. The nature and function of these Tf puncta are unknown; however, co-localization of Tf with EEA.1, and to a lesser extent CD63, suggests the fluorescent probe is transiting normally between early and recycling endosomal compartments. Others have shown that under normal conditions the half-time (t1/2) for Tf internalization is ~2 min and the t1/2 of recycling to the cell surface ~10 min (Hopkins and Trowbridge, 1983; Mayor et al., 1993; Maxfield and McGraw, 2004). For proteins, such as Tf, that cycle between the surface and the interior of the cell, the slow step of recycling results in accumulation within the endocytic recycling center (Maxfield and McGraw, 2004). The enlarged endosomal compartment of C. burnetii-infected cells appears to accommodate greater Tf accumulation before the entry and exit pathways reach equilibrium, resulting in greater steady-state amounts of internalized Tf and longer-lived puncta adjacent to the PV. A small, undetectable change in the rate of Tf entry or exit might also contribute to greater Tf accumulation in C. burnetii-infected cells.
The different endocytic trafficking responses between cells infected with C. trachomatis or C. burnetii reported here are consistent with distinct mechanisms of pathogen vacuole biogenesis. The C. burnetii PV extensively engages the endolysosomal/autophagolysosomal pathways (Larson et al., 2016). Conversely, the chlamydial inclusion is largely disconnected from endocytic pathways, but instead fuses with exocytic vesicles derived from the Golgi apparatus (Fields and Hackstadt, 2002). The total population of cells infected with C. trachomatis does not display significantly greater intracellular Tf than uninfected cells, nor do they upregulate CD63. Only the subset of cells containing large inclusions exhibit increased internalized Tf, a process that correlates with an increase in the apparent rates of internalization. Therefore, C. trachomatis may modulate endocytic transport at later stages of inclusion maturation.
Accumulating evidence indicates C. burnetii modulates endocytic trafficking via the activity of T4BSS effector proteins (Larson et al., 2016). As mentioned above, CvpA and Cig57 target clathrin-mediated endocytosis, suggesting modulation of endocytic trafficking is crucial for generating the large vacuolar replication niche of C. burnetii that appears predominantly composed of late endosomal components (Larson et al., 2013, 2016; Latomanski et al., 2016). Expression of recombinant CvpA lacking domains that interact with clathrin and AP2 fails to rescue impaired growth of a cvpA mutant, a phenotype that mirrors defective PV expansion observed in response to inhibition of receptor-mediated endocytosis by knockdown of clathrin or AP2 (Larson et al., 2013). Intriguingly, ectopic expression of mCherry-tagged CvpA appears to inhibit Tf internalization (Larson et al., 2013), a result that conflicts with the increased intracellular Tf associated with C. burneti-infected cells reported in the current study. A possible explanation for these opposing results is that translocation of CvpA across the PV membrane by C. burnetii during native infection confers spatiotemporal regulation that is absent when CvpA is ectopically expressed in uninfected cells, a condition that may lead to non-specific effector activity within the cell. Indeed, spatiotemporal constraints are known to influence the activity of effectors secreted by other intracellular bacterial pathogens (Galán, 2009). An intriguing area of future investigation is the mechanism(s) of C. burnetii expansion of the endosomal system.
A previous report described a general increase in expression of Tf receptor by C. burnetii-infected J774.1 murine macrophages, but no distinction between surface and intracellular location was made (Howe and Mallavia, 1999). We do not detect differences in total or surface Tf receptor in response to C. burnetii infection, which is consistent with the rate data as internalization rates are dependent upon receptor availability (Hirschmann et al., 2015). Indeed, up-regulation of Tf-mediated endocytosis of Fe3+ may not benefit C. burnetii, as high concentrations of intracellular Fe3+ appear to inhibit C. burnetii growth (Briggs et al., 2008).
Tools commonly employed for examining the role of plasma membrane endocytosis, including treatment of cells with pharmacological inhibitors or ectopic expression of constitutively-active or dominant-negative proteins, can have off target effects that complicate interpretation of results (Ivanov, 2008). Additionally, many of these treatments have limited usefulness for examining cellular responses to C. burnetii infection because of cell toxicity issues that arise when treatments are maintained over time scales required to observe the lengthy process of PV biogenesis. Fluorescence microscopy coupled with computational analysis enables quantification of multiple cell features over large sample sizes, thereby revealing subtle changes in cell biology in the absence of potentially harsh treatments used to perturb cell physiology. Thus, high-content imaging represents a powerful approach to acquire new understanding of virulence mechanisms employed by intracellular pathogens.
Author contributions
Conceived, designed, and performed the experiments: CL. Analyzed the data and wrote the paper: CL and RH.
Funding
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health #ZAI AI000931 to RH.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Drs. David Logan and Anne Carpenter at the Broad Institute for help with CellProfiler analysis.
References
- Awad O., Sarkar C., Panicker L. M., Miller D., Zeng X., Sgambato J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. 10.1093/hmg/ddv297 [DOI] [PubMed] [Google Scholar]
- Beare P. A., Gilk S. D., Larson C. L., Hill J., Stead C. M., Omsland A., et al. (2011). Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2, e00175–e00111. 10.1128/mBio.00175-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braulke T., Bonifacino J. S. (2009). Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793, 605–614. 10.1016/j.bbamcr.2008.10.016 [DOI] [PubMed] [Google Scholar]
- Briggs H. L., Pul N., Seshadri R., Wilson M. J., Tersteeg C., Russell-Lodrigue K. E., et al. (2008). Limited role for iron regulation in Coxiella burnetii pathogenesis. Infect. Immun. 76, 2189–2201. 10.1128/IAI.01609-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell H. D., Kromhout J., Schachter J. (1981). Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 31, 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campoy E. M., Mansilla M. E., Colombo M. I. (2013). Endocytic SNAREs are involved in optimal Coxiella burnetii vacuole development. Cell. Microbiol. 15, 922–941. 10.1111/cmi.12087 [DOI] [PubMed] [Google Scholar]
- Campoy E. M., Zoppino F. C., Colombo M. I. (2011). The early secretory pathway contributes to the growth of the Coxiella-replicative niche. Infect. Immun. 79, 402–413. 10.1128/IAI.00688-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey K. L., Newton H. J., Lührmann A., Roy C. R. (2011). The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 7:e1002056. 10.1371/journal.ppat.1002056.t001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman S. A., Fischer E. R., Howe D., Mead D. J., Heinzen R. A. (2004). Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186, 7344–7352. 10.1128/JB.186.21.7344-7352.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldin C., Melenotte C., Mediannikov O., Ghigo E., Million M., Edouard S., et al. (2017). From Q Fever to Coxiella burnetii infection: a paradigm change. Clin. Microbiol. Rev. 30, 115–190. 10.1128/CMR.00045-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields K. A., Hackstadt T. (2002). The chlamydial inclusion: escape from the endocytic pathway. Annu. Rev. Cell Dev. Biol. 18, 221–245. 10.1146/annurev.cellbio.18.012502.105845 [DOI] [PubMed] [Google Scholar]
- Galán J. E. (2009). Common themes in the design and function of bacterial effectors. Cell Host Microbe 5, 571–579. 10.1016/j.chom.2009.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez M. G., Vázquez C. L., Munafó D. B., Zoppino F. C., Berón W., Rabinovitch M., et al. (2005). Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell. Microbiol. 7, 981–993. 10.1111/j.1462-5822.2005.00527.x [DOI] [PubMed] [Google Scholar]
- Hirschmann D. T., Kasper C. A., Spiess M. (2015). Quantitative analysis of transferrin cycling by automated fluorescence microscopy. Methods Mol. Biol. 1270, 365–378. 10.1007/978-1-4939-2309-0_25 [DOI] [PubMed] [Google Scholar]
- Hopkins C. R., Trowbridge I. S. (1983). Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell. Biol. 97, 508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe D., Mallavia L. P. (1999). Coxiella burnetii infection increases transferrin receptors on J774A. 1 cells. Infect. Immun. 67, 3236–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe D., Shannon J. G., Winfree S., Dorward D. W., Heinzen R. A. (2010). Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect. Immun. 78, 3465–3474. 10.1128/IAI.00406-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J., Helenius A. (2011). Endosome maturation. EMBO J. 30, 3481–3500. 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I. (2008). Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol. Biol. 440, 15–33. 10.1007/978-1-59745-178-92 [DOI] [PubMed] [Google Scholar]
- Kohler L. J., Reed S. R., Sarraf S. A., Arteaga D. D., Newton H. J., Roy C. R. (2016). Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. mBio 7, e01127–e01116. 10.1128/mBio.01127-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson C. L., Beare P. A., Howe D., Heinzen R. A. (2013). Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc. Natl. Acad. Sci. U.S.A. 110, E4770–E4779. 10.1073/pnas.1309195110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson C. L., Beare P. A., Voth D. E., Howe D., Cockrell D. C., Bastidas R. J., et al. (2015). Coxiella burnetii effector proteins that localize to the parasitophorous vacuole membrane promote intracellular replication. Infect. Immun. 83, 661–670. 10.1128/IAI.02763-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson C. L., Martinez E., Beare P. A., Jeffrey B., Heinzen R. A., Bonazzi M. (2016). Right on Q: genetics begin to unravel Coxiella burnetii host cell interactions. Future Microbiol. 11, 919–939. 10.2217/fmb-2016-0044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latomanski E. A., Newton P., Khoo C. A., Newton H. J. (2016). The effector Cig57 hijacks FCHO-mediated vesicular trafficking to facilitate intracellular replication of Coxiella burnetii. PLoS Pathog. 12:e1006101. 10.1371/journal.ppat.1006101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina J. A., Diab H. I., Lishu L., Jeong A. L., Patange S., Raben N., et al. (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7, ra9. 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Allombert J., Cantet F., Lakhani A., Yandrapalli N., Neyret A., et al. (2016). Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc. Natl. Acad. Sci. U.S.A. 113, E3260–E3269. 10.1073/pnas.1522811113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Cantet F., Fava L., Norville I., Bonazzi M. (2014). Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog. 10:e1004013. 10.1371/journal.ppat.1004013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield F. R., McGraw T. E. (2004). Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5, 121–132. 10.1038/nrm1315 [DOI] [PubMed] [Google Scholar]
- Mayor S., Presley J. F., Maxfield F. R. (1993). Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J. Cell. Biol. 121, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough J. A., Newton H. J., Klum S., Swiss R., Agaisse H., Roy C. R. (2013). Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. MBio 4, e00606–00612. 10.1128/mBio.00606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore E. R., Fischer E. R., Mead D. J., Hackstadt T. (2008). The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin-containing exocytic vacuoles. Traffic 9, 2130–2140. 10.1111/j.1600-0854.2008.00828.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton H. J., Kohler L. J., McDonough J. A., Temoche-Diaz M., Crabill E., Hartland E. L., et al. (2014). A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 10:e1004286. 10.1371/journal.ppat.1004286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton H. J., McDonough J. A., Roy C. R. (2013). Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS ONE 8:e54566. 10.1371/journal.pone.0054566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols M. S., Klumperman J. (2009). Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 315, 1584–1592. 10.1016/j.yexcr.2008.09.020 [DOI] [PubMed] [Google Scholar]
- Romano P. S., Gutierrez M. G., Berón W., Rabinovitch M., Colombo M. I. (2007). The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9, 891–909. 10.1111/j.1462-5822.2006.00838.x [DOI] [PubMed] [Google Scholar]
- Saftig P., Klumperman J. (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell. Biol. 10, 623–635. 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- Shannon J. G., Heinzen R. A. (2007). Infection of human monocyte-derived macrophages with Coxiella burnetii. Methods Mol. Biol. 431, 189–200. [DOI] [PubMed] [Google Scholar]
- Stein A., Louveau C., Lepidi H., Ricci F., Baylac P., Davoust B., et al. (2005). Q fever pneumonia: virulence of Coxiella burnetii pathovars in a murine model of aerosol infection. Infect. Immun. 73, 2469–2477. 10.1128/IAI.73.4.2469-2477.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xinhan L., Matsushita M., Numaza M., Taguchi A., Mitsui K., Kanazawa H. (2011). Na+/H+ exchanger isoform 6 (NHE6/SLC9A6) is involved in clathrin-dependent endocytosis of transferrin. Am. J. Physiol. Cell. Physiol. 301, C1431–C1444. 10.1152/ajpcell.00154.2011 [DOI] [PubMed] [Google Scholar]