

Abstract
Hyperlipidemia is a well-established risk factor for developing cardiovascular disease (CVD). The recent American College of Cardiology and American Heart Association guidelines on lipid management emphasize treatment of individuals at increased risk for developing CVD events with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) at doses proven to reduce CVD events. However, there are limited options for patients who are either intolerant to statin therapy, develop CVD despite being on maximally tolerated statin therapy, or have severe hypercholesterolemia. Recently the Food and Drug Administration approved two novel medications for low-density lipoprotein (LDL)-cholesterol reduction: Evolocumab and Alirocumab. These agents target and inactivate proprotein convertase subtilsin-kexin type 9 (PCSK9), a hepatic protease that attaches and internalizes LDL receptors into lysosomes hence promoting their destruction. By preventing LDL receptor destruction, LDL-C levels can be lowered 50%-60% above that achieved by statin therapy alone. This review explores PCSK-9 biology and the mechanisms available to alter it; clinical trials targeting PCSK9 activity, and the current state of clinically available inhibitors of PCSK9.
Keywords: Hyperlipidemia, Statins, Proprotein convertase subtilsin-kexin type 9
Core tip: Hyperlipidemia is a well-established risk factor for developing cardiovascular disease (CVD). However, there are limited options for patients who are either intolerant to statin therapy, develop CVD despite being on maximally tolerated statin therapy, or have severe hypercholesterolemia. The Food and Drug Administration has approved two novel medications for low-density lipoprotein (LDL)-cholesterol reduction in this patient population: Evolocumab and Alirocumab. These agents target and inactivate proprotein convertase subtilsin-kexin type 9 (PCSK9), a hepatic protease that attaches and internalizes LDL receptors into lysosomes hence promoting their destruction. By preventing LDL receptor destruction, LDL-C levels can be lowered 50%-60% above that achieved by statin therapy alone. PCSK9 inhibitors are an exciting agent for reducing LDL-C and have ushered in a new era of lipid lowering therapy.
INTRODUCTION
Hyperlipidemia is a well-established risk factor for developing cardiovascular disease (CVD)[1]. Multiple double blind placebo controlled trials have shown that treatment with HMG CoA Reductase inhibitors (statins) lowers low-density lipoprotein (LDL)-C levels and reduces CVD events in individuals with CVD or those at high risk for developing it[2,3]. However, CVD events continue to occur in some patients on statins, despite receiving maximal tolerated therapy. Other patients develop side effects from statins that limit their use. Hence, newer modalities of treatment to lower LDL-C are needed in clinical practice. Recently the Food and Drug Administration (FDA) approved two medications which target a novel pathway to reduce LDL-C. They are monoclonal antibodies that inactivate proprotein convertase subtilsin-kexin type 9 (PCSK9). This review will explore the biology of PCSK 9, clinical trials targeting PCSK9 activity, and the current state of clinically available inhibitors of PCSK9.
LDL-CHOLESTEROL METABOLISM
LDL-C has been the target of therapy for improving outcomes in patients at high risk for developing CVD and has been considered a surrogate endpoint for clinical events by the FDA[1]. Reviewing LDL cholesterol metabolism is therefore important in understanding therapeutic approaches to treat hyperlipidemia.
The lipid cycle begins with the release of immature very low-density lipoprotein (VLDL) or nascent VLDL from the liver. Nascent VLDL contains apolipoprotein-B100 (apoB-100), apolipoprotein E (apoE), apolipoprotein C1 (apoC1), cholesteryl esters, cholesterol, and triglycerides. While circulating in blood, high-density lipoprotein (HDL) donates apolipoprotein C-II (apoC-II) to nascent VLDL that leads to its maturation. Mature VLDL interacts with lipoprotein lipase (LPL) in the capillary beds of adipose tissues, cardiac muscle and skeletal muscle cells, which leads to extraction of triglycerides from VLDL for storage or energy production in these tissues. VLDL combines with HDL again and an interchange occurs where apoC-II is transferred back to HDL along with phospholipids and triglycerides in exchange for cholesteryl esters via cholesterylester-transfer protein (CETP). This exchange and removal of triglycerides leads to conversion of VLDL to intermediate-density lipoprotein (IDL)[4]. Half of IDLs are recognized and endocytosed by liver cells due to apoB-100 and apoE. The remaining IDL lose apoE, and with an increased concentration of cholesterol compared to triglyceride, transform into low-density lipoproteins (LDL). LDL particles thus formed contain apoB-100, which acts as a ligand for binding to LDL receptors (LDLR). Once LDL binds to LDLR, LDL/LDLR complex is internalized by endocytosis into clathrin coated vesicles. In the cytosol, LDL and LDLR separate with recycling of LDLR to the cell surface. LDLR recycling is a continuous process and each receptor recycles up to 150 times after which they are endocytosed and metabolized[5]. Statins act by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which is involved in intracellular production of cholesterol. This lowers the levels of intracellular cholesterol leading to increased expression of LDLR on cell surfaces causing a reduction in serum LDL-cholesterol[6].
Seidah and colleagues discovered that proprotein convertase subtilisin/kexin type 9 (PCSK9) regulates LDLR degradation and could potentially be a target for modulating LDLR expression and consequently LDL-C levels[7,8]. PCSK9 is a hepatic protease that attaches to and internalizes LDLR into lysosomes hence promoting their destruction[9]. Clinical studies have shown that PCSK9 gain of function mutation is associated with familial hypercholesterolemia and premature CVD[10,11]. Conversely, individuals with loss of function mutations in PCSK9 have been observed to have lower lifetime levels of LDL-C and lower prevalence of CVD[12,13].
Since the discovery of PCSK9, results from preclinical mice studies demonstrated that sterol regulatory element binding protein-2 (SREBP-2) plays a key role in regulating cholesterol metabolism. Low level of intracellular cholesterol activates SREBP-2 and leads to LDLR gene expression. This increases LDLR concentration thus enhancing LDL clearance from circulation[8,14]. At the same time SREBP-2 also induces the expression of PCSK9, which promotes LDLR degradation. Thus, the coordinated interplay of SREBP-2 induced transcription of both LDLR and PCSK9 regulates circulating LDL levels[15,16]. These discoveries resulted in the exploration and development of therapeutic agents to lower LDL levels by targeting PCSK9 activity.
FUNCTIONAL MECHANICS OF PCSK9
Hepatocytes are the predominant site for PCSK9 production, with other sites being intestines and kidneys[17,18]. PCSK9 reduces the number of LDLR in hepatocytes by promoting their metabolism and subsequent degradation[14]. PCSK9 has been shown to act both intracellularly (playing a role as a chaperone) as well as a secreted factor promoting LDLR internalization from the hepatocellular surface. Under normal circumstances, the LDL/LDLR complex is endocytosed by endosomes. The acidic pH of the endosome reduces the affinity of LDL for LDLR with rearrangement of the LDLR’s extracellular domain into a hairpin structure, aiding in its recycling back to plasma membrane. PCSK9 binding inhibits this change and locks the LDLR in an open conformation which prevents its recycling. The LDLR is then routed to lysosomes for degradation (Figure 1)[19,20]. The secreted form of PCSK9 circulates in the bloodstream and can be inactivated by cleavage from proprotein convertase. At a molecular level, the secretion of prodomain and catalytically inactive PCSK9 promotes regular degradation of LDLR implying that PCSK9 acts as a chaperone protein rather than an active catalytic enzyme[21,22].
Figure 1.
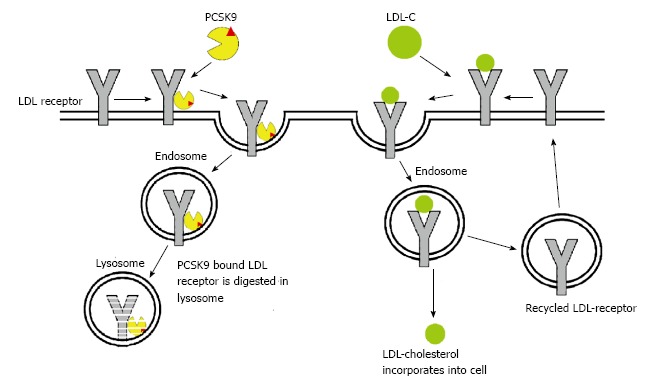
Mechanism and role of PCK9 in low-density lipoprotein-cholesterol metabolism. LDL: Low-density lipoprotein.
As described above, hepatic expression of PCSK9 and LDLR are closely regulated by SREBP-2 and intracellular levels of cholesterol[23,24]. Lipid lowering therapy with statins[25-27], ezetimibe[28] and bile acid binding resins[29] cause induction of SREBP-2 and hence co-induces both PCSK9 and LDLR. The slight increase in PCSK9 activity seen with statins does not negate their therapeutic effectiveness.
OTHER FUNCTIONS AND LOCATIONS OF PCSK9
Apart from hepatocytes, PCSK9 is also expressed in intestine, central nervous system, and mesenchymal cells of the kidney. In vitro studies on human intestinal epithelium have reported recombinant PCSK9 to enhance cholesterol uptake in the human intestinal epithelial cells (Caco-2/15 cell line) via the up regulation of the protein expression of NPC1L1 and CD36 (involved in cholesterol absorption in intestinal cells) along with an increased expression of cholesterol transporters[30,31] and reduced cholesterol synthesis (by reducing HMG-CoA reductase activity)[32]. PCSK9 has been shown to have a role in the metabolism of triglycerides and their accumulation in visceral adipose tissue[33]. It also promotes chylomicron secretion and helps regulate enterocyte cholesterol balance[32]. Studies have evaluated PCSK9 and their association with increased susceptibility to hepatitis C viral infection. Labonté et al[34] demonstrated a reduced expression of CD81 (CD81 is a co-receptor for Hepatitis C virus infection) by PCSK9 leading to protection against infection by hepatitis C. Therefore, PCSK9 inhibitors (Alirocumab) could increase CD81 expression resulting in greater infectivity. However, in vitro and in vivo studies in mice showed that PCSK9 did not reduce CD81 expression and had no effect on HCV infectivity. Hence, the literature remains inconclusive about this potential effect of PCSK9 inhibition[34]. Mbikay et al[35] demonstrated that PCSK9 inhibition in mouse pancreatic islet β cells led to hypoinsulinemia, hyperglycemia and glucose-intolerance. Additionally the islet cells exhibited signs of malformation, apoptosis and inflammation. Current clinical data has failed to show this as a complication with PCSK9 inhibition.
PCSK9 INHIBITION STRATEGIES
Preclinical studies demonstrate that statin-induced LDL reduction occurs through increased LDLR expression on hepatocytes along with increased LDL turnover, which leads to its enhanced clearance from the circulation. However, statins also induce PCSK9 expression, which dampens the effective LDL clearing by promoting LDLR degradation[23]. Since PCSK9 is expressed both intracellularly and in the circulation, multiple potential targets exist for its inhibition. Various modalities to inhibit PCSK9 have been studied including: (1) inhibition of production by gene silencing through antisense oligonucleotides[36] or small interfering RNA[37]; (2) prevention of PCSK9 binding to LDLR using monoclonal antibodies[38], epidermal growth factor-like repeat A (EGF-A), mimetic peptides[39] or adnectins; and (3) inhibition of PCSK autocatalytic sites.
Gene silencing via antisense oligonucleotides or small interfering RNA
This approach targets intracellular PCSK9 activity by utilizing antisense oligonucleotide to reduce intracellular expression of PCSK9. Preclinical trials on hyperlipidemic mice evaluating two such compounds were promising with a reduction in LDL by 38% at six weeks of therapy while doubling LDLR expression in the liver[38]. However, the phase I trials evaluating two of these agents were terminated prematurely and further development of the drug (BMS-84442) was not continued (NCT01082562). Two additional drugs: SPC5001 and SPC4061 showed successful reduction in LDL by 50% during preclinical testing in primates[40]. However, the first phase I trial in healthy human subjects and individuals with familial hypercholesterolemia were terminated early (NCT01 350960)[41,42]. SPC5001 was seen to cause mild to moderate injection site reactions and renal tubular toxicity[43]. Further development of SPC4061 was discontinued for undisclosed reasons.
Similarly, small interfering RNA (siRNA) administration has been shown to significantly reduce plasma PCSK9 and LDL levels in cynomolgus monkeys[37,38,43]. ALN-PCS is a siRNA, which was tested by delivery through second-generation lipid nanoparticles. In a study by Fitzgerald et al[44], ALN-PCS demonstrated a dose dependent reduction in PCSK9 and LDL levels with a reduction of up to 70% in PCSK9 levels and 40% in LDL levels at doses of 0.4 mg/kg. This was the first study to demonstrate intracellular PCSK9 inhibition translated into reduction of circulating LDL levels.
Monoclonal antibodies
Utilization of monoclonal antibodies has been the most effective approach thus far in inhibiting PCSK9 and reducing LDL levels. Currently, at least six monoclonal antibodies (mAb) have been or are being developed and tested: Alirocumab (formerly called SAR236553/REGN727), Evolocumab (formerly called AMG145), RG7 652[45], LGT209 (NCT01979601, NCT01859455), 1B20[46] and Bococizumab (formerly called RN316/PF-049 50615). Alirocumab and Evolocumab have recently been approved by the FDA. The major clinical studies leading up to their approval are outlined later in this paper.
Bococizumab is a unique mAb, which utilizes pH sensitive binding to PCSK9 and was developed for a longer serum half-life and duration of action on LDL reduction[47]. In phase I studies, single intravenous or subcutaneous dosages significantly reduced LDL in patients with hypercholesterolemia, both with and without concomitant atorvastatin therapy[48]. In a phase II trial by Gumbiner et al[48], patients on statin therapy not at target LDL-C were enrolled and observed a 60% reduction in LDL-C after 12 wk of therapy. Five phase III trials are ongoing for Bococizumab including SPIRE-HF (evaluating the efficacy of this agent in heterozygous familial hypercholesterolemia NCT01968980); SPIRE-HR (NCT01968954) and SPIRE-LDL (NCT01968967) trials are comparing Bococizumab to statin therapy in patients with high atherosclerotic cardiovascular risk with a follow up period of up to 12 wk. SPIRE-1 (NCT01975376) and 2 (NCT01975389) have a follow up period of up to 5 years collecting data on safety and efficacy of this drug[49]. Recently, the preliminary results of a study of Bococizumab delivery using an auto-injector device (SPIRE-AI) reported successfully meeting co-primary endpoints of percent change from baseline in fasting LDL-C at week 12 and the delivery system success rate, defined as the percent of patients whose attempts to operate the pre-filled pen. SPIRE-AI is a 12-wk, double-blind, placebo-controlled, randomized, parallel-group, multicenter, phase III clinical trial in 299 patients with hyperlipidemia or mixed dyslipidemia receiving statin therapy and whose LDL-C ≥ 70 mg/dL and assessed the efficacy, safety, tolerability and subcutaneous administration of Bococizumab 150 mg and 75 mg with a pre-filled pen[50].
Along similar lines, adnectins (also known as monobody) and small peptide inhibitors have been investigated for LDL reduction in phase I clinical trials. The results in healthy patients and those with hypercholesterolemia demonstrated a maximal dose related reduction of LDL-C by up to 48%[51]. The advantage of developing adnectins is that they are smaller than mAb, making them cheaper and easier to produce. Their pharmacokinetics have been shown to be favorable with a rapid onset of action in preclinical models, further trials are awaited to see the development of this agent[52].
Inhibition of autocatalytic site
This mechanism as a therapeutic target was first proposed after discovering a loss of function mutation in the autocatalytic cleavage site of PCSK9[53,54]. This approach is still under preclinical investigational phase.
FDA APPROVAL STATUS OF PCSK9 INHIBITORS
The FDA approved Alirocumab (Praluent) in July 2015 for adult patients with heterozygous familial hypercholesterolemia or in patients with clinically significant atherosclerotic CVD requiring additional LDL lowering after being on diet control and maximally tolerated statin therapy. Evolocumab (Repatha) was also approved in August, 2015 for use in adult patients with heterozygous familial hypercholesterolemia, homozygous familial hypercholesterolemia, or clinical atherosclerotic CVD requiring additional lowering of LDL cholesterol after being on a controlled diet and maximally-tolerated statin therapy.
Alirocumab (status: FDA approved)
Alirocumab has been studied in three phase I trials. In two of these trials, healthy volunteers were administered Alirocumab intravenously (n = 40) or subcutaneously (n = 32) which reduced LDL-C in a dose-dependent fashion with a reduction of up to 65% at maximal doses[55]. The third trial evaluated patients with non-familial hypercholesterolemia on atorvastatin and LDL > 100 mg/dL or with LDL > 130 mg/dL being managed by diet alone. Alirocumab reduced LDL-C up to 65% in patients on statins and up to 60% in patients being managed on diet alone. It was observed that Alirocumab remained effective for a longer period of time in patients not on statins[55].
Three phase II trials[56-58] evaluated the efficacy of Alirocumab in patients with familial hypercholesterolemia. Stein et al[58] evaluated 77 patients on statin therapy with LDL-C greater than 100 mg/dL and found that Alirocumab reduced LDL-C in a dose-dependent manner by up to 43% with a maximum dosage of 300 mg every 4 wk. However, the reduction was even greater (up to 70%) at a dosage regimen of 150 mg every 2 wk. Additionally, these patients had a significant reduction in apoB levels and non-high density lipoprotein cholesterol and also had increases in HDL. McKenney et al[56], in their study of 183 patients confirmed a dose dependent reduction of LDL-C with the most efficacious regimen being 150 mg every 2 wk (with LDL reduction up to 70%). Interestingly, different doses of atorvastatin did not make a significant difference in LDL-C reduction. These results were further corroborated by Roth et al[57] in their study of 92 patients with LDL-C > 100 mg/dL. They showed that there was significant and comparable LDL reduction irrespective of the dosage of atorvastatin (10 mg vs 80 mg) when added to Alirocumab 150 mg every 2 wk[57].
The phase III randomized, double-blinded ODYSSEY trials were designed to evaluate Alirocumab for long-term safety, efficacy and adverse events (Table 1) and include CHOICE I, CHOICE II, OLE, LONG TERM, COMBO I, COMBO II, FH I, FH II, HIGH FH, MONO, ALTERNATIVE, OPTIONS I and OPTIONS II. The dosage of Alirocumab administered was 150 mg every 2 wk in ODYSSEY LONG TERM and HIGH FH trials and 75 mg (titrated up to 150 mg to reach pre-specified LDL goals) in ODYSSEY ALTERNATIVE, OPTIONS I, OPTIONS II and COMBO I trials.
Table 1.
Summary of phase III ODYSSEY trials with Alirocumab
| Name of trial | Ref. Allocation and blinding | No. of patients | Inclusion criteria | Study arms (with dosing) | Primary end point | Results |
| LONG TERM (NCT01507831) | Seidah et al[7]; Randomized double blinded trial | 2341 | Either 1 or 2 below and who aren’t adequately controlled with their LLT: (1) Patients with heFH with or without CHD or CHD risk equivalents OR (2) Patients with HCL with CHD or CHD risk equivalents | Alirocumab (SC) (n = 1553) vs Placebo (SC) (n = 788) both with background LLT | Percentage change in calculated LDL cholesterol level from baseline to week 24 | -61.0% change with Alirocumab vs +0.8% change with placebo (CI: -64.3 to -59.4; P < 0.001) |
| FH I (NCT01623115) | Kastelein et al[66]; Randomized double blinded | 486 | Patients with heterozygous familial hypercholesterolemia who are not adequately controlled with their lipid-modifying therapy | Alirocumab (SC) vs Placebo (SC) both with background LLT | Percent change in calculated LDL-C at week 24 | -48.8% for Alirocumab compared with 9.1% for placebo (P < 0.0001) |
| FH II (NCT01709500) | Kastelein et al[66]; Randomized double blinded | 249 | Patients with heFH who are not adequately controlled with their LLT | Alirocumab (SC) vs Placebo (SC) both with background LLT | Percent change in LDL-C to week 24 | -48.7% for Alirocumab compared with 2.8% for placebo (P < 0.0001) |
| HIGH FH (NCT01617655) | Kastelein et al[67]; Randomized double blinded | 107 | Patients with heterozygous familial hypercholesterolemia who are not adequately controlled with their lipid-modifying therapy with LDL > 160 | Alirocumab (SC) (n = 72) vs Placebo (SC) (n = 35) both with background LLT | Percent change in calculated LDL-C at week 24 | Percent decrease from baseline was 45.7% vs 6.6%, difference 39.1, P < 0.0001 Absolute difference in values of LDL-C at 24 wk 107 mg/dL vs 182 mg/dL |
| COMBO I (NCT01644175) | Colhoun et al[60]; Randomized double blinded | 316 | Patients with hypercholesterolemia and estbl CHD or CHD risk equivalents; not controlled with a maximally tolerated LLT, both at stable dose for at least 4 to 6 wk prior to screening | Alirocumab (SC) (n = 205) vs Placebo (SC) (n = 106) | Percent change in calculated LDL-C at week 24 | -48.2% with Alirocumab (CI: -52.0% to -44.4%) and -2.3% with placebo (CI: -7.6% to 3.1%) for Alirocumab and placebo, respectively, an estimated mean difference of -45.9% (CI: -52.5% to -39.3%) (P < 0.0001) |
| COMBO II (NCT01644188) | Moriarty et al[65]; Randomized double blind | 720 | Patients with hypercholesterolemia and established CHD or CHD risk equivalents who are not adequately controlled with a maximally tolerated daily dose of statin at stable dose for at least 4 wk prior to the screening visit | Alirocumab (SC) + placebo (for ezetimibe) orally + background statin therapy (n = 467) vs Placebo (SC) + ezetimibe orally + Background statin therapy (n = 240) | Percent change in calculated LDL-C at week 24 | Reductions in LDL-C from baseline were 50.6% ± 1.4% for Alirocumab vs 20.7% ± 1.9% for ezetimibe (difference 29.8% ± 2.3%; P < 0.0001) |
| OPTIONS I (NCT01730053) | Robinson et al[63]; Randomized double-blinded | 355 | Patients with prior CV disease + LDL-C ≥ 70 mg/dL, or CV risk factors + LDL-C ≥ 100 mg/dL | Alirocumab with atorvastatin 20 mg vs Ezetimibe with atorvastatin 20 mg vs Atorvastatin 40 mg | Percent change in calculated LDL-C to week 24 | Percent reduction from baseline 44.1% (Alirocumab) vs 20.5% (ezetimibe) vs 5.0% (atorvastatin 40); P < 0.0001 |
| Alirocumab with atorvastatin 40 mg vs ezetimibe with atorvastatin 40 mg vs atorvastatin 80 mg vs rosuvastatin 20 mg | Percent reduction from baseline 54% (Alirocumab) vs 22.6% (Ezetimibe) vs 4.8% (Atorvastatin 80) vs 21.4% (rosuvastatin 40); P < 0.0001 | |||||
| OPTIONS II | Robinson et al[63]; Randomized double-blinded | 305 | Patients with prior CV disease + LDL-C ≥ 70 mg/dL, or CV risk factors + LDL-C ≥ 100 mg/dL | Alirocumab with rosuvastatin 10 mg vs ezetimibe with rosuvastatin 10 vs rosuvastatin 20 | Percent change in calculated LDL-C to wk 24 | Percent reduction from baseline 50.6% (Alirocumab) vs 14.4% (ezetimibe) vs 16.3% (rosuvastatin 20); P < 0.0001 |
| Alirocumab with rosuvastatin 20 mg vs ezetimibe with rosuvastatin 20 vs Rosuvastatin 40 | Percent reduction from baseline 36.3% (Alirocumab) vs 11.0% (Ezetimibe) vs 20.3% (rosuvastatin 40); P < 0.0001 | |||||
| ALTERNATIVE (NCT01709513) | Moriarty et al[65]; Randomized double-blinded | 314 | Primary heFH with moderate, high or very high CV risk and history of statin intolerance | Alirocumab + oral placebo vs ezetimibe (10 mg/d) + sc placebo vs atorvastatin (20 mg/d) + sc placebo | Percent change in calculated LDL-C to week 24 in intention to treat group | Percent reduction from baseline 45% (Alirocumab) vs 14.6% (Ezetimibe) with a mean difference of -30.4%; P < 0.0001 |
| CHOICE I (NCT01926782) | Stroes et al[78]; Randomized, double-blinded | 803 | Patients not having adequate control of their hypercholesterolemia based on their individual level of CVD risk | Alirocumab at q4 week regimen vs Placebo | Percent change in LDL from baseline to week 24 for Alirocumab q4w vs placebo in patients with hypercholesterolemia at moderate, high, or very high CVD risk with concomitant statin therapy (n = 547) Percent change in LDL from baseline to week 24 for Alirocumab q4w vs placebo in patients with hypercholesterolemia not on concomitant statin therapy (n = 256) | LDL was reduced by 58.7% with Alirocumab in patients on maximally tolerated statins (P < 0.001) |
| CHOICE II (NCT02023879) | Stroes et al[78]; Randomized, double-blinded | 231 | Patients with primary hypercholesterolemia (heFH or non-FH) not adequately controlled with their non statin lipid modifying therapy or diet and statin intolerance | Alirocumab (SC) vs placebo (SC) | The percent change in LDL-C from baseline to week 24 | Alirocumab reduced LDL-C by 56.4% (P < 0.0001) vs placebo |
| LONG TERM (NCT01507831) | Robinson et al[62]; Randomized, double-blinded | 2341 | Either A or B below and who are not adequately controlled with their LLT: (1) Patients with heFH with or without established CHD or CHD risk equivalents OR (2) Patients with hypercholesterolemia together with established CHD or CHD risk equivalents | Alirocumab (SC) 150 mg every 2 wk vs placebo (SC) every 2 wk | Percentage change in calculated LDL cholesterol level from baseline to week 24, analyzed with the use of an intention-to-treat approach | 150 mg Alirocumab every 2 wk had a 62% reduction in LDL as opposed to a 1% increase in LDL with placebo at 24 wk |
SC: Subcutaneous; LLT: Lipid lowering therapy; heFH: Heterozygous familial hypercholesterolemia; CHD: Coronary heart disease; HCL: Hypercholesterolemia; MACE: Major adverse cardiovascular events; MI: Myocardial infarction; UA: Unstable angina; HR: Hazards ratio; CI: Confidence interval.
CHOICE I study evaluated Alirocumab vs placebo in 803 patients with poorly controlled hypercholesterolemia. Alirocumab reduced LDL by 52% in statin-naïve patients and by 59% in patients on maximally tolerated statins as compared to placebo (P < 0.001)[59]. Similarly, COMBO I and II trials evaluated 316 patients and 720 patients respectively with LDL > 70 mg/dL and high cardiovascular risk on maximally tolerated statin therapy. COMBO I showed Alirocumab reduced LDL-C up to 50% (vs 2% with placebo) after 24 wk of treatment. COMBO II compared ezetimibe to Alirocumab in patients on background statin therapy and found a 50% LDL reduction with Alirocumab vs 20% reduction with ezetimibe at 24 wk[60]. OPTIONS I trial[61] published in August 2015, randomized 355 patients with hypercholesterolemia and LDL > 70 and found the addition of Alirocumab to atorvastatin had the greatest reduction in LDL as compared to addition of ezetimibe, doubling atorvastatin dose or switching to rosuvastatin. Atorvastatin at 20 mg and 40 mg/d regimens reduced LDL by 44% and 54% with addition of Alirocumab respectively vs 21% and 23% with addition of ezetimibe respectively vs 5% and 5% with doubling atorvastatin dose respectively compared to 21% with switching to rosuvastatin 40 mg/d. In the OPTIONS II trial, rosuvastatin was studied using a similar protocol and showed that the addition of Alirocumab had the most significant reduction in LDL-C after 24 wk of therapy as opposed to addition of ezetimibe or doubling the dose of rosuvastatin[62,63]. Another study evaluating the efficacy of Alirocumab as monotherapy (MONO study)[64] compared to ezetimibe in patients with hypercholesterolemia and moderate cardiovascular risk to monotherapy with Alirocumab and found that Alirocumab reduced LDL-C 47% vs 16% by ezetimibe after 24 wk of therapy (P < 0.0001).
CHOICE II study evaluated Alirocumab in patients intolerant to statin therapy. Two hundred and thirty one patients with a history of statin intolerance were shown to have a 56% reduction in LDL with Alirocumab (vs placebo; P < 0.001)[59]. Another study evaluating Alirocumab in patients intolerant to statin therapy is the ODYSSEY ALTERNATIVE trial. Three hundred and fourteen patients completed this randomized controlled trial, which compared Alirocumab 75 mg every 2 wk (n = 126) to ezetimibe 10 mg/d (n = 125) and atorvastatin 20 mg/d (n = 63) for 24 wk. At 24 wk, the data showed a 45% reduction in LDL with Alirocumab as opposed to 15% reduction in LDL with ezetimibe. This trial demonstrated fewer skeletal muscle adverse events in the Alirocumab group as compared to atorvastatin arm [32.5% vs 46% respectively, HR = 0.61 (0.38-0.99; P = 0.042)], with no significant difference when compared to the ezetimibe group (41%) [HR 0.71 (0.47 to 1.06; P = 0.09)][65].
Alirocumab has also been shown to be effective in lowering LDL-C in patients with familial hypercholesterolemia. The FH I and FH II studies evaluated a total of 735 patients (n = 486 and 249 respectively) with heterozygous familial hypercholesterolemia inadequately controlled on lipid lowering therapy and found Alirocumab to reduce LDL levels 48.8% (vs a 9.1% increase in placebo: FH I study) and 48.7% (vs 2.8% increase in LDL with placebo: FH II study) from baseline[66]. ODYSSEY HIGH FH trial reported 105 patients with familial hypercholesterolemia on maximally tolerated statin therapy and LDL > 160 demonstrating a 46% reduction of LDL (vs 7% with placebo) at 24 wk (P < 0.001)[67]. OLE trial (NCT01954394) is currently ongoing with results anticipated by June 2017. This trial is recruiting patients with heterozygous familial hypercholesterolemia who have completed one of the other studies and evaluating for safety parameters including adverse events, laboratory data and vital signs.
ODYSSEY LONG TERM trial was recently published and is a 78-wk follow-up of 2341 patients with hypercholesterolemia and LDL > 70 mg/dL on maximally tolerated statins. The patients receiving 150 mg Alirocumab every 2 wk were shown to have a 62% reduction in LDL as opposed to a 1% increase in LDL with placebo at 24 wk. These results persisted at 78 wk. In a post-hoc analysis, the reduction in LDL was also associated with reduction in the combined end-point of death from coronary artery disease, nonfatal myocardial infarction, fatal or nonfatal ischemic stroke or unstable angina requiring hospitalization (1.7% with Alirocumab vs 3.3% with placebo; HR = 0.52; 95%CI: 0.31-0.9; P = 0.02)[68]. The ODYSSEY Outcomes trial (NCT01663402) is ongoing is closed to recruitment, and will assess the effects of Alirocumab on CVD events in 18000 patients on maximally tolerated statin therapy. Results of this trial are expected in February 2018.
Evolocumab (status: FDA approved)
Evolocumab has been studied in two phase I studies. Dias et al[69] evaluated healthy volunteers in phase Ia and showed a short-term dose-dependent reduction in LDL by up to 65% and after 6 to 8 wk of therapy by up to 75% with a maximally administered dose of 420 mg subcutaneously/intravenously. Phase Ib trial similarly demonstrated up to 75% reduction in LDL as compared to placebo over 1 to 4 wk in healthy volunteers.
Four phase II trials were subsequently performed that continued to show the benefits of Evolocumab with a dose-dependent reduction of LDL (maximal dosing up to 420 mg) when added to maximally tolerated statin therapy in patients with hypercholesterolemia (including familial hypercholesterolemia)[70-72]. In LAPLACE-TIMI57 trial[70], Evolocumab was tested at varying doses ranging from 70 to 140 mg every 2 wk or 280 to 420 mg every 4 wk in 631 patients on stable statin therapy and LDL more than 85 mg/dL. LDL reduction up to 65% was observed with the every two-week regimen as compared to approximately 50% LDL reduction with the every 4-wk regimen. Evolocumab has also been studied as monotherapy in 160 patients with hypercholesterolemia and intolerance to statins in the GAUSS trial[73]. At doses of 420 mg every 4 wk, it was shown to reduce LDL by 40% to 50%. Furthermore, addition of ezetimibe reduced LDL by up to 65%. Subsequently, the MENDEL trial[71] showed a similar efficacy in LDL-C lowering when Evolocumab was used as monotherapy in 406 patients with hypercholesterolemia. Based on these trials, the optimal frequency of Evolocumab therapy was determined to be twice monthly to achieve a 50% to 60% reduction in LDL in combination with statins. However, when used as a monotherapy therapy, a frequency of once every 4 wk would be acceptable. Stein et al[74] evaluated 8 patients with homozygous familial hypercholesterolemia, and found Evolocumab (at 420 mg every 2 wk) to reduce LDL by approximately 25% vs 20% when used every 4 wk.
Evolocumab has further been evaluated in PROFICIO (Program to reduce LDL-C and cardiovascular outcomes following inhibition of PCSK9 in different populations) phase III trials (Table 2). The PROFICIO program includes 14 trials where Evolocumab is being evaluated in patients with hyperlipidemia in combination with statins (LAPLACE-2 and YUKAWA-2); hyperlipidemic patients intolerant to statins (GAUSS-2 and GAUSS-3); standalone in hyperlipidemia (MENDEL-2); heterozygous familial hypercholesterolemia (RUTHERFORD-2 and TAUSSIG); homozygous familial hypercholesterolemia (TESLA and TAUSSIG); with primary hyperlipidemia or mixed cholesterol disorder (THOMAS-1 and THOMAS-2: Device trials). Also, long-term safety and efficacy data is being evaluated by the five following studies: DESCARTES; FOURIER; OSLER-2 trial; GLAGOV trial and TAUSSIG study.
Table 2.
Summary of important phase III PROFICIO (Program to Reduce LDL-C and Cardiovascular Outcomes Following Inhibition of PCSK9 In Different Populations) trials with Evolocumab
| Name of trial | Ref. Allocation and blinding | No. of patients | Inclusion criteria | Study arms (with dosing) | Primary end point | Results |
| LAPLACE-2 (NCT01763866) | Robinson et al[75]; Randomized double blinded trial | 1896 | Individuals with LDL > 150 mg/dL (not on statin); or LDL > 100 mg/dL (on non-intensive statin); or LDL ≥ 80 mg/dL (with intensive statin therapy) | Initially randomized to daily moderate or high intensity atorvastatin for 4 wk. Patients were again randomized to Evolocumab (sc) vs ezetimibe vs placebo | Percentage change in calculated LDL cholesterol level from baseline to week 12 | Evolocumab q2w and qmonthly: 63% to 75% reduction in LDL vs placebo Ezetimibe 19% to 32% reduction in LDL vs placebo |
| YUKAWA-2 (NCT01953328) | Kiyosue et al[76]; Randomized double blinded | 404 | Japanese patients with LDL > 70 on stable dose statins for > 4 wk and high cardiovascular risk | Initially randomized to daily atorvastatin of 5 mg or 20 mg for 4 wk. They were further randomized to Evolocumab (sc) at q2 week and qmonthly vs placebo | Percent change in calculated LDL-C from baseline at week 12 | -67.0% to -76% reduction with Evolocumab compared to placebo (P < 0.0001) |
| GAUSS-2 (NCT01763905) | Stroes et al[78]; Randomized double blinded | 307 | Patients with LDL not at goal according to their cardiovascular risk and not on statin or low dose statin due to history of statin intolerance (> 2 statins) with stable LLT > 4 wk | Evolovumab (SC) at q2 week and qmonthly dosing vs Placebo (SC) + Ezetimibe (10 mg/d) daily | Percent change in LDL-C from baseline at the mean of weeks 10 and 12 and at week 12 Change from baseline LDL at week 12 | -55.3% to -56.1% for Evolocumab compared with -16.6% to -19.2% for ezetimibe (P < 0.0001) -103.6 to -105.4 (Evolocumab) vs -33 to -39 (mg/dL) |
| MENDEL-2 (NCT01763827 | Koren et al[77]; Randomized double-blinded | 614 | NCEP ATP III Framingham risk score of < 10% Fasting LDL-C ≥ 100 mg/dL and < 190 mg/dL | Oral placebo to SC placebo; ezetimibe to SC placebo and oral placebo to SC Evolocumab at dosing regimens of 140 mg biweekly and 420 mg monthly | Percent change from baseline in LDL-C level averaged at weeks 10 and 12 | Percent LDL change from baseline averaged at weeks 10 and 12 in the: Once per 2 wk arm: -56.9% (with Evolocumab) vs -17.5 (with ezetimibe) vs -0.4% (placebo) For monthly arm: -58.8% (with evolocumab) vs -19.1 (with ezetimibe) vs -1.4% (placebo) |
| Percent change from baseline in LDL-C level at week 12 | Percent LDL change from baseline averaged at weeks 12: Once per 2 wk arm: -57% (with Evolocumab) vs -17.8 (with ezetimibe) vs 0.1% (placebo) For monthly arm: -56.1% (with Evolocumab) vs -18.6 (with ezetimibe) vs -1.3% (placebo) | |||||
| RUTHERFORD-2 (NCT01763918) | Raal et al[72]; Randomized double blinded | 329 | Patients with heterozygous familial hypercholesterolemia who are on stable LLT for 4 wk and LDL > 100 mg/dL | Evolocumab (SC) at 140 mg q2 weeks vs placebo SC q2w AND Evolocumab SC qmonthly vs Placebo (SC) | Percent change from baseline in LDL-C level averaged at weeks 10 and 12 Percent change from baseline in LDL-C level at week 12 | Percent LDL change from baseline averaged at weeks 12 in the: Once per 2 wk arm: -61.2% (with Evolocumab) vs -1.1% (with placebo) For monthly arm: -63.3% (with evolocumab) vs 2.3% (with placebo) Percent LDL change from baseline averaged at weeks 10 and 12 in the: Once per 2 wk arm: -61.3% (with Evolocumab) vs -2% (with placebo) For monthly arm: -55.7% (with Evolocumab) vs 5.5% (with placebo) |
| TESLA (NCT01588496) | Raal et al[82]; Randomized double-blinded | 50 | Homozygous familial hypercholesterolemia, on stable lipid-regulating therapy for at least 4 wk, LDL cholesterol ≥ 130 mg/dL (3.4 mmol/L); Triglyceride ≤ 400 mg/dL (4.5 mmol/L); Body weight of ≥ 40 kg at screening, and not receiving lipoprotein apheresis | Evolocumab (SC) 420 mg every 4 wk vs placebo (SC) | Percentage change in ultracentrifugation LDL cholesterol from baseline at week 12 compared with placebo, analyzed by intention-to-treat Percent change from baseline in LDL-C at week 52 | Evolocumab significantly reduced ultracentrifugation LDL cholesterol at 12 wk by 30.9% (95%CI: -43.9% to -18.0%; P < 0.0001) vs placebo |
| DESCARTES (NCT01516879) | Blom et al[80]; Randomized, double-blinded | 901 | Fasting LDL ≥ 75 mg/dL and meeting the following on background LLT: (1) < 100 mg/dL for subjects with diagnosed CHD or CHD risk equivalent; (2) < 130 mg/dL for subjects without diagnosed CHD or CHD risk equivalent; (3) on maximal background LLT defined as atorvastatin 80 mg PO QD and ezetimibe 10 mg PO QD Fasting triglycerides ≤ 400 mg/dL | Evolocumab (SC) 420 mg every 4 wk with diet alone vs placebo with diet Evolocumab (SC) 420 mg every 4 wk with diet + atorvastatin 10 mg/d vs placebo with diet and atorvastatin 10 mg/d Evolocumab (SC) 420 mg every 4 wk + atorvastatin 80 mg/d vs placebo + atorvastatin 80 mg/d Evolocumab (SC) 420 mg every 4 wk + atorvastatin 80 mg/d + ezetimibe 10 mg/d vs placebo + atorvastatin 80 mg/d + ezetimibe 10 mg/d | Percent change from baseline in LDL-C at week 52 | Addition of Evolocumab resulted in LDL reduction by: (1) 51% to 60% in diet alone group; (2) 59% to 64% in patients on 10 mg atorvastatin (3) 51% to 62% in patients on 80 mg atorvastatin (4) 43% to 54% in patients with atorvastatin 80 mg/d and ezetimibe 10 mg/d (P < 0.001 for all) |
SC: Subcutaneous; LLT: Lipid lowering therapy; heFH: Heterozygous familial hypercholesterolemia; CHD: Coronary heart disease; HCL: Hypercholesterolemia; MACE: Major adverse Cardiovascular Events; MI: Myocardial infarction; UA: Unstable angina; HR: Hazards ratio; CI: Confidence interval.
In LAPLACE-2 study 1896 patients with fasting LDL ≥ 150 (when not on statins) or LDL ≥ 100 (on non-intense regimen of statins) or LDL ≥ 70 (on intensive statin therapy) were randomized to a daily moderate or high-intensity statin regimen and after 4 wk, further randomized to receive Evolocumab, ezetimibe or placebo. Evolocumab was shown to reduce LDL levels by 66% to 75% (on every 2 wk regimen) and by 63% to 75% (on once monthly regimen) when compared to placebo in moderate- and high intensity statin groups. In moderate and high intensity statin groups, Evolocumab led to significant reduction in absolute LDL values in both regimens of Evolocumab (every 2 wk and monthly). Adverse events reported were comparable in all groups[75]. YUKAWA-2 study showed a similar reduction in LDL in 404 Japanese patients when Evolocumab regimens (140 mg once every 2 wk and 420 mg once a month) were compared to placebo on 2 regimens of low-dose background statin therapy (5 mg/d and 20 mg/d atorvastatin). Interestingly, the reduction in LDL appeared to be similar irrespective of statin dosage (in combination with Evolocumab) and showed a 67% to 76% LDL reduction at 12 wk[76]. MENDEL-2 trial compared the efficacy of Evolocumab with ezetimibe and placebo in 614 patients with LDL between 100 mg/dL and 190 mg/dL and low risk on Framingham scale (≤ 10%). Evolocumab was shown to reduce LDL by up to 57% more than placebo and 40% more than ezetimibe after 12 wk of therapy[77].
GAUSS-2 trial evaluated 307 patients with statin intolerance and compared the 2 regimens of Evolocumab (140 mg once every 2 wk and 420 mg once a month) to daily oral or subcutaneous placebo (both placebo groups on ezetimibe). At 12 wk, Evolocumab group showed a reduction in LDL by 56% vs 39% in the other groups (placebo + ezetimibe arm)[78]. Along similar lines, GAUSS-3 trial evaluated the efficacy of Evolocumab in 218 statin intolerant patients compared to ezetimibe. The initial phase of the study included administration of atorvastatin (20 mg) for 10 wk and placebo randomized in a 1:1 fashion, followed by a 2-wk washout period and crossover to alternate therapy for another 10 wk. The patients who experienced muscle related adverse effects while on statin therapy and not on placebo were further enrolled in the second phase of the study, which was a 24 wk double blinded randomized controlled trial to compare Evolocumab (420 mg/mo divided in 3 doses) with ezetimibe (10 mg/d). At 24 wk, LDL-C was reduced by 53% with Evolocumab compared to 17% with ezetimibe. Muscle-related side effects were reported in 21% patients on Evolocumab compared to 29% with ezetimibe with stoppage of drug administration due to muscle symptoms in 1% of patients in Evolocumab and 7% of patients on ezetimibe[79].
DESCARTES trial[80] evaluated 901 patients with hyperlipidemia, comparing Evolocumab (420 mg once a month subcutaneous) plus background lipid lowering therapy vs placebo plus background lipid lowering therapy for a period of 52 wk. Background lipid lowering therapy included: Diet alone, low intensity atorvastatin (10 mg), high intensity atorvastatin (80 mg) or atorvastatin 80 mg/d. All patients had fasting LDL-C > 75 mg/dL on background lipid lowering therapy. Addition of Evolocumab resulted in LDL reduction by 51% to 60% in diet alone group, 59% to 64% in patients on 10 mg atorvastatin, 51% to 62% in patients on 80 mg atorvastatin and 43% to 54% in patients with atorvastatin 80 mg/d and ezetimibe 10 mg/d (P < 0.001 for all).
Evolocumab has also been shown to be efficacious in patients with heterozygous and homozygous familial hypercholesterolemia. In the RUTHERFORD-2 trial, 329 patients with heterozygous familial hypercholesterolemia were randomized to receive Evolocumab (140 and 420 mg respectively) or placebo at two weekly and monthly regimens. Evolocumab showed a significant reduction in LDL with both regimens: 140 mg every 2 wk led to 59.2% reduction (CI: 53.4% to 65.1%) and 420 mg once a month led to LDL reduction by 61.3% (CI: 53.6% to 69%) as compared to placebo after 12 wk (P < 0.001 for all)[81]. The TESLA trial examined 50 patients with homozygous familial hypercholesterolemia on stable lipid lowering therapy and evaluated monthly Evolocumab (420 mg) vs placebo therapy for 12 wk. Addition of Evolocumab led to a significant reduction in LDL-C by up to 31% (CI: -44% to -18%; P < 0.0001)[82].
In the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) 1 and 2 trials 4465 patients were enrolled who had completed 1 of the phase 2 or phase 3 studies of Evolocumab (MENDEL-1, LAPLACE TIMI 57, GAUSS-1, RUTHERFORD-1, YUKAWA-1, MENDEL-2, LAPLACE-2, GAUSS-2, RUTHERFORD-2, DESCARTES, THOMAS-1 or THOMAS-2) and randomized to receive either Evolocumab (420 mg/mo in OSLER-1 and 140 mg every 2 wk or 420 mg once a month in OSLER-2 trial) plus standard therapy (n = 2976) or standard therapy (n = 1489). The median follow-up was 11.1 mo. This study showed a 61% reduction in LDL-C with Evolocumab compared to standard therapy (95%CI: 59% to 63%; P < 0.001). Overall adverse events were in 69% of patients in Evolocumab group compared to 65% in standard therapy group. Of note, the neurocognitive adverse events were low, but were more frequent in Evolocumab group and appeared to be unrelated to LDL level at the time of treatment. Composite adverse cardiovascular events (all-cause death, coronary events including myocardial infarction, unstable angina requiring hospitalization, or coronary revascularization, cerebrovascular events including stroke or transient ischemic attack, and heart failure requiring hospitalization) were significantly lower in patients with Evolocumab compared to standard therapy (HR = 0.47; 95%CI: 0.28 to 0.78; P = 0.003)[83,84].
The TAUSSIG trial (NCT01624142) is evaluating Evolocumab therapy in 300 patients with severe familial hypercholesterolemia to determine its efficacy and side effect profile. The results of this study are anticipated by March 2020. Preliminary results reported by Stein et al[74] on 8 patients with LDLR-negative or LDLR defective homozygous familial hypercholesterolemia on stable drug therapy when treated with Evolocumab at 420 mg monthly for ≥ 12 wk, followed by 420 mg every 2 wk for another 12 wk showed LDL reduction by 14% to 16% at 12 wk with 2 wk and 4 wk dosing regimens respectively with no serious adverse events reported[75]. Finally, the preliminary results of GLAGOV study (NCT01813422) evaluating 950 patients with coronary artery disease on lipid lowering therapy undergoing cardiac catheterization for changes in percentage atheroma volume after 78 wk of Evolocumab therapy met primary and secondary endpoints and final results are to be reported in American Heart Association (AHA) conference in November, 2016.
PHARMACOKINETICS AND PHARMACODYNAMICS
The pharmacokinetic and pharmacodynamics parameters of PCSK9 inhibitors are described below[85,86].
Alirocumab
The time taken to reach maximum serum concentration is 3-7 d with similar serum concentration - time profiles between abdomen, upper arm or thigh as the sites of injection. Steady state concentrations are reached at an average of 3 to 4 doses. The volume of distribution following intravenous administration is 0.04 to 0.05 L/kg. The median half-life (t1/2) observed was between 17 to 20 d at 75 or 150 mg dosing every 2 wk. Alirocumab is eliminated in two phases depending upon its plasma concentration. The predominant mode of elimination at lower concentrations is via saturation of the targets (PCSK9) bound to the antibodies; however, at higher concentrations it is primarily through proteolytic pathways[81]. There have been no metabolism studies conducted since it has been previously demonstrated that reticuloendothelial system is responsible for metabolizing antibodies to small peptides and amino acids[87]. The maximum reduction in free plasma PCSK9 levels and LDL was observed within 3 and 15 d respectively, after drug administration with no difference noted between different sites. No dose adjustment is needed for patients with mild or moderately impaired renal or hepatic function. No data are available in patients with severe renal and hepatic impairment.
Evolocumab
The pharmacokinetic and pharmacodynamics properties of Evolocumab[83] demonstrate non-linear pharmacokinetics in absorption at doses below 140 mg; however, between doses of 140 to 420 mg linear pharmacokinetics is observed. The time to reach maximum concentration is 3 to 4 d after a single dose. After a single 420 mg dosage, its volume of distribution has been estimated to be 3.3 L ± 0.5 L. A steady state in serum is observed after about 12 wk of dosing. The t1/2 of Evolocumab is between 11 to 17 d. The maximum reduction of LDL after therapy was similar after dosing of 140 mg every 2 wk or 420 mg once a month with effect within 14 d. Clinical studies have not revealed a difference in pharmacokinetics of Evolocumab in mild or moderate renal and hepatic impairment. However, subjects with severe renal and hepatic impairment have not been studied.
ADVERSE EFFECTS AND CONTRAINDICATIONS
The following side effects have been reported by data gathered from over 7000 patients (n = 2476 for Alirocumab and n = 5416 for Evolocumab) evaluated in the clinical trials mentioned above. Major side effects observed for Alirocumab and Evolocumab are described below.
Alirocumab
Alirocumab is contraindicated in patients who develop serious hypersensitivity reactions like hypersensitivity vasculitis or allergic reactions requiring hospitalization with its usage. The most common adverse effects observed with Alirocumab include nasopharyngitis (11.3% vs 11.1% in placebo), injection site reactions (erythema, itchiness, swelling, pain or tenderness) (7.2% vs 5.1% in placebo), influenza (5.7% vs 4.6% in placebo), urinary tract infection (4.8% vs 4.6% in placebo), diarrhea (4.7% vs 4.4% in placebo), bronchitis (4.3% vs 3.8% in placebo), myalgia (4.2% vs 3.4% in placebo), muscle spasms (3.1% vs 2.4% in placebo), sinusitis (3% vs 2.7% in placebo), cough (2.5% vs 2.3% in placebo), contusion (2.1% vs 1.3% in placebo) and musculoskeletal pain (2.1% vs 1.6% in placebo). The most common adverse events that lead to drug discontinuation were allergic reactions (0.6% for Alirocumab vs 0.2% for placebo) and elevated liver enzymes (0.3% in Alirocumab vs < 0.1% in placebo).
Evolocumab
Contraindications for Evolocumab are similar to Alirocumab. The overall incidence of adverse effects with Evolocumab 140 mg every 2 wk as compared to placebo were 43.6% vs 41% respectively. The most common adverse effects were nasopharyngitis (5.9% vs 4.8% in placebo), upper respiratory tract infection (3.2% vs 2.7% in placebo), back pain (3% vs 2.7% in placebo) and nausea (2.1% vs 1.8% in placebo). Of note the most common adverse events leading to drug discontinuation include myalgia, nausea and dizziness. Other serious adverse events noted were cardiac disorders in 2.4% individuals including palpitations (0.6% vs 0.3% in placebo), angina pectoris (0.3% vs 0.2% in placebo), and ventricular extra systoles (0.3% vs 0.1% in placebo).
In addition, data from trials evaluating Evolocumab and Alirocumab have shown a higher incidence of cognitive adverse events in patients treated with PCSK9 inhibitors (0.9% vs 0.3% for Evolocumab compared to standard care[83] and 1.2% vs 0.5% for Alirocumab compared to placebo[69]). It has been suggested that responder and ascertainment bias might have played a role in reporting of adverse cognitive events in the OSLER trial since the adverse events were not related to the degree of LDL-C lowering with no clustering in the LDL-C < 25 mg/dL group relative to the 25-50 mg/dL or > 50 mg/dL groups[88]. However, patients in ODYSSEY LONG TERM trial were blinded to treatment and followed for nearly 18 mo. Also, the neurocognitive adverse events were measured subjectively and not verified by neurocognitive testing. A dedicated study evaluating neurocognitive adverse events with PCSK9 inhibitors is underway: Evaluating PCSK9 Binding anti-Body Influence oN coGnitive HeAlth in High cardiovascUlar Risk Subjects (EBBINGHAUS) (NCT02207634). It is enrolling individuals without dementia or mild cognitive impairment at baseline randomized in a double-blind, placebo-controlled fashion to evaluate Evolocumab on background statin therapy vs statin therapy alone. The primary outcome being measured is Spatial Working Memory test; an assessment of executive function and the results are expected in September 2017.
Another potential and significant complication with drugs that are monoclonal antibodies is the development of anti-drug antibodies that may interfere with clinical efficacy and increase adverse events[89]. This complication has not been reported in the trials to date using PCSK9 inhibitors.
CLINICAL USE OF PCSK9 INHIBITORS
The 2013 American College of Cardiology/American Heart Association recommendations on cholesterol management centered on identifying patients who would have a reduction in CVD events with statin treatment. The focus shifted from treating to a specific LDL-C level, to treating at risk individuals with a treatment (statin) proven to reduce future CVD events. Subsequently, the IMPROVE-IT trial demonstrated that addition of ezetimibe to simvastatin lowers LDL-C more than that achieved by simvastatin alone and that this reduction in LDL-C was associated with a greater reduction in CVD events compared with simvastatin alone[86,90]. This study raises the issue of LDL-C treatment targets with a lower level of LDL-C corresponding to a lower risk of CVD events. The introduction of PCSK9 inhibitors will necessitate re-evaluation of existing cholesterol treatment recommendations.
PCSK9 inhibitors are especially beneficial in the treatment of familial hypercholesterolemic patients who are intolerant to statins or have an elevated LDL-C level despite being on maximally tolerated statin therapy. Intuitively, addition of a PCSK9 inhibitor to low dose statin therapy will be more effective in lowering LDL and avoiding the side effects of statins, since low dose and high dose statin regimens have yielded similar efficacy when combined with PCSK9 inhibitors.
Several potential barriers exist that may impede the widespread use of these medicines. First, statins have a proven effectiveness that has been demonstrated in multiple long-term studies. Statins have been shown to reduce cardiovascular mortality by 30% and incidence of stroke by 20% in multiple long-term studies[91,92]. PCSK9 inhibitors are effective in reducing LDL-C levels but currently lack data demonstrating their use reduces CVD events. Trials evaluating the effect of PCSK9 inhibitors on long-term CVD events, however, are currently underway: FOURIER (Further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk; n = 22500) for Evolocumab (NCT01764633) and ODYSSEY-OUTCOMES (ODYSSEY outcomes: Evaluation of cardiovascular outcomes after an acute coronary syndrome during treatment with alirocumab SAR236553) (NCT01663402) for Alirocumab. However, their data will not be available until December 2017 (for Alirocumab) and February 2018 (for Evolocumab).
Another potential barrier to widespread use of PCSK9 inhibitors is their cost. The Institute for Clinical and Economic Review (ICER) reported that the number needed to treat for 5 years to avoid one major adverse cardiovascular event (NNT5) is 28. However, a list price of $ 14350 per year generates a cost-effectiveness ratio which far exceeds the accepted threshold of $ 100000/quality-affected life-years[93]. To achieve cost-effectiveness at this threshold would require a price reduction by 60% to 65% of the current price. At the conclusion of their report, the ICER suggested a reduction by 85% to an annual cost of $2177 might be required to avoid excessive cost burdens to the health care system[94]. It should be noted that since there are limited data on clinical adverse cardiovascular events, cost effectiveness data might change once results from ongoing CVD endpoint studies are available.
PCSK9 therapy is a welcome treatment option for statin intolerant patients who require treatment of their hyperlipidemia. It will be important that busy practitioners do not under-prescribe statins nor be dissuaded from attempting to find a dose of and statin agent that is tolerated by the patient because PCSK9 inhibitors are available. Despite these obstacles, PCSK9 inhibitors are an exciting agent for reducing LDL-C hyperlipidemia and have ushered in a new era of lipid lowering therapy.
Footnotes
Conflict-of-interest statement: All authors report no conflicts of interest.
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: August 16, 2016
First decision: September 6, 2016
Article in press: December 9, 2016
P- Reviewer: Aronow WS, Hohenegger M, Kasai T, Labonte P, Sun CK S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889–2934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Brugts JJ, Yetgin T, Hoeks SE, Gotto AM, Shepherd J, Westendorp RG, de Craen AJ, Knopp RH, Nakamura H, Ridker P, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ. 2009;338:b2376. doi: 10.1136/bmj.b2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor F, Huffman MD, Macedo AF, Moore TH, Burke M, Davey Smith G, Ward K, Ebrahim S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;(1):CD004816. doi: 10.1002/14651858.CD004816.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shelness GS, Sellers JA. Very-low-density lipoprotein assembly and secretion. Curr Opin Lipidol. 2001;12:151–157. doi: 10.1097/00041433-200104000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annu Rev Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 6.Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med. 2001;5:378–387. doi: 10.1111/j.1582-4934.2001.tb00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A, Chretien M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 9.Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res. 2012;53:2515–2524. doi: 10.1194/jlr.R026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tibolla G, Norata GD, Artali R, Meneghetti F, Catapano AL. Proprotein convertase subtilisin/kexin type 9 (PCSK9): from structure-function relation to therapeutic inhibition. Nutr Metab Cardiovasc Dis. 2011;21:835–843. doi: 10.1016/j.numecd.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Davignon J, Dubuc G, Seidah NG. The influence of PCSK9 polymorphisms on serum low-density lipoprotein cholesterol and risk of atherosclerosis. Curr Atheroscler Rep. 2010;12:308–315. doi: 10.1007/s11883-010-0123-6. [DOI] [PubMed] [Google Scholar]
- 12.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 13.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 14.Denis M, Marcinkiewicz J, Zaid A, Gauthier D, Poirier S, Lazure C, Seidah NG, Prat A. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation. 2012;125:894–901. doi: 10.1161/CIRCULATIONAHA.111.057406. [DOI] [PubMed] [Google Scholar]
- 15.Maxwell KN, Soccio RE, Duncan EM, Sehayek E, Breslow JL. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109–2119. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaid A, Roubtsova A, Essalmani R, Marcinkiewicz J, Chamberland A, Hamelin J, Tremblay M, Jacques H, Jin W, Davignon J, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48:646–654. doi: 10.1002/hep.22354. [DOI] [PubMed] [Google Scholar]
- 19.Lo Surdo P, Bottomley MJ, Calzetta A, Settembre EC, Cirillo A, Pandit S, Ni YG, Hubbard B, Sitlani A, Carfí A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011;12:1300–1305. doi: 10.1038/embor.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto T, Lu C, Ryan RO. A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J Biol Chem. 2011;286:5464–5470. doi: 10.1074/jbc.M110.199042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, et al. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J. 2007;406:203–207. doi: 10.1042/BJ20070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282:20799–20803. doi: 10.1074/jbc.C700095200. [DOI] [PubMed] [Google Scholar]
- 23.Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486–1495. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Dong B, Park SW, Lee HS, Chen W, Liu J. Hepatocyte nuclear factor 1alpha plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem. 2009;284:28885–28895. doi: 10.1074/jbc.M109.052407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49:394–398. doi: 10.1194/jlr.M700437-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Awan Z, Seidah NG, MacFadyen JG, Benjannet S, Chasman DI, Ridker PM, Genest J. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. 2012;58:183–189. doi: 10.1373/clinchem.2011.172932. [DOI] [PubMed] [Google Scholar]
- 27.Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–2721. doi: 10.1194/jlr.M008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davignon J, Dubuc G. Statins and ezetimibe modulate plasma proprotein convertase subtilisin kexin-9 (PCSK9) levels. Trans Am Clin Climatol Assoc. 2009;120:163–173. [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsson LM, Abrahamsson A, Sahlin S, Gustafsson U, Angelin B, Parini P, Einarsson C. Bile acids and lipoprotein metabolism: effects of cholestyramine and chenodeoxycholic acid on human hepatic mRNA expression. Biochem Biophys Res Commun. 2007;357:707–711. doi: 10.1016/j.bbrc.2007.03.196. [DOI] [PubMed] [Google Scholar]
- 30.Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. doi: 10.1146/annurev-physiol-012110-142233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nassir F, Wilson B, Han X, Gross RW, Abumrad NA. CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J Biol Chem. 2007;282:19493–19501. doi: 10.1074/jbc.M703330200. [DOI] [PubMed] [Google Scholar]
- 32.Levy E, Ben Djoudi Ouadda A, Spahis S, Sane AT, Garofalo C, Grenier É, Emonnot L, Yara S, Couture P, Beaulieu JF, Ménard D, Seidah NG, Elchebly M. PCSK9 plays a significant role in cholesterol homeostasis and lipid transport in intestinal epithelial cells. Atherosclerosis. 2013;227:297–306. doi: 10.1016/j.atherosclerosis.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 33.Roubtsova A, Munkonda MN, Awan Z, Marcinkiewicz J, Chamberland A, Lazure C, Cianflone K, Seidah NG, Prat A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol. 2011;31:785–791. doi: 10.1161/ATVBAHA.110.220988. [DOI] [PubMed] [Google Scholar]
- 34.Labonté P, Begley S, Guévin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology. 2009;50:17–24. doi: 10.1002/hep.22911. [DOI] [PubMed] [Google Scholar]
- 35.Mbikay M, Sirois F, Mayne J, Wang GS, Chen A, Dewpura T, Prat A, Seidah NG, Chretien M, Scott FW. PCSK9-deficient mice exhibit impaired glucose tolerance and pancreatic islet abnormalities. FEBS Lett. 2010;584:701–706. doi: 10.1016/j.febslet.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 36.Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, Crooke RM. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–767. doi: 10.1194/jlr.C600025-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, Butler D, Charisse K, Dorkin R, Fan Y, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duff CJ, Scott MJ, Kirby IT, Hutchinson SE, Martin SL, Hooper NM. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J. 2009;419:577–584. doi: 10.1042/BJ20082407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73. doi: 10.1016/j.bbrc.2008.07.106. [DOI] [PubMed] [Google Scholar]
- 40.Lindholm MW, Elmén J, Fisker N, Hansen HF, Persson R, Møller MR, Rosenbohm C, Ørum H, Straarup EM, Koch T. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol Ther. 2012;20:376–381. doi: 10.1038/mt.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hooper AJ, Burnett JR. Anti-PCSK9 therapies for the treatment of hypercholesterolemia. Expert Opin Biol Ther. 2013;13:429–435. doi: 10.1517/14712598.2012.748743. [DOI] [PubMed] [Google Scholar]
- 42.Gupta N, Fisker N, Asselin MC, Lindholm M, Rosenbohm C, Ørum H, Elmén J, Seidah NG, Straarup EM. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS One. 2010;5:e10682. doi: 10.1371/journal.pone.0010682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Poelgeest EP, Hodges MR, Moerland M, Tessier Y, Levin AA, Persson R, Lindholm MW, Dumong Erichsen K, Ørum H, Cohen AF, et al. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): a first-in-human randomized, placebo-controlled trial. Br J Clin Pharmacol. 2015;80:1350–1361. doi: 10.1111/bcp.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Budha NR, Leabman M, Jin JY, Wada DR, Baruch A, Peng K, Tingley WG, Davis JD. Modeling and Simulation to Support Phase 2 Dose Selection for RG7652, a Fully Human Monoclonal Antibody Against Proprotein Convertase Subtilisin/Kexin Type 9. AAPS J. 2015;17:881–890. doi: 10.1208/s12248-015-9750-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, McCabe T, Condra JH, Ni YG, Peterson LB, Wang W, Strack AM, Wang F, Pandit S, Hammond H, et al. An anti-PCSK9 antibody reduces LDL-cholesterol on top of a statin and suppresses hepatocyte SREBP-regulated genes. Int J Biol Sci. 2012;8:310–327. doi: 10.7150/ijbs.3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaparro-Riggers J, Liang H, DeVay RM, Bai L, Sutton JE, Chen W, Geng T, Lindquist K, Casas MG, Boustany LM, et al. Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J Biol Chem. 2012;287:11090–11097. doi: 10.1074/jbc.M111.319764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gumbiner B. Effects of 12 weeks of treatment with RN316 (PF-04950615), a humanized IgG2_a monoclonal antibody binding proprotein convertase subtilisin kexin type 9, in hypercholesterolemic subjects on high and maximal dose statins. Presented at. Am Heart Assoc Sci Sess, Los Angeles. 2012 [Google Scholar]
- 49.Dadu RT, Ballantyne CM. Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol. 2014;11:563–575. doi: 10.1038/nrcardio.2014.84. [DOI] [PubMed] [Google Scholar]
- 50.Pfizer Announces Positive Topline Results from Second Phase 3 Lipid-Lowering Study Evaluating Bococizumab/Pfizer: One of the world's premier biopharmaceutical companies. 2016. [cited. 2016. p. Oct 12]. Available from: http://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_positive_topline_results_from_second_phase_3_lipid_lowering_study_evaluating_bococizumab. [Google Scholar]
- 51.Stein EA, Kasichayanula S, Turner T, Kranz T, Arumugam U, Biernat L, Lee J. LDL cholesterol reduction with bms-962476, an adnectin inhibitor of PCSK9: results of a single ascending dose study. J Am Coll Cardiol. 2014;63(12_S):A1372. [Google Scholar]
- 52.Mitchell T, Chao G, Sitkoff D, Lo F, Monshizadegan H, Meyers D, Low S, Russo K, DiBella R, Denhez F, et al. Pharmacologic profile of the Adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J Pharmacol Exp Ther. 2014;350:412–424. doi: 10.1124/jpet.114.214221. [DOI] [PubMed] [Google Scholar]
- 53.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem. 2013;288:8279–8288. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang Y, Palyha O, Castro-Perez J, Lee A, Ai X, Ha S, Pinto S. Abstract 15853: Identification of Novel Sites Required for PCSK9 Autocatalytic Cleavage. Circulation. 2015;132(Suppl 3):A15853–A15853. [Google Scholar]
- 55.Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 56.McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 57.Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–1900. doi: 10.1056/NEJMoa1201832. [DOI] [PubMed] [Google Scholar]
- 58.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 59.Regeneron and Sanofi Announce Positive Topline Results from First Phase 3 Trials Evaluating Monthly Dosing of Alirocumab in Patients with Hypercholesterolemia. (01/09/205). [accessed. 2015. p. Dec 9]. Available from: http://www.prnewswire.com/news-releases/regeneron-and-sanofi-announce-positive-topline-results-from-first-phase-3-trials-evaluating-monthly-dosing-of-alirocumab-in-patients-with-hypercholesterolemia-300017492.html. [Google Scholar]
- 60.Colhoun HM, Robinson JG, Farnier M, Cariou B, Blom D, Kereiakes DJ, Lorenzato C, Pordy R, Chaudhari U. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc Disord. 2014;14:121. doi: 10.1186/1471-2261-14-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bays H, Gaudet D, Weiss R, Ruiz JL, Watts GF, Gouni-Berthold I, Robinson J, Zhao J, Hanotin C, Donahue S. Alirocumab as Add-On to Atorvastatin Versus Other Lipid Treatment Strategies: ODYSSEY OPTIONS I Randomized Trial. J Clin Endocrinol Metab. 2015;100:3140–3148. doi: 10.1210/jc.2015-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JG, Colhoun HM, Bays HE, Jones PH, Du Y, Hanotin C, Donahue S. Efficacy and safety of alirocumab as add-on therapy in high-cardiovascular-risk patients with hypercholesterolemia not adequately controlled with atorvastatin (20 or 40 mg) or rosuvastatin (10 or 20 mg): design and rationale of the ODYSSEY OPTIONS Studies. Clin Cardiol. 2014;37:597–604. doi: 10.1002/clc.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The American College of Cardiology. Odyssey Options I and II. (11/19/2014). [accessed. 2015. p. Dec 9]. Available from: http://www.acc.org/Latest-in-Cardiology/Clinical-Trials/2014/11/18/15/20/ODYSSEY-OPTIONS-I-and-II. [Google Scholar]
- 64.Roth EM, Taskinen MR, Ginsberg H, Kastelein J, Colhoun HM, Merlet L, Baccara-Dinet MT. A 24-week study of alirocumab as monotherapy versus ezetimibe: the first phase 3 data of a proprotein convertase subtilisin/kexin type 9 inhibitor. J Am Coll Cardiol. 2014:63 (12_S). [Google Scholar]
- 65.Moriarty PM, Thompson PD, Cannon CP, Guyton JR, Bergeron J, Zieve FJ, Bruckert E, Jacobson TA, Kopecky SL, Baccara-Dinet MT, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: The ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9:758–769. doi: 10.1016/j.jacl.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 66.Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003. doi: 10.1093/eurheartj/ehv370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.The American College of Cardiology. Odyssey High FH. (11/19/2014). [accessed. 2015. p. Dec 9]. Available from: http://www.acc.org/latest-in-cardiology/clinical-trials/2014/11/18/15/23/odyssey-high-fh. [Google Scholar]
- 68.Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 69.Dias CS, Shaywitz AJ, Wasserman SM, Smith BP, Gao B, Stolman DS, Crispino CP, Smirnakis KV, Emery MG, Colbert A, et al. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888–1898. doi: 10.1016/j.jacc.2012.08.986. [DOI] [PubMed] [Google Scholar]
- 70.Giugliano RP, Desai NR, Kohli P, Rogers WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald ST, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380:2007–2017. doi: 10.1016/S0140-6736(12)61770-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, Bolognese M, Wasserman SM. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006. doi: 10.1016/S0140-6736(12)61771-1. [DOI] [PubMed] [Google Scholar]
- 72.Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–2417. doi: 10.1161/CIRCULATIONAHA.112.144055. [DOI] [PubMed] [Google Scholar]
- 73.Sullivan D, Olsson AG, Scott R, Kim JB, Xue A, Gebski V, Wasserman SM, Stein EA. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–2506. doi: 10.1001/jama.2012.25790. [DOI] [PubMed] [Google Scholar]
- 74.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 75.Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, Somaratne R, Legg JC, Nelson P, Scott R, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–1882. doi: 10.1001/jama.2014.4030. [DOI] [PubMed] [Google Scholar]
- 76.Kiyosue A, Honarpour N, Xue A, Wasserman S, Hirayama A. Effects of evolocumab (AMG 145) in hypercholesterolemic, statin-treated, japanese patients at high cardiovascular risk: results from the phase iii yukawa 2 study. J Am Coll Cardiol. 2015:65 (10_S). [Google Scholar]
- 77.Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, Kim JB, Scott R, Wasserman SM, Bays H. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–2540. doi: 10.1016/j.jacc.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 78.Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, Bruckert E, Cho L, Dent R, Knusel B, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–2548. doi: 10.1016/j.jacc.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 79.Nissen SE, Stroes E, Dent-Acosta RE, Rosenson RS, Lehman SJ, Sattar N, Preiss D, Bruckert E, Ceška R, Lepor N, et al. Efficacy and Tolerability of Evolocumab vs Ezetimibe in Patients With Muscle-Related Statin Intolerance: The GAUSS-3 Randomized Clinical Trial. JAMA. 2016;315:1580–1590. doi: 10.1001/jama.2016.3608. [DOI] [PubMed] [Google Scholar]
- 80.Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, Burgess L, Ceska R, Roth E, Koren MJ, Ballantyne CM, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–1819. doi: 10.1056/NEJMoa1316222. [DOI] [PubMed] [Google Scholar]
- 81.Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–340. doi: 10.1016/S0140-6736(14)61399-4. [DOI] [PubMed] [Google Scholar]
- 82.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–350. doi: 10.1016/S0140-6736(14)61374-X. [DOI] [PubMed] [Google Scholar]
- 83.Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, Civeira F, Somaratne R, Nelson P, Liu T, et al. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234–243. doi: 10.1161/CIRCULATIONAHA.113.007012. [DOI] [PubMed] [Google Scholar]
- 84.Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 85.Lunven C, Paehler T, Poitiers F, Brunet A, Rey J, Hanotin C, Sasiela WJ. A randomized study of the relative pharmacokinetics, pharmacodynamics, and safety of alirocumab, a fully human monoclonal antibody to PCSK9, after single subcutaneous administration at three different injection sites in healthy subjects. Cardiovasc Ther. 2014;32:297–301. doi: 10.1111/1755-5922.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cicero AF, Colletti A, Borghi C. Profile of evolocumab and its potential in the treatment of hyperlipidemia. Drug Des Devel Ther. 2015;9:3073–3082. doi: 10.2147/DDDT.S67498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- 88.Swiger K. Early Evidence Linking PCSK9 Inhibitors to Neurocognitive Adverse Events: Does Correlation Imply Causation? American College of Cardiology2015. [cited 2016 Oct 12] Available from: http://www.acc.org/latest-in-cardiology/articles/2015/06/01/12/36/early-evidence-linking-pcsk9-inhibitors-toneurocognitive-adverse-events. [Google Scholar]
- 89.Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228:18–28. doi: 10.1016/j.atherosclerosis.2013.01.044. [DOI] [PubMed] [Google Scholar]
- 90.Jarcho JA, Keaney JF. Proof That Lower Is Better--LDL Cholesterol and IMPROVE-IT. N Engl J Med. 2015;372:2448–2450. doi: 10.1056/NEJMe1507041. [DOI] [PubMed] [Google Scholar]
- 91.Minder CM, Blumenthal RS, Blaha MJ. Statins for primary prevention of cardiovascular disease: the benefits outweigh the risks. Curr Opin Cardiol. 2013;28:554–560. doi: 10.1097/HCO.0b013e32836429e6. [DOI] [PubMed] [Google Scholar]
- 92.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. [Google Scholar]
- 93.Anderson JL, Heidenreich PA, Barnett PG, Creager MA, Fonarow GC, Gibbons RJ, Halperin JL, Hlatky MA, Jacobs AK, Mark DB, et al. ACC/AHA statement on cost/value methodology in clinical practice guidelines and performance measures: a report of the American College of Cardiology/American Heart Association Task Force on Performance Measures and Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2304–2322. doi: 10.1016/j.jacc.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 94.PCSK9 Inhibitors for Treatment of High Cholesterol. New England Comparative Effectiveness Public Advisory Council. (n.d.) Available from: http://cepac.icer-review.org/adaptations/cholesterol/