

Abstract
PNAs are emerging as useful synthetic devices targeting natural miRNAs. In particular 3 classes of structurally modified PNAs analogs are herein described, namely α, β and γ, which differ by their backbone modification. Their mode and binding affinity for natural nucleic acids and their use in medicinal chemistry as potential miRNA binders is discussed.
Keywords: antisense, backbone modification, chirality, miRNA, PNA
Abbreviations
- Aeg
amino ethyl glycine
- AMO
anti-miRNA antisense oligonucleotide
- ASO
antisense oligonucleotide
- CCPs
cell penetrating peptides
- FAM
fluorescein
- GPNA
guanidine-modified PNA
- Hys
histidine
- LNA
Locked nucleic acid
- Lys,K
Lysine
- NLS
nuclear localization signal peptide
- PLGA
lactic-co-glycolic acid
- PNA
peptide nucleic acid
- Phe
phenylalanine
- Q
glutamine
- R
arginine
- SPS
solid phase synthesis
- TAT
Trans-Activator of Transcription
- Tm
melting temperature
- TMR
tetramethylrhodamine
- Trp
tryptophan
- Tyr
tyrosine
- Val
valine.
Introduction
Peptide nucleic acid (PNA) is a synthetic polymer, analog to DNA and RNA, in which the sugar phosphate backbone has been replaced by a unit of N-(2 aminoethyl) glycine (aeg).1 Each of the 4 natural bases is then connected to the backbone via a methylene carbonyl linker. PNA has been designed by Nielsen and coworkers in 1991 as a synthetic ligand that recognizes double stranded DNA's via Hoogsteen like-base pairs in the major groove.1 The design of PNA was conceived to match the relative distance at which the sugar backbone holds the important structural units, namely the 3′-OH, the 5′OH and the nucleobase. As it could be seen from (Fig. 1), the PNA backbone is made of a linear chain containing six σ-bonds, to which the nucleobase is connected via a three σ-bonds (i.e. a methylene carbonyl) linker. Therefore in a DNA backbone six σ-bonds separate each nucleobase unit, while the distance between the backbone and the nucleobase is three bonds. This precise dimension of the backbone is indispensable to obtain polymers that recognize DNA complementary strands with high affinity and this will be discussed in more details throughout this article. It should be noted that modification of the backbone also involved substitution at the positions α, β and γ (Fig. 1). Although modifications at α, β and γ position did not alter the main chain length, they provided compounds with remarkable diverse ability to bind which it will be discussed in details (vide infra).
Figure 1.
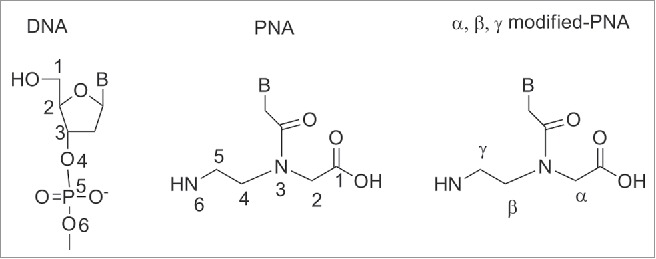
Chemical structure of DNA, PNA and backbone modified-PNA.
By convention, PNA is always represented with the N-terminus upward (or side to the left) and the C-terminus downward (or side to the right) as it would occur for peptides (Fig. 2).The name “PNA” was created to highlight their synthesis (SPS) as well as their analogy to nucleic acids. However, it should be noted that PNA is not a peptide or a nucleic acid in the formal sense.2-3
Figure 2.
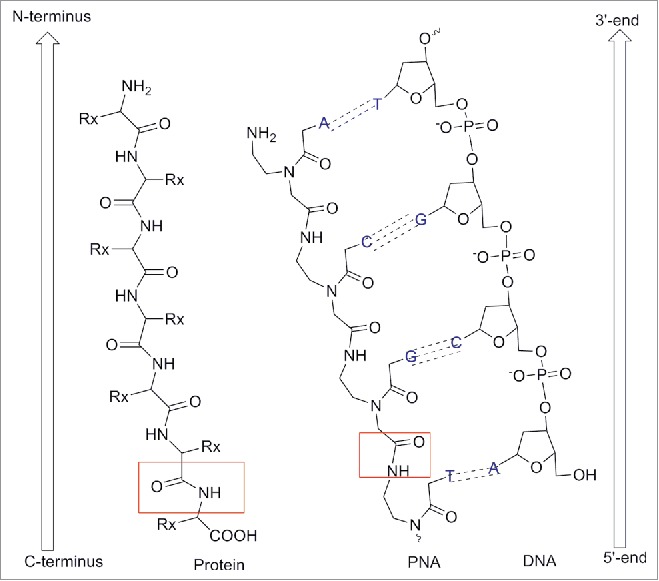
Chemical structure of PNAs compared to DNA and protein. Adapted from European Journal of Human Genetics (2004) 12, 694–700. doi:10.1038/sj.ejhg.5201226.
In addition, PNA has an achiral and neutral backbone which makes it very different from DNAs that possess a chiral and cyclic moiety. The main consequence of lack of chirality lays in the ability of PNA to bind to complementary nucleic acids in both parallel and antiparallel orientation, a property that is forbidden to natural DNA and RNA (Fig. 3) .
Figure 3.
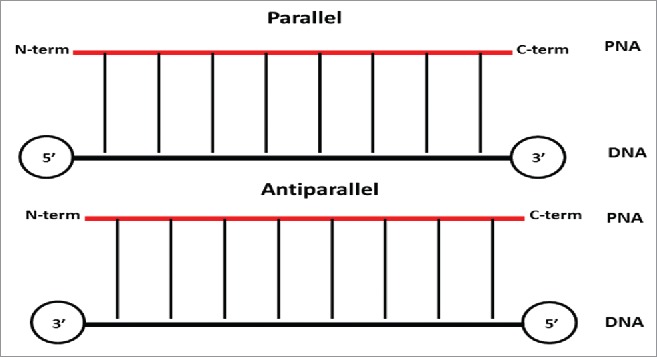
PNA:DNA duplex binding orientation.
In the antiparallel hybridization, PNA's N-terminus binds to DNA's 3′ end, whereas its C-terminus hybridize to DNA's 5′ end. In contrast, in the parallel hybridization PNA's N-terminus binds to DNA's 5′ end, whereas the C-terminus hybridize to DNA's 3 ’end (Fig. 3). It should be noted that, although both parallel and antiparallel mode of binding are permitted, the antiparallel orientation has been found to be more stable and, therefore, favored at some extent.3 Conversely to DNA, PNA possesses a neutral, not charged, backbone which impact on its water solubility and the stability of their duplexes formed with DNAs and RNAs. This is due to the absence of electrostatic repulsion between the PNA strand and the DNA/RNA one.3 As a consequence, the thermal stability of PNA–DNA and PNA-RNA duplexes is higher when compared to DNA-DNA or DNA-RNA ones.4-5 Peptide nucleic acids could also make binding to complementary sequences of PNA to form PNA-PNA duplexes that possess a higher stability when compared with PNA/DNA or PNA/RNA complementary duplexes.6 It should be noted that not only the PNA/PNA duplexes possess higher affinity, but also their specificity was found to be higher. The Watson Crick base pairing rules are strictly observed in hybrids of PNA and nucleic acids.5,6 However, a mismatch in a PNA/DNA duplex is generally more destabilizing than a mismatch in a DNA/DNA pair.5 The average ΔTm of a single mismatch in PNA/DNA duplex was of 15°C compared to 11°C for DNA/DNA duplex, when this was measured on a 15-mer.7 A unique property of PNAs lays in the ability to invade DNAs duplexes to furnish (PNA)2: DNA triplexes or other higher order complexes.1,2,8
For example it has been found that PNA built on a homopyrimidine scaffold, i.e., containing only pyrimidine type nucleobases, bound to DNA's double helices via 2 competing modes: (a) triple helix (PNA:DNA 1:2); (b) strand invasion, where PNA displaces one DNA's strands followed by the formation of a triplex (PNA:DNA, 2:1).5
PNA binds to double-stranded DNA in 4 diverse modes which includes (Fig. 4): Triplex (1), Triplex Invasion (2) Duplex Invasion (3) Double Duplex Invasion (4). Triplex (1) and triplex invasion (2) require a homopurine DNA target and thus could be only elicited by a complementary homopyrimidine PNA. The duplex invasion mode (3) can, in principle, be formed form with any PNAs sequence, although they elicit appreciable stability in particular when targeting homopurines DNAs. The Double Duplex Invasion mode (4), is the most stable, however it is only viable when using PNAs containing modified nucleobases, i.e. pseudo-complementary nucleobases.5
Figure 4.
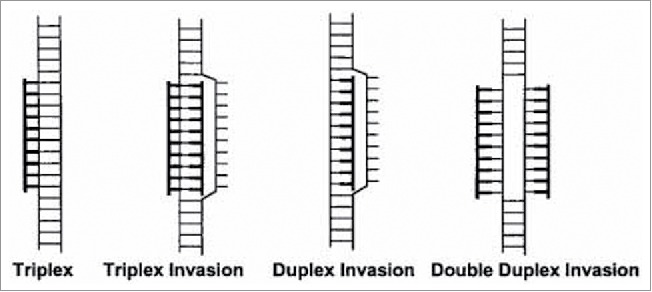
Complexes formed between PNA and double-stranded DNA. Duplex invasion (PNA-DNA/DNA), double duplex invasion (PNA-DNA/PNA-DNA), triplex (PNA·DNA-DNA), triplex invasion (PNA·DNA-PNA/DNA), PNA moieties are drawn in bold. Source: PNAs 1999 vol. 96 no. 21, Method in Molecular Biology, vol 208, Peptide Nucleic Acids, Methods and Protocols (Ed) PE Nielsen.
The stability of a given PNA-DNA duplexes is independent from the ionic strength of the medium,9 only when the DNA (or RNA) target does not possess a secondary structure. Since any relevant DNA or RNA targeted by PNAs would have a competing structure (i.e., duplex, stem-loop, quadruplex) which stability depends on ionic strength, the affinity of a PNA for its target will also be ionic strength dependent. In the context of designing an opportune PNA targeting miRNAs, it should be considered that pri-miRNAs, pre-miRNAs or miRNAs loaded on a RISC complex constitutes competing structures that would explain the significant ionic strength dependence of PNA while eliciting its antagomir activity.
PNAs are not easily recognized by either nucleases or proteases, for which reason they are resistant to enzymatic degradation.10 PNAs are stable across a wide range of temperatures and pHs, unlike DNA, which undergo loss of purine under acidic conditions. Rearrangement of the PNAs might occur, however, under strong alkaline conditions (pH >11).11
Since its discovery, PNAs has attracted interest of chemists and biologists for their chemical, physical, and biological properties and their potential application in diagnostic and pharmaceutics. PNAs are currently employed for: 1) studies related to molecular biology and biotechnology,12 2) the development of gene-targeted drugs in antigene or antisense strategy,13,14 3) the development of diagnostics tools,15 and biosensors.16 Studies are currently undergoing to improve the PNA properties (i.e. solubility, parallel antiparallel discriminative ability) which will result in a number of subsequent generations of PNAs.17
Targeting MIRNA with PNAs
The development of new therapeutics via targeting of relevant microRNA's (miRNAs) is one of the up and coming fields of bio-medicine. miRNAs are a family of small (19–23 unit) non coding RNAs that regulate gene expression by sequence-selective targeting of mRNAs,18-20 leading to a translational repression or mRNA degradation depending on the degree of complementarity between miRNAs and target sequences.19
Essential for miRNA binding to the mRNA is the seed region. The seed region is a conserved sequence which spans bases 2–8 from the 5′-end of the miRNA. Even though base pairing of miRNA and its target mRNA does not match perfectly, the “seed sequence” must be fully complementary to observe translational repression.19
A short single strand non-coding RNA is first charged into the relevant miRNA-induced silencing complex (miRISC). In turn, miRISC make a complex with a target mRNA impairing its normal function as template for protein biosynthesis. This, in cascade, may lead to impairment of highly regulated biological functions such as differentiation, cell cycle and apoptosis.(Fig. 5).20
Figure 5.
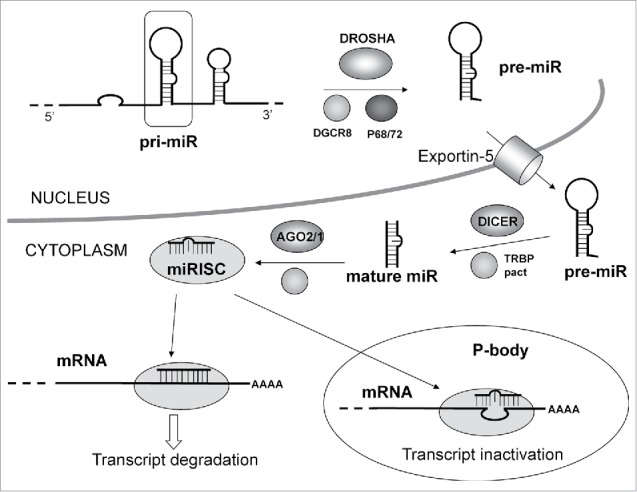
MicroRNA pathway: The primary miRNA transcript (pri-miR) is processed by DROSHA–DGCR8 (Pasha) complex in the nucleus. The maturation includes the production of the primary miRNA transcript (pri-miRNA) by RNA polymerase II or III and cleavage of the pri-miRNA by the complex Drosha–DGCR8 (Pasha) in the nucleus. The resulting precursor hairpin, the pre-miRNA, is exported from the nucleus by Exportin-5–Ran-GTP. In the cytoplasm, the RNase Dicer in complex with the double-stranded RNA-binding protein TRBP cleaves the pre-miRNA hairpin to its mature length. The functional strand of the mature miRNA is loaded together with Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides RISC to silence target mRNAs through mRNA cleavage, translational repression or deadenylation, whereas the passenger strand (black) is degraded. Original Source “Targets in Gene Therapy (Intech) DOI: 10.5772/21081.
Two divergent pathways arise depending on the level of miRNA complementarity to the mRNA target. When perfect complementarity occurs, the mRNA is cleaved and degraded.19 Conversely when imperfect complementarity exists between the miRNA and its target the mRNA translation result impeded, but no mRNA degradation occurs.20 In this latter case, which is the most represented in human cells, the miRNA modulates the rate at which mRNA is expressed leaving the amount of mRNA functioning in the cell intact.
Considering their role in the regulation of protein expression, miRNAs have been proposed as potential template for drug design.21 In addition it has been shown that targeting of miRNAs biogenesis had a vast impact on specific phenotypes and on pathological conditions as well.22
There are at present 25,141 identified mature miRNAs in 193 species, including 2,042 mature miRNAs in humans. However, only some specific targets have been validated.23 A single miRNA, as predicted via bioinformatics analysis, could potentially target many mRNAs through imperfect base complementarity.
It has been shown in specific diseases that miRNAs dysregulation can also alter gene expression. In this context, a therapy aimed at miRNA replacement or anti-sense inhibition of miRNAs offer the potential to restore gene expression. The specific ability of miRNAs to target gene networks, and in particular those controlling key cellular processes, offers the opportunity of carrying out pharmaceutical intervention by modulating gene pathways rather than a single target as in antisense strategy.23,24 When designing antisense oligonucleotides to target mRNA, site selection could be challenging as the DNA targets contains hundreds to thousands bases. MicroRNAs instead are very short, usually only 21-23 base long, so anti-miRNA oligonucleotides (AMOs) are usually designed to be a perfect reverse complement intended to base pair with the full miRNA, avoiding therefore the ‘site selection’. Moreover, in the antisense approach only a few complementary oligonucleotides can successfully hybridize to a targeted mRNA. This is probably due to target accessibility, which in turn may be due to secondary or tertiary mRNA structure and/or to the proteins bound to the RNA.25
Target site splicing is a therapeutic approach to correct disease-associated aberrant splicing. In this context, antisense oligonucleotides showed some promising results.26 Pre-mRNA splicing into mature mRNA is an essential step for expression of most genes in higher eukaryotes.27 Defects in this process typically affect cellular function and can have pathological consequences. Pre-mRNA splicing mutations account for at least 15% of disease-causing mutations28 with up to 50% of all mutations described in some genes.29,30
Rationalizing the effects of variants on splicing is not easy; natural and regulatory splice sites display considerable sequence variation. Furthermore, to verify the consequences of each variant is necessary to perform in vitro experiments; which is time consuming, costly and not-practical. The employment of the in silico prediction approach have become essential to analyze these variants. However even this approach has a drawback: the type of nucleotides that are common in functional splice could be misinterpreted leading to false-positive mutation predictions.
Conversely, the targeting of miRNA by synthetic oligonucleotides is easy to design and effect. miRNAs are fully complementary to the guide strand. There is no need for gene-specific sequences like in conventional antisense oligonucleotide, where target prediction and site selection may be challenging, or concerns may arise about the nature of the secondary structure of a given target fragment.
miRNAs have been targeted via two approaches: (a) inhibition of miRNA biogenesis by targeting enzymatic active protein complexes Drosha, and Dicer, or other miRNA pathways components,31,32 (b) targeting of mature miRNA with modified oligonucleotides.33
Antisense oligonucleotides (ASO) have been used for over 30 years to target mRNAs and now are being used to study miRNAs. Different ASO designs can work through different mechanism of action, for example providing steric inhibition and then impeding translation or degradation of the target RNA.34 Natural oligonucleotides are not suitable as antisense agents because they are rapidly degraded by exo and endonucleases, present in the serum and in the intracellular environment.34,35 Modifications able to confer nuclease stability and binding affinity improve AMOs performance.33 Hence, the same modified oligonucletides used in antisense technology can be used to improve the performance, and the potency, of synthetic agents targeting miRNAs (AMOs or anti-miRNA). In Figure 6 are reported the most common oligonucleotide modifications used for the preparation of anti-miRNA agents. Some representative example will be discussed below. For a more in depth description see references.33,36,37
Figure 6.
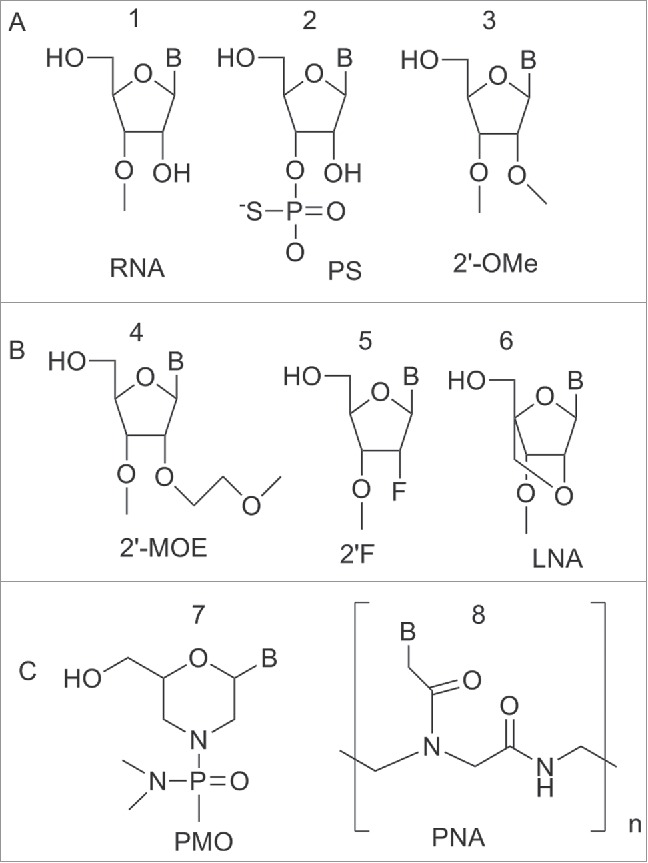
Structure of nucleic acid variants used in miRNA targeting. (A) Phosphorothioate backbone, 2′-OMe (ribose modified) (B) Ribose variants: 2′-MOE, 2′F, LNA (C) Non-ribose backbones: PMO and PNA.
The Phosphorothioate (PS) modified backbone 2 (Fig. 6) in which the sulfur atom substitutes a non-bridging oxygen, has been employed as a building block to prepare anti-miRNA. This modification reduced the affinity of the ASO to target miRNA but conferred a significant increase in nuclease resistance, a property fundamental for long-term assays in cell culture.38,39
The most common sugar modifications used to increase the duplex melting temperature (Tm) and improve nuclease resistance of anti-miRNA include: the 2′-O-methyl (2′-OMe) 3, 2′-O-methoxy-ethyl (2′-MOE) 4, 2′-deoxy-2′-fluoro 5 and the bicyclic locked nucleic acid (LNA) 6 (Fig. 6).40-45
All the 2′-modifications increased the affinity of the ASO to its target RNA. Additionally, the 2′-modifications extended the half-life of the ASO in serum from 3-5 min (unmodified ASO) to 2-3 days. This improved stability translated into the observed higher efficacy in vivo.46
2′-O-methyl RNA (2′-OMe) oligonucleotides 3 (Fig. 6) were employed for the first time as AMOs in 2004.47 Meister et al. described the use of 2′-OMe oligonucleotides to reduce the expression of miRNA-21 in HeLa cells.47 Hutvagner et al.48 used 2′-OMe oligonucleotide to prepare a 31-mer which inhibited let-7 and synthetic siRNA specific for luciferase in HeLa cells and in Caenorhabditis elegans.
The 2′-MOE 4 (Fig. 6) modification improved the nuclease resistance. This was again reflected in the increased Tm. 2′-MOE 4 was demonstrated to provide more potent compounds compared to 2′-OMe in traditional anti-mRNA antisense.39
The replacement of the OH at 2′ in the ribose with a fluorine provided 2′F 5 (Fig. 6). The introduction of a fluorine increased the binding affinity toward complementary RNAs. This was explained considering that fluorine is able to enhance base-pairing and stacking interaction due to its ability to localize electronic charge.49 LNA 6 (Fig. 6) exhibited the highest affinity toward complementary RNAs. Typically there is an increase of 2–8°C in the Tm per each LNA unit introduced.50
Chan et al. first described the application of LNA constructs in anti-miRNA strategy: an anti-miRNA-21 sequence bringing a central core of 8 LNA units flanked on each side by DNA oligomers demonstrated to work slightly better than a previously tested 2′-OMe AMO.51
Lennox and Behlke studied the relative potency of different AMO designs using a miR-21 reporter system. This study identified in LNA/2′-OMe mixmers, the most potent type of AMO. However, in spite of increased binding, the LNA/2′-OMe mixmers showed decreased specificity.42
Obad et al. reported the use of short 8-mer built on a LNA-PS scaffold called tiny LNA, designed to bind solely the seed region. The tiny LNA effectively inhibited the function of both individual miRNA-21 as well as entire family of miRNAs (miRNA-221/222 and let-79) in cell cultures using a luciferase reporter system.52
LNA built AMOs are more effective as miRNA inhibitors than the correspondent 2′-OMe ones.38,53 However, LNA-modified ASOs present a significant risk of hepatotoxicity.53
All the AMOs described until now were based on the modification on the ribose or nucleic acid backbone but other non natural nucleic acid analog such as phosphorodiamidate morpholino oligonucleotide (PMO)54 and peptide nucleic acid can be employed to inhibit miRNAs.14,55-58
PNAs constitutes good alternative as they have high stability to both chemical and enzymatic degradation.10 PNAs form very stable PNA:RNA duplexes, which can efficiently disrupt the dsRNA duplex.7 The first example in this field was reported by Fabani et al.14 who used PNAs and PNA-peptide conjugates to target miRNA-122, obtaining compounds of superior binding and activity compared to 2′-O-methyl oligonucleotides (Table 1). The inhibition ability of PNA based anti-miRNAs was demonstrated without the use of electroporation or any transfection agent but only by conjugating the PNA to cell penetrating peptide (R6-Penetratin) or to 4 lysines residue. The PNA oligomers and the LNA/2′-OMe mixmers were found more effective than 2′O-methyl oligomers.14
Table 1.
PNAs as anti-miRNA agents.
| Target | PNA modification | System Cellular/animal | Effect | Ref |
|---|---|---|---|---|
| miR-122 | K-PNA-K3 | -human hepatocellular carcinoma cells -primary rat hepatocytes | Decrease in miR-122 and mRNA of its target genes. (Aldolase A) | 14 |
| miR-16 | RRRQRRKKR-O-PNA | HeLa cells | Upregulation of bcl-2 mRNA levels | 55 |
| miR-21 | RRRQRRKKR-O-PNA | HeLa cells | Upregulation of mRNA targets measured by luciferase assay | 55 |
| miR-24 | RRRQRRKKR-O-PNA | HeLa cells | Upregulation of mRNA targets measured by luciferase assay | 55 |
| miR-155 | K-PNA-K3 | LPS-activated primary B cells and mice | Up-regulation of 724 transcripts | 56 |
| miR-155 | PLGA NP coated with R9 -PNA | KB cells | Decrease in miR-155 and mRNA | 57 |
| miR-155 | RQIKIWFQNRRMKWKK-PNA | Mouse model of Lymphoma | Decrease in miR-155 SHIP1 repression | 58 |
| miR-210 | R8-PNA | K562 chronic myelogenous leukemia cells | Alterations of erythroid differentation | 62 |
| miR-221 | R8-PNA | Breast cancer cells | Upregulation of p27kip1 gene expression | 63 |
| miR-375 | RRRQRRKKR-PNA | Limb mesenchymal cell | Increase Migration of chondrogenic progenitors Expression level of Cadherin 7 | 64 |
| miR-210 | γ-GPNA | K562 leukemic cells | Alterations of erythroid differentation | 123 |
In parallel, a series of CPPs such as R6 pen (RQIKIWFQNRRMKWKK), Tat (GRKKRRQRRRPPQ), 4 lysines sequence, transportan (AGYLLGKINLKALA-ALAKKIL) conjugated with PNA were found effective for miRNA targeting.55 In particular a Tat cationic modified peptide RRRQRRKKRR showed the best result. The inhibition of miRNA-16 and miRNA-21 by PNAs, monitored by gene luciferase assay, resulted to be more effective compared to LNA and 2′-OMe analogs, showing no cytoxicity at the concentration used, whereas LNAs showed a reduced cell viability. PNAs were more resistant to degradation compared to LNAs, suggesting a better potential as drug candidate.55
Oh et al.55 compared the effect of PNA based AMO with other modified analogs including 2′-OMe and LNA. The PNA-based anti miRNA-24 showed an inhibitory effect 2-fold greater than LNA or 2′-OMe-modified ones. Transfection of LNA-modified AMO into cultured cell also decreased cell viabilities. The results obtained, confirmed that the LNA-modified AMO could be cytotoxic.53 Furthermore, Oh et al.55 compared the activity of other PNA-based anti-miRNA sequences such as let7a, let7b, 10a, 16, 21, 20a and 31 with other modified-AMOs (LNA, 2′-OMe). The PNA sequences exhibited similar or better miRNA inhibition effects than others.55
Since inhibitory activity of AMOs decrease as a function of storage and time, the shelf life of PNA was also tested. PNA sequences stored at room temperature and at 4°C showed no decrease in inhibitory activity for up to 14 weeks. The sequences based on 2′-OMe-modified and LNA-modified analogs, conversely, decreased their miRNA inhibitory activity. Additionally PNA-based sequences showed a long lasting effect on miRNA inhibition in cell also in the presence of serum. Thus, these results demonstrated the advances of PNA-based antisense strategy to target miRNA over other analogs employed for similar application.55
PNAs efficiently blocked miRNA-155, expressed in the haematopoietic system, in both cultured B cells and in mice. Remarkably, miRNA-155 inhibition by PNA in primary B cells was achieved in the absence of any transfection agent.56 The high efficiency of PNA built anti-miRNA-155 was confirmed by the fact that global gene expression of anti-miRNA-155 PNA treated B cells was superimposable to the one of miRNA-155-deficient mice. Interestingly, in this case, anti-miRNA-155 PNA induced additional changes in gene expression.56
The delivery of anti-miRNA PNAs molecules was achieved by using a lactic-co-glycolic acid (PLGA) polymer nanoparticles. The interaction between nanoparticles and PNAs which is neutral, occurred via hydrophobic effect. This is a remarkable difference from nucleic acid analogs which, being charged, interact with nanaoparticles via strong polar interactions. Saltzman et al. reported the use of (PLGA) polymer nanoparticles (100-200nm) coated with a nona-arginine (R9) cell penetrating peptide to deliver neutral anti-miRNAs (PNA and PMO) to inhibit miRNA-155.57
The inhibition of miRNA-155 was monitored by a dual luciferase reporter system in which the target binding sequence for miRNA-155 was introduced at 3′UTR of Renilla Luciferase. KB cells transfected with this sensor system showed the inhibition of miRNA-155 transfected by anti-miRNA PNA and PMO.57
Babar et al.58 employed PLGA nanoparticles coated with penetratin (ANTP-NP), a different CPP. The delivery of antisense peptide nucleic acids encapsulated in ANTP-NP inhibited miRNA-155 and slowed the growth of pre-B-cell tumors in vivo, demonstrating these constructs as promising therapeutics for lymphoma/and leukemia.
The reduction of miRNA-155 level indicated effective delivery and binding of complementary PNAs, resulting in loss of endogenous miRNA activity. The anti-tumor potency of anti-miRNA-155 was also investigated in vivo. Tail-vein delivery of 1.5mg/kg anti-miRNA-155 PNAs encapsulated in ANTP-NP, 2 treatments over 3 days, significantly delayed tumor growth. After 5 days, 50 % decrease in growth was observed, compared to control-untreated tumors. Babar demonstrated that therapeutic response was proportional to the anti-miRNA-155 dose delivered to the tumor site, hypothesizing, then, an improved efficacy for systemic treatment at high doses.58 It should be noted that the systemic dose used in this study was approximately 25-fold less compared to anti-miRNA doses used by other group.59-61
PNA has also been exploited to target miRNA-210, a microRNA associated with hypoxia and involved in erythroid differentiation of leukemic K562 cells.62 The PNA conjugated with a polyarginine peptide (R-pep-PNA-a210) entered efficiently into the cells, strongly inhibited the miRNA-210 activity, and altered the expression of raptor γ-globin genes. A single administration was sufficient to obtain high effect.62
Polyarginine-PNA conjugate (Rpep-PNA-a221) was employed to target miRNA-221. The construct showed high affinity for RNA target. Efficient cellular uptake occurred without a transfection agent and target miRNA-221 resulted strongly inhibited.63 This study showed that anti-miRNA could be effectively delivered using a suitable peptide carrier (RpepPNA-a221), altering the expression of miRNA-221-regulated functions in breast cancer cells.
A PNA-based antisense oligonucleotide was employed to study the role of miRNA-375 on chondrogenic differentiation.64 The RRRQRRKKR CPP was used to allow the PNA sequence to enter into the cells. In this study, miRNA-375 was identified as a negative regulator of chondrogenic differentiation in limb mesenchymal cells through targeting of cadherin-7.64
PNA based molecules demonstrated to be more effective miRNA inhibitor than other DNA-based AMOs and did not show cytotoxicity at concentration up to 1μM. The effects of PNA-based AMOs were shown to persist for 9 days. Also, PNA-based AMOs showed considerable stability at storage temperature.55
Two principal properties should be kept in mind when designing an AMO: potency and specificity. In general, chemical modifications that increase binding affinity also increase potency. Concluding, there is no single best modification pattern or design to employ in AMOs; several potent AMO design strategies exist. The design strategy of choice should be tailored to the experimental aim33 and PNA proved to be a good candidate to exert this function.
PNAs Backbone Modifications
PNA is amenable to be used in several biomedical contexts. However, it is not a fully developed tool yet with some issues still being outstanding. These are: 1) low water solubility, 2) poor cell permeability, 3) ambiguity in nucleic acids recognition (parallel/antiparallel). For this reason, many researchers have focused their attention on developing new monomers to be used in the construction of second generation PNAs lacking the abovementioned issues.65-82
Out of the above 1-3, water solubility is the least severe, with the exception of PNA's sequences rich in adenine and guanine. In addition, it should be noted that most of the cellular experiments are carried out at sub-micromolar or nanomolar.66 concentrations, at which most of PNAs are soluble.13,65
Several cellular types, i.e., neurons and astrocytes, were shown to be permeable to PNAs without the use of a delivery system.67,68
The use of CPPs such as pTAT (RRRQRRKKR), transportan,69 Antennapedia,70 for example, allowed delivering PNA inside the cells as well as polyarginine or arginine rich peptides.71,72
This approach has been extended to the use of cationic peptide-lipid conjugates73 and to the newly discovered peptide carriers.74 Nuclear localization signal peptide (NLS) has been shown to deliver the PNAs into the nucleus, but the cellular uptake is not enhanced as in other peptide cases.69 Therefore “bioshuttle,” complex peptide carriers including more than one peptide have been proposed to allow both cellular and nuclear permeation.75,76
In the last few years several improvements have been achieved by modifying original PNAs structure to improve their performances in term of affinity and specificity toward complementary oligonucleotides. Modifications introduced in the PNAs structure can impact on their biologic potency via 3 diverse effect: a) improvement of DNA/RNA binding affinity; b) improvement of sequence specificity, in particular for directional preference (anti-parallel vs parallel) and mismatch recognition; c) improvement of bioavailability (i.e. cell internalization, pharmacokinetics).
Excellent reviews described chemically modified PNA,77 their structure activity relationship78 as well as the introduction of functional groups with different charges/polarity, flexibility in the backbone.79
Preorganization of conformation, i.e., the ability to adopt a conformation most suitable for DNA binding, has been used to improve the antisense and antigene properties of PNAs. It has been reasoned that an opportunely pre-organized ligand would display a higher binding affinity for DNA or RNA, thus minimizing the loss of the entropy due to the binding process. The preorganization can be obtained by cyclization of the PNA backbone or by adding substituents to the backbone.80,81
The presence of chirality in the backbone has also been found to influence the affinity of PNAs to complementary DNA. In the absence of stereogenic centers, 2 achiral complementary PNAs will form an equimolar mixtures of left-handed and right-handed helices. Whereas, the insertion of at least one stereogenic center in one of the PNA strands, leads to the predominance of one orientation of the helices.82 Introducing one chiral PNA monomer in a PNA strand resulted in an increased binding affinity, when the side chain was positively charged. The duplex stability was found to be dependent from the stereochemistry: PNAs carrying a D chiral monomer at C2 bound to complementary antiparallel DNA strand with higher affinity compared to the PNAs having a L-chiral monomer at the same position.82 Thus, the affinity of PNAs for complementary DNAs was function of the following factors: electrostatic interactions, steric hindrance and chirality, i.e. preference for the R-configuration at C2 in the monomer, S- configuration for position C5. In this review we will focus on chiral PNAs obtained by the insertion of a chiral center on the original N-(2-aminoethyl) glycine backbone at C2 (α-PNAs), C5 (γ-PNAs) and C4 (β-PNAs) their binding properties and their potential use in medicinal chemistry (Fig. 1).
α-modified PNA monomer
The α-modified, hence chiral, PNAs represents the most investigated class. Typical modification includes introduction of side chains belonging to proteinogenic amino acids, sugar, and carbocycles. The introduction of these additional groups at the α position imparted chirality to the PNAs. The analysis of the melting temperature of PNA-DNA duplex containing one α-modified PNA monomer showed a slight destabilization of the resulting duplex. This result, consistent across a vast number of examples, has been interpreted considering the steric hindrance of the side chain. The absolute configuration of the monomer has also an influence on the stability of DNA/PNA duplexes and D aminoacids perturbed the duplex stability at a less extent compared to the L series. It has also established that introduction of positive charges has a positive influence on the duplex stability.83
Nielsen et al. first reported a monomer in which the glycine moiety was replaced by alanine.84 Both the D and L enantiomers were synthesized and inserted into different oligomers. The thermal stability of the PNA-DNA duplexes containing the L-form alanine showed reduced stability compared to PNA-DNA duplex containing glycine. Conversely, the use of monomers containing D-alanine produced PNAs able to bind to cDNAs with similar stability.84 Hence, PNA containing the D-form of 1 (Fig. 7) hybridized to complementary DNA with higher affinity than PNA containing the L-form of 1 (Fig. 7).84
Figure 7.
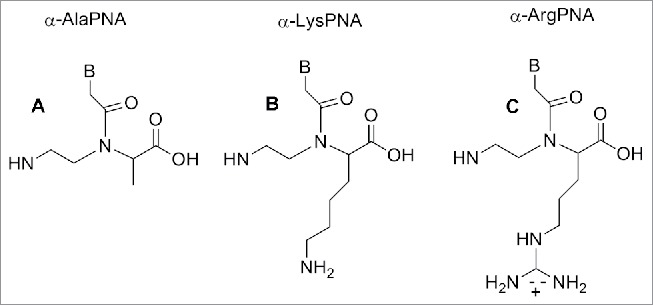
α-Ala, α-Lys, α-Arg modified PNA. Structure adapted from refs. 84,86,90
Lysine, arginine and serine were used in place of glycine to produce relevant PNA monomers bringing an α-amino functionality on the backbone.85 The lysine containing monomer possesses a positive charge which is known to be beneficial in the formation of PNA-DNA duplex.83,85 In this case also, the stability of the resulting duplex is influenced by the chirality present on the monomer backbone; the Tm of PNA-DNA duplexes in which the PNA contains a D-lysine is comparable to the achiral PNA. Conversely, when the PNA contains an L- monomer a reduction of Tm is observed.83,85
Hence, the D-lysine containing PNA showed a higher Tm compared to the unmodified PNA due to a constructive electrostatic interaction formed by the ammonium group of lysine and the phosphate of DNA.
The introduction of 3 consecutive D-lysine in PNAs produced an increase of the construct specificity of recognition and enhanced the ability to discriminate between parallel and antiparallel complementary strand.86
The X-ray analysis of the structure of the D-lysine modified PNA/DNA duplex showed these constructs are rigid to the point they form only a P helix conformation which is more stable, in this case, over other possible conformations.86 Additionally, the presence of chiral centers contributed to increasing the PNA selectivity in the recognition of complementary DNA.86
Modified PNAs containing chiral lysine-PNA backbone were used in the detection of DNA mutation in cystic fibrosis. A PNA oligomer containing a chiral box of 3 consecutive Lys -PNA was employed to detect a DNA mutation (R553X) related to cystic fibrosis, using capillary electrophoresis experiments.87 A Lys modified PNA probe was employed to recognize the CF W1282X mutation using surface plasmon resonance.88 The α-lysine side chain can be employed to introduce other functional molecules in the PNA oligomer. Seitz et al. reported a PNA oligomer bearing 2 fluorofores (FAM and TMR) in the middle of the PNA sequences.89 The introduction of a D-arginine PNA moiety in a PNA sequence (Fig. 7C) behave in different fashion compared to the D-lysine constructs. When one monomer containing of D-arginine was inserted into a PNA decamer, destabilization of the duplex PNA-DNA was observed, although the guanidium group is positively charged as the lysine amino functionality. However, introducing a D-arginine containing moiety in every other position of an α-GPNAs, produced constructs with increased DNA's affinity.90 This result was explained considering that the electrostatic attraction between the guanidinium group and the phosphate overcame the steric repulsion in the latter cases.90 PNA containing arginine residues (GPNA) were more easily taken up by mammalian cell and resulted in being less toxic compared to PNA and PNA conjugated with polyarginine.91
GPNAs were successfully used to target the transcriptional start site of E-cadherin gene producing a sequence-specific antisense effect.91 GPNAs were also employed for PNA microarray analysis and their higher sequence selectivity in this context was demonstrated.92
PNAs modified with bulky amino acids, such as His, Tyr, Trp, Phe and Val, furnished lower Tm compared to the un-substituted ones, due to their substantially larger steric hindrance.93 Sugars were also introduced at the α position of PNAs: 1) to allow the interaction of the oligomers with those receptors recognizing glycosylated substrates; 2) to enhance their biodistribution (Fig. 8A-E).94 UV melting experiments demonstrated that a galactose in the α-position in the backbone induced slight reduction of the Tm in the modified PNA/DNA duplex compared to the unmodified one. A C-linked glycosylated α-PNA (Fig. 8F) was synthesized and incorporated in a PNA oligomers exploiting the Fmoc chemistry.95
Figure 8.
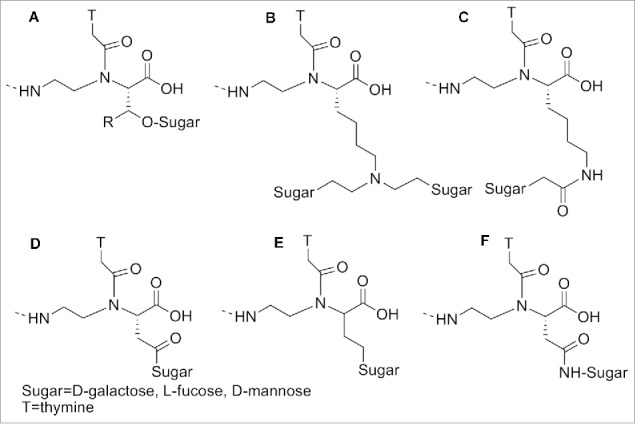
α -modified sugar PNA. Structure adapted from refs. 94,95
Some α-modified PNAs were used to study the interaction between PNA/DNA duplex and small molecules. For this purpose PNAs oligomers containing L or D-leucine-based α-PNA monomers (Fig. 9), were synthesized and the effect of the α-side chain on cyanine dye aggregation was evaluated.96
Figure 9.
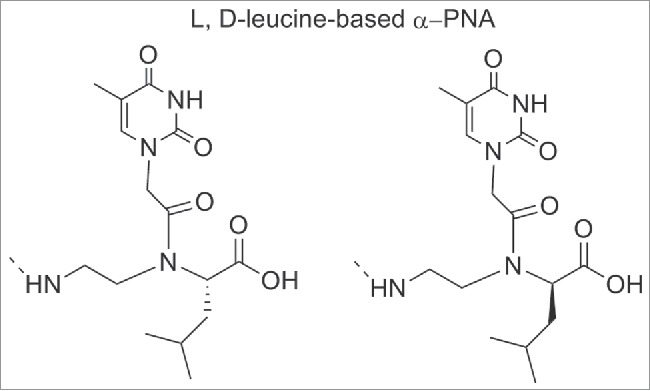
Structure of L, D-leucine-based α-PNA. Structure adapted from ref. 96
When L-leucine based unit was inserted, the cyanine dye aggregation was significantly reduced whereas the insertion of L-leucine one was less inhibitory. It has been shown that in the duplex, the isobutyl group of L-leucine was directed into the minor groove; the effective blockage of the dye access suggested the dye binds to the minor groove. The minor groove of PNA-DNA duplexes served as a template for the mechanism by which the cyanine dye assembled into helical aggregates.
Recently, Rozners et al.97 discovered that short PNAs bind, in a strong and sequence selective fashion, to a homopurine tract of double-helical RNA via formation of a triple helix. Combining this newly discovered PNA's property and the known α-guanidine-PNAs enhanced cellular uptake they tested the potential of GPNA in molecular recognition of double-helical RNAs (Fig. 10, 1). RNAs duplexes are an attractive target for molecular recognition since many noncoding RNAs play important roles in the control of gene expression.21
Figure 10.
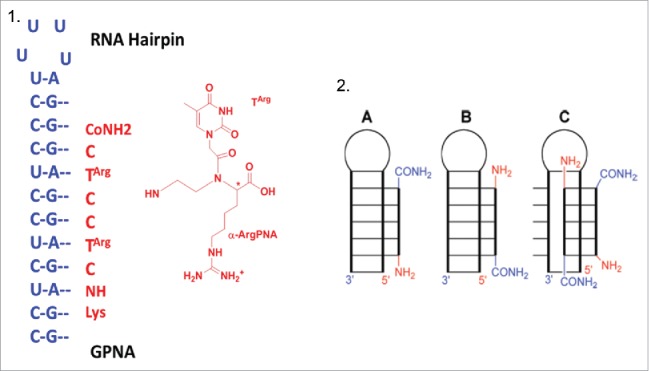
1) RNA hairpin structure and Targ monomer; 2) Representation of binding modes: (A) parallel triple helix, (B) antiparallel triple helix, (C) strand invasion triplex. Source: Biochemistry 2012; 51: 63-73.
Isothermal titration calorimetry (ITC) experiments run between guanidine-modified PNA (GPNA) and RNA hairpins showed that GPNA possessed a reduced affinity and sequence selectivity for triple-helical recognition of RNA. The stoichiometry of binding resulted of 2:1 PNA-RNA complex, thus suggesting a strand invasion triplex binding mode combining antiparallel PNA via Watson-Crick hydrogen bonds and parallel PNA binding via Hoogsteen hydrogen bonds (Fig. 10, 2C). In contrast the unmodified PNA formed a 1:1 PNA-RNA triple helix (Fig. 10, 2B)
Nevertheless, promising results were obtained for recognition of biological relevant double-helical RNA. The α-GPNA derived from D-arginine recognized the transactivation response element of HIV-1 with high affinity and sequence selectivity, presumably via Watson-Crick duplex formation. On the other hand, strong and sequence selective triple helixes were formed by unmodified and nucleobase-modified PNA and the purine-rich strand of the bacterial A-site RNA. The results obtained showed that PNAs chemical modifications may enhance molecular recognition of non-coding RNAs.97 The α-D-GPNA can be employed for strand invasion recognition of biologically relevant RNAs (pri-miRNA, pre-miRNA etc) possessing hairpin structures that are thermally weaker because of noncanonical base pairs, bulges, and internal loops.
β-modified PNA monomer
Cyclic PNA analogs containing a chiral center at the β-position such as cyclopropane (cpr), cyclopentyl (cp) and cyclohexyl (ch) were prepared to impart PNA a pre-organized structure which would then translate in a higher selectivity for DNA/RNA.98-107 (Fig. 11).
Figure 11.
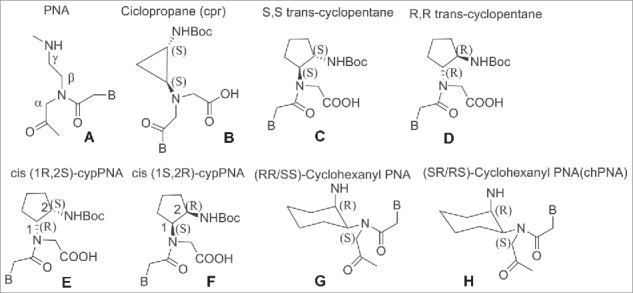
β-ciclic PNA analogs. Structures adapted from Refs. 98–107
In these cases, the effect of the cyclic structure overrun the effect of the β substitution. PNAs containing a cyclopropane have an embedded rigid 3-member ring (Fig. 11B).98 The introduction of a single unit of trans cyclopropane (tcpr) PNA(S,S)-cyclopropane in an aeg-PNA sequence resulted in destabilization of the complexes with DNA and RNA compared to the unmodified PNA. In the case of tcprPNA–RNA complexes, a well-defined melting transition was visible in both the heating and cooling experiments whereas in the DNAs one PNA sequences was able to form a 1 : 2 complex.98
Trans-cyclopentane-derived PNAs monomer (tcypPNAs) was obtained replacing the ethylenediamine portion of aminoethylglycine of aegPNAs with a (S,S)-trans-cyclopentane diamine unit (Fig. 11C). Trans-cyclopentane-derived PNAs (tcypPNAs) was introduced in PNAs strands at several positions and in varying number within PNA backbones containing mixed-base sequences.
Compared to unmodified PNA, tcypPNA showed an improved binding affinity, higher Tm and higher sequence specificity to cDNA, indicating that tcypPNA could be used as probes in DNA diagnostics. Introduction of cyclopentanes into the PNA backbone afforded PNAs having improved binding affinity to complementary DNAs.99,100 Ganesh et al. reported a cis-cyclopentane analog in which the 5-member ring was obtained by bridging Cβ and Cγ of the aminoethyl moiety. The cp-PNAs obtained showed higher affinity toward RNA rather than DNA; in particular the 1R2S PNA's enantiomer (cp-PNA) (Fig. 11E) formed more stable hybrids compared to the 1S2R enantiomers. Both enantiomers stabilized PNA2:DNA and PNA2:RNA triplexes.101,102 PNAs containing a cyclohexyl moiety in the backbone in either (S,S) or (R,R) configuration 7 were also prepared (Fig. 11).103 Introducing (S,S)-cyclohexyl monomer in PNAs oligomers had little effect on the thermal stability (ΔTm = − 1 °C) of their duplexes with DNA and RNA. In contrast, introduction of (R,R)-cyclohexyl units dramatically decreased the thermal stability of the PNA-DNA (RNA) complexes (ΔTm = − 8 °C for each (R,R) residue). In PNA-PNA duplexes the introduction of (R,R) and (S,S) residue only exerted a slight effect on the stability and the duplexes containing the 2 isomers assumed opposite handedness.103 Chiral oligomers containing cis(1S,2R/1R,2S)-aminocyclohexylglycyl thymine monomers in the center and at the N-terminus were prepared. The hybridization of these constructs with cDNA indicated that the (1S,2R)-cyclohexyl moiety enhanced selective binding with RNA over DNA. This arose from comparing the Tm values of the duplexes formed by these molecules and their targets with aegPNA as a control (Fig. 11H).104,105 The (1S,2R)-cyclohexyl PNA possess a dihedral angle β of 65°C which is compatible with the geometry required to form either PNA:DNA or PNA:RNA complexes.106 It was only recently that a single substituent in β-position was introduced by Sugiyama et al.107 who installed a methyl group at the β-position (Fig. 12). This position corresponds to the C4′ of the deoxyribose moiety of the DNA which is a chiral carbon. The incorporation of a substituent at the β-position was expected to influence the conformation and the binding properties of PNA toward DNA.
Figure 12.
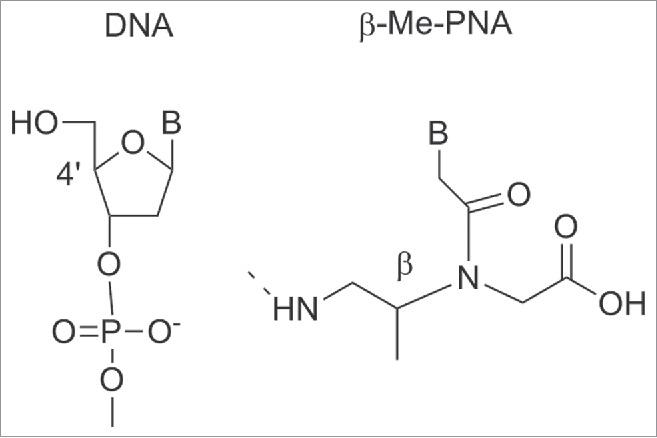
β–methyl PNA analog. Structures adapted from Ref. 107
Both the S- and the R- monomers of β–Me PNA were synthesized and incorporated individually into a 10-residue mixed PNA sequences. PNA containing the S-forms of β–Me PNA showed similar Tm values compared to unmodified PNA. In contrast, the enantiomer of β PNA bearing 3 R- β–Me monomers did not bind to DNA. The stereochemistry of the β-carbon of the PNA backbone was fundamental to the hybridization ability of PNA and strictly limited to the S-configuration. CD spectra analysis suggested that the β–(S)-PNA adopted a right-handed helix whereas β–(R)-PNA a left handed one. The right handed structure of β–(S)-PNA seems not to contribute to the total stability of PNA-DNA duplexes. Unfavorable steric interactions brought by the β–methyl groups in the PNA-DNA duplex may counteract the benefit of the induced structure.107 It should be noted that the development of β-PNAs is a new and unexplored field, therefore it is likely that future expansion involving more sophisticated designs will be reported.
γ-modified PNA monomer
Among all the backbone modification, the introduction of a stereogenic center at γ-position is the most promising one. Compared to unmodified PNAs the γ-modified have several advantages such us improved solubility, better permeability to cells (in particular the γ-GPNAs), increased stability of PNA-DNA duplexes and opportunities for further functionalization.108-118
The first example of a chiral γ modified PNA monomer (Fig. 13A) was reported in 1994 by Lyang et al.108 but they started to gain attention only from 2005. Appella et al.109 synthesized a PNA monomer carrying the L-lysine side chain at γ-position (Fig. 13B), useful to incorporate in a PNA oligomer functional groups such as a fluorofore.110
Figure 14.
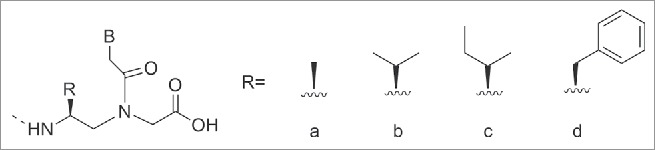
γ-modified PNAs with different amino acid. Structures adapted from Ref. 117
Figure 13.
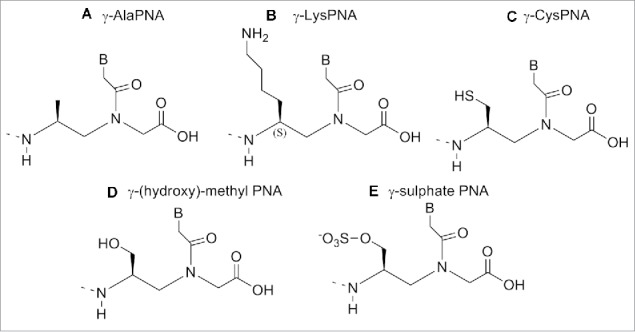
γ-modified PNA analogs. Structures adapted from Refs. 109−114
Incorporation of γ modified Lys monomer showed a slightly increased stability of the duplex PNA/DNA compared to the unmodified one. Furthermore a superior ability of γ-PNAs to discriminate single base mismatch was observed.109 Being the chirality of the backbone crucial to PNA/DNA duplex formation, the stereochemistry of Kγ-PNA was examined. UV melting experiments carried out on duplex PNA/DNA containing L or D isomer showed that D-Lys based monomer induced a large destabilization of the duplex whereas the L-Lys one stabilized the duplex.109
The side chain with L configuration oriented along the periphery of the duplex whereas the D configuration directed to the interior of the duplex. Using a similar procedure, a cysteine based modified PNA monomers was also synthesized. The introduction of a thiol group in a PNA chain.111 (Figure 13C) is suitable for a post assembly conjugation. Indeed PNAs oligomers carrying Cys-based monomer at the N-terminus could be used for native chemical ligation with thioesters PNA or peptides to furnish longer PNA or peptide-PNA conjugates.112
A γ-(hydroxy)-methyl PNA was reported by Romanelli et al.113 The new analog (Fig. 13D) was introduced at a different position (N-termini, C-termini, M middle, C+M) in a PNA oligomer and its binding ability was evaluated in: a) to double stranded and b) inhibiting DNA-protein interactions.113
With the exception of the duplex formed by PNA at the N terminus, all the duplexes obtained with complementary PNAs possessed a very similar secondary structure. The PNA(N)/DNA duplex resembled the circular dichroism (CD) spectrum of an antiparallel PNA/DNA duplex. Whereas hybrids formed by PNA (C), PNA (M) and PNA (C+M), possess a structure very similar to that of PNA/RNA duplexes. The position of the chiral monomer and the oligomers length influenced the structure of the single strand and of the hybrids with DNA.113 Among the sequences designed, the PNA one carrying the modified monomer at C-terminus was the most promising in the inhibitory effect to the generation of Sp1/DNA complex.113
Romanelli et al.114 developed a novel γ-sulfate PNA analog (Fig. 13E), designed to be similar to DNA in terms of polarity, charge and solubility. The sulfate and the phosphate group have similar geometry, steric properties and polarity; a sulfate monoester carries the same charge as the phosphate diester present in oligonucleotides.
Three consecutive sulfate monomers were introduced into homopyrimidine PNA sequence CTCCTCCTC, in which all the thymines were replaced by the γ-modified sulfate. PNA2•DNA triplex formed by sulfate-modified PNA was less stable (ΔTm=−5.6 °C) than the Nielsen's PNAs. The destabilization was explained considering the electrostatic repulsion between the negatively charged phosphate of DNA and sulfate present on the modified PNA. The γ-sulfate PNA modified sequence could be lipofected into human breast cancer (SKB3) cells due to its negative charge and exhibited antigene activity against ErbB2 gene.114
The α-Lys PNA's thermal stability was compared to the one of γ-Lys PNAs. This study revealed that the γ-modification was more effective at improving the DNA binding ability; additionally the stereochemical effect on DNA binding was more evident in γ-PNA rather than the α-PNA.115,116
Randomly folded, single stranded PNAs can be pre-organized into either a right-handed or left-handed helix by inserting an appropriate stereogenic center at the γ-backbone position.
In particular γ-PNAs derived from L-amino acids adopted a right handed conformation whereas those derived from D-amino acids adopted a left-handed one; however only the right handed γ-PNAs were able to hybridize to natural counterparts (DNA/RNA) with high affinity and sequence selectivity.116 A study to determine the extent of steric accommodation for the side-chain modification at the γ position within in PNA was performed.117 Along with these studies, a structural characterization of the γ-modified PNAs was completed to provide insights into the origin of γPNA preorganization. PNA thymine units possessing sterically hindered side-chains of alanine, valine, isoleucine, and phenylalanine (Fig. 14) were developed.117
The PNA oligomers, containing a single monomer modification were hybridized with antiparallel complementary DNA strand and melting temperatures Tm were measured. The incorporation of a single γ-PNA monomer based on each of the 4 amino acids placed in the middle of the sequence furnished an increased Tm value for the duplex of 4°C (ΔTm=+4°C).117 γPNAs resulted in being particularly suitable for targeting B-form duplex DNA, a mixed-sequence of L-alanine based γPNAs decamer was able to invade a B-form DNA with the assistance of terminal acridine or modified nucleobase (G-clamp).118,119 The incorporation of G-clamp nucleobases allowed the invasion at physiological ionic strength which does not normally happen. The invasion normally occurs under low salt conditions (Fig. 15C).118
Figure 15.
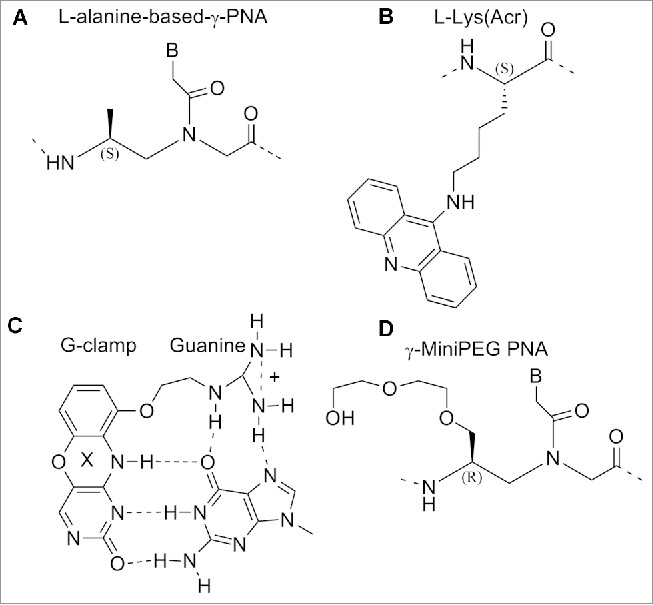
Structure of L-alanine-based-γ-PNA, acridine-linked-L-lysine, G-clamp base pair, γ-MiniPEG PNA. Structures adapted from Refs. 118–121
The use of miniPEG modified γPNAs (MP γPNAs) (Fig. 15D) as an antisense agent for the invasion of the structured secondary microRNA pre-let-7 target has been reported.120
The results provided evidenced that the MP γPNAs were able to invade the pre-let-7 microRNA in a sequence-specific manner. In the previous section we have described the ability of α-GPNAs to pass the cell membrane and to target RNAs in living cells without significant toxicity. The high cost for the synthesis of the monomer discouraged the use in clinical trials. The γ-GPNAs, second generation GPNA (Fig. 16C) could be prepared from a cheaper source (Boc-L-Lysine) bearing an homoarginine side chain at γ -position.121
Figure 16.
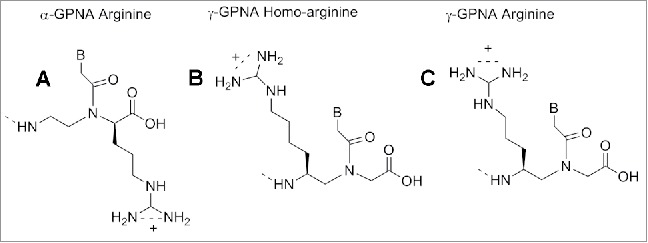
Comparison of the structure of γ-GPNAs.
The stability of PNA-DNA duplexes containing γ-GPNAs and α-GPNA were compared: γ-GPNAs were significantly more stable compared to α-GPNA.121 The particular design of the sequence also influenced the thermal stability; alternating the unit in the backbone enhanced the thermal stability to a higher degree than a consecutive arrangement. This enhancement was attributed to a conformational preorganization and not to an electrostatic effect between guanidinium and phosphate groups, because of the lack of salt dependence of ΔTm. The cellular uptake of γ-GPNA was higher, probably due to their helical conformation. Indeed, a fully alternate γ-PNA decamer was taken up by HeLa cells with an efficiency comparable to that of TAT transduction domain. Also, designing PNAs containing a peptide mimic can allow recognition by specific receptors as recently demonstrated by a short PNA mimicking the function of a nuclear localization signal peptide (NLS). The cellular uptake of this construct (Fig. 17) was examined in rhabdomyosarcoma cells (RH30 cells) and localized in the cell nuclei, whereas unmodified PNA was not detected.122 NLS peptide is known to be transported to the nuclei by an importin-mediated mechanism and based on the similarity of the localization patterns, it was assumed that modified PNAs could interact with importin performing in this case as a potential peptide mimic rather than a nucleic acid one.
Figure 17.
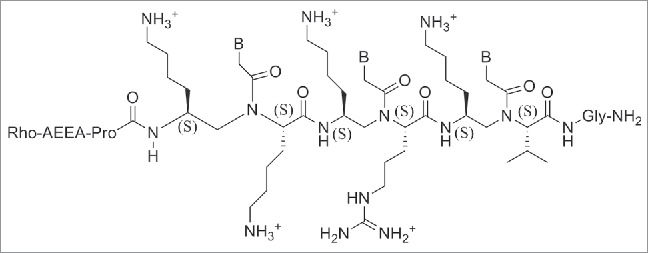
Modified PNA containing NLS sequences. Structure adapted from Ref. 123
The same strategy was applied to design and synthesize PNAs with both peptide properties and RNA-binding ability.123 A series of 18-mer GPNAs were employed as anti-miRNA-210 in leukemic K562 cells.123 Unmodified PNAs, R8-conjugated PNAs and modified PNAs containing 8 arginine residues on the backbone, either at C2-modified (R) or C5-modified (S) monomers, all with the same sequence, were compared. Two different templates were employed for the modified PNAs: one with alternated chiral and achiral monomers and one with a stretch of chiral monomers at the N terminus. All the modified PNAs efficiently entered into the cells and the fluorescent PNAs were detected into the cytoplasm. The γ-GPNA with consecutive arrangement showed the best anti-miRNA-210 activity. The good overall anti-miRNA activity of γ-GPNA resulted from a combination of cellular uptake and RNA binding property. The presence of substituents in the PNA backbone seems to play a relevant role in both cellular uptake and in the mechanism of miRNA recognition and activation.123
Conclusions
The high affinity of PNAs for RNA and the strong chemical and enzymatic stability of these compounds (unmodified and the backbone-modified version) have made them a good candidates as anti-miRNAs. PNA based AMO showed to be more effective miRNA inhibitor compared to other DNA-based AMOs (2′-OMe, LNA), no cytotoxic at concentration up to 1μM and to possess a long lasting effect.
The effects of PNA-based AMOs were shown to persist for 9 days. Also, PNA-based AMOs showed considerable stability at storage temperature. Data indicates that PNA is an emerging technology likely to succeed, despite the limited number of targets studied so far. Introducing a substituent at different positions (α,β,γ) of the PNA backbone has demonstrated to be useful at improving DNA binding and sequence selectivity. The α-GPNA, MiniPEG- γPNAs and γ-GPNA modified PNAs showed promising results in miRNA targeting. In particular the α-GPNA was employed to target double-helical RNAs via a strand invasion triplex binding mode. The transactivation response element of HIV-1 and bacterial A site RNA were also successful targeted. The mini PEG modified γPNAs (MP γPNAs) was employed as an antisense agent to invade microRNA pre-let7 sequence and the γ-GPNA modified sequence was used as anti-miRNA-210 in leukemic K562 cells. Among all the analogs the γ-GPNAs showed an improved cellular uptake, compared to the unmodified PNAs, higher bio-stability and a good anti-miRNA activity therefore making them a good candidates for the next generation of drugs which will be able to modulate genes.
Disclosure of Potential Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Nielsen PE, Engholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991; 254(5037):1497-500; PMID:1962210; http://dx.doi.org/ 10.1126/science.1962210 [DOI] [PubMed] [Google Scholar]
- 2.Engholm M, Buchardt O, Nielsen PE, Berg RH. Peptide nucleic acids (PNA). Oligonucleotide analogues with an achiral peptide backbone. J Amer Chem Soc 1992; 114:1895-7; http://dx.doi.org/ 10.1021/ja00031a062 [DOI] [Google Scholar]
- 3.Nielsen PE, Egholm M, Berg RH, Buchardt O. Peptide nucleic acids (PNA). DNA analogues with a polyamide backbone. Antisense Research and Application. Crook S, Lebleu B (eds.). CRC Press, Boca Raton, 1993; 363-373 [Google Scholar]
- 4..Egholm M, Buchardt O, Nielsen PE, Berg RH. Recognition of guanine and adenine in DNA by cytosine and thymine containing peptide nucleic acids (PNA). J Amer Chem Soc 1992; 114:9677-8; http://dx.doi.org/ 10.1021/ja00050a068 [DOI] [Google Scholar]
- 5.Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993; 365:566-8; PMID:7692304; http://dx.doi.org/ 10.1038/365566a0 [DOI] [PubMed] [Google Scholar]
- 6.Wittung P, Nielsen PE, Buchardt O, Egholm M, Norden B. DNA like double helix formed by peptide nucleic acid. Nature 1994; 368:561-3; PMID:8139692; http://dx.doi.org/ 10.1038/368561a0 [DOI] [PubMed] [Google Scholar]
- 7.Nielsen PE. Peptide nucleic acids-protocol and applications. Norfolk: Horizon Bioscience; 2014 [Google Scholar]
- 8.Lohse J, Dahl O, Nielsen PE. Double duplex invasion by peptide nucleic acid: a general principle for sequence-specific targeting of double-stranded DNA. Proc Natl Acad Sci USA 1999; 96(21):11804-08; PMID:10518531; http://dx.doi.org/ 10.1073/pnas.96.21.11804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomac S, Sarkar M, Ratilainen T, Wittung P, Nielsen PE, Nordén B, Gräslund A. Ion effect on the stability and conformation of peptide nucleic acid complexes. J Am Chem Soc 1996; 118:5544-52; http://dx.doi.org/ 10.1021/ja960495l [DOI] [Google Scholar]
- 10.Demidov VV, Potaman VN, Kamenetskii MDF, Egholm M, Buchart O, Sonnischsen SH, Nielsen P E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol 1994; 48:1310-3; PMID:7945427; http://dx.doi.org/ 10.1016/0006-2952(94)90171-6 [DOI] [PubMed] [Google Scholar]
- 11.Eriksson M, Christensen L, Schmidt J, Haaima G, Orgel L, Nielsen PE. Sequence dependent N-terminal rearrangement and degradation of peptide nucleic acid (PNA) in aqueous solution. New J Chem 1998; 22:1055-9; http://dx.doi.org/ 10.1039/a803214i [DOI] [PubMed] [Google Scholar]
- 12.Ray A, Norden B. Peptide nucleic acid (PNA): its medical and biotechnical applications and promise for the future. Faseb J 2000; 14, 1041-60; PMID:10834926 [DOI] [PubMed] [Google Scholar]
- 13.Lundin K E, Good L, Stomberg R, Graslund A, Smith CIE. Biological activity and biotechnological aspects of peptide nucleic acid. Adv Genet 2006; 16:1-51; http://dx.doi.org/ 10.1016/S0065-2660(06)56001-8 [DOI] [PubMed] [Google Scholar]
- 14.Fabani MM, Gait M J. miR-122 targeting with LNA-2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA 2008; 14:336-46; PMID:18073344; http://dx.doi.org/ 10.1261/rna.844108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang N, Appella H. Advantages of Peptide Nucleic Acids as diagnostic platform for the detection of nucleic acids in resource limited settings. J Infect Dis 2010; 201:S42-S45; PMID:20225945; http://dx.doi.org/ 10.1086/650389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Briones C, Moreno M. Applications of peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) in biosensor development. Anal Bioanal Chem 2012; 402:3071-89; PMID:22297860; http://dx.doi.org/ 10.1007/s00216-012-5742-z [DOI] [PubMed] [Google Scholar]
- 17.Pensato S, Saviano M, Romanelli A. New peptide nucleic acid analogues: synthesis and applications. Expert Opin Biol Ther 2007; 7(8):1219-32; PMID:17696820; http://dx.doi.org/ 10.1517/14712598.7.8.1219 [DOI] [PubMed] [Google Scholar]
- 18.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11(9):597-610; PMID:20661255 [DOI] [PubMed] [Google Scholar]
- 19.He L, Hannon G J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2010; 5(7):522-31; http://dx.doi.org/ 10.1038/nrg1379 [DOI] [PubMed] [Google Scholar]
- 20.Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human diseases. Development 2005; 132:4643-62; http://dx.doi.org/ 10.1242/dev.02073 [DOI] [PubMed] [Google Scholar]
- 21.Brown B D, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental application. Nat Rev Genet 2009; 10:578-85; PMID:19609263; http://dx.doi.org/ 10.1038/nrg2628 [DOI] [PubMed] [Google Scholar]
- 22.Kota SK, Balasubramanian S. Cancer therapy via modulation of micro RNA levels: a promising future. Drug Discov Today 2010; 15:733-40; PMID:20692360; http://dx.doi.org/ 10.1016/j.drudis.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 23.Hennessy EJ, Moore KJ. Using microRNA as an alternative treatment for hyperlipidemia and Cardiovascular Disease: cardio-miRs in the pipeline. Cardiovasc Pharmacol 2013; 62(3):247-54; http://dx.doi.org/ 10.1097/FJC.0b013e31829d48bf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garzon R, Marcucci G, Carlo M, Croce CM. Targeting MicroRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010. 9(10):775-89; PMID:20885409; http://dx.doi.org/ 10.1038/nrd3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dias N; Stein C A. Antisense Oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther 2002; 1(5):347-55; PMID:12489851 [PubMed] [Google Scholar]
- 26.Havens MA, Duelli DM, Hastings ML. Targeting RNA Splicing for Disease Therapy. Wiley Interdiscip Rev RNA. 2013; 4(3):247-66; PMID:23512601; http://dx.doi.org/ 10.1002/wrna.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglas AGL, Wood MJW. RNA splicing: disease and therapy. Brif Funct Genomics 2011; 10(3):51-164 [DOI] [PubMed] [Google Scholar]
- 28.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single basepair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 1992; 90(1-2)41-54; PMID:1427786; http://dx.doi.org/ 10.1007/BF00210743 [DOI] [PubMed] [Google Scholar]
- 29.Ars E, Serra E, García J, Kruyer H, Gaona A, Lazaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet 2000; 9(2):237-47; PMID:10607834; http://dx.doi.org/ 10.1093/hmg/9.2.237 [DOI] [PubMed] [Google Scholar]
- 30.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P. Splicing defects in the ataxiatelangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999; 64(6):1617-31; PMID:10330348; http://dx.doi.org/ 10.1086/302418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science 2004; 303:5654:95-8; PMID:14631048; http://dx.doi.org/ 10.1126/science.1090599 [DOI] [PubMed] [Google Scholar]
- 32.Soifer HS, Rossi JJ, Saetrom P, MicroRNAs in disease and potential therapeutic application. Mol Ther 2007; 15(12):2070-1079; PMID:17878899; http://dx.doi.org/ 10.1038/sj.mt.6300311 [DOI] [PubMed] [Google Scholar]
- 33.Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Therapy, 2011; 18:1111-20; PMID:21753793; http://dx.doi.org/ 10.1038/gt.2011.100 [DOI] [PubMed] [Google Scholar]
- 34.Lebedeva I, Stein CA. Antisense oligonucleotides:promise and reality. Annu Rev Pharmacol Toxicol 2001; 41:403-19; PMID:11264463; http://dx.doi.org/ 10.1146/annurev.pharmtox.41.1.403 [DOI] [PubMed] [Google Scholar]
- 35.Rayburn ER, Zhang R. Antisense, RNAi and gene silencing strategies for therapy: mission possible or impossible?. Drug Discov Today 2008; 13(11-12):513-21; PMID:18549978; http://dx.doi.org/ 10.1016/j.drudis.2008.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. Embo Mol Med 2014; 6(7):851-64; PMID:24935956; http://dx.doi.org/ 10.15252/emmm.201100899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhen L, Alhousseynou S, Yang D. MicroRNA: and Emerging Therapeutic target and Intervention Tool. Int J Mol Sci 2008; 9:978-99; PMID:19325841; http://dx.doi.org/ 10.3390/ijms9060978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods 2008; 44:55-60; PMID:18158133; http://dx.doi.org/ 10.1016/j.ymeth.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 39.Lennox KA, Sabel JL, Johnson MJ, Moreira BG, Fletcher CA, Rose SD, Behlke MA, Laikhter AL, Walder JA, Dagle JM. Characterization of modified antisense oligonucleotides in Xenopus laevis embryos. Oligonucleotides 2006; 16(1):26-42; PMID:16584293; http://dx.doi.org/ 10.1089/oli.2006.16.26 [DOI] [PubMed] [Google Scholar]
- 40.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res 2006; 34:2294-304; PMID:16690972; http://dx.doi.org/ 10.1093/nar/gkl183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stenvang J, Kauppinen S. MicroRNAs as targets for antisense-based therapeutics. Expert Opin Biol Ther 2008; 8:59-81; PMID:18081537; http://dx.doi.org/ 10.1517/14712598.8.1.59 [DOI] [PubMed] [Google Scholar]
- 42.Lennox KA, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res 2010; 27:1788-99; PMID:20424893; http://dx.doi.org/ 10.1007/s11095-010-0156-0 [DOI] [PubMed] [Google Scholar]
- 43.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 2012; 11:860-72; PMID:23080337; http://dx.doi.org/ 10.1038/nrd3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012; 3(1):1; PMID:22230293; http://dx.doi.org/ 10.1186/1758-907X-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U, et al.. LNA-mediated microRNA silencing in non-human primates. Nature 2008b; 452:896-9; http://dx.doi.org/ 10.1038/nature06783 [DOI] [PubMed] [Google Scholar]
- 46.Watts JK, Deleavey GF, Damha MJ. Chemically modified siRNA: tools and applications. Drug Discov. Today 2008; 13:842-55 [DOI] [PubMed] [Google Scholar]
- 47.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA 2004; 10:544-50; PMID:14970398; http://dx.doi.org/ 10.1261/rna.5235104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol 2004; 2:E98; PMID:15024405; http://dx.doi.org/ 10.1371/journal.pbio.0020098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pallan PS, Greene EM, Jicman PA, Pandey RK, Manoharan M, Rozners E, Egli M. Unexpected origins of the enhanced pairing affinity of 2′-fluoro-modified RNA. Nucleic Acids Res. 2011; 39(8) 3482-95; PMID:21183463; http://dx.doi.org/ 10.1093/nar/gkq1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen M, Wengel J. LNA: a versatile tool to therapeutics and genomics. Trends Biotechnol. 2003; 21(2):74-81; PMID:12573856; http://dx.doi.org/ 10.1016/S0167-7799(02)00038-0 [DOI] [PubMed] [Google Scholar]
- 51.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005; 65:6029-33; PMID:16024602; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0137 [DOI] [PubMed] [Google Scholar]
- 52.Obad S, Dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C; Fu C, Lindow M, et al.. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 2011; 43:371-8; PMID:21423181; http://dx.doi.org/ 10.1038/ng.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyzykiewicz TK, Hung G, Monia BP. Bennett CF. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 2007; 35; 687-700; PMID:17182632; http://dx.doi.org/ 10.1093/nar/gkl1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet 2007; 39:259-63; PMID:17220889; http://dx.doi.org/ 10.1038/ng1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh SY, Ju YS, Park H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol Cells 2009; 28:341-5; PMID:19812898; http://dx.doi.org/ 10.1007/s10059-009-0134-8 [DOI] [PubMed] [Google Scholar]
- 56.Fabani MM, Abreu-Goodger C, Williams D, Lyons PA, Torres AG, Smith KG, Enright AJ, Gait MJ, Vigorito E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res 2010; 38(13):4466-75; PMID:20223773; http://dx.doi.org/ 10.1093/nar/gkq160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng CJ, Saltzman WM. Polymer nanoparticle–mediated delivery of microRNA inhibition and alternative splicing. Mol Pharm 2012; 9(5):1481-8; PMID:22482958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, Slack FJ. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci USA 2012; 109(26):E1695-704; http://dx.doi.org/ 10.1073/pnas.1201516109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG; Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005; 438:685-9; PMID:16258535; http://dx.doi.org/ 10.1038/nature04303 [DOI] [PubMed] [Google Scholar]
- 60.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol 2010; 28(4):341-7; PMID:20351690; http://dx.doi.org/ 10.1038/nbt.1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V, Giacomini P, et al.. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One 2008; 21;3(5):e2236; http://dx.doi.org/ 10.1371/journal.pone.0002236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fabbri E, Manicardi A, Tedeschi T, Sforza S, Bianchi N, Brognara E, Finotti A, Breveglieri G, Borgatti M, Corradini R, et al.. Modulation of the biological activity of microRNA-210 with peptide nucleic acids (PNAs). Chem Med Chem 2011; 6(12):2192-202; PMID:22012891; http://dx.doi.org/ 10.1002/cmdc.201100270 [DOI] [PubMed] [Google Scholar]
- 63.Brognara E, Fabbri E, Aimi F, Manicardi A, Bianchi N, Finotti A, Breveglieri G, Borgatti M, Corradini R, Marchelli R, et al.. Peptide nucleic acid targeting miR-221 modulate p27kip1 expression in breast cancer MDA-MB-231 cells. Int J of Onc 2012; 41:2119-27 [DOI] [PubMed] [Google Scholar]
- 64.Song J, Kim D, Chun CH, Jin EJ. MicroRNA-375, a new regulator of cadherin-7, suppresses the migration of chondrogenic progenitors. Cell Signal. 2013; 25(3):698-706; PMID:23178988; http://dx.doi.org/ 10.1016/j.cellsig.2012.11.014 [DOI] [PubMed] [Google Scholar]
- 65.Bonham MA, Brown S, Boyd AL, Brown PH, Bruckenstein DA, Hanvey JC, Thomson SA, Pipe A, Hassman F, Bisi JE, et al.. An assessment of the antisense properties of RNaseH competent and steric-blocking oligomers. Nucleic Acids Res 1995; 23:1197-203; PMID:7537874; http://dx.doi.org/ 10.1093/nar/23.7.1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shiraishi T, Hamzavi R, Nielsen PE. Subnanomolar antisense activity of phosphonate-peptide nucleic acid (PNA) conjugates delivered by cationic lipids to HeLa cells. Nucleic Acids Res 2008; 36:4424-32; PMID:18596083; http://dx.doi.org/ 10.1093/nar/gkn401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adlerz L, Soomets U, Holmlund L, Viirlaid S, Langel U Iverfeldt K. Down-regulation of amyloid precursor protein by peptide nucleic acid oligomer in cultured rat primary neurons and astrocytes. Neurosci Lett 2003; 336:55-9; PMID:12493601; http://dx.doi.org/ 10.1016/S0304-3940(02)01219-3 [DOI] [PubMed] [Google Scholar]
- 68.Pession A, Tonelli R, Fronza R, Sciamanna E, Corradini R, Sforza S, Tedeschi T, Marchelli R, Montanaro L, Camerin C, et al.. Targeted inhibition of NMYC by peptide nucleic acid (PNA) in N-myc amplified human neuroblastoma cells: cell-cycle inhibition with induction of neuronal cell differentiation and apoptosis. Int J Oncol 2004; 24:265-72; PMID:14719101 [PubMed] [Google Scholar]
- 69.Bendifallah N, Rasmussen FW, Zachar V, Ebbesen P, Nielsen PE, Koppelhus U. Evaluation of cell-penetrating peptides (cpps) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconj Chem 2006; 17:750-8; http://dx.doi.org/ 10.1021/bc050283q [DOI] [PubMed] [Google Scholar]
- 70.Pooga M, Soomets U, Hallbrink M, Valkna A, Saar K, Rezaei K, Kahl U, Hao JX, Xu XJ, Wiesenfeld-Hallin Z, et al.. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol 1998; 16:857-61; PMID:9743120; http://dx.doi.org/ 10.1038/nbt0998-857 [DOI] [PubMed] [Google Scholar]
- 71.Rothbard JB, Jessop TC, Lewis RS, Murray B A, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc 2004; 126:9506-8; PMID:15291531; http://dx.doi.org/ 10.1021/ja0482536 [DOI] [PubMed] [Google Scholar]
- 72.Abes R, Arzumanov A, Moulton H, Abes S, Ivanova G, Gait MJ, Iversen P, Lebleu B. Arginine-rich cell penetrating peptides: Design, structure-activity, and applications to alter premRNA splicing by steric-block oligonucleotides. J Pept Sci 2008; 14:455-60; PMID:18236382; http://dx.doi.org/ 10.1002/psc.979 [DOI] [PubMed] [Google Scholar]
- 73.Koppelhus U, Shiraishi T, Zachar V, Pankratova S, Nielsen PE. Improved cellular activity of antisense peptide nucleic acids by conjugation to a cationic peptide-lipid (CatLip) domain. Bioconjugate Chem 2008; 19:1526-34; http://dx.doi.org/ 10.1021/bc800068h [DOI] [PubMed] [Google Scholar]
- 74.El-Andaloussi S, Johansson HJ, Holm T, Langel Ü. A Novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther 2007; 15:1820-6; PMID:17622242; http://dx.doi.org/ 10.1038/sj.mt.6300255 [DOI] [PubMed] [Google Scholar]
- 75.Braun K, Peschke P, Pipkorn R, Lampel S, Wachsmth M, Waldeck W, Friendrick E, Debus JA. Biological transporter for the delivery of peptide ncleic acids (PNAs) to the nuclear compartment of living cells. J Mol Biol 2002; 318:237-43; PMID:12051833; http://dx.doi.org/ 10.1016/S0022-2836(02)00031-1 [DOI] [PubMed] [Google Scholar]
- 76.Ivanova G D, Arzumanov A, Abes R, Yin H, Wood MJA, Lebleu B, Gait MJ. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res 2008; 36:6418-28; PMID:18842625; http://dx.doi.org/ 10.1093/nar/gkn671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ganesh KN, Nielsen PE. Peptide Nucleic Acids: analogs and derivatives. Curr Org Chem 2000; 4:931-43; http://dx.doi.org/ 10.2174/1385272003375969 [DOI] [Google Scholar]
- 78.Nielsen PE, Haaima G. Peptide Nucleic acid (PNA). A DNA mimic with a pseudopeptide backbone. Chem Soc Rev 1997; 26:73-8; http://dx.doi.org/ 10.1039/cs9972600073 [DOI] [Google Scholar]
- 79.Corradini R, Sforza S, Tedeschi T, ToTsingan F, Manicardi A, Marchelli R. Peptide nucleic acids with a structural biased backbone. Updated review and emerging challenges. Curr Top Med 2011; 11:1535-54; http://dx.doi.org/ 10.2174/156802611795860979 [DOI] [PubMed] [Google Scholar]
- 80.Kumar VA, Ganesh K. Conformationally constrained PNA analogues: Structural evolution toward DNA/RNA binding selectivity. Acc Chem Res 2005; 38:404-12; PMID:15895978; http://dx.doi.org/ 10.1021/ar030277e [DOI] [PubMed] [Google Scholar]
- 81.Corradini R, Sforza S, Tedeschi T, Totsingan F, Manicardi A, Marchelli R. Peptide nucleic acids with a structurally biased backbone. Updated review and emerging challenges. Curr Top Med Chem 2011; 11:1535-54; PMID:21510833; http://dx.doi.org/ 10.2174/156802611795860979 [DOI] [PubMed] [Google Scholar]
- 82.Tedeschi T, Sforza S, Dossena A, Corradini R, Marchelli R. Lysine-based peptide nucleic acids (PNAs) with strong chiral constraint: control of helix handedness and DNA binding by chirality. Chirality 2005; 17:S196-204; PMID:15952136; http://dx.doi.org/ 10.1002/chir.20128 [DOI] [PubMed] [Google Scholar]
- 83.Sforza S, Corradini R, Ghirardi S, Dossena A, Marchelli R. DNA binding of a D-lysine-based chiral PNA: direction control and mismatch recognition. Eur J Org Chem 2000; 16:2905-13; http://dx.doi.org/ 10.1002/1099-0690(200008)2000:16<2905::AID-EJOC2905>3.0.CO;2-D [DOI] [Google Scholar]
- 84.Duelholm KL, Petersen KH, Jensen DK, Egholm M, Nielsen PE, Buchardt O. Peptide Nucleic acid (PNA) with a chiral backbone based on alanine. Bioorg Med Chem Lett 1994; 4:1077-80; http://dx.doi.org/ 10.1016/S0960-894X(01)80684-3 [DOI] [Google Scholar]
- 85.Katritzky AR, Narindoshvili T. Chiral peptide nucleic acid monomers (PNAM) with modified backbones. Org Biomol Chem 2008; 6:3171-6; PMID:18698477; http://dx.doi.org/; http://dx.doi.org/ 10.1039/b806141f [DOI] [PubMed] [Google Scholar]
- 86.Menchise V, De Simone G, Tedeschi T, Corradini R, Sforza S, Marchelli R, Capasso D, Saviano M, Pedone C. Insights into peptide nucleic acid (PNA) structural features: the crystal structure of a D-lysine-based chiral PNA-DNA duplex. Proc Natl Acad Sci USA 2003; 100(21):12021-6; PMID:14512516; http://dx.doi.org/ 10.1073/pnas.2034746100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tedeschi T, Chiari M, Galaverna G, Sforza S, Cretich M, Corradini R, Marchelli R. Detection of the R553X DNA single point mutation related to cystic fibrosis by a “chiral box” D-lysine-peptide nucleic acid probe by capillary electrophoresis. Electrophoresis 2005; 26(22):4310-6; PMID:16287174; http://dx.doi.org/ 10.1002/elps.200410390 [DOI] [PubMed] [Google Scholar]
- 88.Corradini R., Feriotto G, Sforza S, Marchelli R, Gambari R. Enhanced recognition of cystic fibrosis W1282X DNA point mutation by chiral peptide nucleic acid probes by a surface plasmon resonance biosensor. J Mol Recognit 2004; 17(1):76-84; PMID:14872540; http://dx.doi.org/ 10.1002/jmr.646 [DOI] [PubMed] [Google Scholar]
- 89.Dose C, Seitz O. Single nucleotide specific detection of DNA by native chemical ligation of fluorescence labeled PNA-probes. Bioorg Med Chem 2008; 16:65-77; PMID:17499998; http://dx.doi.org/ 10.1016/j.bmc.2007.04.059 [DOI] [PubMed] [Google Scholar]
- 90.Zhou P, Dragulescu-Andrasi A, Bhattacharya B, O’Keefe H, Vatta P, Hyldig-Nielsen JJ, Ly DH. Synthesis of cell-permeable peptide nucleic acids and characterization of their hybridization and uptake properties. Bioorg Med Chem Lett 2006; 16:4931-5; PMID:16809033; http://dx.doi.org/ 10.1016/j.bmcl.2006.06.052 [DOI] [PubMed] [Google Scholar]
- 91.Dragulescu-Andrasi A, Rapireddy S, He G, Bhattacharya B, Hyldig-Nielsen JJ, Zon G, Ly DH. Cell-permeable peptide nucleic acid designed to bind to the 5'-untranslated region of E-cadherin transcript induces potent and sequence-specific antisense effects. J Am Chem Soc 2006; 128:16104-12; PMID:17165763; http://dx.doi.org/ 10.1021/ja063383v [DOI] [PubMed] [Google Scholar]
- 92.Rossi S, Calabretta A, Tedeschi T, Sforza S, Arcioni S, Baldoni L, Corradini R, Marchelli R. Selective recognition of DNA from olive leaves and olive oil by PNA and modified-PNA microarrays. Artificial DNA:PNA XNA 2012; 3:63-72; http://dx.doi.org/ 10.4161/adna.20603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Püschl A, Sforza S, Haaima G, Dahl O, Nielsen PE. Peptide nucleic acids (PNAs) with a functional backbone. Tetrahedron Lett 1998; 39:4707-10; http://dx.doi.org/ 10.1016/S0040-4039(98)00862-4 [DOI] [Google Scholar]
- 94.Hamzavi R, Dolle F, Tavitian B, Dahl O, Nielsen PE. Modulation of the pharmacokinetic properties of PNA: Preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers. Bioconjug Chem 2003; 14:941-54; PMID:13129397; http://dx.doi.org/ 10.1021/bc034022x [DOI] [PubMed] [Google Scholar]
- 95.Hamzavi R, Meyer C, Metzler-Nolte N. Synthesis of a C-linked glycosylated thymine-based PNA monomer and its incorporation into a PNA oligomer. Org Biomol Chem 2005; 4:3648-51; http://dx.doi.org/ 10.1039/b607463d [DOI] [PubMed] [Google Scholar]
- 96.Dilek I, Madrid M, Singh R, Urrea CP, Armitage BA. Effect of PNA backbone modifications on cyanine dye binding to PNA-DNA duplexes investigated by optical spectroscopy and molecular dynamics simulations. J Am Chem Soc 2005; 127:3339-45; PMID:15755150; http://dx.doi.org/ 10.1021/ja045145a [DOI] [PubMed] [Google Scholar]
- 97.Gupta P, Muse O, Rozners E, Recognition of Double-Stranded RNA by Guanidine-Modified Peptide Nucleic Acids, Biochemistry 2012; 51:63-73; PMID:22146072; http://dx.doi.org/ 10.1021/bi201570a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pokorski JK, Myers MC, Appella DH. Cyclopropane PNA: Observable triplex melting in a PNA constrained with a 3-membered ring. Tetrahedron Lett 2005; 46:915-7; http://dx.doi.org/ 10.1016/j.tetlet.2004.12.061 [DOI] [Google Scholar]
- 99.Myers MC, Witschi MA, Larionova NV, Frank JM, Haynes RD, Hara T, Grajkowski A, Appella DH. A cyclopentane conformational restraint for a peptide nucleic acid: Design, asymmetric synthesis, and improved binding affinity to DNA and RNA. Org Lett 2003; 5:2695-8; PMID:12868892; http://dx.doi.org/ 10.1021/ol0348811 [DOI] [PubMed] [Google Scholar]
- 100.Pokorski JK, Witschi MA, Purnell BL, Appella DH. (S,S)-trans-Cyclopentane-constrained peptide nucleic acids. A general backbone modification that improves binding affinity and sequence specificity. J Am Chem Soc 2004; 126:15067-73; PMID:15548003; http://dx.doi.org/ 10.1021/ja046280q [DOI] [PubMed] [Google Scholar]
- 101.Govindaraju T, Kumar VA, Ganesh KN. cis-Cyclopentyl PNA (cpPNA) as constrained chiral PNA analogues: Stereochemical dependence of DNA/RNA hybridiation. Chem Commun 2004; 860-1; http://dx.doi.org/ 10.1039/b317000d [DOI] [PubMed] [Google Scholar]
- 102.Govindaraju T, Kumar VA, Ganesh KN. (1S,2R/1R,2S)-cis-cyclopentyl PNAs (cpPNAs) as constrained PNA analogues: synthesis and evaluation of aeg-cpPNA chimera and stereopreferences in hybridization with DNA/RNA. J Org Chem 2004; 69:5725-34; PMID:15307746; http://dx.doi.org/ 10.1021/jo049442+ [DOI] [PubMed] [Google Scholar]
- 103.Lagriffoule P, Wittung P, Ericksson M, Jensen DK, Norden B, Buchardt O, Nielsen PE. Peptide nucleic acids with a conformationally constrained chiral cyclohexyl-derived backbone. Chem Eur J 1997; 3:912-9; http://dx.doi.org/ 10.1002/chem.19970030613 [DOI] [Google Scholar]
- 104.Govindaraju T, Gonnade RG, Bhadbhade MM, Kumar VA, Ganesh KN. (1S,2R/1R,2S)-Aminocyclohexyl Glycyl Thymine PNA: Synthesis, Monomer Crystal Structures, and DNA/RNA Hybridization Studies. Org Lett 2003; 5:3013-6; PMID:12916969; http://dx.doi.org/ 10.1021/ol034933m [DOI] [PubMed] [Google Scholar]
- 105.Govindaraju T, Kumar VA, Ganesh KN. Synthesis and Evaluation of (1S,2R/1R,2S)-Aminocyclohexylglycyl PNAs as Conformationally Preorganized PNA Analogues for DNA/RNA Recognition. J Org Chem 2004; 69:1858-65; PMID:15058930; http://dx.doi.org/ 10.1021/jo035747x [DOI] [PubMed] [Google Scholar]
- 106.Govindaraju T, Kumar VA, Ganesh KN. (SR/RS)-Cyclohexanyl PNAs: Conformationally Preorganized PNA Analogues with unprecedented preference for duplex formation with RNA. J Am Chem Soc, 2005; 127(12):4144-5; PMID:15783176; http://dx.doi.org/ 10.1021/ja044142v [DOI] [PubMed] [Google Scholar]
- 107.Sugiyama T, Imamura Y, Demizu Y, Kurihara M, Takano M, Kittaka A. β-PNA: peptide nucleic acid (PNA) with a chiral center at the β-position of the PNA backbone. Biorg Med Chem Lett 2011; 21:7317-20; http://dx.doi.org/ 10.1016/j.bmcl.2011.10.017 [DOI] [PubMed] [Google Scholar]
- 108.Kosynkina L, Wang W, Liang TC. A convenient synthesis of chiral Peptide Nucleic Acid (PNA) monomers. Tetrahedron Lett 1994; 35(29):5173-6; http://dx.doi.org/ 10.1016/S0040-4039(00)77056-0 [DOI] [Google Scholar]
- 108.Englund EA, Appella DH. γ -Substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew Chem Int Ed 2007; 46:1414-141; http://dx.doi.org/ 10.1002/anie.200603483 [DOI] [PubMed] [Google Scholar]
- 110.Englund EA, Appella DH. Synthesis of γ-substituted peptide nucleic acids: A new place to attach fluorophores without affecting DNA binding. Org Lett 2005; 7:3465-7; PMID:16048318; http://dx.doi.org/ 10.1021/ol051143z [DOI] [PubMed] [Google Scholar]
- 111.De Koning MC, Petersen L, Weterings JJ, Overhand M, Van der Marel GA, Filippov DV. Synthesis of thiol-modified peptide nucleic acids designed for post-assembly conjugation reactions. Tetrahedron 2006; 62:3248-58; http://dx.doi.org/ 10.1016/j.tet.2006.01.065 [DOI] [Google Scholar]
- 112.Dose C, Seitz O. Convergent Synthesis of Peptide Nucleic Acids by Native Chemical Ligation. Org Lett 2005; 7:4365-8; PMID:16178534; http://dx.doi.org/ 10.1021/ol051489+ [DOI] [PubMed] [Google Scholar]
- 113.Pensato S, Saviano M, Bianchi N, Borgatti M, Fabbri E, Gambari R, Romanelli A. A γ-hydroxymethyl PNAs: Synthesis, interaction with DNA and inhibition of protein/DNA interaction. Bioorg Chem 2010; 38:196-201; PMID:20643471; http://dx.doi.org/ 10.1016/j.bioorg.2010.06.002 [DOI] [PubMed] [Google Scholar]
- 114.Avitabile C, Moggio L, Malgieri G, Capasso D, Gaetano S.D, Saviano M, Pedone C, Romanelli A. γ-sulphate PNA (PNA S): Highly selective DNA binding molecule showing promising antigene activity. PLoS One 2012; 7(5):e35774; PMID:22586450; http://dx.doi.org/ 10.1371/journal.pone.0035774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sforza S, Tedeschi T, Corradini R, Marchelli R. Induction of helical handedness and DNA binding properties of peptide nucleic acids (PNAs) with two stereogenic centres. Eur J Org Chem 2007; 5879-85; http://dx.doi.org/ 10.1002/ejoc.200700644 [DOI] [Google Scholar]
- 116.Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure. J Am Chem Soc 2006; 128:10258-67; PMID:16881656; http://dx.doi.org/ 10.1021/ja0625576 [DOI] [PubMed] [Google Scholar]
- 117.Crawford MJ, Rapireddy S, Bahal R, Sacui I, Ly DH. Effect of steric constraint at the γ-backbone position on the conformations and hybridization properties of PNAs. J Nucleic Acids 2011; 2011:652702; PMID:21776375; http://dx.doi.org/ 10.4061/2011/652702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chenna V, Rapireddy S, Sahu B, Ausin C, Pedroso E, Ly DH. A simple cytosine to G-clamp nucleobase substitution enables chiral γ -PNAs to invade mixed-sequence double-helical B-form DNA. ChemBioChem 2008; 9:2388-91; PMID:18816545; http://dx.doi.org/ 10.1002/cbic.200800441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rapireddy S, He G, Roy S, Armitage BA, Ly DH. Strand invasion of mixed-sequence B-DNA by acridine-linked, γ-peptide nucleic acid (γ-PNA). J Am Chem Soc 2007; 129:15596-600; PMID:18027941; http://dx.doi.org/ 10.1021/ja074886j [DOI] [PubMed] [Google Scholar]
- 120.Bahal R, Sahu B, Rapireddy S, Lee CM, Ly DH. Sequence-unrestricted, Watson–Crick recognition of double helical B-DNA by (R)-MiniPEG-γPNAs. ChemBioChem 2012; 13:56-60; PMID:22135012; http://dx.doi.org/ 10.1002/cbic.201100646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sahu B, Chenna V, Lathrop KL, Thomas SM, Zon G, Livak KJ, Ly DH. Synthesis of Conformationally Preorganized and Cell-Permeable Guanidine-Based γ-Peptide Nucleic Acids (γGPNAs). J Org Chem 2009; 74(4):1509-16; PMID:19161276; http://dx.doi.org/ 10.1021/jo802211n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sforza S, Tedeschi T, Calabretta A, Corradini R, Camerin C, Tonelli R, Pession A, Marchelli R. A peptide nucleic acid embedding a pseudopeptide nuclear localization sequence in the backbone behaves as a peptide mimic. Eur J Org Chem 2010; 2010:2441-44; http://dx.doi.org/ 10.1002/ejoc.201000123 [DOI] [Google Scholar]
- 123.Manicardi A, Fabbri E, Tedeschi T, Sforza S, Bianchi N, Brognara E, Gambari R, Marchelli R, Corradini R. Cellular uptakes, biostabilities and anti-miR-210 activities of chiral arginine-PNAs in leukaemic K562 cells. ChemBioChem 2012; 13:1327-37; PMID:22639449; http://dx.doi.org/ 10.1002/cbic.201100745 [DOI] [PMC free article] [PubMed] [Google Scholar]