

Abstract
Objective:
To determine the average age of MS onset vs the age at which Epstein-Barr infection has previously occurred and stratify this analysis by sex and the blood level of Epstein-Barr nuclear antigen 1 (EBNA1) antibody.
Methods:
Using infectious mononucleosis (IM) as a temporal marker in data from the Swedish epidemiologic investigation of MS, 259 adult IM/MS cases were identified and then augmented to account for “missing” childhood data so that the average age of MS onset could be determined for cases binned by age of IM (as stratified by sex and EBNA1 titer level).
Results:
Mean age of IM vs mean age of MS reveals a positive time correlation for all IM ages (from ∼5 to ∼30 years), with IM-to-MS delay decreasing with increased age. When bifurcated by sex or EBNA1 blood titer levels, males and high-titer subpopulations show even stronger positive time correlation, while females and low-titer populations show negative time correlation in early childhood (long IM/MS delay). The correlation becomes positive in females beyond puberty.
Conclusions:
IM/MS time correlation implies causality if IM is time random. Alternative confounding models seem implausible, in light of constraints imposed by time-invariant delay observed here. Childhood infection with Epstein-Barr virus (EBV) in females and/or those genetically prone to low EBNA1 blood titers will develop MS slowly. Males and/or high EBNA1-prone develop MS more rapidly following IM infection at all ages. For all, postpubescent EBV infection is critical for the initiation and rapid development of MS.
The association of Epstein-Barr virus (EBV) with MS has been recognized for many years. Investigation of the association between MS and a large number of infectious agents, including EBV, indicated that EBV presented by far the greatest risk (risk ratio) for MS among all of the agents studied.1 Adult MS cases are rarely EBV-negative (seronegative), and among all such cases, the incidence of EBV negativity is 16 times lower than that in control populations.1 In the most thorough study showing “EBV before MS,” 305 MS cases were serologically tracked before onset, and all showed seroconversion.2 Prior studies had indicated that “late-onset” EBV (adolescent years infection) presented a particularly high risk, especially, but not exclusively, associated with infectious mononucleosis (IM), a prominent clinical syndrome following infection with EBV.
IM had also been strongly associated with MS for many years, but in 2007, the first full population study was reported for the Danish population.3 That study of 104 IM/MS cases reported a standardized incidence ratio (SIRIM) for MS among those with IM of 2.27. The study cross-referenced IM cases confirmed with the Paul-Bunnell test for heterophilic antibody with reported MS cases. The data were accumulated over several decades and inclusive of MS diagnosis at all ages.
Despite this significant progress, definitive proof of EBV as a causative agent for MS has remained elusive. Recent evidence of a different type for a causal relationship is the observation of “interaction” between MS ORs for IM, and ORs for the MS risk allele human leukocyte antigen(HLA) DRB1*15.4 Equally important is the interaction between the OR for this same allele and the OR for high Epstein-Barr nuclear antigen-antibody level (specifically, the Epstein-Barr nuclear antigen 1 [EBNA1] blood titer level).5 Together, these 2 studies provide added evidence for a strong, perhaps causative, relationship between both the IM and non-IM forms of EBV and MS.
However, DRB1*15 may be “interactive” in only ∼43% of (Swedish) MS cases. We speculate here that (IM and non-IM) late EBV may be the more universal MS trigger for most MS, regardless of specific MS genetic (HLA or other) susceptibility. The objectives of this article are twofold. First, using the recently developed Swedish epidemiologic investigation of MS (EIMS) data, we analyzed the 259 IM/MS case subpopulation using IM as a tag on age at EBV to show time correlation. We further investigated the data set bifurcated by sex and by the EBNA1 titer level. The purpose of this objective is to characterize the nature of the temporal development of the disease while also providing evidence for detailed IM/MS time correlation. The strength of the evidence for consideration of EBV as causal then rests on whether evidence exists that EBV (IM) infection can be shown to be predominantly time random; that is, particularly in the physical sciences, if “B” is shown to always occur at a predictable time after “A,” and if “A” is shown to be time random, then this is considered very strong evidence for causation of “B” by “A.”
The second objective of this article is to examine the EIMS distribution data of non–IM MS cases to determine whether the implied distribution of age of initiating (non-IM) EBV is similar to or significantly different from the known distribution of age of IM; that is, is non-IM EBV (in MS cases) also occurring primarily in the teen years?
METHODS
We detail here 3 different methodologies used in this study.
Methodology of the EIMS program and restoration of childhood MS data.
The Swedish EIMS program was established in 2005 with a goal of tracking all identifiable MS cases in Sweden and of gathering specific data on this full population of MS cases. These data include the age at first symptom and the age at diagnosis of IM (if it has occurred) through retrospective questionnaires at the time of diagnosis. Serologic data, including EBNA1 titers,5 are also taken at diagnosis. Till date, 2,600 cases and 5,200 controls have accrued under this study.
The EIMS program is ongoing, but as of 2013, the study had accumulated 259 IM/MS cases and 1526 total MS cases that we had accessed. The IM/MS subpopulation retrospectively determined MS onset age, and that is always the age reported in this article. The full (IM/non-IM) EIMS population data record diagnostic age only, as it is impractical to determine onset for this larger number. One case in the 259 IM/MS subpopulation showed MS onset before IM, and it was deleted from our study. This deleted case (atypical) was diagnosed for MS and IM at the same age, 44 years, the oldest IM case by 4 years. MS onset, for this case was retrospectively, that is, through recollection at diagnosis (age 44 years) and examination of prior medical records, to have occurred at age 28 years.
The strength of the EIMS program is in its ability to generate an accurate sampling of the full population. Weakness includes the need to use retrospective questionnaires in gathering data. Two additional issues, as they relate to aspects of this study, include the exclusion of data for minors (younger than 18 years) from the collected data owing to privacy laws and the indirect exclusion of possible IM after MS from the data owing to those cases necessarily being classified as EBV (not IM) negative. Restorations of these data are critical to the accuracy of our present undertaking. We may briefly summarize these restorations here while deferring detail to appendix e-1 at Neurology.org/nn. For pediatric restoration, and based on the literature, we assume that 5% of total MS cases (Sweden) experience diagnosis before age 18 years, thus introducing 13.5 pediatric cases to the EIMS total of 258. These 13.5 added cases are then allocated by sex, age at MS onset, and by implication, age at EBV, according to the onset and sex data reported in recent studies of pediatric MS.6,7 Conclusions in this article are insensitive to these allocations. Restoration level for IM-negative cases is shown to be insignificant based on the extremely low incidence of EBV– cases observed by EIMS.
Methodology for treating IM-MS delay.
We introduce here, as detailed in appendix e-2, a simple concept and formalism that enables quantitative definition of the relationships between the distribution of onset of MS (termed OMS), the delay distribution of the time from EBV infection to MS onset (termed DMS), and the distribution of initiating EBV (IM) infection (termed OEB). With this formalism, it is reasonably argued that those distributions are reproducible and meaningful within a large homogeneous population. It can also be argued that over short periods of EBV infection through puberty (≃2–3 years) or longer periods after age 20 (∼5–10) years, DMS (delay) will remain approximately unchanged.
The principal metric that we report here in figures 1 and e-2.1 is the plot of average age of MS onset vs average age of IM, binned by 5-year increments of age at IM. For each such IM bin, we also generate a delay distribution, DMS (delay), such that, for example:
Figure 1. Average age at MS vs average binned age at infectious mononucleosis, stratified by sex (epidemiologic investigation of MS).

Bracketed numbers indicate case number for datum. Fractions result from pediatric correction (ΔP). EBV = Epstein-Barr virus; IM = infectious mononucleosis; P = pediatric.
In the event <DMS>IM, j were constant for all EBV infection ages then <OMS>j vs binned <OEB>j would be a straight line at 45° angle, indicating what in the physical sciences is referred to as time invariant delay (TID).
We cite the idealized case, “time invariance,” because that is the strongest indicator that we have of strong time correlation. In this regard, standard statistical metrics are ambiguous in dealing with this requirement. For example, the scatter plot of all 258 EIMS IM/MS cases has a time correlation coefficient of 0.2 (Pearson; per A.K. Hedström, Karolinska). But what does that mean (figure e-2.3)? Statistical uncertainty in the average <DMS> values may be approximated by 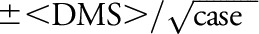 and that is presented in a tabular form (appendix e-2, table e-2.1). Yet it is conformance to the TID ideal that is of great interest, and none of these other statistical metrics speak to that qualitative metric.
and that is presented in a tabular form (appendix e-2, table e-2.1). Yet it is conformance to the TID ideal that is of great interest, and none of these other statistical metrics speak to that qualitative metric.
The ability to use the IM/MS population to generate a family of delay distributions, DMSIM bin (delay), presented an opportunity to derive OEBfull (age) for a full (IM and non-IM) population. This methodology may be summarized. Assuming that the delay distributions of EBV and IM forms of initiation are approximately the same, we follow an iterative procedure of varying OEBfull, bin by bin, until the convolution of these bin populations with the delay distributions yields OMSfull (age).
The derivation of OEBfull (age) also required a “conversion,” of the EIMS “diagnostic” distribution to an “onset” distribution. This and associated methodology detail are presented in appendices e-2 and e-3.
Standard protocol approvals, registrations, and patient consents.
Institutional review board approval was not required for this study.
RESULTS
Figure 1 shows average age of MS onset vs average age of IM, binned in 5-year IM increments, and stratified by sex. The combined-sex plot is shown in figure e-2.1, and a plot of all 258 individual EIMS cases vs age at IM and at MS is given in figure e-2.3. Fractional case numbers for these data result from our approximation that pediatric cases missing from EIMS represent exactly 5% of total cases (258 EIMS + 13.5 pediatric). The male plot of <MS> vs <IM> is matched to linear and almost time invariant over all infection ages. The female data are noteworthy because they reflect a rapid drop in disease development time (<DMS>), starting with extreme (25.4 years) delay for early (5 years) infection, dropping down to only an 8-year delay for adult (∼25 years) infection, and indicating a uniquely severe transition through puberty for females. Statistical uncertainty in delay  is shown for the most uncertain stratifications (male, figure 1, and both EBNA1, figure 2).
is shown for the most uncertain stratifications (male, figure 1, and both EBNA1, figure 2).
Figure 2. Age at MS vs average binned age of infectious mononucleosis, stratified by Epstein-Barr virus nuclear antigen 1 titer level (epidemiologic investigation of MS).

Bracketed numbers indicate case number for datum. IM = infectious mononucleosis.
Figure 2 shows the average age of MS onset vs average age of IM, stratified by the EBNA1 titer level. These data could not be pediatric corrected. EIMS EBNA1 immunoglobulin G (IgG) (total) measurements significantly predated other data reported here. Consequently, a smaller sample of only 99 measurements exists, randomly distributed among our 259 cases. “High” and “low” titers refer to whether the measurement was above or below the median value measured for controls,5 with a high/low case number ratio of approximately 2.4:1. The “high-titer” data show a similar disease development behavior vs time of infection as seen in males. The “low-titer” disease development is similar to that seen in females. The EBNA1 blood titer level may be, like sex, primarily a genetic marker; that is, titer level variation may reflect genetic-based variation in response to EBV infection, with a high EBNA1 level suggesting a deficient immune response. However, it is also possible that the EBNA1 titer level variance reflects EBV variants.
The IM-to-MS delay distribution (DMS) is shown in figure 3 for select IM age ranges (<10 years and >20 years) and for males and females. “Black” cases represent pediatric cases (correction). Notable is detail on the large drop in IM-MS delay from childhood infection to adult infection. This effect is particularly strong in females. Note that F/M ratio for infection under age 10 years is comparable (at ×2.64) to adult values. However, this F/M ratio peaks dramatically at 4.6, IM age 11–15 years (figure e-2.3). This peak may be modulated by social factors affecting male, female ages at infection. Of note, the “low EBNA1” female cases for early IM show extreme delay.
Figure 3. Infectious mononucleosis to MS delay distribution (DMS) for childhood, adulthood, male, and female.

Childhood: (A) female and (B) male. Childhood female to male ratio = 35.2/13.3 = 2.64. Adulthood: (C) female and (D) male. ΔP = pediatric correction. EBNA 1 = Epstein-Barr nuclear antigen 1; IM = infectious mononucleosis; P = pediatric.
Figure 4 summarizes prior data on a single graph that emphasizes a significant biologic transition in response to EBV infection that occurs in early puberty (age 10–15 years). The transition is strongest for females and results in a twofold increase in MS incidence vs that in males (for that peak period) and greater than a twofold drop in IM-to-MS delay time from childhood through adolescence.
Figure 4. Summary of data suggests immunologic transition for Epstein-Barr virus infections, age 10 to 15 years.

EBV = Epstein-Barr virus; D (#) = delay (at # years); IM = infectious mononucleosis; Yrs = years.
Our primary objective is exploration of whether there is a strong time correlation between age at IM (EBV) and MS onset, while also investigating potential differences in disease development with the age of infection. A secondary objective has been to use these same EIMS data (including MS cases without prior IM) to derive a best estimate of the EBV infection profile vs age for all MS cases. This iterative deconvolution, detailed in the appendix, resulted in the EBV infection distribution (OEBfull) of figure 5A. OEBIM is also shown for comparison. The OEBfull distribution, like the OEBIM distribution, is suppressed at early ages (<10 years), serving to both quantify and thus better define the environmental risk factor, “late EBV.” In this sense, IM may be thought of as a temporal or biologic tag on late EBV. The similarity between OEBIM and OEBfull should not be a surprise. Significant EBV serologic evidence has accrued in recent years that IM and non–IM-MS cases are very similar. The EIMS program has shown that mean EBNA1 IgG titer levels for IM and non–IM-MS cases (155, 150) are indistinguishable but significantly higher than those for non-MS controls (128, 127) (absorbance units, unpublished). The interaction between ORs for the EBNA1 titer level and HLA DRB1*15 status, applicable to both IM and non-IM cases, was also previously noted.5
Figure 5. Infection and delay distributions.

(A) OEBfull and OEBIM. (B) DMSIM, EIMS, and Denmark.3 EIMS = epidemiologic investigation of MS; EBV = Epstein-Barr virus; IM = infectious mononucleosis; P = pediatric; ΔP = pediatric correction (5% of total MS).
Figure 5B provides the full delay distribution (DMStotal) for all IM bins so as to allow comparison with the prior Danish study.3 Differences noted are thought to be primarily due to the diagnosis (Denmark) vs onset (EIMS) definitions of MS initiation.
DISCUSSION
These results suggest that MS disease development reflects immune system development in a dynamic way for both IM and non-IM forms of EBV initiation. While males and those with high EBNA1 blood titer levels (measured at diagnosis) show gradual reduction in IM-to-MS delay, from childhood to adult infection, low-titer cases and females show a marked acceleration in disease development, from childhood to adulthood. This is quite consistent with the many who have observed that estrogen and female puberty are particularly strong risk factors for MS. The overall association of infection during puberty with accelerating MS development is made most clear in figure 4 which shows the early teen years (particularly for females) as a period of very rapid change (in this regard, it is interesting that the late teens appear to reflect a period of relaxation of this phenomenon for females as the sex ratio subsides). All these are consistent with our hypothesis here that late EBV is the common environmental risk factor giving rise to enhanced MS risk (e.g., the SIRIM), whether from the IM or non-IM forms of EBV. We have attempted, in figure 5A, to quantify late EBV for both IM and non-IM forms, and by doing so, show their distributive similarity.
An illuminating model positing CD8+ T-cell deficiency stands today as the sole substantive example for what might explain the unique risk of “late EBV infection.”8 This model of “CD8+ deficiency” describes that this “deficiency” becomes worse through the teen years and is exacerbated by higher levels of estrogen. It is also consistent with the observation of high EBNA1 titer levels (implying ineffective CD8+ cells) as an MS risk factor. Finally, it is consistent with similar observations regarding the role of CD8+ in the EBV-causal form of lymphoma.9
The low-titer curve (figure 2) provides an important result with IM-MS delay dropping from 28 years to 7.5 years in only 17 years. The data are, however, compromised by very low sample size and an inability to do pediatric correction. Low titer would not be reflective of CD8+ deficiency, as presumably, these individuals have relatively low infected B-cell counts. Yet these cases do get MS: slowly for early infection, rapidly when infected in the late teens. MS is a complex autoimmune disease with first-order differences in development based on the HLA DRB1*15 status, A*02 status, sex status, and the related EBNA1 titer levels. It is feasible that low-titer cases reflect individuals who are susceptible to MS despite an absence of serious CD8+ deficiency. Possible explanations for these low-titer cases include increased reactivity to myelin mimics or a blood-brain barrier that is more susceptible to passage of immune cells (increased vascular cell adhesion molecule on the endothelium). Whatever these added speculative susceptibilities, as the CD8+ count drops through the teen years, these cases succumb, and the rapid MS development may reflect these added vulnerabilities. We make 2 assumptions in the preceding discussion. Titer level at diagnosis correlates with the titer level at initial infection, and second, titer level at infection is likely the strongest indicator that MS will ensue. Support for these hypotheses may be found in 2 recent studies.10,11
The discussion has focused, thus far, on disease development assuming MS is triggered by EBV. We return now to that primary objective and question. The most basic metric for time correlation, “no MS before EBV,” that is, “immunity studies”2 have been the sole means to date for detecting such correlation. The most thorough of these immunity studies is perhaps the 2010 study which used U.S. Army–archived blood samples to provide longitudinal data on serologic history for 305 MS cases.2 All cases were EBV positive before MS, with 10 seroconverting within the time range accessed by available blood samples. This investigation provided strong evidence supporting the premise that “EBV occurs before MS” while also providing the first data on EBV-MS delay. We have introduced here a distinctly different (from immunity studies) yet complementary methodology, TID detection. Differences between these metrics are quantified in appendix e-4.12–14 Both methods are vulnerable to noncausal “confounding” hypotheses (regarding immunity studies, a common confounding phenomenon causes MS and is hypothesized to also cause early EBV susceptibility). We make here and in detail in appendix e-4 2 counter arguments.15–17 First, beneficial to either method, EBV is inherently time random. Second, TID detection demands a qualitatively more complex confounding, hypothesized to cause a weak form (both MS and EBV occur later) and strong (MS and EBV occur early). This seems highly unlikely. We, therefore, view TID detection in all males and high EBNA1 titer level cases and in postpubescent females as providing significant evidence for a causal EBV-MS relationship.
If IM is time random (as argued in appendix e-4) and we detect TID, then it follows that random delay in IM to very old age results in no MS; absent IM, no MS occurs. We summarize here our strongest argument for “random” IM and a causal relationship. EBV infection is triggered by saliva transfer in a manner that age of infection is heavily modulated in a statistical (i.e., time random) manner by social norms and ages at various forms of socialization. Data support increased infection at periods of initial schooling or dating. The very strong time correlation between age of IM and age of MS for all males and for all postpubescent females can thus only be simply and easily explained by hypothesizing an IM/MS causal relationship if, as appears to be the case, the initial IM infections are occurring in a time-random fashion.
Generation, here, of IM-MS delay distributions has enabled determination, through deconvolution, of the IM, non-IM EBV distribution, OEBfull. Like OEBIM, OEBfull is late EBV, as both distributions experience >75% of their infections beyond age 10 years; that is, in a pubescent or postpubescent timeframe. By contrast, less than 45% of a general population (e.g., white non-Hispanic Americans)17 show infection after age 10 years. This commonality of “late” infection and the large body of similar EBV serologic data for IM and non–IM-MS cases argues that the evidence for IM as causal should logically be extended to all late ([post] pubescent) EBV infections as causal; that is, as necessary.
This article accomplishes 4 things. First, it strengthens the argument for IM (EBV) as causal by qualitatively increasing the demands on confounding explanations for EBV-MS time correlation. Absent evidence for TID: confounding models need only claim earlier EBV infection in all the MS susceptible in noncausal explanations for time correlation seen in immunity studies. With TID, these models must also explain later EBV in late-developing MS and stay in synchronicity with MS development variation at all ages. Second, cited evidence for IM as time random obviates all confounding counterarguments for causality. Third, this article gives specific insight into disease development time, stratified by sex, age at infection, and EBNA1 titer level, from IM infection to MS onset. Finally, this article provides evidence that its conclusions are equally applicable to both IM and non-IM forms of EBV.
Although incidental to our primary objectives, the EIMS EBV− risk ratio data presented here (table e-1.4) serve to reinforce results of the most comprehensive prior “immunity study,”1 while largely explaining recently reported discrepancies12 from that study.
The perspective of this study uses analytic strategies common to engineering and the physical sciences and stands distinct from treatment of this issue with the tools of epidemiology. While epidemiology generally shuns the notion of causality, the physical and engineering scientists do not shy away from this issue. It is illuminating to contrast these nuances, if not polarities, in analysis of a complex medical issue.
ACKNOWLEDGMENT
This article would not have been possible without access to the Swedish EIMS data. The authors thank Professors Hans Wigzell and Lars Alfredsson for their roles in that decision and its implementation. They also thank Professor Alberto Ascherio for first making them aware of the significant epidemiologic studies being conducted on MS and IM, as that suggestion served to initiate this work.
GLOSSARY
- EBNA1
Epstein-Barr nuclear antigen 1
- EBV
Epstein-Barr virus
- EIMS
epidemiologic investigation of MS
- HLA
human leukocyte antigen
- IgG
immunoglobulin G
- IM
infectious mononucleosis
- SIR
standardized incidence ratio
- TID
time-invariant delay
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
J.E. and L.S. contributed to the conception and design of the study. J.E., P.P.H., and L.S. contributed to the drafting of the manuscript and figures.
STUDY FUNDING
No targeted funding.
DISCLOSURE
J. Endriz and P.P. Ho report no disclosures. L. Steinman served on the scientific advisory boards for Novartis, Receptos, Atreca, Tolerion, Teva, and AbbVie; received travel funding and/or speaker honoraria from Celgene and AbbVie; served on the editorial boards for MS Journal and Proceedings of the National Academy of Sciences; has a patent pending on cytokines and type 1 interferons; holds multiple patents on antigen-specific tolerance; received research support from Atara, Celgene, and Biogen; and holds stock options and board membership in Tolerion Board of Directors Bioatla. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis: part I: the role of infection. Ann Neurol 2007;61:288–299. [DOI] [PubMed] [Google Scholar]
- 2.Levin LI, Munger KL, O'Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol 2010;67:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen TR, Rostgaard K, Nielsen NM, et al. Multiple sclerosis after infectious mononucleosis. Arch Neurol 2007;64:72–75. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen TR, Rostgaard K, Askling J, et al. Effects of infectious mononucleosis and HLA-DRB1*15 in multiple sclerosis. Mult Scler 2009;15:431–436. [DOI] [PubMed] [Google Scholar]
- 5.Sundqvist E, Sundstrom P, Linden M, et al. Epstein-Barr virus and multiple sclerosis: interaction with HLA. Genes Immun 2012;13:14–20. [DOI] [PubMed] [Google Scholar]
- 6.Ghezzi A, Deplano V, Faroni J, et al. Multiple sclerosis in childhood: clinical features of 149 cases. Mult Scler 1997;3:43–46. [DOI] [PubMed] [Google Scholar]
- 7.Banwell B, Krupp L, Kennedy J, et al. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol 2007;6:773–781. [DOI] [PubMed] [Google Scholar]
- 8.Pender MP. CD8+ T-cell deficiency, epstein-barr virus infection, vitamin D deficiency, and steps to autoimmunity: a unifying hypothesis. Autoimmune Dis 2012;2012:189096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hjalgrim H, Rostgaard K, Johnson PC, et al. HLA-A alleles and infectious mononucleosis suggest a critical role for cytotoxic T-cell response in EBV-related Hodgkin lymphoma. Proc Natl Acad Sci USA 2010;107:6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munger KL, Levin LI, O'Reilly EJ, Falk KI, Ascherio A. Anti-Epstein-Barr virus antibodies as serological markers of multiple sclerosis: a prospective study among United States military personnel. Mult Scler 2011;17:1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salzer J, Nystrom M, Hallmans G, Stenlund H, Wadell G, Sundstrom P. Epstein-Barr virus antibodies and vitamin D in prospective multiple sclerosis biobank samples. Mult Scler 2013;19:1587–1591. [DOI] [PubMed] [Google Scholar]
- 12.Pakpoor J, Disanto G, Gerber JE, et al. The risk of developing multiple sclerosis in individuals seronegative for Epstein-Barr virus: a meta-analysis. Mult Scler 2013;19:162–166. [DOI] [PubMed] [Google Scholar]
- 13.Pohl D, Krone B, Rostasy K, et al. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology 2006;67:2063–2065. [DOI] [PubMed] [Google Scholar]
- 14.Deuschle K, Hofmann J, Otto C, et al. Are there Epstein-Barr virus seronegative patients with multiple sclerosis? Mult Scler 2013;19:1242–1243. [DOI] [PubMed] [Google Scholar]
- 15.Rubicz R, Yolken R, Drigalenko E, et al. A genome-wide integrative genomic study localizes genetic factors influencing antibodies against Epstein-Barr virus nuclear antigen 1 (EBNA-1). PLoS Genet 2013;9:e1003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorner M, Zucol F, Berger C, et al. Distinct ex vivo susceptibility of B-cell subsets to epstein-barr virus infection according to differentiation status and tissue origin. J Virol 2008;82:4400–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balfour HH Jr, Sifakis F, Sliman JA, Knight JA, Schmeling DO, Thomas W. Age-specific prevalence of Epstein-Barr virus infection among individuals aged 6–19 years in the United States and factors affecting its acquisition. J Infect Dis 2013;208:1286–1293. [DOI] [PubMed] [Google Scholar]