

Abstract
Polymorphisms in the PTPN11 gene encoding for the tyrosine phosphatase SHP‐2 were described in patients with ulcerative colitis. We have recently demonstrated that mice with an intestinal epithelial cell‐specific deletion of SHP‐2 (SHP‐2IEC‐KO) develop severe colitis 1 month after birth. However, the mechanisms by which SHP‐2 deletion induces colonic inflammation remain to be elucidated. We generated SHP‐2IEC‐KO mice lacking Myd88 exclusively in the intestinal epithelium. The colonic phenotype was histologically analyzed and cell differentiation was determined by electron microscopy and lysozyme or Alcian blue staining. Microbiota composition was analyzed by 16S sequencing. Results show that innate defense genes including those specific to Paneth cells were strongly up‐regulated in SHP‐2‐deficient colons. Expansion of intermediate cells (common progenitors of the Goblet and Paneth cell lineages) was found in the colon of SHP‐2IEC‐KO mice whereas Goblet cell number was clearly diminished. These alterations in Goblet/intermediate cell ratio were noticed 2 weeks after birth, before the onset of inflammation and were associated with significant alterations in microbiota composition. Indeed, an increase in Enterobacteriaceae and a decrease in Firmicutes were observed in the colon of these mice, indicating that dysbiosis also occurred prior to inflammation. Importantly, loss of epithelial Myd88 expression inhibited colitis development in SHP‐2IEC‐KO mice, rescued Goblet/intermediate cell ratio, and prevented NFκB hyperactivation and inflammation. These data indicate that SHP‐2 is functionally important for the maintenance of appropriate barrier function and host‐microbiota homeostasis in the large intestine. J. Cell. Physiol. 231: 2529–2540, 2016. © 2016 The Authors. Journal of Cellular Physiology published by Wiley Periodicals, Inc.
Crohn's disease (CD) and ulcerative colitis (UC) are multifactorial inflammatory bowel diseases, involving various interactions among genetic, luminal, and environmental factors that lead to dysregulated inflammation (Kaser et al., 2010). Recent genome‐wide association studies have highlighted the important contribution of genetic susceptibility in development of these diseases. These studies have identified 163 independent loci for IBD including 110 loci linked to both CD and UC. This suggests common pathways in CD and UC pathogenesis, although differences in clinical phenotypes remain (Cho and Brant, 2011; Coskun, 2014). Thirty gene loci have been classified as CD specific and 23 as UC specific. CD is associated with abnormal intracellular processing of bacteria, autophagy, and innate immunity, whereas UC is associated with epithelial barrier dysfunction.
Recently, tyrosine phosphatase (PTP) variants in the PTPN22, PTPN2, and PTPN11 genes were associated with IBD onset (Spalinger et al., 2015). In particular, intronic polymorphisms in the PTPN11 gene encoding for the tyrosine phosphatase SHP‐2 were described in Japanese patients with UC (Narumi et al., 2009). However, the impact of these polymorphisms on SHP‐2 function was not elucidated. The authors speculated that PTPN11 polymorphisms may change the expression, activity, or binding of SHP‐2 to receptors in T and B cells. However, this phosphatase is not only expressed in immune cells but also in intestinal epithelial cells (IECs). Importantly, IECs are critical in the maintenance of immune homeostasis in the intestine. Indeed, they form a chemical and physical barrier separating luminal microbes and immune cells, and participate in local inflammation response following a mucosal insult (Peterson and Artis, 2014). We thus recently analyzed the role of SHP‐2 in this tissue by generating mice with an IEC‐specific deletion of SHP‐2 expression. These mice rapidly develop inflammation 1 month after birth, with histopathological features typical of UC (Coulombe et al., 2013). Of note, inflammation was not detected in the small intestine. Additionally, we found reduced SHP‐2 expression in intestinal biopsies from patients with active UC, emphasizing the inverse correlation between SHP‐2 levels and colonic inflammation (Coulombe et al., 2013). However, the exact molecular mechanisms by which SHP‐2 epithelial deletion induces chronic inflammation in the colon remain to be elucidated.
Our objective in this study was to further characterize the mechanisms by which SHP‐2 epithelial deletion induces chronic colonic inflammation in mice. We observed that 2 weeks after birth, SHP‐2IEC‐KO neonates feature reduced Goblet cell numbers associated with increased expression of several antimicrobial peptides (α‐defensins, Reg3γ, Reg3β, and lysozyme) as well as expansion of Paneth cells in their small intestine and of intermediate cells in the colon. Microbiota composition was changed in SHP‐2IEC‐KO mice. Specifically, an increase in Enterobacteriaceae and a reduction in Firmicutes were observed in mutant mice, indicating that dysbiosis develops before the appearance of inflammation. Interestingly, epithelial Myd88 deletion inhibits colitis development and secretory cell fate alterations in SHP‐2‐deficient mice. Our results suggest that dysfunction in SHP‐2 signaling severely impairs colonic epithelial barrier function resulting in microbiota‐driven inflammation as observed in patients with IBD (Swidsinski et al., 2005; Fava and Danese, 2011). Hence, epithelial SHP‐2 is a genetic factor that influences secretory cell fate, microbiota composition and therefore, intestinal homeostasis.
Materials and Methods
Animals
Shp‐2flox/flox mice (F3) were backcrossed with C57BL/6 mice for nine generations. All experiments were performed with F12 mice. Myd88flox/flox mice were purchased from The Jackson Laboratory (Bar Harbor, MA). The C57BL/6 12.4KbVilCre transgenic line was provided by Dr. Deborah Gumucio (University of Michigan, Ann Arbor, MI) (Madison et al., 2002). Mutations were genotyped according to manufacturer's instructions or the published protocols (Madison et al., 2002). All experiments were approved by the Animal Research Ethics Committee of the Université de Sherbrooke.
Microarray analysis
RNA was isolated from total colon extracts of three controls and three SHP‐2IEC‐KO newborn mice using the RNeasy mini kit (Qiagen, Toronto, ON, Canada). RNA were sent to the McGill University and Génome Québec Innovation Centre and a GeneChip Mouse Genome 430 2.0 Array (Affymetrix, Cleveland, OH) was performed. Data analysis, normalization, average difference, and expression for each feature on the chip were performed using Flexarray 1.6.1 with default parameters (Microarray platform, McGill University and Genome Quebec Innovation Centre).
RNA isolation and quantitative PCR
RNA was isolated from total colon extracts in newborn mice or from scraped colonic mucosa in 2‐week‐old mice using the RNeasy mini kit (Qiagen). Reverse transcription were performed using AMV‐RT (Roche Diagnostics, Laval, QC, Canada) according to the manufacturer's instructions and qPCR were performed by the RNomics Platform at the Université de Sherbrooke (QC, Canada). Target expression was quantified relatively to Pum1, Sdha, and Txnl4b. Primer sequences and conditions are available upon request.
Histological staining and immunohistochemistry
Murine tissues were fixed in 4% paraformaldehyde overnight at 4°C, then dehydrated and embedded in paraffin. Sections of 5 μm were applied to Probe‐On Plus slides (Fisher Scientific Canada, Nepean, ON, Canada) and kept at room temperature. Tissue preparation, embedding, and hematoxylin and eosin (H&E) coloration were described previously (Langlois et al., 2009) and were performed by the Electron Microscopy & Histology platform at the Université de Sherbrooke. For Paneth/Goblet staining, immunohistochemistry against lysozyme (Dako, Copenhagen, Danemark) was performed followed by an Alcian blue staining.
Electron microscopy
Tissues intended for electron microscopy analysis were fixed with 2.8% glutaraldehyde in 0.1 M cacodylate buffer and processed as previously described (Boudreau et al., 2007).
Gut microbiota analysis
Bacterial DNA from the cecal content of mice was extracted through mechanical lysis by bead beating (BioSpec, Bartlesville, OK), followed by enzymatic‐lysis and phenol:choloroform extraction. qPCR was used to quantify the different bacterial groups of the cecal microbiota using genus‐ and group‐specific primers as described (Matsuki et al., 2002; Malinen et al., 2003). Briefly, PCR amplification and detection were performed with iCycler iQ5 detection system using SsoFastTM Evagreen supermix (Bio‐Rad, Hercules, CA). The results are expressed as means ± SD of cycle threshold values (Ct). Ct values indicate the number of cycles required for the fluorescent signal to cross the threshold and are inversely proportional to the amount of target nucleic acid in the sample.
Deep sequencing analysis of 16S rRNA with Illumina
The V3 region of the 16S rRNA gene was amplified as described with modifications (Whelan et al., 2014). Products were separated from primers and primer dimers by electrophoresis on a 2% agarose gel. PCR products of the correct size were recovered using a QIAquick gel extraction kit (Qiagen). A total of 13,728,024 reads before quality filtering (an average of 292,085.617 reads per sample with a range of 120,066–401,952), 4,281,616 reads after quality filtering (an average of 84,652 reads per sample with a range of 516–147,427), and 10,219 OTUs (an average of 211.5 OTUs per sample, after quality filtering) were obtained from the 45 samples sequenced (6 control and 6 SHP‐2IEC‐KO mice aged of 4 days; 9 control and 7 SHP‐2IEC‐KO mice aged of 2 weeks; 8 control and 7 SHP‐2IEC‐KO mice aged of 1 month). Custom, in‐house Perl scripts were developed to process the sequences after Illumina sequencing (Whelan et al., 2014). Cutadapt (Martin, 2011) was used to trim any over‐read, and paired‐end sequences were aligned with PANDAseq (Masella et al., 2012) with a 0.7 quality threshold. If a mismatch in the assembly of a specific set of paired‐end sequences was discovered, they were culled. Additionally, any sequences with ambiguous base calls were also discarded. Operational taxonomic units (OTUs) were picked using AbundantOTU+ (Ye, 2011), and sequences were clustered to 97% sequence identity operational taxonomic units (OTUs). Taxonomy was assigned at a 0.8 threshold using the Ribosomal Database Project (RDP) (Caporaso et al., 2010) classifier v.2.2 trained against the Greengenes SSU database (February, 2011 release). For all downstream analyses, we filtered the obtained OTU table excluding “Root” and excluding any sequence that was not present at least three times across the entire dataset. Calculations of within‐community diversity (α‐diversity) and between‐community diversity (β‐diversity) were run using QIIME (Caporaso et al., 2010), and the phyloseq package (version 1.8.2) implemented in R (version 3.1.0) (McMurdie and Holmes, 2013). Alpha diversity index (Shannon) and richness (observed species) of each category were compared using the script compare_alpha_diversity.py in QIIME. The distance matrix calculated with Bray Curtis method on normalized data (all samples were subsampled to the same depth) was analyzed with PERMANOVA analysis using the script compare_categories.py in QIIME in order to assess the strength and statistical significance of sample groupings. Statistical analyses on the taxonomic composition of the colonic samples were done with the script group_significance.py in QIIME, which determines with Kruskal–Wallis test whether OTU relative abundance is different between categories. Before running the script, the OTU table was filtered, removing OTUs not present in at least 25% of samples. The P values obtained from the comparison between categories were then corrected with the “False discovery rate” (FDR) method. Secondly, we analyzed the taxonomic composition of the samples with multiple t‐tests on the table L6 (the highest assigned taxonomic level) generated by running the script: summarize_taxa_through_plots.py in QIIME. The P values obtained from the comparison between groups were corrected with the Holm–Sidak method, with α = 5.000%.
Disease activity index (DAI)
SHP‐2IEC‐KO mice and control littermates were scored based on a scale from 0 to 4 for rectal bleeding, stool consistency, blood loss, and colon hardness. Then, a cumulative DAI was calculated as previously detailed (Cooper et al., 1993).
Histological scoring
Cumulative histological scores were calculated as described (McCafferty et al., 1999) with a maximum possible score of 11. Briefly, features were graded as follows: extent of destruction of mucosal architecture (0, 1, 2, and 3, normal, mild, moderate, and extensive damage, respectively), presence, and degree of cellular infiltration (0, 1, 2, and 3, normal, mild, moderate, and transmural infiltration, respectively), extent of muscle thickening (0, 1, 2, and 3, normal, mild, moderate, and extensive thickening, respectively), presence or absence of Goblet cell depletion (0, absent; 1, present), and the presence or absence of crypt abscesses (0, absent; 1, present).
Western blot analysis
Proteins were isolated from scraped mucosa (1‐month‐old mice) in RIPA buffer and Western blots were performed as done before (Langlois et al., 2009). Antibodies against phosphorylated RelA (S536) (Cell Signaling, Danvers, MA) and β‐actin (Millipore, Billerica, MA) were used for Western blot analyses. Horseradish peroxidase‐anti‐mouse and anti‐rabbit antibodies were purchased from Amersham Biosciences (Pittsburg, PA).
Statistical analysis
Statistical analyses were calculated with GraphPad Prism 5 software (Irvine, CA). Student's two‐tailed t‐test was used if a normal distribution could be assumed. Mann–Whitney U‐test was performed elsewhere. Differences were considered significant at *P ≤ 0.05, **P ≤ 0.01, or ***P ≤ 0.001.
Results
Colonic expression of antimicrobial factors including Paneth cell markers in SHP‐2IEC‐KO neonates
We have previously reported that SHP‐2 ablation in the intestinal epithelium (SHP‐2IEC‐KO mice) does not compromise intestinal morphogenesis as observed by neonatal body weight, intestinal tissue architecture and cell differentiation (Coulombe et al., 2013). However, 1 month after birth, SHP‐2IEC‐KO mice spontaneously develop severe colitis which resembles to UC (Coulombe et al., 2013; Heuberger et al., 2014; Yamashita et al., 2014).
To identify the early genes altered by the loss of SHP‐2 in colonic crypts, gene expression profiling was performed three days after birth. Data were analyzed by the Database for Annotation, Visualization, and Integrated Discovery (DAVID) software to generate protein networks under SHP‐2 regulation. Intriguingly, some defense response genes including Defα (α‐defensins), Ido‐1, Leap‐2, and Reg3β emerge as the highest up‐regulated genes induced in SHP‐2‐deficient colons (Table I). To confirm the expression of antimicrobial genes, we performed several qPCR analyses instead of Western blots because many of these genes encode peptides that require proteolytic processing before secretion and activation and therefore, they are not easy to detect by Western blotting. Defensin group, for example, contains numerous diverse peptides and therefore we used primers to amplify the α‐defensin subtype. As shown in Figure 1A, qPCR analyses confirmed the significant induction of antimicrobial genes including Defα (9.9‐fold), Lyz1 (lysozyme 1) (1.7‐fold), and Reg3β (2.7‐fold) three days after birth in the colon of SHP‐2IEC‐KO neonates (Fig. 1A). In addition, increased expression of matrix metalloproteinase‐7 (Mmp7), an enzyme that cleaves the inactive precursors pro‐α‐defensins into active α‐defensins was also observed (Fig. 1A). Of note, 2 weeks after birth, the increase in the expression of these genes was even more pronounced: Defα by 5,865‐fold, Lyz1 by 140‐fold, Reg3β by 39‐fold, Reg3γ by 81‐fold, and Mmp7 by 32‐fold (Fig. 1B). Increased expression of these antimicrobial factors was also found in the small intestine of newborn and two week‐old SHP‐2IEC‐KO mice (Fig. S1A and B, Supporting Information).
Table I.
Up‐regulated genes induced in SHP‐2IEC‐KO colons (n = 3 mice per group)
| Gene | Identification | Relative expression | P |
|---|---|---|---|
| Defensin. alpha. 24 (Defa24) | 1450631_x_at | 4.15 | 2.80−6 |
| Indoleamine 2.3‐dioxygenase 1 (Ido1) | 1420437_at | 2.80 | 0.014 |
| Interleukin 1 receptor‐like 1 (Il1rl1) | 1422317_a_at | 2.57 | 0.021 |
| Chemokine (C‐ X‐C motif) ligand 5 (Cxcl5) | 1419728_at | 2.39 | 0.047 |
| Liver‐expressed antimicrobial peptide 2 (Leap2) | 1427480_at | 2.34 | 2.75−4 |
| Regenerating islet‐derived 3 beta (Reg3β) | 1416297_s_at | 2.12 | 3.50−5 |
| Leukocyte cell‐derived chemotaxin 2 (Lect2) | 1449492_a_at | 2.04 | 9.81−5 |
| Chemokine (C‐C motif) ligand 20 (Ccl20) | 1422029_at | 1.93 | 3.53−3 |
| Complement component 2 (C2) | 1416051_at | 1.65 | 9.99−5 |
| Chemokine (C‐C motif) ligand 25 (Ccl25) | 1418777_at | 1.65 | 3.10−4 |
| Regenerating islet‐derived 3 gamma (Reg3γ) | 1448872_at | 1.63 | 0.014 |
| Alpha‐2‐HS‐ glycoprotein (Ahsg) | 1455093_a_at | 1.58 | 0.041 |
| Interleukin 18 receptor 1 (Il18r1) | 1421628_at | 1.55 | 1.77−3 |
| Radical S‐ adenosyl methionine domain containing 2 (Rsad2) | 1436058_at | 1.54 | 0.025 |
| Interferon regulatory factor 7 (Irf7) | 1417244_a_at | 1.51 | 7.75−4 |
Figure 1.
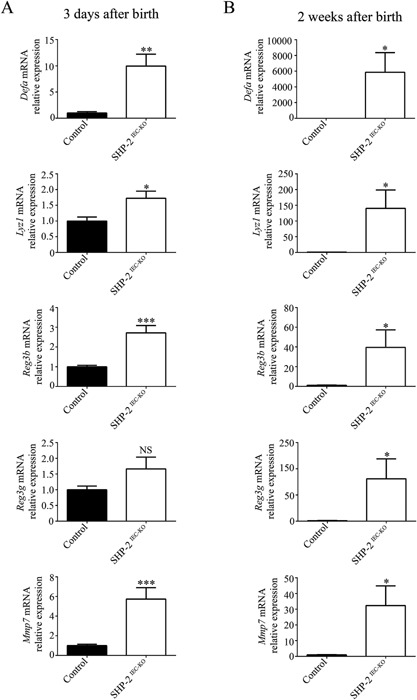
Expression of antimicrobial peptides in the colon of SHP‐ 2IEC‐KO mice. qPCR of Defα, Reg3β, Reg3γ, Lyz1, and Mmp7 mRNAs in total extracts from 3‐day‐old (A) and in enriched mucosal extracts from 2‐week‐old (B) SHP‐2IEC‐KO mice versus controls. Relative expression was normalized with housekeeping genes (n ≥ 6 per group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, NS: not significant. The error bars indicate SEM.
The increased expression of Defα and Lyz1 in the colon of SHP‐2‐deficient mice was surprising as in mice, the production of these mRNAs normally occurs in Paneth cells exclusively located in the small intestine (Bevins and Salzman, 2011). We thus decided to verify the putative presence of Paneth‐like cells in the colon of our mutant mice by using lysozyme staining and/or H&E coloration revealing cytoplasmic eosinophilic granules of these cells. As expected, in control mice, Paneth cells were restricted at the base of the small intestinal crypts (Fig. S2, see arrows, Supporting Information) and these cells were absent from colonic crypts (Fig. 2A and B). By contrast, 2 weeks after birth, SHP‐2IEC‐KO mice exhibited cells with eosinophilic granules and lysozyme staining not only in the small intestine (Fig. S2, see arrows and data not shown, Supporting Information) but also in the colon (Fig. 2A and B, see arrows). Thus, Paneth cell specific genes and Paneth‐like cells were observed in the colonic crypt of mutant mice, respectively, 3 days and 2 weeks after birth and then, before the appearance of histopathological manifestations of inflammation (Coulombe et al., 2013). Concomitantly, reduced mucin staining and Goblet cell number were observed in colons of 2‐week‐old and 1‐month‐old SHP‐2IEC‐KO mice (Fig. 2C), as we previously reported (Coulombe et al., 2013).
Figure 2.
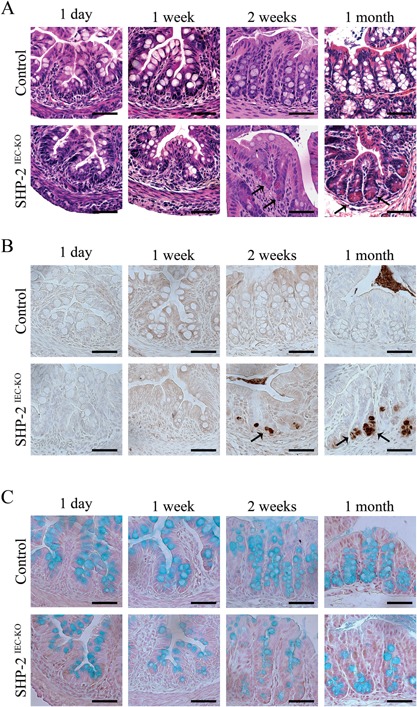
Lysozyme‐positive cells in the colon of young SHP‐2IEC‐ KO mice. H&E staining (A), immunohistochemistry against lysozyme (B), and Alcian blue staining (C) were performed on 1‐day‐old, 1‐week‐old, 2‐week‐old, and 1‐month‐old control and SHP‐2IEC‐ KO murine colons. Scale bars, 50 μm.
Expansion of intermediate cells in the colon of SHP‐2IEC‐KO mice
The phenotype of lysozyme‐positive cells observed in the colon tissue from SHP‐2IEC‐KO mice was further analyzed by electron microscopy. Interestingly, as shown in Figure 3A, colonic lysozyme‐positive cells of SHP‐2IEC‐KO mice exhibited a different morphology compared to typical small intestinal Paneth cells. Indeed, Paneth‐like cell granules in SHP‐2‐deficient colons were strikingly different, with the granules being irregular in size, less dense with a lattice‐like appearance at periphery (Fig. 3A, see arrows). This phenotype was reminiscent to the phenotype of intermediate cells which were previously found in the small intestine, at the junctions of the crypt‐villus axis (Calvert et al., 1988). These cells presumably represent the common precursors of the Paneth and Goblet cell lineages in the small intestine (Kamal et al., 2001), expressing both Paneth cell and Goblet cell markers. Accordingly, co‐staining for lysozyme and Alcian blue in control small intestines confirmed that dual‐positive cells were rare and that Alcian blue‐positive cells were distinct from lysozyme‐positive cells (King et al., 2013) (Fig. 3B). In contrast, in the colon of SHP‐2IEC‐KO mice, we easily detected the presence of both lysozyme‐negative/Alcian blue‐positive cells and also lysozyme‐positive/Alcian blue‐positive cells within the crypts (Fig. 3C, see arrows). Thus, these data suggest that the expansion of the lysozyme‐positive cells in the colon of SHP‐2IEC‐KO mice is due to an increased number of intermediate cells that exhibit some characteristics typical of both Goblet and Paneth cells. These data suggest that SHP‐2‐dependent signaling directs Goblet cell differentiation in the colon following progenitor specification.
Figure 3.
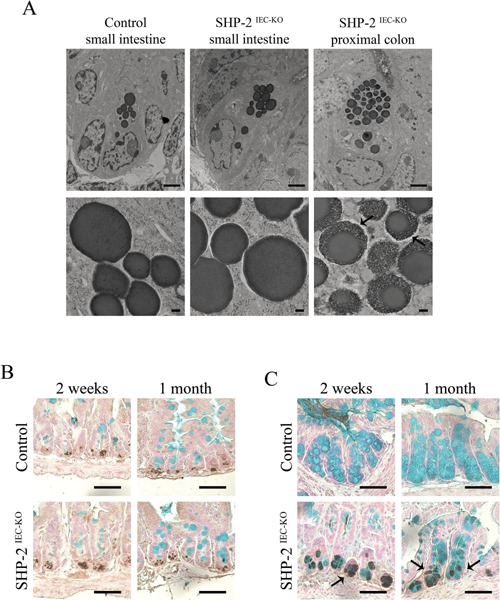
Expansion of intermediate cells in the colon of SHP‐ 2IEC‐KO mice. (A) Electron microscopy of Paneth‐like cells from the small intestine and colon of 2‐week‐old control and SHP‐2IEC‐KO mice. Scale bars, 2 μm (top panels) and 200 ηm (bottom panels). Co‐staining of lysozyme and Alcian blue was performed on small intestine (B) and colon (C) tissues of 2‐week‐old and 1‐month‐old control and mutant mice. Scale bars, 50 μm.
Loss of IEC‐specific SHP‐2 affects microbiota composition
Antimicrobial peptides including defensins not only protect the intestinal mucosa against pathogens but also shape the composition of the resident microbiota (Salzman et al., 2010). We thus evaluated whether loss of SHP‐2 in IECs led to intestinal microbiota changes. We observed that neonatal control and SHP‐2IEC‐KO mice had similar gut microbiota composition (Fig. 4A). However, at 2 weeks of age, we observed significant changes in the microbiota that continued to be present at 1 month after birth (Fig. 4B–E). Specifically, at 2‐week old, SHP‐2IEC‐KO mice had a reduction of both bacterial richness (measured as observed species) and diversity (measured with the Shannon's diversity index) in the cecal microbiota when compared to control mice (Fig. 4F). We observed an increase in the relative abundance of total Proteobacteria (Fig. 4B) and bacteria belonging to the genus Bacteroides and Escherichia in SHP‐2IEC‐KO in comparison to control mice (all corrected P < 0.05) (Fig. 4H). However, SHP‐2IEC‐KO mice had lower relative abundance of unclassified Lachnospiraceae and bacteria of the genus Oscillospira, both belonging to the order Clostridiales, than control mice (all corrected P < 0.05) (Fig. 4H). Similarly, we observed a reduction in bacterial richness and diversity in the cecal microbiota in 1‐month‐old SHP‐2IEC‐KO mice when compared to control mice (Fig. 4G). Moreover, SHP‐2IEC‐KO mice had higher relative abundance of total Proteobacteria and lower relative abundance of total Firmicutes when compared to control mice (all corrected P < 0.05) (Fig. 4C). In particular, SHP‐2IEC‐KO mice had higher relative abundance of Bacteroides and Escherichia genera, but lower of unclassified Lachnospiraceae (all corrected P < 0.05) (Fig. 4H). We then targeted changes in specific groups using qPCR, confirming the increases in Enterobacteriaceae and reductions in Firmicutes (Clostridium leptum and Clostridium coccoides) between controls and SHP‐2IEC‐KO mice (Table II). Thus, microbiota alterations in this model occur prior the appearance of clinical signs of colitis and thus it is possible that the dysbiosis contributes to the induction of inflammation observed in SHP‐2IEC‐KO mice.
Figure 4.
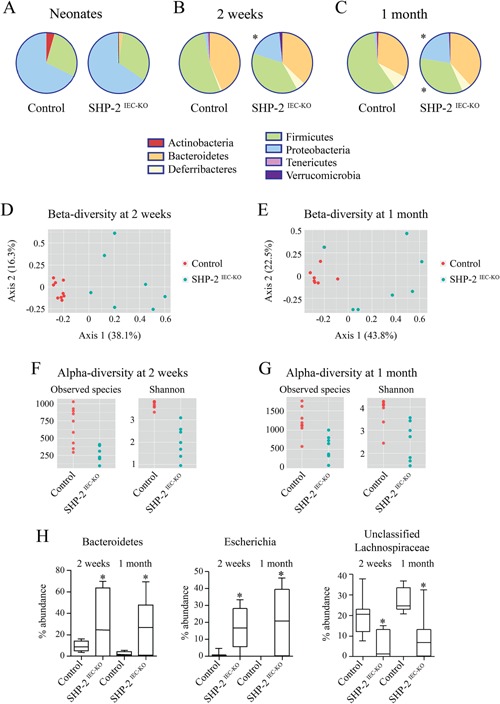
Altered microbiota composition in SHP‐2IEC‐KO mice. Taxonomic microbiota composition at phylum level in SHP‐ 2IEC‐KO and control littermates at the age of 4 days (A), 2 weeks (B), and 1 month (C) was determined by deep sequencing analysis. PCoA ordination plot constructed on the Bray Curtis distance matrix calculated on normalized data from SHP‐2IEC‐KO and control littermates at the age of 2 weeks (D) and 1 month (E). Alpha‐diversity (within community) measures (richness, measured as observed species and diversity measured as Shannon's diversity index) in SHP‐ 2IEC‐KO and control littermates at the age of 2 weeks (F) and 1 month (G). (H) Changes at genus level in SHP‐2IEC‐KO and control littermates at the age of 2 weeks and 1 month (n ≥ 7 per group). *P ≤ 0.05. The error bars indicate SEM.
Table II.
Microbiota composition in SHP‐2IEC‐KO mice compared to control littermates (n ≥ 5 mice per group)
| 2 weeks | 4 weeks | ||||||
|---|---|---|---|---|---|---|---|
| Mean (Ct) | Standard deviation | P‐value | Mean (Ct) | Standard deviation | P‐value | ||
| Target bacterial group | |||||||
| – | |||||||
| Total bacteria | Control | 15.32 | 1.65 | 0.132 | 13.09 | 0.97 | 0.257 |
| SHP‐2IEC‐KO | 16.57 | 1.64 | 15.04 | 2.27 | |||
| Lactobacillus | Control | 21.22 | 1.68 | 0.063 | 21.13 | 1.27 | 0.063 |
| SHP‐2IEC‐KO | 23.70 | 1.80 | 25.82 | 3.11 | |||
| Bacteroides fragilis | Control | 20.70 | 3.26 | 0.762 | 18.50 | 2.32 | 0.999 |
| SHP‐2IEC‐KO | 23.01 | 7.46 | 18.75 | 0.99 | |||
| Clostridium leptum | Control | 17.41 | 1.98 | 0.065 | 16.18 | 0.56 | 0.006 |
| SHP‐2IEC‐KO | 20.73 | 2.82 | 19.72 | 2.83 | |||
| Clostridium coccoides | Control | 16.80 | 1.84 | 0.24 | 13.09 | 0.60 | 0.006 |
| SHP‐2IEC‐KO | 21.57 | 5.54 | 18.67 | 4.00 | |||
| Enterobacteriaceae | Control | 22.85 | 3.47 | 0.029 | 29.02 | 2.64 | 0.012 |
| SHP‐2IEC‐KO | 18.21 | 1.79 | 20.84 | 4.19 | |||
Epithelial Myd88 deletion inhibits colitis development in SHP‐2‐deficient mice
One might speculate that different bacterial microbiote composition and diminished functionality of mucus barrier observed early in SHP‐2‐deficient mice causes the onset of inflammation. Indeed, we previously demonstrated that treatment of SHP‐2IEC‐KO mice with antibiotics inhibited colitis development (Coulombe et al., 2013). Stimulation of IEC Toll‐like receptors (TLR) by luminal commensals provides cytoprotective signals to the host (Rakoff‐Nahoum et al., 2004). However, mice with defective immune regulation do not develop colitis in germ‐free conditions indicating that commensal bacteria can trigger inflammation (Nell et al., 2010). To, therefore, investigate the role of TLR activation, we crossed SHP‐2IEC‐KO mice with mice lacking epithelial Myd88, an adaptor protein required for signaling by the majority of TLRs (Warner and Nunez, 2013). Of importance, epithelial Myd88 deletion impaired the development of colitis in 1 month‐old SHP‐2IEC‐KO mice, as illustrated by the partial restoration of body weight (Fig. 5A), the marked DAI reduction (Fig. 5B), the maintenance of histological integrity (Fig. 5C and D) as well as RelA (p65 regulatory subunit of NFκB) phosphorylation (Fig. 5E). This suggests that TLR‐mediated bacterial recognition is involved in colitis initiation in SHP‐2IEC‐KO mice. Importantly, epithelial deficiency of Myd88 alone did not significantly alter Goblet cell number in mice aged of 2 weeks. In addition, no lysozyme‐positive cell was detected in the colon of these mice. However, epithelial Myd88 deletion in SHP‐2IEC‐KO mice significantly increased Goblet cell number (Fig. 6A and B) and markedly reduced the number of intermediate cells (lysozyme‐positive cells) in comparison to SHP‐2IEC‐KO mice (Fig. 6C). Thus, TLR‐Myd88 signaling contributes to the observed early cell fate alterations that precede inflammation in SHP‐2IEC‐KO mice.
Figure 5.
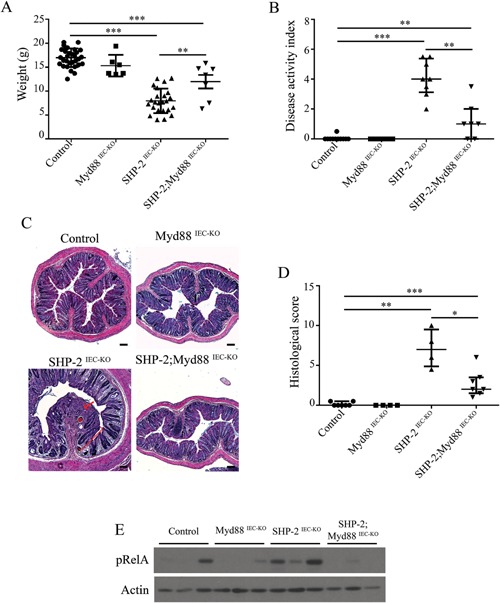
Loss of epithelial Myd88 signaling inhibits colitis in SHP‐2‐deficient mice. (A) Body weight of control, Myd88IEC‐KO, SHP‐2IEC‐KO, and SHP‐2;Myd88IEC‐KO mice at 1 month (n ≥ 6 per group). **P ≤ 0.01, ***P ≤ 0.001; two‐tailed Student's t‐test. Error bars indicate SEM. (B) DAI of 1‐month‐old control, Myd88IEC‐KO, SHP‐2IEC‐KO, and SHP‐2; Myd88IEC‐KO mice littermates were calculated by scoring stool softness, occult fecal blood, rectal bleeding, and colon hardness (n ≥ 7 per group). **P ≤ 0.01, ***P ≤ 0.001; Mann–Whitney U‐test. Error bars indicate interquartile range. (C) H&E staining from 1‐month‐old control, Myd88IEC‐KO, SHP‐2IEC‐KO, and SHP‐2; Myd88IEC‐KO murine colons were performed. Scale bars: 100 μm. (D) Histological score from 1‐month‐old mice was calculated as described in the section Material and Methods (n ≥ 4 per group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; Mann–Whitney U‐test. Error bars indicate interquartile range. (E) Mucosal enrichments from control, Myd88IEC‐KO, SHP‐2IEC‐KO, and Myd88IEC‐KO; SHP‐2IEC‐ KO colons aged of 1 month were analyzed by Western blot for the expression of phosphorylated RelA (S536). β‐actin served as loading control.
Figure 6.
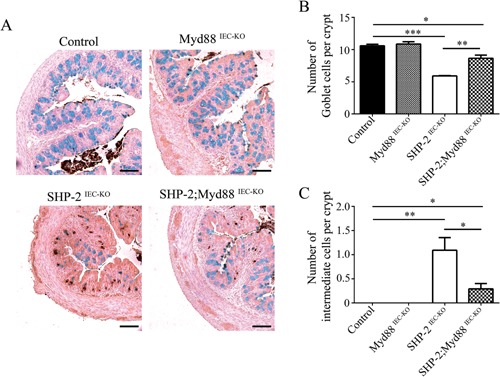
Loss of epithelial Myd88 increases the number of Goblet cells and decreases the number of intermediate cells in SHP‐2IEC‐ KO mice. (A) Co‐staining for lysozyme and Alcian blue cells were performed on colon tissues of 2‐week‐old control, Myd88IEC‐ KO, SHP‐2IEC‐KO, and Myd88IEC‐KO; SHP‐ 2IEC‐KO mice. Scale bars, 100 μm. (B) Epithelial cells from 2‐week‐old murine proximal colon stained positively for Alcian blue were counted in 15 crypts (n ≥ 3 per group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; two‐tailed Student's t‐test. The error bars indicate the SEM. (C) Epithelial cells from 2‐week‐old murine proximal colon stained positively for lysozyme were counted in 15 crypts (n ≥ 3 per group). **P ≤ 0.01, ***P ≤ 0.001; Mann–Whitney U‐test. Error bars indicate interquartile range.
Discussion
Mice with a deletion of SHP‐2 expression specifically in intestinal epithelium spontaneously develop colonic inflammation which resembles to UC (Coulombe et al., 2013; Heuberger et al., 2014; Yamashita et al., 2014). Herein, we observed that one of the earliest physiological modifications observed upon epithelial deletion of SHP‐2 in the intestine was the increased expression of many antimicrobial peptides in the colon. Indeed, our results showed that Reg3β, Defα, and Lyz1 were markedly induced in the colon of neonate mutants. The induction of Defα and Lyz1 in the colon of mutant mice was surprising as these peptides are mainly produced by Paneth cells that are normally confined at the bottom of the crypts in the small intestine and not in the colon. However, Paneth cell metaplasia has been frequently observed in various sites of inflammation, including the colon (Hiemstra and Zaat, 2013). This metaplastic response is usually viewed as a process which provides additional antimicrobial protection at sites of inflammation (Shi, 2007; Hiemstra and Zaat, 2013). However, herein, expression of Paneth cell genes was detected in newborn mutants and therefore, well before the onset of inflammation detected 1 month after birth. We have also noticed a distinct lysozyme‐positive cell population in the colon of our mutant mice. Intriguingly, these lysozyme‐positive cells were only detected 2 weeks after birth. These observations are reminiscent of changes that occur during the development and maturation of small intestine after birth. Indeed, in newborn mice, some Paneth cell products including defensins and UEA1 lectin are expressed by epithelial cells of the immature small intestine, well before crypt morphogenesis and formation of Paneth cells (Falk et al., 1994; Darmoul et al., 1997). For instance, mRNA and protein expression of some defensins was detected in P1 mouse intestine, despite the absence of recognizable Paneth cells (Darmoul et al., 1997). Hence, one might speculate that a similar phenomenon occurs in the colon of SHP‐2IEC‐KO neonates. Importantly, these distinct cells were also positive for Alcian blue staining indicating an intermediate phenotype between Paneth and Goblet cells. Such intermediate cells were previously described in the small intestine and presumably represent the common progenitors of the Paneth and Goblet cell lineages (Kamal et al., 2001). Indeed, these particular cells were stained positively for both lysozyme and Alcian blue. Of note, expansion of these intermediate cells in the colon of mutant mice was associated with a marked decrease in the number of mature Goblet cells. Thus, these observations suggest that SHP‐2‐dependent signaling directs cells to the Goblet cell fate and prevents Paneth cell expansion in the colon. Dysregulation of epithelial cell fate or differentiation in the colon may have serious consequences for the host as exemplified with mice deficient for Muc2 that rapidly develop severe colitis (Van der Sluis et al., 2006).
Notably, antimicrobial peptides have been involved not only in establishing the barrier between microbes and the epithelial cells but also in shaping microbiota composition (Salzman et al., 2010). For example, Paneth cell α‐defensins directly regulate microbiota composition as previously demonstrated with mice deficient in Mmp7 (which lack mature α‐defensins) and mice expressing human HD5 (which exhibit more α‐defensins). Indeed, the lack of mature α‐defensins shifts the dominant bacterial phyla present in the small intestine, decreasing the relative abundance of Bacteroidetes, and increasing the relative abundance of Firmicutes. Also, expression of the human defensin HD5 in murine Paneth cells increases the relative abundance of Bacteroidetes, in particular of Bacteroides genus, and decreases Firmicutes (Salzman et al., 2010). These changes in microbiota composition were observed without changes in microbial loads suggesting that Paneth cell α‐defensins are not involved in regulating the total number of bacteria in the intestine. Of note, at 4 weeks of age, SHP‐2 mutant mice had decreased Firmicutes (uncl.Lachnospiraceae and the genus Oscillospira), while Proteobacteria (Escherichia genus) and Bacteroidetes (Bacteroides genus) were increased. Similar changes in microbial composition were observed at 2 weeks, but no significant differences were observed among neonates, suggesting that the diversification of the microbiota occurs postnatally in SHP‐2IEC‐KO mice. It is of interest that the changes in microbiota observed in SHP‐2IEC‐KO mice are reminiscent of those reported in IBD patients (Lupp et al., 2007; Kostic et al., 2014). Thus, the results suggest that changes in the expression of a single epithelial phosphatase can have a significant effect on key gut microbial groups important for homeostasis. Interestingly, microbiota alterations in the model occur prior the appearance of clinical signs of colitis and thus it is possible that they play a role in colitis development.
Several studies indicate that the gut microbiota is required for triggering inflammation and IBD pathogenesis; indeed, short‐term treatment with antibiotics dramatically reduces inflammation and exhibits some efficacy in IBD (Casellas et al., 1998). Furthermore, various mouse models failed to develop intestinal inflammation in germ‐free setting. Likewise, we previously demonstrated that antibiotic treatment prevented colitis development in SHP‐2IEC‐KO mice (Coulombe et al., 2013). Additionally, herein we observed that deletion of epithelial Myd88 attenuated colitis severity in this model. Therefore, TLR/Myd88‐mediated bacterial recognition by the epithelium may be critical in inducing intestinal inflammation in SHP‐2IEC‐KO mice. By contrast, a protective function of Myd88 signaling in intestinal epithelium was previously reported (Rakoff‐Nahoum et al., 2004). For instance, deletion of Myd88 specifically in IECs triggers spontaneous intestinal inflammation (Gong et al., 2010). This concept is also supported by the severe colitis rapidly observed in mice with IEC‐specific deletion of NEMO/IKKγ, a downstream effector of TLRs and Myd88 (Nenci et al., 2007). Interestingly, in these models, diminished expression of antimicrobial peptides and translocation of commensals into the intestinal mucosa were associated with chronic inflammation (Gong et al., 2010; Frantz et al., 2012). Although these studies indicate that epithelial Myd88 signaling mediates mucosal protective functions, we cannot exclude that overactivation of epithelial Myd88 signaling may also induce inflammation. In line with this, previous studies demonstrated that TLR4 deficiency impairs mucosal healing resulting in bacterial translocation (Fukata et al., 2007) while too much TLR4 signaling in the intestinal epithelium (as in villin‐TLR4 mice) induces inflammation and tumorigenesis (Fukata et al., 2011). Such dual signaling function has been also observed for NFκB and STAT3 which can drive or attenuate inflammation depending on the specific challenges and pathophysiological conditions (Pasparakis, 2009; Hruz et al., 2010).
In summary, our studies shed new light on the cellular roles of SHP‐2 in the colon epithelium, which regulates secretory cell fate, crucial for the maintenance of mucosal barrier function and for the control of host defense. How SHP‐2 exactly impacts on cell fate is not totally clear but may rely on its ability to activate the MAP Kinase pathway. Indeed, Heuberger et al. recently reported in the small intestine that SHP‐2‐dependent ERK signaling controls the choice between Goblet and Paneth cell fate. In fact, they have shown that inhibition of ERK signaling in small intestinal organoids and cultured cells promoted Wnt/β‐catenin transcriptional activity through the control of relative abundance of Tcf4 isoforms (Heuberger et al., 2014). Although we observed that such cell fate regulation by SHP‐2 also occurs in the colon, our data suggest that the molecular mechanisms involved are more complicated and may implicate the contribution of microbiota since epithelial Myd88 deletion rescued Goblet/intermediate cell ratio and inhibits colitis induction. Future studies are needed to specify which TLRs and Myd88 downstream signaling effectors are involved in cell fate regulation and colitis development in SHP‐2IEC‐KO mice. Interestingly, Myd88 expressed in Paneth cells has been demonstrated to promote the expression of a complex antimicrobial program in the small intestine (Vaishnava et al., 2008). Furthermore, IEC‐specific TLR4 deletion markedly increased Goblet cell number in the small intestine (Sodhi et al., 2012). Hence, one might speculate that TLR4/Myd88 signaling may regulate secretory cell fate and differentiation, both in the small and large intestine. Conversely, the high abundance of gram‐negative bacteria in SHP‐2IEC‐KO deficient colons might also point toward a role for TLR4 in colitis initiation. Nonetheless, IECs express other TLRs (such as TLR2, TLR5, TLR9) which are also dependent of Myd88 for their signaling and which can trigger innate immune responses and inflammation in the intestine (Rosenstiel, 2013; Elia et al., 2015; Yu and Gao, 2015). Interestingly, TLR2, TLR4, and TLR5 have been reported to be phosphorylated on tyrosine after stimulation (Arbibe et al., 2000; Sarkar et al., 2003; Ivison et al., 2007; Medvedev et al., 2007); in some cases, this phosphorylation is required for full activation of the downstream signaling. On the other hand, Myd88 protein can also undergo tyrosine phosphorylation after stimulation. However, tyrosine phosphorylation of Myd88 promotes its degradation leading to downregulated TLR signaling (Mansell et al., 2006; Han et al., 2010). Thus, it is possible that SHP‐2 regulates TLR/Myd88 signaling in IECs through tyrosine dephosphorylation. Of note, SHP‐2 generally phosphorylates tyrosines surrounded by two or more acidic amino acids on the N‐terminal side and one or more acidic amino acids on the C‐terminal side. SHP‐2 also prefers the acidic residue aspartic acid (D) at pY −2 position with no basic residue on the C‐terminal side (Ren et al., 2011; Bunda et al., 2015). We did not find such general SHP‐2 recognition consensus motif on Myd88, TLR2, TLR4, TLR5, and TLR9 mouse sequences. Nevertheless, additional experiments such as immunoprecipitations with anti‐phosphotyrosine antibodies are needed to firmly determine if phosphorylation of Myd88 and/or certain TLR(s) is increased in SHP‐2 deficient IECs.
In conclusion, our results suggest that SHP‐2 maintains barrier function in the colon and thereby, helps to prevent spontaneous microbiota driven inflammation as seen in patients with IBD (Swidsinski et al., 2005; Fava and Danese, 2011). Hence, our study highlights SHP‐2 as a potential new target for therapeutic interventions aimed at improving barrier function in IBD patients.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Expression of antimicrobial peptides in the small intestine of SHP‐2IEC‐KO mice.
Figure S2. Paneth cells in the small intestine of SHP‐2IEC‐ KO mice.
Supporting Figure S1.
Supporting Figure S2.
Acknowledgments
We thank Anne Vézina for technical assistance and Sébastien A. Roy for fruitful discussions. We thank the Electron Microscopy & Histology Research Core of the FMSS at the Université de Sherbrooke for histology, electron microscopy, and phenotyping services. This research was supported by grant from Crohn and Colitis Canada Foundation and Canadian Institutes of Health Research to NR. Geneviève Coulombe is a NSERC Alexander Graham Bell student scholar. Ariane Langlois was supported by the 2014 Crohn's and Colitis Canada Summer Studenship award. Nathalie Rivard, François Boudreau, and Nathalie Perreault are members of the FRSQ‐Funded Centre de Recherche du CHUS. Nathalie Rivard is a recipient of a Canada Research Chair in colorectal cancer and inflammatory cell signaling. Elena F. Verdu holds a CRC and is funded by a Crohn's and Colitis Canada grant. Giada De Palma holds a PDF by CIHR/ CAG, and Justin McCarville a Canadian Celiac Association student award.
Literature Cited
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. 2000. Toll‐like receptor 2‐mediated NF‐kappa B activation requires a Rac1‐dependent pathway. Nat Immunol 1:533–540. [DOI] [PubMed] [Google Scholar]
- Bevins CL, Salzman NH. 2011. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9:356‐ 368. [DOI] [PubMed] [Google Scholar]
- Boudreau F, Lussier CR, Mongrain S, Darsigny M, Drouin JL, Doyon G, Suh ER, Beaulieu JF, Rivard N, Perreault N. 2007. Loss of cathepsin L activity promotes claudin‐1 overexpression and intestinal neoplasia. FASEB J 21:3853–3865. [DOI] [PubMed] [Google Scholar]
- Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, Raught B, Zhang ZY, Zadeh G, Ohh M. 2015. Inhibition of SHP2‐mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 6:8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert R, Bordeleau G, Grondin G, Vezina A, Ferrari J. 1988. On the presence of intermediate cells in the small intestine. Anat Rec 220:291–295. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas F, Borruel N, Papo M, Guarner F, Antolin M, Videla S, Malagelada JR. 1998. Antiinflammatory effects of enterically coated amoxicillin‐clavulanic acid in active ulcerative colitis. Inflamm Bowel Dis 4:1–5. [DOI] [PubMed] [Google Scholar]
- Cho JH, Brant SR. 2011. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 140:1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HS, Murthy SN, Shah RS, Sedergran DJ. 1993. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 69:238–249. [PubMed] [Google Scholar]
- Coskun M. 2014. Intestinal epithelium in inflammatory bowel disease. Front Med 1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe G, Leblanc C, Cagnol S, Maloum F, Lemieux E, Perreault N, Feng GS, Boudreau F, Rivard N. 2013. Epithelial tyrosine phosphatase SHP‐2 protects against intestinal inflammation in mice. Mol Cell Biol 33:2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmoul D, Brown D, Selsted ME, Ouellette AJ. 1997. Cryptdin gene expression in developing mouse small intestine. Am J Physiol 272:G197–G206. [DOI] [PubMed] [Google Scholar]
- Elia PP, Tolentino YF, Bernardazzi C, de Souza HS. 2015. The role of innate immunity receptors in the pathogenesis of inflammatory bowel disease. Mediators Inflamm 2015:936193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk P, Roth KA, Gordon JI. 1994. Lectins are sensitive tools for defining the differentiation programs of mouse gut epithelial cell lineages. Am J Physiol 266:G987–1003. [DOI] [PubMed] [Google Scholar]
- Fava F, Danese S. 2011. Intestinal microbiota in inflammatory bowel disease: Friend of foe? World J Gastroenterol 17:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz AL, Rogier EW, Weber CR, Shen L, Cohen DA, Fenton LA, Bruno ME, Kaetzel CS. 2012. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin‐2, and antibacterial peptides. Mucosal Immunol 5:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. 2007. Toll‐like receptor‐4 promotes the development of colitis‐associated colorectal tumors. Gastroenterology 133:1869–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, Espana C, Ungaro R, Harpaz N, Cooper HS, Elson G, Kosco‐Vilbois M, Zaias J, Perez MT, Mayer L, Vamadevan AS, Lira SA, Abreu MT. 2011. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis‐associated tumorigenesis. Inflamm Bowel Dis 17:1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J, Xu J, Zhu W, Gao X, Li N, Li J. 2010. Epithelial‐specific blockade of MyD88‐dependent pathway causes spontaneous small intestinal inflammation. Clin Immunol 136:245–256. [DOI] [PubMed] [Google Scholar]
- Han C, Jin J, Xu S, Liu H, Li N, Cao X. 2010. Integrin CD11b negatively regulates TLR‐triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl‐b. Nat Immunol 11:734–742. [DOI] [PubMed] [Google Scholar]
- Heuberger J, Kosel F, Qi J, Grossmann KS, Rajewsky K, Birchmeier W. 2014. Shp2/MAPK signaling controls goblet/paneth cell fate decisions in the intestine. Proc Natl Acad Sci USA 111:3472–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemstra PS, Zaat SA. 2013. Antimicrobial peptides and innate immunity. Basel: Springer. [Google Scholar]
- Hruz P, Dann SM, Eckmann L. 2010. STAT3 and its activators in intestinal defense and mucosal homeostasis. Curr Opin Gastroenterol 26:109–115. [DOI] [PubMed] [Google Scholar]
- Ivison SM, Khan MA, Graham NR, Bernales CQ, Kaleem A, Tirling CO, Cherkasov A, Steiner TS. 2007. A phosphorylation site in the Toll‐like receptor 5 TIR domain is required for inflammatory signalling in response to flagellin. Biochem Biophys Res Commun 352:936–941. [DOI] [PubMed] [Google Scholar]
- Kamal M, Wakelin D, Ouellette AJ, Smith A, Podolsky DK, Mahida YR. 2001. Mucosal T cells regulate Paneth and intermediate cell numbers in the small intestine of T. spiralis‐infected mice. Clin Exp Immunol 126:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. 2010. Inflammatory bowel disease. Annu Rev Immunol 28:573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SL, Mohiuddin JJ, Dekaney CM. 2013. Paneth cells expand from newly created and preexisting cells during repair after doxorubicin‐induced damage. Am J Physiol Gastrointest Liver Physiol 305:G151–G162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Xavier RJ, Gevers D. 2014. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 146:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois MJ, Roy SA, Auclair BA, Jones C, Boudreau F, Carrier JC, Rivard N, Perreault N. 2009. Epithelial phosphatase and tensin homolog regulates intestinal architecture and secretory cell commitment and acts as a modifier gene in neoplasia. FASEB J 23:1835–1844. [DOI] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. 2007. Host‐mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2:204. [DOI] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. 2002. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 277:33275–33283. [DOI] [PubMed] [Google Scholar]
- Malinen E, Kassinen A, Rinttila T, Palva A. 2003. Comparison of real‐time PCR with SYBR Green I or 5'‐nuclease assays and dot‐blot hybridization with rDNA‐targeted oligonucleotide probes in quantification of selected faecal bacteria. Microbiology 149:269–277. [DOI] [PubMed] [Google Scholar]
- Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O'Neill LA, Hertzog PJ. 2006. Suppressor of cytokine signaling 1 negatively regulates Toll‐like receptor signaling by mediating Mal degradation. Nat Immunol 7:148–155. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17:10–12. [Google Scholar]
- Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. 2012. PANDAseq: Paired‐end assembler for illumina sequences. BMC Bioinformatics 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Watanabe K, Fujimoto J, Miyamoto Y, Takada T, Matsumoto K, Oyaizu H, Tanaka R. 2002. Development of 16S rRNA‐gene‐targeted group‐specific primers for the detection and identification of predominant bacteria in human feces. Appl Environ Microbiol 68:5445–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty DM, Miampamba M, Sihota E, Sharkey KA, Kubes P. 1999. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut 45:864–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. 2013. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev AE, Piao W, Shoenfelt J, Rhee SH, Chen H, Basu S, Wahl LM, Fenton MJ, Vogel SN. 2007. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem 282:16042–16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumi Y, Isomoto H, Shiota M, Sato K, Kondo S, Machida H, Yanagihara K, Mizuta Y, Kohno S, Tsukamoto K. 2009. Polymorphisms of PTPN11 coding SHP‐2 as biomarkers for ulcerative colitis susceptibility in the Japanese population. J Clin Immunol 29:303‐ 310. [DOI] [PubMed] [Google Scholar]
- Nell S, Suerbaum S, Josenhans C. 2010. The impact of the microbiota on the pathogenesis of IBD: Lessons from mouse infection models. Nat Rev Microbiol 8:564–577. [DOI] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, Gumucio D, Neurath MF, Pasparakis M. 2007. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446:557–561. [DOI] [PubMed] [Google Scholar]
- Pasparakis M. 2009. Regulation of tissue homeostasis by NF‐kappaB signalling: Implications for inflammatory diseases. Nat Rev Immunol 9:778–788. [DOI] [PubMed] [Google Scholar]
- Peterson LW, Artis D. 2014. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol 14:141‐ 153. [DOI] [PubMed] [Google Scholar]
- Rakoff‐ Nahoum S, Paglino J, Eslami‐Varzaneh F, Edberg S, Medzhitov R. 2004. Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell 118:229–241. [DOI] [PubMed] [Google Scholar]
- Ren L, Chen X, Luechapanichkul R, Selner NG, Meyer TM, Wavreille AS, Chan R, Iorio C, Zhou X, Neel BG, Pei D. 2011. Substrate specificity of protein tyrosine phosphatases 1B, RPTPalpha, SHP‐1, and SHP‐2. Biochemistry 50:2339–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstiel P. 2013. Stories of love and hate: Innate immunity and host‐microbe crosstalk in the intestine. Curr Opin Gastroenterol 29:125–132. [DOI] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson‐Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. 2010. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Smith HL, Rowe TM, Sen GC. 2003. Double‐stranded RNA signaling by Toll‐like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J Biol Chem 278:4393–4396. [DOI] [PubMed] [Google Scholar]
- Shi J. 2007. Defensins and Paneth cells in inflammatory bowel disease. Inflamm Bowel Dis 13:1284–1292. [DOI] [PubMed] [Google Scholar]
- Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T, Jr. , Russo AM, Afrazi A, Good M, Brower‐Sinning R, Firek B, Morowitz MJ, Ozolek JA, Gittes GK, Billiar TR, Hackam DJ. 2012. Intestinal epithelial Toll‐like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 143:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalinger MR, McCole DF, Rogler G, Scharl M. 2015. Role of protein tyrosine phosphatases in regulating the immune system: Implications for chronic intestinal inflammation. Inflamm Bowel Dis 21:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidsinski A, Weber J, Loening‐Baucke V, Hale LP, Lochs H. 2005. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43:3380–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. 2008. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host‐microbial interface. Proc Natl Acad Sci USA 105:20858–20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Buller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. 2006. Muc2‐deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131:117‐ 129. [DOI] [PubMed] [Google Scholar]
- Warner N, Nunez G. 2013. MyD88: A critical adaptor protein in innate immunity signal transduction. J Immunol 190:3–4. [DOI] [PubMed] [Google Scholar]
- Whelan FJ, Verschoor CP, Stearns JC, Rossi L, Luinstra K, Loeb M, Smieja M, Johnstone J, Surette MG, Bowdish DM. 2014. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann Am Thorac Soc 11:513–521. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Kotani T, Park JH, Murata Y, Okazawa H, Ohnishi H, Ku Y, Matozaki T. 2014. Role of the protein tyrosine phosphatase Shp2 in homeostasis of the intestinal epithelium. PLoS ONE 9:e92904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. 2011. Identification and quantification of abundant species from pyrosequences of 16S rRNA by consensus alignment. Proc IEEE Int Conf Bioinformatics Biomed 2010:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Gao N. 2015. Compartmentalizing intestinal epithelial cell toll‐like receptors for immune surveillance. Cell Mol Life Sci 72:3343–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Expression of antimicrobial peptides in the small intestine of SHP‐2IEC‐KO mice.
Figure S2. Paneth cells in the small intestine of SHP‐2IEC‐ KO mice.
Supporting Figure S1.
Supporting Figure S2.