

Abstract
Human Cellular Retinol Binding Protein II (hCRBPII), a member of the intracellular Lipid binding protein family, is a monomeric protein responsible for the intracellular transport of retinol and retinal. Herein we report that hCRBPII forms an extensive domain swapped dimer during bacterial expression. The domain swapped region encompasses almost half of the protein. The dimer represents a novel structural architecture with the mouths of the two binding cavities facing each other, producing a new binding cavity that spans the length of the protein complex. Though wild-type hCRBPII forms the dimer, the propensity for dimerization can be substantially increased via mutation at Tyr60. The monomeric form of the wild type protein represents the thermodynamically more stable species, making the domain swapped dimer a kinetically trapped entity. Hypothetically, the wild type protein has evolved to minimize dimerization of the folding intermediate through a critical hydrogen bond (Tyr60-Glu72) that disfavors the dimeric form.
eTOC Blurb
hCRBPII, a member of the iLBP family that carries lipophilic molecules through the cytosol, is found to form an extensive domain swapped dimer during overexpression. No interconversion is observed between monomer and dimer, indicating that two distinct protein folds can be obtained from the same polypeptide. Structures of several mutant dimers suggest a folding pathway involving an open monomer intermediate, with the resultant fold dependent on the structure of this intermediate.

Introduction
Three-dimensional (3D) domain swapping, first discovered by Eisenberg and coworkers in 1994 to account for the dimerization mechanism of diphtheria toxin (DT), is a process in which two or more monomeric protein molecules exchange identical structural elements to form dimers or higher-order oligomers.(Bennett et al., 1995; Liu and Eisenberg, 2002; Liu et al., 1998) In most cases the exchanged region is located at the N-or C-terminus of the protein, although in a few instances it is found in the middle of the protein sequence.(Bax et al., 1990; Hakansson et al., 2001; Rousseau et al., 2012) Domain swapping, combined with gene duplication is responsible for the evolution of a number of larger protein domains from smaller fragments.(Szilagyi et al., 2012) Others have postulated that domain swapping can also lead to aggregation, amyloid and fibril formation.(Stroud et al., 2012) Inappropriate protein aggregation is implicated in a number of maladies including Parkinson’s disease, Alzheimer disease, diabetes, Huntington’s disease and many others.(Cleary et al., 2005; Fandrich et al., 2001; Gronenborn, 2009; Schlunegger et al., 1997; Selkoe, 2004; Shtilerman et al., 2002; Sunde and Blake, 1997) So far about 40 distinctly different proteins are reported to form domain swapped oligomers.(Carey et al., 2007; Koharudin et al., 2013) Triggers of domain-swapping include lyophilization, non-physiological pH,(Bennett et al., 1994) unusually high protein concentration,(Ellis, 2001; Liu et al., 2002; Minton, 2000) temperature, mutation and even ligand binding.(Frank et al., 2002; O’Neill et al., 2001) For example a single site mutation in protein L from Peptostreptococcus magnus leads to domain swapping.(O’Neill et al., 2001) In addition, as expected for an oligomerization process, it is typically favored by protein folding at high protein concentration,(Ellis, 2001; Minton, 2000) suggesting that folding intermediates at elevated levels may be involved in the process.(Bennett et al., 1995; Koharudin et al., 2013)
Intracellular lipid binding proteins (iLBPs) are small, relatively stable, cytosolic proteins responsible for the transport of a variety of insoluble hydrophobic molecules, such as retinoids and fatty acids, throughout the cell. Though relatively divergent in sequence, all those structurally characterized contain a 10-stranded beta barrel and two alpha helices located at the mouth of the relatively large internal binding cavity. Structures of a number of iLBPs have been determined and all share this monomeric fold. Numerous studies of the folding mechanism of members of the iLBP family have been conducted, most notably those of the Gierasch lab, who have primarily focused on human Cellular Retinoic Acid Binding Protein I (hCRABPI). In an elegant series of experiments, using a variety of interrogation techniques, the Gierasch lab has concluded that early beta barrel closure is a hallmark of the pathway, thus preventing off pathway aggregation and potential amyloid formation in this protein.(Gershenson et al., 2014) Investigations into the folding mechanism of other members of the family have also been conducted, all of which are consistent with a relatively rugged energy landscape, indicating a number of possible stable or meta-stable intermediates, a situation that is generally characteristic of proteins that are largely beta sheet in structure.(Gershenson et al., 2014)
Human Cellular Retinol Binding Protein II (hCRBPII), a 133 residue member of the iLBP family, plays an important role in the metabolism of vitamin A and is responsible for intracellular retinol and retinal transport (Figure S1A).(Nossoni et al., 2014; Ong and Page, 1987) The ligand is buried deeply within the relatively large binding cavity (1113 Å3).(Cowan et al., 1993; Nossoni et al., 2014; Wang et al., 2012) Both hCRBPII and other members of this family are remarkably resilient to mutation.(Berbasova et al., 2013; Berbasova et al., 2016; Crist et al., 2006; Huntress et al., 2013; Lee et al., 2012; Nossoni et al., 2014; Vaezeslami et al., 2008; Vaezeslami et al., 2006; Vasileiou et al., 2009a; Vasileiou et al., 2007; Vasileiou et al., 2009b; Wang et al., 2014; Wang et al., 2012; Yapici et al., 2015) For example more than 150 structurally stable mutants of hCRBPII have been characterized in our lab in the course of our studies of protein-chromophore interactions.(Wang et al., 2012) The robustness of this family of proteins, in particular hCRBPII, to mutations was the central feature that led us to use them as templates to develop rhodopsin mimics for the purpose of investigating factors that contribute to wavelength regulation.(Crist et al., 2006; Huntress et al., 2013; Lee et al., 2012; Nossoni et al., 2014; Vaezeslami et al., 2008; Vaezeslami et al., 2006; Vasileiou et al., 2009a; Vasileiou et al., 2007; Vasileiou et al., 2009b; Wang et al., 2014; Wang et al., 2012; Yapici et al., 2015) During the preparation of mutants targeted for these studies, we observed protein bands that eluted separately on ion-exchange chromatography, but nonetheless, were the same species on SDS-PAGE. This led us to the discovery of a set of hCRBPII protein mutants capable of domain swapped dimerization.
Herein we report domain swapped structures of hCRBPII. The extent of domain swapping is large, involving almost 50% of the protein sequence and occurs during routine protein bacterial over-expression at 16–30 °C. hCRB PII domain swapped dimerization is favored by high expression levels in E. coli and high concentrations in vitro. Protein stability studies indicate the domain swapped dimer to be a less stable, kinetically trapped species, depending on the mutant. The resultant dimer structure represents a fold not identified in the SCOP or CATH protein fold databases. Together, the data suggest that the folding of hCRBPII occurs via a structurally ordered “open monomer,” with the relative orientation of the N- and C- terminal structural units determining the propensity for dimerization. The domain swapped dimer may therefore represent the kinetic trapping of a structurally well-ordered “open monomer” folding intermediate. The existence of the domain swapped dimer of hCRBPII suggests a folding pathway distinct from that characterized for hCRABPI, indicating that evolutionary related members of the same structural family can follow different folding pathways. Furthermore, the domain swapped dimer may represent a new, biologically relevant structural fold for the iLBP protein family, though it remains to be seen if they form in vivo, and have distinct functionality relative to their parent, monomeric forms.
Results and Discussion
Our original interest in hCRBPII stemmed from our development of rhodopsin mimics to study the mechanism of wavelength tuning. The binding site was redesigned to bind all-trans retinal and form a Protonated Schiff Base (PSB) via the Q108K:K40L (KL) double mutant.(Wang et al., 2012) The Tyr60 position, located in beta-strand D, approximately 4 Å from the center of the polyene chain of all-trans-retinal (Figure S1A, and S1B), was then interrogated, resulting in the generation of the Q108K:K40L:Y60W (KLY60W) mutant protein.(Folli et al., 2001; Gunasekaran et al., 2004; Nossoni et al., 2014; Wang et al., 2012) The second purification step, Source-Q ion exchange chromatography, unexpectedly gave two KLY60W hCRBPII-containing protein fractions, one eluting at 40 mM NaCl, where hCRBPII mutants typically elute, and a second eluting at 80 mM NaCl (Figure S2A). Interestingly, the UV-Vis spectra of each protein fraction bound to all-trans-retinal as a PSB complex produced distinct spectra (40 mM fraction, λmax = 496 nm; 80 nm fraction, λmax of 514 nm, Figure S2C), suggesting that the two species differ in protein conformation (Figure S2B). Size-exclusion chromatography showed that the 40 mM protein eluted as a 15 kD monomer, and the 80 mM fraction eluted as a 30 kD dimer (Figure S2D and S2E).
Dimer formation of other hCRBPII variants
Since many hCRBPII proteins mutated at the 40 and 108 positions have been purified in monomeric form in our lab,(Wang et al., 2012) we hypothesized that mutation of the 60 position in the KLY60W series was most likely responsible for the formation of the dimeric species and made several single mutants at this position (Table S1). As shown, Y60W gave monomer/dimer ratios identical to that of KLY60W, confirming the importance of position 60. In fact most mutations resulted in increased dimer formation, including Y60F, Y60L and Y60I, while the Y60H mutant produced no dimeric protein, with the Y60L and Y60I mutants both producing 80–100% dimeric protein (Table S1A and Figure S2F–I). Together, this data indicate that mutation at position 60 can significantly favor the formation of dimeric species. Position 60 lies near the N-terminal end of strand 4, and is not a position conserved in iLBP family members, though it is a Tyr in most retinol binding proteins.(Gunasekaran et al., 2004)
To better understand the nature of the dimerization phenomenon, in vitro refolding experiments were conducted on the WT and several of the mutants (Table S1B). In each case, dimer formation was favored by higher refolding concentrations while monomer formation was favored at lower refolding concentrations. This agreed with our E. coli-expression results, where expression at higher temperatures, which leads to higher levels of expression and therefore higher concentrations of pre-folded proteins, (30 °C versus 25 °C) favored dimer formation while lower temperature expression favored the monomer (Table S1B). Taken together, these experiments are consistent with the dimer/monomer ratio being kinetically controlled, with higher concentrations favoring the bi-molecular dimer formation. Melting experiments show the dimer to be less stable than the monomer for two of the three variants (WT, KLY60W), indicating the dimer to be the kinetic product (the melting curve for the Y60L monomer was not readily interpretable (Figure S4).
Intrigued at the relative ease of producing the dimeric species, we next investigated the possibility that WT hCRBPII could also form a dimeric species by overexpressing hCRBPII at the dimer favoring induction temperature of 30 °C, resulting in both monomeric and dimeric protein (Figure S2I). To our knowledge, this is the first example of a domain swapped system where both monomer and dimer of the wild-type species are expressed without monomer/dimer exchange. Binding assays show that the monomer and dimer have similar affinity for retinal and retinol as ligands (Figure S1E–H).
Structural analysis reveals an extensive domain swapped dimer
In an effort to understand the nature of the dimeric form of hCRBPII, the dimeric forms of the wild type and several mutants (WT hCRBPII, KLY60W, Y60W and Y60L) were crystallized and their structures determined (Table 1). The same extensive, domain swapped dimer was observed for all four of these structures (Figures 1A, 1B, 1C, 2A and 2B). The domain swapping is extensive, involving residue 1–56, which includes 3 beta strands and two helices (Figures 1C, 2A, and 2B). This corresponds to an 83% extent of swapping as defined in the domain swapping 3D knowledge database (http://caps.ncbs.res.in/3dswap/index.html).(Shameer et al., 2011) The two domains in the dimer are each highly similar to that of the monomer hCRBPII structure with RMSD ranging from 0.382–0.397 between monomeric hCRBPII and a single domain of the dimer (Figures 1D and S1D). The four domain swapped dimer structures so far determined have in common a large rotation about the psi angle of Thr 56. While the psi angle of Thr56 is about −12° in monomeric hCRBPII, it ranges from 132 ° – 169° in the domain swapped dimers (Figure S5A). This 150°+ rotation about the psi angle of Thr56 represents the hinge motion required for domain swapping (Figure 1D). Though mutation of residue 60 substantially affects the monomer/dimer ratio, it lies not in the hinge loop region, but is instead 3 amino acids away, near the middle of β-strand D (Figure S1A). In fact substantial deviations in torsion angles from the monomer structure can be seen in residues 55 – 60, which encompasses the hinge loop region and the first 3 amino acids of the 4th beta sheet (Figures S5A–E). The domain swapping orients the opening of the two binding sites to face one another, creating a continuous internal cavity that stretches from one side of the dimer to the other (nearly 40 Å in length, Figure S1C). A search of the various structural fold databases (SCOP 1.75, CATH 3.5) reveals no other protein of similar architecture.(Sillitoe et al., 2013)
Table 1.
X-ray crystallography data and refinement statistics of the dimer structures
| Q108K:K40L:Y60W (dimer) | Y60W (dimer) | Y60L (dimer) | E72A (dimer) | WT (dimer) | |
|---|---|---|---|---|---|
| Space group | P21212 | P21212 | P21212 | P21212 | P21212 |
| a(Å) | 62.77 | 62.62 | 63.603 | 62.238 | 64.021 |
| b(Å) | 109.22 | 109.77 | 60.424 | 72.901 | 60.544 |
| c (Å) | 36.39 | 36.32 | 36.776 | 60.487 | 36.851 |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β(°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| δ (°) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| Molecules per Asymmetric Unit | 2 | 2 | 1 | 2 | 1 |
| Total reflection | 288797 | 345311 | 301085 | 380739 | 33891 |
| Unique Reflection | 20048 | 55151 | 21293 | 27242 | 4455 |
| Completeness (%) | 99.9(100.0)a | 98.6(100.0)a | 100(99.81)a | 100(100)a | 100(99.70)a |
| Average I/σ | 78.16(6.23)a | 66.13(7.71)a | 82.03(3.18)a | 75.13(2.76)a | 34.63(2.41)a |
| Rmerge(%) | 4.7(46.5)a | 5.2(31.7)a | 4.1(69.5)a | 5.5(84.7)a | 5.9(73.1)a |
| Resolution (Å) (Last Shell) | 50.00 – 1.91(1.94 – 1.91)a | 50.00 – 1.70(1.73 – 1.70)a | 50.00 – 1.55(1.58 – 1.55)a | 50 – 1.77(1.8 – 1.77)a | 50.00 – 2.66(2.71 – 2.66)a |
| Rwork/Rfree (%) | 18.00/21.84 | 18.17/23.94 | 19.91/24.57 | 17.94/22.41 | 17.57/26.28 |
| RMSD from ideal values | |||||
| Bond Length (Å) | 0.008 | 0.023 | 0.007 | 0.013 | 0.002 |
| Bond Angle (°) | 1.11 | 2.11 | 1.03 | 1.28 | 0.51 |
| Average B factor | 28.71 | 19.92 | 34.31 | 34.17 | 36.71 |
| Number of water molecules | 352 | 217 | 147 | 462 | 50 |
| PDB ID | 4ZR2 | 4ZCB | 4ZH6 | 5DPQ | 4ZH9 |
Values in the parenthesis refer to the last resolution shell.
Figure 1.
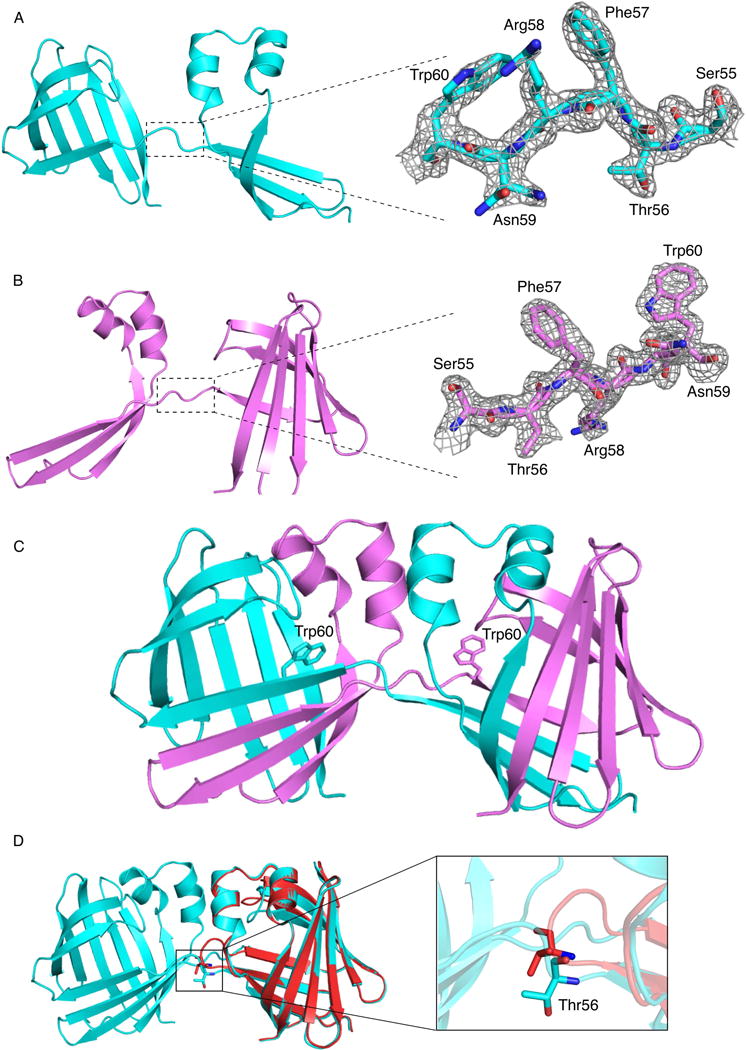
The crystal structures of hCRBPII dimer species and comparison with their monomeric structures. A. Chain A of KLY60W (where only the Thr56 psi angle differs from the closed monomer form) with the electron density, contoured at 1.0 σ, of the hinge loop region encompassing amino acids 55–60, Residues 55–60 are highlighted. Atoms colored by type, N, blue, O, red, C, cyan. Color scheme retained unless otherwise noted. B. Chain B of the KLY60W mutant, showing the electron density, contoured at 1.0 σ, of the hinge loop region encompassing amino acids 55–60. Residues 55–60 are highlighted. C. The structure of the KLY60W dimer: In chain A (cyan) Trp60 is pointing inside the binding pocket, while in chain B (pink) Trp60 is pointing toward the solvent. D. An overlay of the dimeric (cyan) and monomeric (red) KLY60W mutant structures. Res56 for both structures is shown in sticks. The hinge loop region in the monomer and dimer is encircled, showing the dramatic difference in the two structures.
Figure 2.
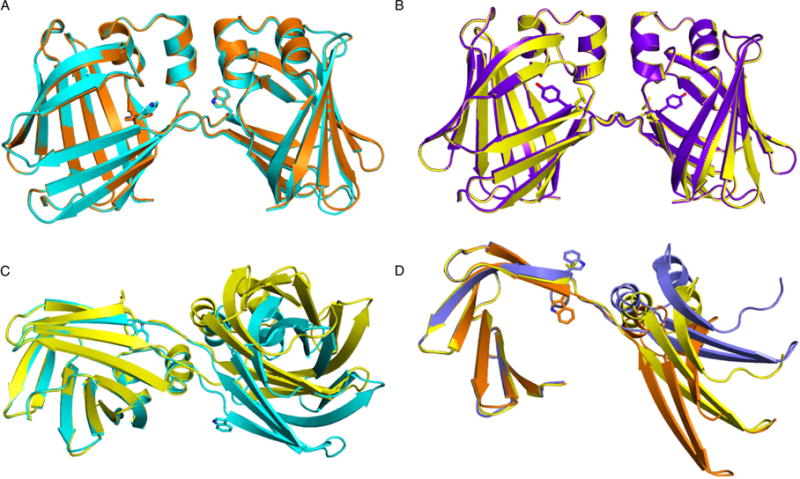
A comparison of symmetric and asymmetric hCRBPII dimers. Res60 in all structures is shown as a stick model A. An overlay of the asymmetric dimers: KLY60W (cyan) and Y60W (orange), Showing that they are virtually identical. B. An overlay of the symmetric WT hCRBPII (purple) and Y60L (yellow) dimers. C. A comparison of the symmetric and asymmetric dimers. The C-terminal 76 amino acids of the Y60L (yellow), and chain A of the KLY60W (cyan), dimers are overlaid (on the left side of the figure). Both chains of the dimers are shown. D. The C-terminal 76 amino acids of the symmetric Y60L (yellow) and chain A (orange) and chain B (blue) of the Y60W dimers are overlaid, showing the large deviation of the N-terminal regions of the three chains.
Though all four dimer structures share the same domain swapped interface, there are substantial differences in the structures. They can be classified as either symmetric (WT hCRBPII and Y60L), where the two chains that make up the dimer are essentially identical, or asymmetric (KLY60W and Y60W), where the two chains are distinct from one another (Figure 2). As shown in Figure 2B the two symmetric structures are similar, with the relative orientation of the two domains in the dimer essentially identical. However, there are significant differences in the torsion angles around residue 60, with a concomitant repositioning of the side chain (Figure 3). Tyr60 is buried inside of the protein, making a hydrogen bond with Glu72 in both the WT monomer and dimer, while Leu60 is flipped out of the interior and solvent exposed in the Y60L dimer (Figure 3A). This “flipped out” conformation is the result of a large deviation of the residue 59 phi angle relative to its monomer value (about −121° in the monomer versus −8 0° in the Y60L dimer) (Figure 3C).
Figure 3.
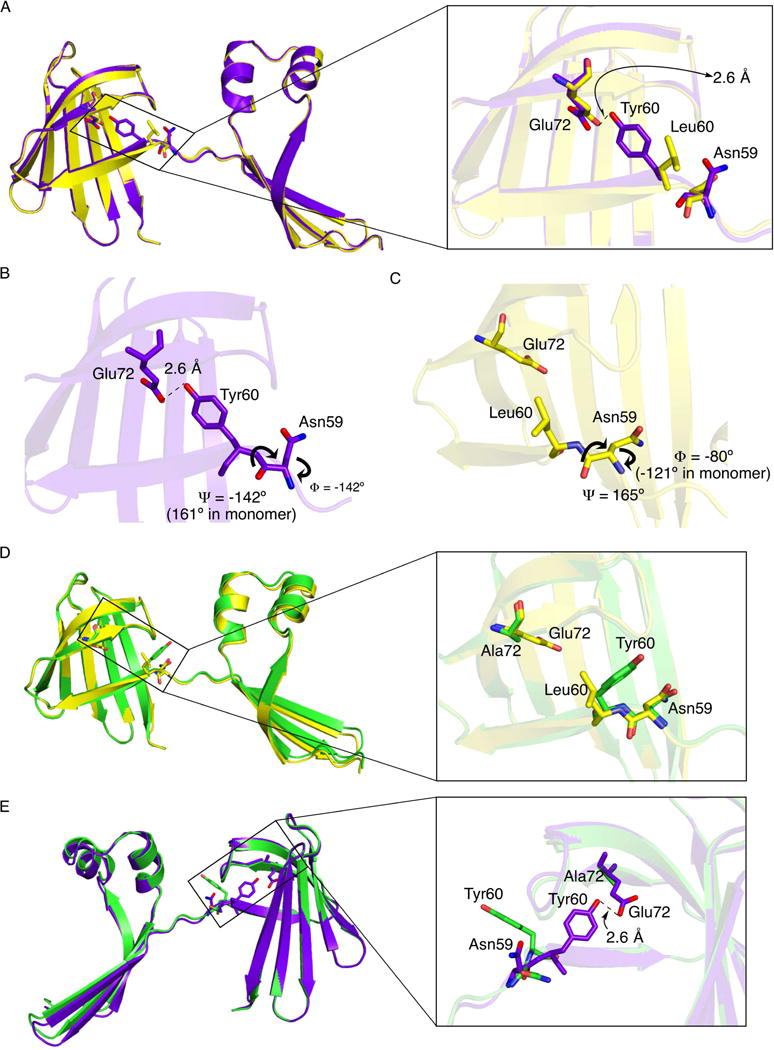
Torsional deviations outside the hinge-loop region define the relative orientation of the two domains of the dimer, with the highest deviation seen in Asn59. The loss of the E72/Y60 hydrogen bond leads to a dimer without Rhamachandran outliers. A. The WT (purple) and Y60L (yellow) dimers are overlaid. Inset, the hydrogen bond between Tyr60 and Glu72 compared to the “flipped out” conformation of Leu60. B. The critical Tyr60 and Asn59 region in the WT hCRBPII dimer, showing the key phi/psi angles. C. The same region in the Y60L dimer, showing the “flipped out” Leu60 and the key phi/psi angles. Comparison of the two shows how the large difference in the phi angle is compensated for in the psi angle, keeping the main chain of the two on a similar trajectory. This necessitates the large Ramachandran deviation in the psi angle of the WT CRBPII dimer. D. Overlay of chain A of the E72A dimer (green) and the Y60L dimer (yellow). Note that both show residue 60 pointing toward the solvent. E. Overlay of the E72A chain A (purple) and WT (green) dimers. Inset shows that without the E72/Y60 hydrogen bond, residue 60 is free to flip out, leading to a more relaxed dimer structure.
In contrast, the phi torsion angle of Asn59 in the WT dimer is −142°, which leads to the “flipped in” conformation (Figures 3A and 3B). To compensate for the resulting change in trajectory of the main chain, the Asn59 psi angle is radically rotated relative to all of the other structures (−142° in the WT dimer versus 161° in the monomer and 165 in the dimer Y60L, see Figures S5D and S5E in the SI), resulting in a substantial Ramachandran outlier at Asn59. Nonetheless it results in the main chains of the two symmetric structures once again closely tracking each other (Figure 3A).
In contrast to the symmetrical dimers, both Y60W and KLY60W structures are similar to one another and asymmetric (Figures 2A). As shown there is a significant deviation in the position of the N-terminal region when the C-terminal regions are overlaid (Figures 2C and 2D). The positional deviation is as large as 14 Å between the two conformations. This represents a radical change in the relative orientation of the two domains relative to the symmetric dimer.
The source of this large deviation can be seen in the torsion angles of the residues between 56–60 (Figures S5A–E). While in chain A of both KLY60W and Y60W dimers, all of the torsion angles, except the psi angle of Thr56, are similar to those of a monomer, in chain B a number of other torsion angles, namely the psi angles of residues Thr56 and Phe57, and the phi angle of Phe57, deviate from their monomer values (Figures 2 and S5A–E). Presumably, these differences in torsion angles are required to orient the two halves of the chain properly for dimer formation.
A possible mechanism for domain swapping in hCRBPII
The prevalence of domain swapped hCRBPII species was a complete surprise, especially the existence of the WT domain swapped dimer. Our working hypothesis is that the two halves of hCRBPII (the N-terminal and C-terminal halves) are capable of at least partially folding independently, initially as an extended “open monomer.” The path to monomer or dimer then depends on the ability of the two halves of the protein to either close on each other, which must involve at least rotation of the psi angle of Thr56, to form the monomer, or for the two halves of the protein to orient themselves for dimer formation. As one would expect, this is a rate-governed process, thus factors such as temperature and concentration should affect the outcome. As discussed previously, higher concentrations lead to more dimers, while lower concentrations reduce the opportunity for two “open monomers’ to encounter each other. One, but not the only, low energy pathway for a “dimer friendly” orientation appears to involve the flipping out of the residue at position 60, as seen in both the Y60L symmetric dimer and molecule B of both asymmetric dimers (Figures 2B and 2D). In contrast WT hCRBPII is “spring loaded” against this conformation by the hydrogen bond made between Glu72 and Tyr60, which serves to hold Tyr60 in the “flipped in” position (Figures 3A and 3B). Formation of the WT symmetric dimer then requires an almost 90° rotation of the A sn59 psi angle resulting in a substantial Ramachandran outlier (Figure S5D). This can be thought of as a “twist against the spring”. This explains the relatively small quantities of WT dimer and its relative instability. This hypothesis suggests that the folding of hCRBPII involves an open monomer intermediate that can be trapped in a dimeric form. One strategy of avoiding dimerization is for the open monomer to resist conformations required for dimerization. The conformation where only the Thr56 psi angle differs from the closed monomer represents such a state, as it can only dimerize with a molecule having the very different conformation seen in molecule B of the asymmetric structures (Figures 2D and 4). It is then expected that the ease with which residue 60 adopts the flipped out conformation can affect the relative amounts of each folded species. A Tyr at position 60 is resistant to flipping out due to the hydrogen bond it forms with Glu72, “spring loading” it against dimerization, and therefore requiring an unfavorable psi angle in Asn59 to achieve the relative orientation required for dimerization (Figure 3B). This hypothesis leads to the prediction that mutation of E72 to a residue incompetent for hydrogen bonding will “loosen the spring” resulting in an increase in dimer formation (Figure S5F). It should be noted that the open monomer is anticipated to be relatively flexible, especially when compared to the fully formed dimer.
Figure 4.
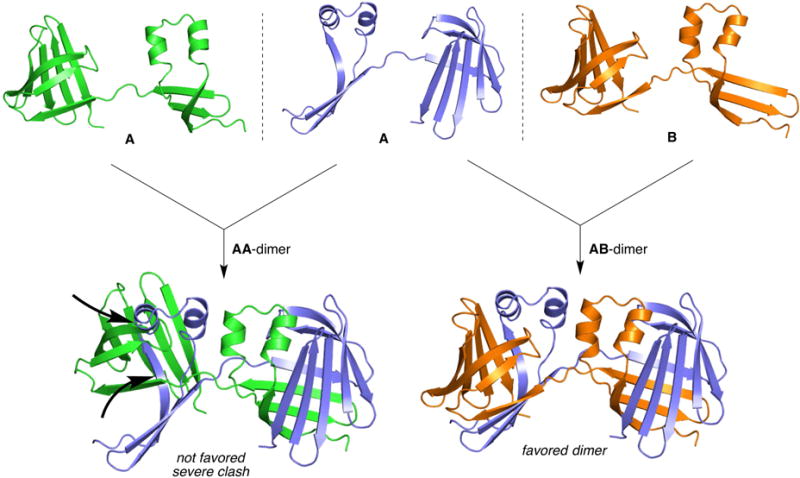
Model of folding in hCRBPII, which hypothesizes that an open monomer folding intermediate leads to domain swapped dimerization and requires proper relative orientation of the two halves of the open monomer: Left, the two A chains of an asymmetric dimer cannot dimerize due to severe steric clashes. Right, the A and B chains of the asymmetric dimer fit perfectly to form a stable, asymmetric dimer.
Dimerization and structure of the E72A mutant
To this end, the E72A mutant was created, expressed and purified. As predicted this mutation led to a dramatic increase in dimerization relative to WT hCRBPII. While WT hCRBPII produced only 10% dimer, the E72A mutant produced 50% dimer (Table S1, Figure S3G, S3H). The E72A mutant was then crystallized and its structure determined.
Though a crystallographic two-fold axis does not relate the two monomers in this dimer, the relative orientation of the N- and C-terminal halves of the protein are essentially identical to that of the symmetric dimers (Figures 3D and 3E). However, the two monomers that make up the dimer are not identical due to deviations in the torsion angles at position 57 (phi angles of −85° versus −133) and most notabl y, position 60 (phi angles of 35° versus −72° and psi angles of 92° versus 127) (Figures 4 and S5F). The result is that Tyr 60 is in the “flipped out” conformation, similar to that seen in both the Y60L dimer and molecule B of the KLY60W and Y60W asymmetric dimers as expected. This demonstrates that release of the E72/Y60 hydrogen bond increases the flexibility along the strand linking the C- and N- terminal ends, allowing an increase in dimerization, as predicted (Figure S5F). Together the data are consistent with the hypothesis that the interaction between Y60 and E72 in the open monomer folding intermediate effectively holds the two domains in a conformation unfavorable to dimerization, promoting the formation of the physiologically relevant monomeric species.
Conclusions
We have discovered and characterized an extensive, domain swapped dimer of hCRBPII, a member of the iLBP family, during routine protein expression in E. coli. with no evidence of interconversion between dimeric and monomeric forms, even over many days of incubation at room temperature. It therefore appears that the dimeric form may represent a new protein fold of hCRBPII that cannot interconvert to its native monomeric fold. A confluence of data implies that the monomer/dimer ratio is kinetically controlled, and that the dimeric form represents the kinetic trapping of a relatively well-structured “open monomer” folding intermediate. The relative amounts of the two folds is then determined by the concentration of the folding intermediate, and the ease at which the two halves of the protein can adopt relative orientations favorable for dimer formation (Figure 4). This represents a significant departure from the folding pathway reported for hCRABPI, where it is observed that beta barrel collapse, involving elements of both halves of the sequence, occurs early in the folding pathway, thus preventing the formation of off pathway species such as the dimer we have identified for hCRBPII.(Gershenson et al., 2014) This indicates that evolutionarily and structurally related members of the same protein family can have substantially different folding pathways to obtain relatively similar 3D structures.
Further, our data suggest that the WT sequence has built into it a resistance to dimerization caused by an interaction between Tyr60 and Glu72 (Figures 3 and S5F), which promotes a relative orientation of the two halves of the protein that is inconsistent with dimer formation. Consistent with this hypothesis, mutation of Glu72 to Ala results in a substantial increase in the dimeric form of the protein, even though it is not in the strand connecting the two halves of the dimer.
Given this proposed mechanism, we would predict that there are other mutational “hot spots” for dimerization, both located along the strand (including residues 52–64) that connects the two halves of the dimer and therefore serves to control the relative orientation of the two halves, and perhaps the residues that interact with them (Figure S5F). The physiological role of a putative dimer is unclear, but it is clear that neither its presence, nor its potential biology can be ignored. The ease with which the dimeric fold can become dominant in WT hCRBPII suggests that other members of the iLBP family may also produce domain swapped dimers similar to that seen for hCRBPII. Indeed a domain swapped dimer of FABP5 was recently reported.(Sanson et al., 2013) Clearly, investigation of other members of the family are warranted as it seems quite possible that the domain swapped dimer investigated here may be the physiologically relevant form for some members of the iLBP protein family.
Experimental Procedures
Construction, expression and purification of of the mutants
The cloning, expression and purification of hCRBPII mutants were performed as previously described (Wang et al., 2012), save the modifications described in the Supporting Information.
Crystallization, data collection and refinement
Crystallization, data collection and refinement of the structures were performed as previously described (Wang et al., 2012) save the modifications described in the Supporting Information. Crystallographic, atomic coordinates and structure factors have been deposited in the Protein Data Bank, [PDB ID codes: 4ZCB (Y60W, dimer); 4ZH6 (Y60L, dimer); 4ZH9 (WT dimer); 4ZJ0 (Q108K:K40L:Y60W, monomer); 4ZGU (Y60W, monomer); 4ZR2 (Q108K:K40L:Y60W, dimer); 5DG4 (Y60L, monomer); 5DPQ (E72A, dimer)].
Protein Refolding
The monomeric protein fraction was denatured by addition of urea to 8 M at 4 °C and dialyzed against 10 mM Tris pH = 8.0 with a Slide-A-Lyzer® Dialysis Cassette (3,500 MWCO, Thermo Fisher Sci.). The renatured protein was concentrated and analyzed by size exclusion chromatography.
Thermal Melting Curves for monomer and dimer mutants
10 μM protein (in 50 mM phosphate buffer, pH = 7.5) was heated and monitored by CD spectroscopy using a JASCO (J-810) Spectropolarimeter connected to a NESLAB RTE-111 bath circulator. The temperature was increased 5 °C for each CD measurement. The curves were fitted based on the Levenberg-Marquardt formula using OriginLab (Northampton, MA).(Greenfield, 2006; Ramos et al., 2008)
Kd determination
The dissociation constant (Kd) of both monomer and dimer species with all-trans-retinal and all-trans-retinol were determined by fluorescence titration as previously reported.(Vasileiou et al., 2007)
Supplementary Material
HIGHLIGHTS.
An extensive domain swapped dimer of hCRBPII is formed when overexpressed.
No interconversion between monomer and dimer is observed at room temperature.
Structures of mutant dimers suggest a folding pathway with an open monomer intermediate.
It is proposed that folding is governed by the structure of the folding intermediate.
Acknowledgments
This work was funded by the NIH (Grant No. GM101353). The crystallography data were collected at Advanced Photon Source, operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the DOE under contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The authors would like to thank Hadi Gholami for assistance with the CD experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
The project was conceived based on experimental observations by WW. Overall direction and administration of the project was the responsibility of BB and JHG. ZA and ZN contributed equally to the experimental portion. All authors contributed to the discussion and analysis of data, and design of experiments. BB and JHG were responsible for writing the bulk of the manuscript.
Accession Numbers: 4ZCB, 4ZH6, 4ZH9, 4ZJ0, 4ZGU, 4ZR2, 5DG4, 5DPQ
References
- Bax B, Lapatto R, Nalini V, Driessen H, Lindley PF, Mahadevan D, Blundell TL, Slingsby C. X-ray analysis of beta-B2-crystallin and evolution of oligomeric lens proteins. Nature. 1990;347:776–780. doi: 10.1038/347776a0. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Choe S, Eisenberg D. Rlefined strucutre of dimeric diphtheria-toxin at 2.0-angstrom resolution. Protein Sci. 1994;3:1444–1463. doi: 10.1002/pro.5560030911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping - A mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbasova T, Nosrati M, Vasileiou C, Wang WJ, Sing K, Lee S, Yapici I, Geiger JH, Borhan B. Rational Design of a Colorimetric pH Sensor from a Soluble Retinoic Acid Chaperone. J Am Chem Soc. 2013;135:16111–16119. doi: 10.1021/ja404900k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbasova T, Santos EM, Nosrati M, Vasileiou C, Geiger JH, Borhan B. Light-Activated Reversible Imine Isomerization: Towards a Photochromic Protein Switch. ChemBioChem. 2016;17:407–414. doi: 10.1002/cbic.201500613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey J, Lindman S, Bauer M, Linse S. Protein reconstitution and three-dimensional domain swapping: Benefits and constraints of covalency. Protein Sci. 2007;16:2317–2333. doi: 10.1110/ps.072985007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cowan SW, Newcomer ME, Jones TA. Crystallographic studies on a family of cellular lipophilic transport proteins - refinement of P2 myelin protein and the structure determination and refinement of cellular retinol-binding protein in complex with all-trans-retinol. J Mol Biol. 1993;230:1225–1246. doi: 10.1006/jmbi.1993.1238. [DOI] [PubMed] [Google Scholar]
- Crist RM, Vasileiou C, Rabago-Smith M, Geiger JH, Borhan B. Engineering a rhodopsin protein mimic. J Am Chem Soc. 2006;128:4522–4523. doi: 10.1021/ja058591m. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: an important but neglected aspect of the intracellular environment (vol 11, pg 114, 2001) Curr Opin Struct Biol. 2001;11:500–500. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- Fandrich M, Fletcher MA, Dobson CM. Amyloid fibrils from muscle myoglobin - Even an ordinary globular protein can assume a rogue guise if conditions are right. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- Folli C, Calderone V, Ottonello S, Bolchi A, Zanotti G, Stoppini M, Berni R. Identification, retinoid binding, and x-ray analysis of a human retinol-binding protein. Proc Natl Acad Sci. 2001;98:3710–3715. doi: 10.1073/pnas.061455898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MK, Dyda F, Dobrodumov A, Gronenborn AM. Core mutations switch monomeric protein GB1 into an intertwined tetramer. Nat Struct Biol. 2002;9:877–885. doi: 10.1038/nsb854. [DOI] [PubMed] [Google Scholar]
- Gershenson A, Gierasch LM, Pastore A, Radford SE. Energy landscapes of functional proteins are inherently risky. Nat Chem Biol. 2014;10:884–891. doi: 10.1038/nchembio.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nature Protocols. 2006;1:2527–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronenborn AM. Protein acrobatics in pairs - dimerization via domain swapping. Curr Opin Struct Biol. 2009;19:39–49. doi: 10.1016/j.sbi.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran K, Hagler AT, Gierasch LM. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins-Structure Function and Genetics. 2004;54:179–194. doi: 10.1002/prot.10520. [DOI] [PubMed] [Google Scholar]
- Hakansson M, Svensson A, Fast J, Linse S. An extended hydrophobic core induces EF-hand swapping. Protein Sci. 2001;10:927–933. doi: 10.1110/ps.47501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntress MM, Gozem S, Malley KR, Jailaubekov AE, Vasileiou C, Vengris M, Geiger JH, Borhan B, Schapiro I, Larsen DS, et al. Toward an Understanding of the Retinal Chromophore in Rhodopsin Mimics. J Phys Chem B. 2013;117:10053–10070. doi: 10.1021/jp305935t. [DOI] [PubMed] [Google Scholar]
- Koharudin LMI, Liu L, Gronenborn AM. Different 3D domain-swapped oligomeric cyanovirin-N structures suggest trapped folding intermediates. Proc Natl Acad Sci. 2013;110:7702–7707. doi: 10.1073/pnas.1300327110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KSS, Berbasova T, Vasileiou C, Jia X, Wang W, Choi Y, Nossoni F, Geiger JH, Borhan B. Probing Wavelength Regulation with an Engineered Rhodopsin Mimic and a C15-Retinal Analogue. Chempluschem. 2012;77:273–276. [Google Scholar]
- Liu Y, Eisenberg D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YS, Gotte G, Libonati M, Eisenberg D. Structures of the two 3D domain-swapped RNase A trimers. Protein Sci. 2002;11:371–380. doi: 10.1110/ps.36602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YS, Hart PJ, Schlunegger MP, Eisenberg D. The crystal structure of a 3D domain-swapped dimer of RNase A at a 2.1-angstrom resolution. Proc Natl Acad Sci. 1998;95:3437–3442. doi: 10.1073/pnas.95.7.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton AP. Implications of macromolecular crowding for protein assembly. Curr Opin Struct Biol. 2000;10:34–39. doi: 10.1016/s0959-440x(99)00045-7. [DOI] [PubMed] [Google Scholar]
- Nossoni Z, Assar Z, Yapici I, Nosrati M, Wang W, Berbasova T, Vasileiou C, Borhan B, Geiger J. Structures of holo wild-type human cellular retinol-binding protein II (hCRBPII) bound to retinol and retinal. Acta Crystallogr Sect D Biol Crystallogr. 2014;70:3226–3232. doi: 10.1107/S1399004714023839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill JW, Kim DE, Johnsen K, Baker D, Zhang KYJ. Single-site mutations induce 3D domain swapping in the B1 domain of protein L from Peptostreptococcus magnus. Structure. 2001;9:1017–1027. doi: 10.1016/s0969-2126(01)00667-0. [DOI] [PubMed] [Google Scholar]
- Ong DE, Page DL. Cellular retinol-binding protein (type-2) is abundant in human small-intestine. J Lipid Res. 1987;28:739–745. [PubMed] [Google Scholar]
- Ramos CRR, Oyama S, Jr, Sforca ML, Pertinhez TA, Ho PL, Spisni A. 2POA: Schistosoma mansoni Sm14 Fatty Acid-Binding Protein: improvement of protein stability by substitution of the single Cys62 residue. Worldwide Protein Data Bank 2008 [Google Scholar]
- Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. In: Mattews JM, editor. Protein Dimerization and Oligomerization in Biology. 2012. pp. 137–152. [DOI] [PubMed] [Google Scholar]
- Sanson B, Wang T, Sun J, Kaczocha M, Ojima I, Deutsch D, Li H. 4AZM: Human epidermal fatty acid-binding protein (FABP5) in complex with the inhibitor BMS-309413. Worldwide Protein Data Bank 2013 [Google Scholar]
- Schlunegger MP, Bennett MJ, Eisenberg D. Oligomer formation by 3D domain swapping: A model for protein assembly and misassembly. In: Wetzel R, editor. Advances in Protein Chemistry, Vol 50: Protein Misassembly. 1997. pp. 61–122. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways (vol 426, pg 900, 2003) Nature. 2004;428:445–445. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Shameer K, Shingate PN, Manjunath SCP, Karthika M, Pugalenthi G, Sowdhamini R. 3DSwap: curated knowledgebase of proteins involved in 3D domain swapping. Database- the Journal of Biological Databases and Curation. 2011 doi: 10.1093/database/bar042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtilerman MD, Ding TT, Lansbury PT. Molecular crowding accelerates fibrillization of alpha-synuclein: Could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- Sillitoe I, Cuff AL, Dessailly BH, Dawson NL, Furnham N, Lee D, Lees JG, Lewis TE, Studer RA, Rentzsch R, et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 2013;41:D490–D498. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud JC, Liu C, Teng PK, Eisenberg D. Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc Natl Acad Sci. 2012;109:7717–7722. doi: 10.1073/pnas.1203193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M, Blake C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. In: Wetzel R, editor. Advances in Protein Chemistry, Vol 50: Protein Misassembly. 1997. pp. 123–159. [DOI] [PubMed] [Google Scholar]
- Szilagyi A, Zhang Y, Zavodszky P. Intra-Chain 3D Segment Swapping Spawns the Evolution of New Multidomain Protein Architectures. J Mol Biol. 2012;415:221–235. doi: 10.1016/j.jmb.2011.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaezeslami S, Jia XF, Vasileiou C, Borhan B, Geiger JH. Structural analysis of site-directed mutants of cellular retinoic acid-binding protein II addresses the relationship between structural integrity and ligand binding. Acta Crystallogr Sect D Biol Crystallogr. 2008;64:1228–1239. doi: 10.1107/S0907444908032216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaezeslami S, Mathes E, Vasilelou C, Borhan B, Geiger JH. The structure of apo-wild-type cellular retinoic acid binding protein II at 1.4 A and its relationship to ligand binding and nuclear translocation. J Mol Biol. 2006;363:687–701. doi: 10.1016/j.jmb.2006.08.059. [DOI] [PubMed] [Google Scholar]
- Vasileiou C, Lee KSS, Crist RM, Vaezeslami S, Goins SA, Geiger JH, Borhan B. Dissection of the critical binding determinants of cellular retinoic acid binding protein II by mutagenesis and fluorescence binding assay. Proteins: Struct Funct Bioinform. 2009a;76:281–290. doi: 10.1002/prot.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou C, Vaezeslami S, Crist RM, Rabago-Smith M, Geiger JH, Borhan B. Protein design: Reengineering Cellular Retinoic Acid Binding Protein II into a rhodopsin protein mimic. J Am Chem Soc. 2007;129:6140–6148. doi: 10.1021/ja067546r. [DOI] [PubMed] [Google Scholar]
- Vasileiou C, Wang W, Jia X, Lee KSS, Watson CT, Geiger JH, Borhan B. Elucidating the exact role of engineered CRABPII residues for the formation of a retinal protonated Schiff base. Proteins: Struct Funct Bioinform. 2009b;77:812–822. doi: 10.1002/prot.22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Geiger JH, Borhan B. The photochemical determinants of color vision. Bioessays. 2014;36:65–74. doi: 10.1002/bies.201300094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Nossoni Z, Berbasova T, Watson CT, Yapici I, Lee KSS, Vasileiou C, Geiger JH, Borhan B. Tuning the Electronic Absorption of Protein-Embedded All-trans-Retinal. Science. 2012;338:1340–1343. doi: 10.1126/science.1226135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapici I, Lee KSS, Berbasova T, Nosrati M, Jia X, Vasileiou C, Wang W, Santos EM, Geiger JH, Borhan B. “Turn-On” Protein Fluorescence: In Situ Formation of Cyanine Dyes. J Am Chem Soc. 2015;137:1073–1080. doi: 10.1021/ja506376j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.