

Abstract
Two synthetic routes to the briarane stereotetrad have been investigated. The first route employed a boron aldol reaction to establish the stereogenic all-carbon quaternary carbon (C1). In this case, it was found that torsional steering in the transition state led to the formation of the undesired configuration at this position. The second route makes use of a highly diastereoselective acetylide conjugate addition/β-ketoester alkylation sequence to construct the vicinal C1 and C10 stereocenters with the correct relative configuration. Originally, it was proposed that torsional steering in the transition state for the ketoester alkylation step was the primary factor responsible for generating the major product. DFT calculations reveal that while torsional steering does play a role, larger conformational factors must also be considered. Tthese calculations also reveal that an unusual C–H•••π (alkyne) interaction may contribute to lowering the energy of one transition state that leads to the observed stereoisomer. Ultimately, this strategy leads to a concise synthesis (under 10 steps) of the stereotetrad core common to the briarane diterpenoids.
Introduction
The briarane skeletal system represents one of the more common natural product families that have been isolated from the gorgonian octocorals.1 The wide distribution of gorgonian corals has led to the isolation of an extremely large number of briarane-type diterpenoids.2 Several examples have also been reported from non-gorgonian sources such as the sea pansy R. reniformis,3 the sea pen octocoral S. tentaculatum,4 and the Mediterranean nudibranch mollusk Armina maculata.5 The briaranes are thought2a,d to be biosynthetically related to the cembrane diterpenoids, another natural product family found in several marine gorgonian species.6 The briarane skeleton consists of a trans-fused bicyclo[8.4.0]tetradecane ring system. Most members also contain a γ-lactone comprising the C7-C8-C17-C19 carbons. Another common feature is a congested set of four contiguous stereogenic centers comprising C1-C2-C10-C14. Oxidation can be present at nearly every carbon in the system as evidenced by the representative family members shown in Figure 1.
Figure 1.

The briarane skeleton and representative family members.
The first member of the family, briarein A, was reported by Cierezco and co-workers in 1977.7 Since that time, over 600 unique briarane diterpenoids have been characterized.2 Given the size and structural diversity associated with the briarane family, it is not surprising that many family members have demonstrated activity in numerous areas, including: anti-inflammatory, antiviral, antifungal, immunomodulatory, insect control, antifouling, and ichthyotoxicity. Others, like brianthein W,8 excavatolide M,9 and briareolate ester L10 have promising activity against several cancer cell lines.11 Unfortunately, the difficulties associated with collecting marine natural products, together with the scarcity of the natural material has hampered further investigations into the biological activity of these compounds and others in the family.
Despite the wide structural diversity and interesting biological activity associated with the briaranes, relatively little synthetic effort has been expended on their synthesis.12 Only a few groups have reported synthetic progress towards any member of the briaranes. To date, no completed total synthesis of any family member has been reported; a fact that illustrates the significant challenge posed by these compounds.
Recent synthetic efforts have focused on accessing what is arguably the most challenging portion of the briarane framework – the C1-C2-C10-C14 stereotetrad core. In 2006 Ito and Iguchi13 reported the first efforts in this area (Scheme 1). Key steps in this synthesis include the use of a radical cyclization to construct the all-carbon quaternary stereocenter (C1) and a diastereoselective addition of vinylmagnesium bromide to generate the C2 stereocenter. The later was rationalized with a chelation model in which the C10 alkyl group adopts an equatorial position and blocks the Re face of the aldehyde. In 2010, Bates and co-workers reported the use of an Eschenmoser–Claisen rearrangement to generate the C1 stereocenter, en route to the completed stereotetrad (Scheme 2).14 More recently, Crimmins and co-workers reported the construction of the C1-C2-C10 stereotriad using a dianionic Ireland–Claisen rearrangement (Scheme 3).15 Crimmins has yet to report the advancement of this material to the completed stereotetrad core.
Scheme 1.
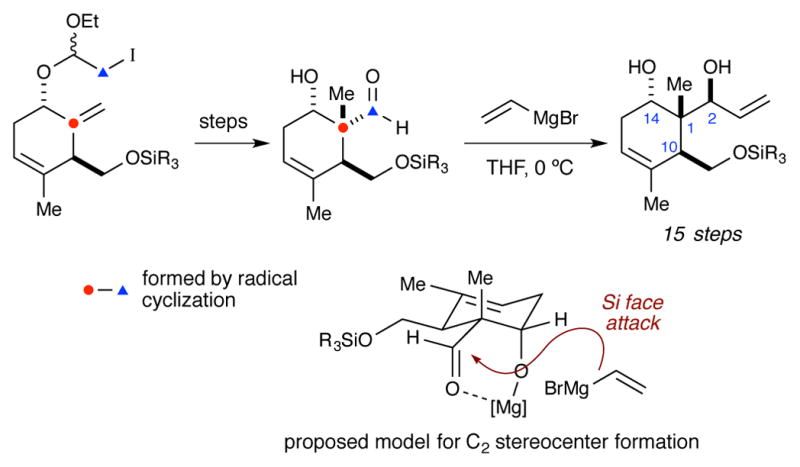
Ito and Iguchi’s synthesis of the briarane stereotetrad.
Scheme 2.
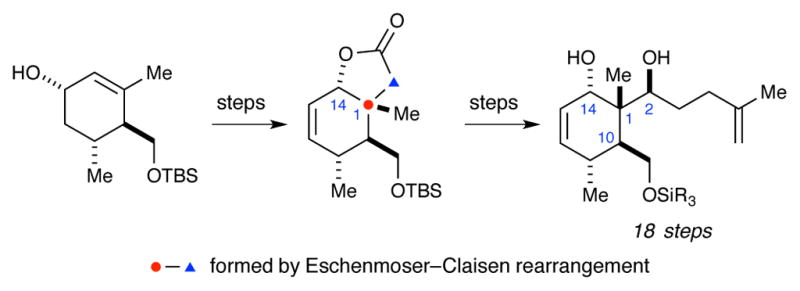
Bates’ synthesis of the briarane stereotetrad.
Scheme 3.
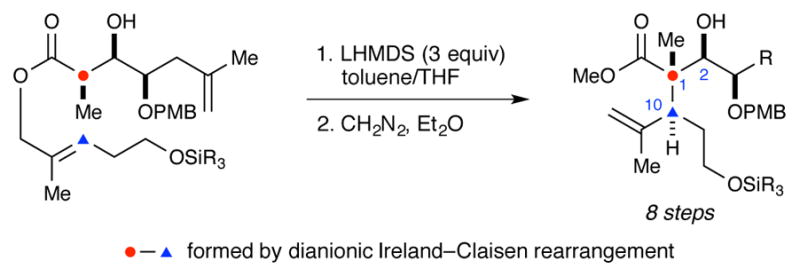
Crimmins’ construction of the C1, C2, and C10 stereocenters.
The work reported by Ito/Iguchi, Bates, and Crimmins demonstrate the challenges associated with accessing the briarane stereotetrad. They provide a foundation for future synthetic efforts, but also offer room for improvement. In broad terms, it is clear that in order to realize a successful synthesis of even the simplest members of the family an efficient synthesis of the stereotetrad is required. With this in mind, we set out to devise a synthetic route to the briaranes that harnesses the dense functionality and rich reactivity profile of 2,5-cyclohexadienones. Herein, we report a more complete account of our efforts aimed at developing a concise synthetic route to the briarane stereotetrad.16 In the course of our studies, we encountered several problems related to reactivity and stereoselectivity that can be traced to the profound and unexpected influence of torsional steering17 in the relevant transition states.
Results and discussion
Retrosynthetic analysis
Inspired by the work of Ito/Iguchi and Bates, we identified briarane stereotetrad fragment 1 as our initial target (Scheme 4A). This fragment contains the congested C1-C2-C10-C14 stereotetrad as well as a C11–C12 alkene. The later would be critical for accessing the myriad of family members that are oxygenated at C11 and C12. Furthermore, the remaining functionality contained within intermediate 1 could, in principle, be used as a basis for closing the 10-membered ring and installing the butyrolactone moiety. Thus, we believed this intermediate would prove useful as a common building block for synthesizing numerous briarane family members as well as non-natural analogues; provided it could be prepared in relatively few synthetic steps.
Scheme 4.

Initial synthetic strategy.
Several early attempts at using a bicyclic lactone similar to 5 as the starting material were made. Unfortunately, the C1 position in these compounds proved to be quite recalcitrant to many of the reactions needed to functionalize this position (e.g., alkylation and aldol reactions).18 We hypothesized that the rigid nature of this bicyclic system might prevent any C1 enolates from achieving a conformation necessary to successfully engage the electrophiles in question. With this in mind, we thought that moving to a less rigid monocyclic enolate might alleviate these reactivity problems.
Thus, a new approach was devised in which building block 1 would be accessed from cyclohexenone 2 through reductive cleavage of the C11 alkoxide and stereoselective reduction of the C14 ketone. The C1 and C2 stereocenters in 2 would be generated concurrently by using an aldol variant. Although such a reaction was not successful on bicyclic substrates (i.e., those derived from 5), we hoped that the enolate derived from 3 would be more stable and sterically accessible. Furthermore, it was expected that the C10 stereocenter would control the approach of the aldehyde partner (vida infra). Ultimately, cyclohexadieneone 4 would serve as the starting material for this route. However, performing a conjugate addition on dienone 4 was expected to be challenging, as reagents known to typically promote conjugate additions (i.e., cuprates) often lead to rearomatization of 2,5-cyclohexadienones.19 We were also aware that the enolate resulting from a conjugate addition could also undergo rearomatization under basic conditions.20 Nevertheless, we were optimistic that suitable conditions could be found that would enable the isolation of the conjugate addition product.
An important consideration made at the outset was the identity of the fragment (R1) attached to the C10 stereocenter. This fragment must be able to be converted into the carbonyl-containing fragment present in building block 1, but must also be compatible with the chemistry needed to forge cyclohexenone 2. With this in mind, two possibilities were considered: an allyl group and a TMS-protected alkyne. An allyl group could easily be converted into an aldehyde through an oxidative cleavage reaction (Scheme 4B), and would be quite resistant to the reactions proposed for the early stages of our synthesis. Similarly, a TMS-alkyne would likely survive the early stage chemistry and could be converted into a carboxylic acid through hydroboration-oxidation (Scheme 4C).21
Synthesis of an aldol precursor
Our synthetic work began by investigating the installation of an allyl fragment to C10. Initially, we were inspired by a report by Taber, in which a formal conjugate addition of an allyl fragment could be accomplished in two steps by first performing a 1,2-addition of allylmagnesium bromide followed by an anionic oxy-cope rearrangement.22 Thus, addition of allylmagnesium bromide into cyclohexadienone 6 gave tertiary alcohol 7 as a 1:1 mixture of diastereomers23 (Scheme 5). Treating 7 with KH and 18-crown-6 in THF affected the desired anionic oxy-cope rearrangement, but this was accompanied by a significant amount of decomposition. As a result, the desired enone (8) was isolated in only 10% yield. Unfortunately, all attempts at improving upon this promising result only resulted in general decomposition or aromatization to the phenol. The use of a Sakurai reaction (allyltrimethylsilane, TiCl4) to install the C10 allyl fragment only returned aromatized products.
Scheme 5.
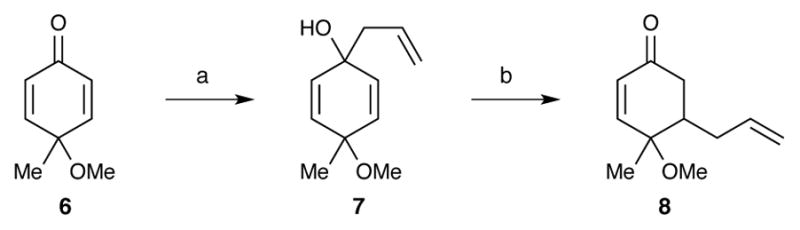
(a) allylMgBr, THF, −78 °C, 75%, 1:1 dr; (b) KH, 18-crown-6, THF, rt, 10%.
We then investigated different conditions for affecting the conjugate addition of acetylide nucleophiles. In our hands, the use of aluminium24 and nickel promoters25 failed to add TMS acetylene into methoxydienone 6. Our attention then turned toward the possibility of using quinol 9 in a ligand-assisted nucleophilic addition (LANA) reaction.26 We were delighted to find that the reaction of three equivalents of the Mg acetylide of trimethylsilylacetylene with quinol 9 produced cyclohexenone 10 as the only observed product (Scheme 6). Notably, neither conversion of 9 into the lithium alkoxide prior to acetylide addition nor the addition of HMPA was necessary in this case.26 Incomplete conversion was observed with two equivalents of the magnesium acetylide. A similar LANA reaction was attempted using allylmagnesium bromide, but only the 1,2-addition product was observed.
Scheme 6.
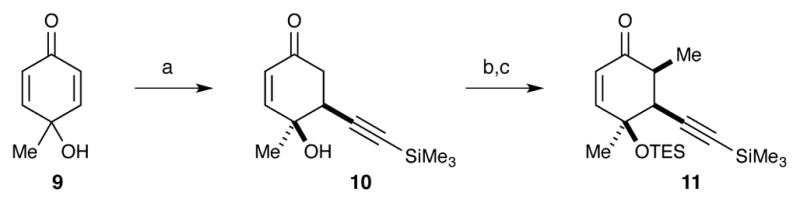
(a) TMS acetylene, EtMgBr, THF, −78 °C to rt, 72%; (b) Et3SiCl, imidazole, DMF, rt, 66%; (c) LDA, MeI, 10:1 THF/HMPA, −78 °C to rt, 63%.
Following the successful LANA reaction, the tertiary alcohol was protected as a TES ether. α-Methylation of the ketone was achieved using LDA in the presence of HMPA. Good conversion (~76%) to 11 could be achieved without HMPA, but we were unable to separate the methylated product from the unreacted starting material. Addition of HMPA led to complete consumption of starting material, simplifying purification. The use of alternate amide bases (LHMDS, LiNEt2) led to inconsistent results or poor performance.
Optimization and stereochemical analysis of the aldol reaction
With ketone 11 in hand, we were in a position to investigate the key aldol reaction needed to forge the C1–C2 bond. Unfortunately, the literature provided little guidance with respect to which aldol conditions might provide the best selectivity in this cases, or even which diastereomer to expect as the major product.27 The present situation was further complicated by uncertainty regarding the conformational preference of the polysubstituted enolate (Figure 2). There was some literature support for an axial preference of alkoxides in 3-substituted cyclohxenes (i.e., conformer II).28 However, such a conformer would introduce an A1,2 interaction between the enolate methyl group and pseudoequitorial alkyne. The allylic strain can be alleviated by placing the alkyne in a more preferred29 pseudoaxial position (conformer I), but this would require the silyloxy group to move into a, seemingly, less favorable equitorial position. However, when the approach of the aldehyde to each conformer is considered, one finds that both may give rise to the desired diastereomer as the major product. In other words, the approach of the aldehyde to the Si face of conformer I will be blocked by the axial alkyne, and approach to the Si face of conformer II should be disfavored due to the axial silyl ether.
Figure 2.
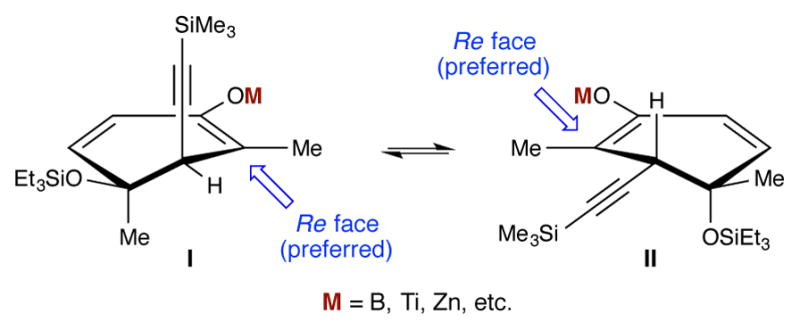
Initial stereochemical analysis of the planned aldol reaction.
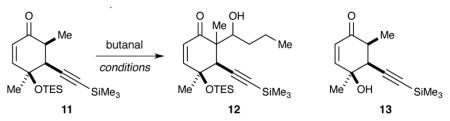
When ketone 11 was subjected to standard boron-aldol conditions (Bu2BOTf, i-Pr2NEt),30 we were excited to isolate a small amount of material that was identified by mass spectrometry to be aldol adduct 12. But, starting ketone 11 and desilylated starting material (13) were also isolated. Encouraged by this result, we investigated other boron Lewis acids (Cy2BOTf or PhBCl2 31), but these also resulted in very low conversion (Table 1, entry 1). Importantly, these experiments revealed that adding the Lewis acid to a mixture of the substrate and base could minimize desilylation of ketone 11.
Table 1.
Attempted aldol reactions with ketone 25.
| |||
|---|---|---|---|
| entry | enolization conditionsa | % conversion (yield) | drb |
| 1 | Bu2BOTf, Cy2BOTf, or PhBCl2, DIPEA, CH2Cl2, −78 °C | trace | – |
| 2 | LDA, THF, −78→0 °C | 50 | 10:6.2:3.9:1 |
| 3 | LDA, THF, −78→0 °C, then Ti(Oi-Pr)3Cl, −78 °C | 70 | 3.2:1 |
| 4 | Et3N, CH2Cl2, −78 °C→0 °C, then Bu2BOTf | 100 (67) | >20:1 |
In all cases, the aldehyde was added at −78 °C.;
Determined by 1H NMR. Diastereomers were inseparable. Relative configurations were not assigned for entries 2 and 3.
At this time, it was unclear whether the low conversion of ketone 11 was due simply to incomplete enolization or the unreactivity of the resulting sterically congested enolate. In order to probe this question, a brief deuterium incorporation study was performed (Scheme 7). Deprotonation of 11 with LDA at −78 °C for 1h led to only 30% observed deuterium incorporation. However, complete deuterium incorporation was observed by allowing the reaction to warm to 0 °C for 30 minutes. Importantly, no decomposition was observed at these warmer temperatures.
Scheme 7.
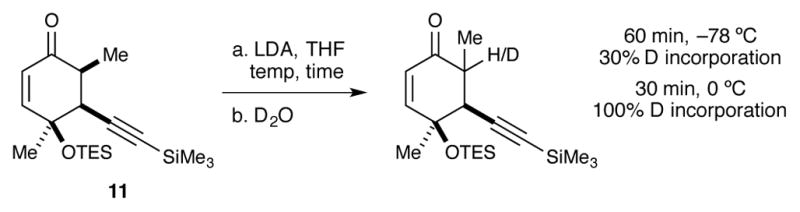
Deuterium incorporation study.
We then returned to the investigation of Li, B, and Ti enolates using our improved deprotonation conditions. We were able to achieve 50% conversion from the Li enolate (Table 1, entry 2). However, this resulted in an inseparable mixture of all four possible diastereomers in a 10:6.2:3.9:1 ratio. Due to significant spectral overlap, we were unable to assign the relative configuration of these products. Formation of a Ti enolate32 (Table 1, entry 3) resulted in both improved conversion (70%) and diastereoselectivity (dr = 3.2:1). We attributed the improved selectivity to a closed transition state during the reaction with the Ti enolate. The poor diastereoselectivity during observed with the Li enolate may be due to competing open and closed transition states. To our delight, performing a boron aldol reaction (Table 1, entry 4) utilizing elevated deprotonation temperatures and a less-bulky base (NEt3) led to full consumption of the starting material. The resulting aldol adduct 12 could be obtained in 67% yield as a single diastereomer.
Several 1D and 2D NOE experiments were performed to assign the relative configuration of boron aldol product 12 (Figure 3). For the desired diastereomer (12a) we expected to observe NOE interactions between the C10 proton (blue) and both the methyl group on C11 (red) and the methyne proton (pink) formed during the aldol. An NOE interaction between the C10 proton and the methyl group on C1 (green) would be unlikely in this diastereomer, due to the trans relationship between these groups. However, a very clear NOE correlation was observed between the C10 proton and both methyl groups, indicating that the product contained the undesired configuration at C1 (i.e., 12b).
Figure 3.

1D NOE analysis of aldol product 12.
The high selectivity observed for the formation of diastereomer 12b appears to run counter to the steric considerations that were made during our original analysis (Figure 2). Previously, our group observed similar results during the alkylation of a bicyclic malonate in support of a total synthesis of sorbicillactone A.33 Subsequent computational work revealed those observations could be attributed to torsional steering in the competing SN2 transition states.34 Torsional steering is a stereoselectivity phenomenon that has been advanced by Houk over the past several decades.17 Essentially, this model states that when two diastereomeric transition state can compete, the one that gives rise to favorable torsional effects (staggering) is favored over the one that gives rise to unfavorable torsional effects (eclipsing).35 Because of this history, we reevaluated the different aldol transition states to see if torsional steering could be operating in this scenario. Indeed, if one considers the enolate conformation in which there is an axial silyloxy group and an equitorial alkyne (e.g., conformer II in Figure 2) to be the reactive conformer, then it is clear from the Newman projections in Figure 4 that approach of the aldehyde to the Re face of the enolate will lead to eclipsing interactions in III. In contrast, approach to the Si face will lead to a more staggered transition state (IV) and the formation of the observed diastereomer (12b). Although this outcome was surprising at the time, we did note that it is quite consistent with the stereoselectivity observed during the formation of ketone 11 (cf., Scheme 6). There the C1 methyl group was installed syn to the alkyne (JCH1–CH10 = 4.4 Hz in 11).
Figure 4.

Revised stereoselectivity model during boron aldol reaction of ketone 11. Coordination between aldehyde and Lewis acid not shown for clarity.
Revised synthetic plan
The high level of stereocontrol observed during the boron aldol reaction indicated to us that disconnecting the C1 stereocenter at C1–C2 bond will be quite challenging with the C10 stereocenter in place.§ However, we reasoned that the same torsional strain arguments that thwarted our efforts in constructing the C1–C2 bond, might allow us to introduce the C15 methyl group after the C1–C2 bond was in place. In order to accomplish this though, a significant revision to our synthetic approach would be required (Scheme 8).
Scheme 8.
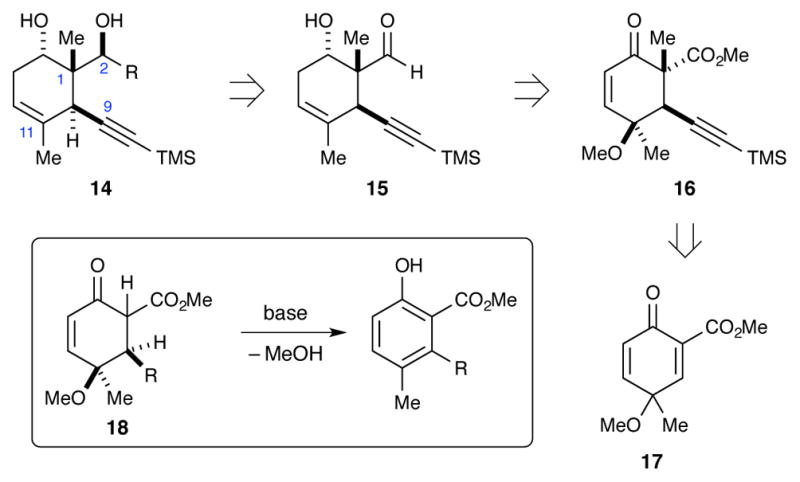
Revised synthetic plan to accommodate torsional steering during installation of the C15 methyl group.
Our revised route to briarane building block 14 involved using a diastereoselective addition to hydroxyaldehyde 15. Ito and Iguchi’s success with a similar transformation13 provided strong support for the feasibility of this disconnection. We envisioned hydroxyaldehyde 15 could be formed by reduction of a C14 ketone and reductive cleavage of the methoxy group in cyclohexenone 16. In turn, β-ketoester 16 would be formed through conjugate addition of an acetylide into cyclohexadienone 17 followed by stereoselective methylation. Much like our previous route, aromatization after the conjugate addition would be a potential problem. In this case though, the problem would be exacerbated by the enhanced acidity of the β-ketoester intermediate (18, Scheme 8 inset) and the basic conditions needed for the alkylation. An initial report of how this was accomplished has been reported.16 Some of the following discussion is a summary of this work.
Formation of the C1 and C10 stereocenters
Dienone 17 was prepared by oxidative dearomatization of salicylate 19 (Scheme 9). Prior to our work there were not many examples of such oxidations being performed on o-hydroxybenzoates.36 We found that a good yield of dienone 17 could be achieved by using μ-oxo-bridged diiodide 21 as the oxidant.37 Several conditions for the addition of TMS-acetylides into the more electron-deficient double bond were evaluated.16 Eventually, we found that an aluminum acetylide38 offered the best compromise of conversion, yield, and product cleanliness.39 In order to minimize rearomatization, the methylation reaction was performed without purifying the conjugate addition product. With the C1 and C10 stereocenters established, we then performed the reductive cleavage of the unnecessary C11 methoxy group. This was accomplished in good yield by treating ketoester 16 with Zn in AcOH.40
Scheme 9.

(a) 21, MeOH, 0 °C, 61%; (b) i. TMS acetylene, BuLi, Et2O, −78 °C, then Et2AlCl (in hexane), 0 °C, then 17 in toluene; ii. K2CO3, 10 mol% 18-crown-6, CH3I, toluene, rt, 43%; (c) Zn dust (10 equiv), AcOH, rt, 71%.
Computational investigation of C1 alkylation
With regard to the formation of the C1 stereocenter, our hypothesis at the outset was that torsional steering would direct the incoming electrophile to the same face of the nucleophile as the C10 substituent; in a similar manner to what was observed during the aldol reaction discussed above. In an effort to better understand the how the formation of the C1 stereocenter is being formed, a series of DFT calculations (B3LYPD3/6-311++G(d,p), SMDtoluene) were performed. Several choices were made in order to simplify the calculations. First, the remote TMS alkyne was replaced with a terminal alkyne. It was assumed that this change would not have a significant impact on the relative energetics of the various species being considered. Secondly, the effect of 18-crown-6 used in the experimental work would be difficult to model explicitly and may not be experimentally relevant.41,42 Consequently, the potassium salt of β-ketoester 18 was modelled without other ligands.
The first task was to evaluate the conformational preference of the β-ketoester nucleophile (Figure 5). Calculations revealed the favored conformation (22b) to be the one in which the methoxy group is located in an equitorial position. This can be rationalized in terms of the strong axial preference of the alkyne (A value 0.18),29 but closer examination of the optimized structures reveals another source for this conformational preference. In the more stable conformer (22b), the extended π-system that runs from C12 through to the ester carbonyl oxygen is rather flat, as one would expect for a resonance-stabilized system. However, this same π-system is more twisted in the higher energy conformer (22a). The twisting in 22a is most evident in the dihedral encompassing the ketoester enolate (ϕ2), where the angle is almost 23° larger in 22a than in 22b. As a consequence of the twisting in 22a, there will be less resonance stabilization in this conformer. Evidence to support this idea was found by performing a Natural Bond Orbital (NBO) analysis43,44 of the two conformers (see ESI). The origin of the twisting in conformer 22a becomes clear when one considers the placement of the equitorial alkyne with respect to the methoxyl group of the ester. Placing the ester in a more planar orientation would bring the lone pairs of the oxygen atom closer to the π-cloud of the alkyne. Apparently, any increase in resonance stabilization the molecule would gain (~3 kcal, see ESI) is not sufficient to overcome this destabilizing interaction.
Figure 5.

Conformational analysis of the ketoester nucleophile. The dihedral formed by C12–C13–C14–C1 is ϕ1. The dihedral formed by C14–C1–C2–O(carbonyl) is ϕ2. Briarane numbering is used for atom labels.
Although conformer 22b is energetically favored, this does not necessarily need to be the most reactive conformer, as this alkylation could be governed by Curtin–Hammett kinetics.45,46 Indeed, the transition state from 22b that leads to observed product 16 (Me syn to alkyne) would, to a first approximation, involve severe eclipsing interactions between the axial alkyne and incoming CH3 group. Consequently, calculations were performed to identify the diastereomeric transition states arising from both conformers. For these calculations, the CH3I used in the experiment was replaced with CH3Cl. Previous work in our laboratory has validated this modification.34,47
TS-1 was found to be significantly higher in energy than the other structures (Figure 6). Part of this energy discrepancy can be attributed to the severe torsional strain (Figure 7) present in this structure.17 It is also interesting to note that in TS-1 the Cl–CH3–C coordinate is close to linear (~177°), but is noticeably more acute (~160°) in all other transition structures (Figure 6). The non-linearity was accompanied by a close contact between the chloride leaving group and the potassium counterion (~3.5 Å) that is smaller than the sum of the van der Wall radii.48 It is possible that this interaction has a stabilizing affect on TS-2–TS-4 as it would lead to diminished charge separation in the non-polar medium (toluene).
Figure 6.

Calculated transition structures. Relative energies (kcal/mol) are in parenthesis.
Figure 7.
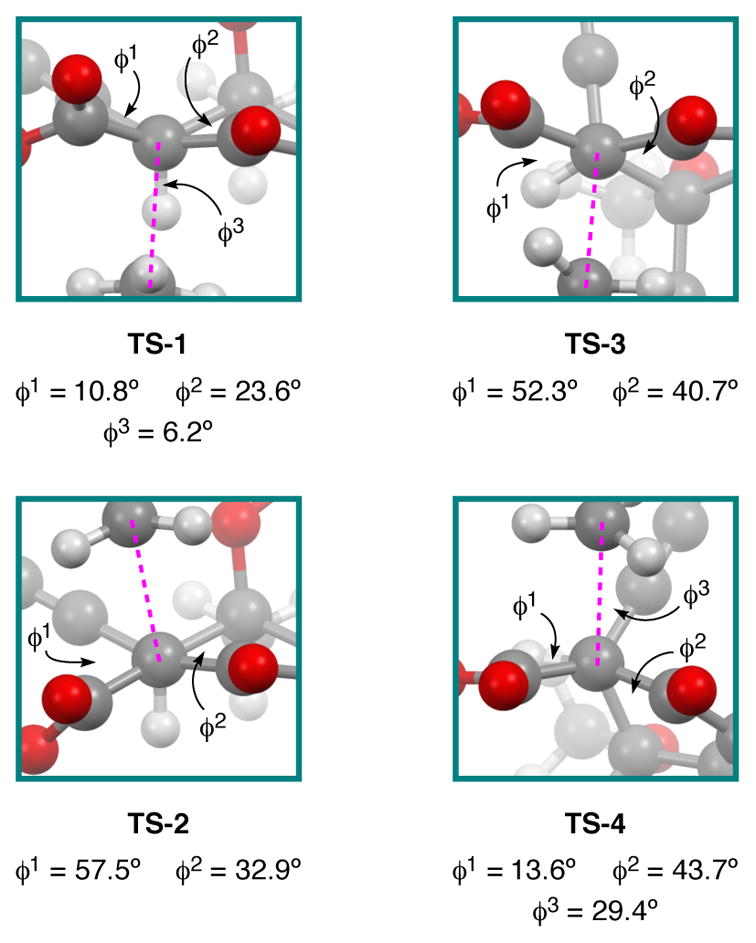
Comparison of dihedral angles measured in TS-1–TS-4. K atom removed for clarity.
It was surprising to see that TS-2, TS-3, and TS-4 were all energetically similar. While TS-2 and TS-4 would both lead to the experimentally observed stereoisomer, TS-3 would give rise to a stereoisomer with the opposite configuration at C1. The energy difference between TS-2 and TS-3 would be consistent with the observed stereoselectivity, however, these transition structures arise from different nucleophile conformations. The low energy conformer (22b) leads to TS-3, whereas the high energy conformer (22a) leads to TS-2. If we only compare those structures derived from conformer 22b, the overall preference still favors the observed stereoisomer, but the energy difference (~0.6 kcal/mol) suggests that the reaction should not be as selective as it is. In the end, both of the lowest energy transition states (TS-2 and TS-4) involve approach of the methyl group to the same face of the nucleophile (syn addition relative to alkyne). There is only one low energy transition state involving addition of the methyl group anti to the alkyne.
Upon closer inspection, TS-4 appears to be lower in energy than expected. One should expect TS-4 to have an eclipsed geometry and therefore be higher in energy than TS-3, which has a staggered geometry. Additionally, the steric penalty that must be paid to have the methyl group approach the nucleophile syn to the alkyne in TS-4 should be greater than that needed to have the methyl approach syn to the methine proton in TS-1. Even a qualitative comparison between TS-1 and TS-4 would likely conclude that TS-1 appears to be sterically less crowded than TS-4. Moreover, TS-1 appears to be the least sterically crowded of all the transition structures, but is the highest in energy. Closer inspection of the measured dihedral angles (Figure 7) reveals that TS-4 appears to have a geometry that is intermediate between eclipsed (TS-1) and staggered (TS-2 and TS-3). In order to explain why TS-4 would adopt this geometry, we looked for structural features that might provide additional stabilization to TS-4.
First, we noticed that TS-4 has the shortest K–Cl separation among all of the calculated structures. Although this will likely impart some stabilization to TS-4, it is difficult to say how much a 0.1 Å shortening of an ionic interaction will affect the energy of the system; the benefit is probably far short of 7 kcal/mol (the energy difference between TS-1 and TS-2). A potentially more important contributor was found when inspecting through-space interactions involving the planar CH3 group in the transition state (Figure S4, ESI). In TS-1, the closest contacts were observed between two protons on the CH3 and the vicinal methine proton (2.434 A, 2.469 Å). But, these are just outside the sum of the van der Waals radii for the atoms in question.48 In TS-3 there are two H•••H interactions (2.290 A, 2.359 Å) that are closer than the sum of the van der Waals radii that may impart some degree of destabilization.
In contrast to the non-existent or destabilizing interactions found in TS-1 and TS-3, some potentially stabilizing interactions were identified in TS-2 and TS-4 (Figure 8). In TS-2 there is a short contact (2.406 Å) between the reacting CH3 and the oxygen atom of the axial methoxy group. It has been known for some time that C–H•••O hydrogen bonds and interactions can impart some degree of stabilization.49,50 More interesting was a potential C–H•••π(alkyne) interaction in TS-4. Such interactions have been observed in the solid state with alkyne-containing organometallic complexes.51 In an analysis of the Cambridge Structural Database, Steiner and Tamm52 found that C–H•••π(alkyne) distances were typically 2.6–2.9 Å and occasionally even shorter.53 In TS-4 the C–H is 2.645 Å from the centroid of the alkyne. In addition, the C–H is positioned at an angle (83.3°) that is close to the idealized T-shape geometry for a C–H•••π(alkyne) interaction.53 Evidence for both the C–H•••O hydrogen bond in TS-2 and the C–H•••π(alkyne) interaction in TS-4 was found by performing an NBO analysis on these transition structures.
Figure 8.
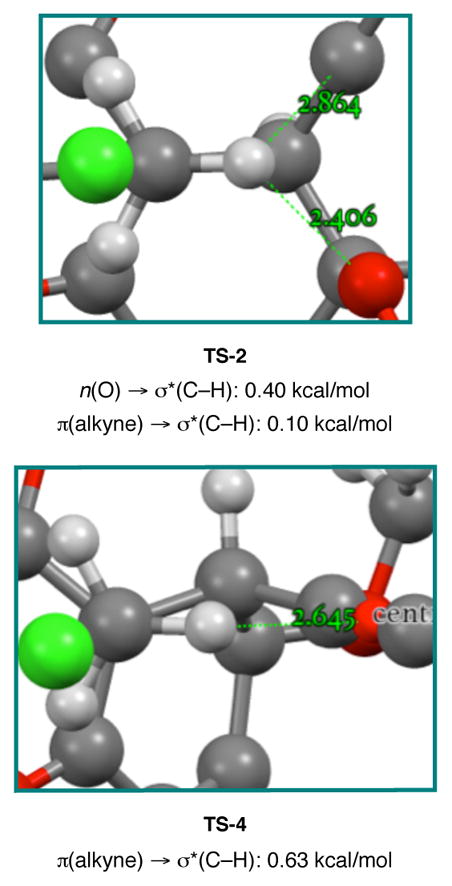
Measured close contacts to the planar CH3 in TS-2 and TS-4. C11 methyl group removed from TS-4 for clarity. Donor-acceptor energies obtained by NBO second order perturbation analysis.
Completion of stereotetrad
With ketoester 20 in hand, we turned our attention to installing the C14 and C2 stereocenters needed for the briarane stereotetrad. The details of this effort have been previously reported16 and are only summarized here.
The stereoselective reduction of the C14 ketone was much more difficult than anticipated. In the end, we found that performing the reduction with NaBH4 in the presence of Y(OTf)3 provided a nearly 1:1 ratio of 23a and 23b (Scheme 10). For comparison, NaBH4 alone favored the formation of the undesired diastereomer 23b (1:16.2 ratio). The triflate counterion appears to be important, as the use of YCl3 was a little more selective toward alcohol 23b (1:2.2 ratio).
Scheme 10.
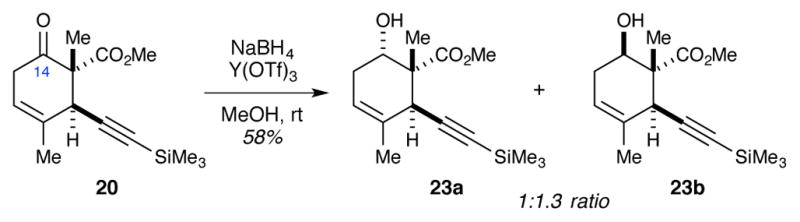
Stereoselective reduction of the C14 ketone.
DFT calculations were performed to determine the conformational preference of ketoester 20 and better understand its reactivity (Figure 9). Calculations were done using B3LYPD3/6-311++G(d,p) and the SMD solvation model for THF and methanol. There was a clear energetic preference for the conformational isomer with an axial alkyne (20AX) over that with an equitorial alkyne (20EQ). This finding is in line with what would be predicted based on A values for methyl, ester, and alkyne groups.29 The favored conformer (20AX) is also the one with the smaller dipole moment. Given this preference, it is easy to imagine how an axial alkyne would prevent approach of a hydride nucleophile to the C14 ketone from an axial direction. At this time it is not entirely clear why yttrium salts are so special, but it is possible that Y(+3) is more effective at coordinating the carbonyl groups of the ketoester. This binding event may result in a greater population of a conformer with an equitorial alkyne (e.g., 20EQ).
Figure 9.
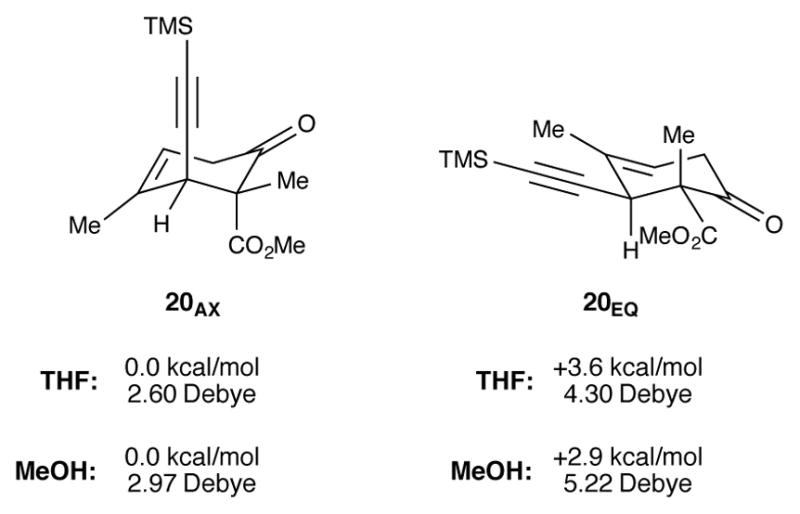
Conformational analysis of ketoester 20. Relative free energies are given.
With alcohol 23a in hand, the final steps to the stereotetrad could be completed (Scheme 11). A two-step procedure converted the ester into aldehyde 15 in acceptable yield. In contrast to the high selectivity Ito and Iguchi observed for addition of vinylmagnesium bromide into a similar β-hydroxy aldehyde (cf., Scheme 1), we observed poor selectivity during the addition of allylmagnesium bromide into 15. The best results (dr of 4.2:1 favoring desired diastereomer) were obtained when the reaction was carried out in Et2O. Several factors could be affecting the selectivity in our case, but we believe it is, once again, the axial preference for the alkyne that is the most significant contributor. In order to achieve the same reactive conformation proposed by Ito and Iguchi, the alkyne must occupy an equitorial position as shown by conformer V. But, if the alkyne remains in its energetically preferred axial position, as shown by VI, then the proposed chelation between the aldehyde and the magnesium alkoxide could be disrupted. Breaking this chelation may allow the aldehyde to adopt several reactive conformations that could give rise to different stereoisomers. Even if chelation is present in conformer VI, it is possible that both faces of the aldehyde reside in similar steric environments. Nevertheless, the formation of acetonide 25a represents the completion of a rapid (9 step) synthesis of a protected briarane stereotetrad fragment.
Scheme 11.
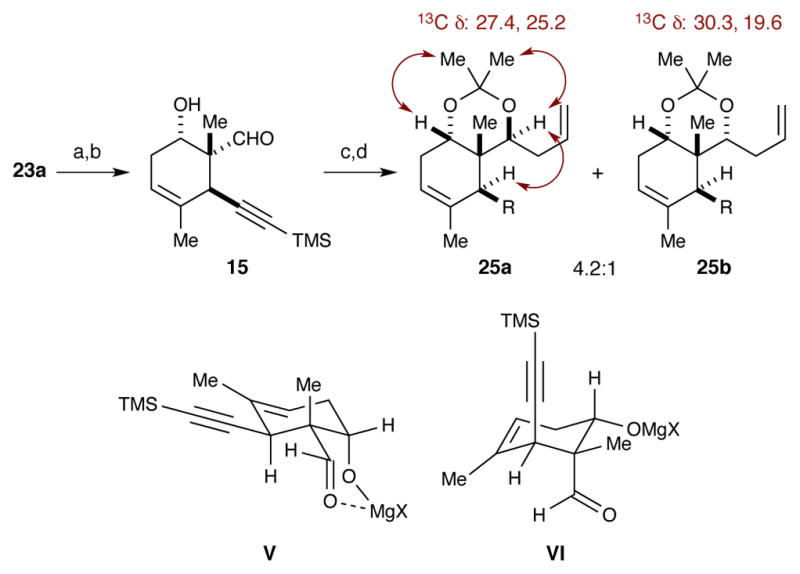
(a) LiAlH4, Et2O, 0 °C; (b) TEMPO, PhI(OAc)2, DCM, rt, 57% (2 steps); (c) allylMgBr, Et2O, −78 °C; (d) Me2C(OMe)2, PPTS, acetone, rt, 34% (2 steps).
Conclusions
In conclusion, we have reported two routes to access the stereotetrad core of the briarane diterpenoids that take advantage of the inherent reactivity and dense functionality associated with cyclohexadienones. In both cases, the configuration of the key all-carbon quaternary stereocenter (C1) was controlled by torsional steering in the transition state. When an aldol reaction was used to generate this stereocenter, the lowest energy pathway (staggered transition state) is the one that results in the undesired diastereomer. However, torsional steering can be harnessed in a productive manner by using a β-ketoester alkylation to generate the C1 stereocenter. After this adjustment to our strategy we were able to access a protected briarane stereotetrad fragment in only 9 steps from salicylate 19. This is a significant improvement in efficiency compared to previous efforts.13–15
Despite our success, there is room for improvement with our approach. In particular, the stereoselective reduction of the C14 ketone and the Grignard addition to C2 both suffer from poor stereoselectivity. Fortunately, both problems can be traced back to our strategic choice of using an acetylide nucleophile for forming the C9–C10 bond and the axial preference for the resulting alkyne sidechain. We believe that if the acetylide is replaced with an sp3 hybridized nucleophile we will be able to better control the conformational preference of the substrates in question. In turn, this should improve the stereoselectivity observed when forming the C14 and C2 stereocenters. We are currently exploring several options to accomplish this and will report our results, as well as our efforts to elaborate the stereotetrad core into a completed briarane natural product, in due course.
Experimental
Computational Details
All calculations were performed using the Gaussian 09 suite of electronic structure programs.54 All geometries optimized at the B3LYPD355,56 level with no frozen coordinates. Initial optimizations were performed using the 6–31+G(d)57 basis set. Final optimizations were performed using the 6–311++G(d,p) basis set. An ultrafine grid density was used for final numerical integrations.58 In order to account for solvation effects, the SMD solvation model59 for toluene, tetrahydrofuran, and methanol was employed during geometry optimizations. Energy minima and transition states were identified through frequency analysis. The NBO 3.1 program, as implemented by Gaussian 09 was used to evaluate the energies of donor and acceptor orbitals. The Gibbs energies for all relevant species can be found in the supporting information.
Transition structures TS-1, TS-2, and TS-3 were located by manually constructing an initial guess based on the classical SN2 transition state geometry. The initial guess was then optimized using the Berny algorithm60 as implemented by Gaussian 09. In the case of TS-4, this approach was not successful. In this case, the initial guess was constructed by using a relaxed scan to locate an approximate transition structure. Briefly, the carbon atom of CH3Cl was placed 2.9 Å away from the nucleophilic carbon and the distance was gradually shortened in 0.1 Å increments. The structure corresponding to the energy maximum during this scan was then used as the initial guess for further optimization (see ESI for details).
Materials and Methods
Unless otherwise stated, reactions were performed in flame- or oven-dried glassware under an argon or nitrogen atmosphere using anhydrous solvents. Tetrahydrofuran (THF) was distilled from sodium/benzophenone or purchased in anhydrous form from Sigma Aldrich. Unless otherwise stated, reactions were monitored using thin-layer chromatography (TLC) using plates precoated with silica gel with UV254 (250 mm) and visualized by UV light or KMnO4, phosphomolybdic acid, or anisaldehyde stains, followed by heating. Silica gel (particle size 32–63 mm) was used for flash column chromatography. 1H and 13C NMR spectra are reported relative to the residual solvent peak, or tetramethylsilane when the residual solvent peak is obscured. Data for 1H NMR spectra are reported as follows: chemical shift (ppm) (multiplicity, coupling constant (Hz), integration). Multiplicity is described using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, bs = broad singlet, app = apparent. FTIR samples were prepared on NaCl plates either neat or by evaporation from CHCl3 or CH2Cl2. Procedures and characterization data for compounds 16, 17, 19, 20, 21, 23a, 23b, 24a, 24b, 15, 25a have been reported.16
4-Hydroxy-4-methylcyclohexa-2,5-dien-1-one (9)
p-Cresol (3.00 g, 27.7 mmol, 1 eq.) was dissolved in 140 mL of a mixture of MeCN:H2O (3:1). PIDA (9.4 g, 29.1 mmol, 1.05 eq.) was added and the reaction was stirred for 1 hour. When the reaction was complete by TLC, the mixture was partially concentrated by rotary evaporation and quenched with 50 mL 10 wt.% Na2S2O3 solution and 200 mL of H2O. The aqueous layer was extracted with 3 x 100 mL EtOAc. The combined organic layers were washed with brine, dried with Na2SO4, filtered, and concentrated. Purification by flash chromatography (1:4 to 1:1 EtOAc:Hexane) gave a yellow solid (1.49 g, 39%). The 1H NMR spectrum was consistent with a literature report.61 1H NMR (500 MHz, CDCl3) δ 6.90–6.86 (m, 2H), 6.14–6.11 (m, 2H), 2.23 (s, 1H), 1.48 (s, 3H).
(4S*,5S*)-4-Hydroxy-4-methyl-5-((trimethylsilyl)ethynyl)cyclohex-2-en-1-one (10)
TMS acetylene (2.13 g, 21.7 mmol, 3 eq.) was dissolved in 50 mL of anhydrous THF and cooled to 0 °C. EtMgBr (3M in Et2O, 7.23 mL, 21.7 mmol, 3 eq.) was added. The solution was stirred for 1 hour and cooled to −78 °C. In a separate flask, quinol 9 (1.00 g, 7.24 mmol, 1 eq.) was dissolved in 25 mL of anhydrous THF. The quinol solution was added to the cold acetylide solution via cannula and stirred at −78 °C for 15 min. The solution was then warmed to ambient temperature for 2 hours. The reaction was quenched by addition of 100 mL saturated NH4Cl solution. The organic layer was extracted with 3 x 50 mL EtOAc. The combined organic layers were washed with brine, dried with Na2SO4, filtered, and concentrated. The product was purified by flash chromatography (1:4 EtOAc:Hexane) to give the product (1.17 g, 72%). IR (thin film) 3455, 2961, 2899, 2175, 1681, 1250, 1129, 843 cm™1. 1H NMR δ 6.67 (dd, J = 0.6, 10.2 Hz, 1H), 5.92 (d, J = 10.2 Hz, 1H), 3.08 (ddd, J = 0.3, 4.6, 8.0 Hz, 1H), 2.75 (dd, J = 8.1, 16.7 Hz, 1H), 2.61 (bs, 1H), 2.59 (dd, J = 4.7, 16.8Hz, 1H), 1.50 (s, 3H), 0.14 (s, 9H). 13C NMR (125 MHz, CDCl3, DEPT) δ 196.64 (C), 152.18 (CH), 128.48 (CH), 103.46 (C), 90.85 (C), 68.38 (C), 40.70 (CH), 39.84 (CH2), 26.64 (CH3), 0.03 (CH3). HRMS (ESI+) 245.0968 calc’d for C12H18O2SiNa+, 245.0987 found.
(4S*,5S*,6S*)-4,6-Dimethyl-4-((triethylsilyl)oxy)-5-((trimethylsilyl)ethynyl)cyclohex-2-en-1-one (11)
Enone 10 (1.19 g, 5.37 mmol, 1 eq.) and imidazole (1.46 g, 21.48 mmol, 4 eq.) were dissolved in DMF (18 mL). Et3SiCl (1.62 g, 10.74 mmol, 2 eq.) was added and the solution was stirred at rt for 3 hours. The reaction mixture was quenched with water (100 mL) and extracted with 3 x 20 mL EtOAc. The combined organic layers were washed with brine, dried with Na2SO4, and concentrated. The residue was purified by flash chromatography (1:20 EtOAc:Hexane) to give the product silyl ether (1.25 g, 66%) as a colorless oil. IR (thin film) 2958, 2912, 2877, 2178, 1693, 1458, 1412, 1249, 1116, 1013, 842, 728 cm™1. 1H NMR (500 MHz, CDCl3) δ 6.71 (d, J = 10.1 Hz, 1H), 5.86 (d, J = 10.1 Hz, 1H), 2.86–2.93 (m, 2H), 2.51–2.57 (m, 1H), 1.55 (s, 3H), 0.96 (t, J = 7.9 Hz, 9H), 0.57–0.66 (m, 6H), 0.15 (s, 9H). 13C NMR (125 MHz, CDCl3, DEPT) δ 198.30 (C), 152.80 (CH), 127.95 (CH), 105.09 (C), 87.87 (C), 69.87 (C), 41.76 (CH), 39.82 (CH2), 28.01 (CH3), 7.12 (CH3), 6.84 (CH2), 0.90 (CH3). HRMS (ESI+) 359.1833 calc’d for C18H32O2Si2Na+, 359.1840 found.
The obtained silyl ether (200 mg, 0.59 mmol, 1 eq.) was dissolved in 6 mL of a 10:1 THF:HMPA solution. The solution was cooled to −78 °C and 0.76 mL of a 1M solution of LDA (freshly prepared in THF, 0.71 mmol, 1.2 eq.) was added. The solution was stirred at −78 °C for 1 hour and MeI (419 mg, 2.95 mmol, 5 eq.) was added. The reaction was allowed to warm to room temperature and stirred for 2 h at which time it was quenched with 25 mL of a saturated NH4Cl solution. The organic layer was extracted with 3 x 10 mL EtOAc. The combined organic extracts were washed with brine, dried with Na2SO4, and concentrated. Purification by flash chromatography (1:10 EtOAc:Hexane) gave compound 11 as a colorless oil (129 mg, 63%). IR (Thin Film) 2957.64, 2788, 2176, 1690, 1249, 1106 cm™1. 1H NMR (500 MHz, CDCl3) δ 6.62 (dd, J = 1.6, 10.3 Hz, 1H), 5.83 (d, J = 10.3 Hz, 1H), 3.00 (dd, J = 1.6, 4.4 Hz, 1H), 2.61(dq, J = 4.4, 6.7 Hz, 1H), 1.52 (s, 3H), 1.27 (d, J = 6.8 Hz, 3H), 0.99 (t, J = 7.9 Hz, 9H), 0.66 (q, J = 7.9 Hz, 6H), 0.09 (s, 9H). 13C NMR (125 MHz, CDCl3, DEPT) δ 199.58 (C), 153.13 (CH), 126.59 (CH), 103.84 (C), 89.61 (C), 73.36 (C), 49.14 (CH), 43.60 (C), 27.74 (CH3), 13.82 (CH3), 7.26 (CH3), 6.95 (CH2), 0.06 (CH3). HRMS (ESI+) 373.1990 calc’d for C19H34O2Si2Na+, 373.1993 found.
(4S*,5S*,6R*)-6-(1-Hydroxybutyl)-4,6-dimethyl-4-((triethylsilyl)oxy)-5-((trimethylsilyl)ethynyl)cyclohex-2-en-1-one (12b)
Compound 11 (20 mg, 0.057 mmol, 1 eq.) was dissolved in 0.6 mL of CH2Cl2. Et3N (17 mg, 0.17 mmol, 3 eq.) was added. The solution was cooled to −78 °C and Bu2BOTf (70 μL of a 1M sol’n in CH2Cl2, 0.07 mmol, 1.2 eq.) was added. The solution was warmed to 0 °C for 30 min and cooled to −78 °C. Butanal (21 mg, 0.29 mmol, 5 eq.) was added. The solution was stirred at −78 °C for 1.5 hours and quenched with 1 mL saturated NaHCO3. The organic layer was extracted with 3 x 1 mL CH2Cl2. The combined organic layers were dried with Na2SO4 and concentrated. The residue was purified by flash chromatography (1:4 EtOAc:Hexane) to give the product as a colorless oil (16.1 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 6.51 (d, J = 10.2 Hz, 1H), 6.00 (d, J = 10.2 Hz, 1H), 4.72 (t, J = 2.32 Hz, 1H), 4.08 (td, J = 2.2, 10.5 Hz, 1H), 2.92 (s, 1H), 1.63 (s, 3H), 1.27–1.59 (m, 8H), 1.17 (s, 3H), 1.01 (t, J = 7.0 Hz, 9H), 0.89–0.96 (m, 3H), 0.70–0.79 (m, 6H), 0.19 (s, 9H). 13C NMR (500 MHz, CDCl3) δ 200.74, 147.63, 129.99, 102.32, 91.41, 72.86, 70.43, 52.57, 52.27, 33.03, 29.81, 21.28, 19.76, 13.89, 7.03, 6.84, 0.10. HRMS (ESI+) 445.2565 calc’d for C23H42O3Si2Na+, 445.2615 found.
Supplementary Material
Acknowledgments
Financial support for this work provided by Texas Tech University and the National Science Foundation (1361604). NMR data was recorded in a facility supported by The National Institutes of Health (S10 OD011952). We thank Dr. Rodolfo Tello-Aburto (New Mexico Tech) and Dr. Kyle Kalstabakken (3M Corporation) for performing initial experiments related to this project when in our group. Profs. William Hase and David Birney (Texas Tech) are thanked for helpful discussions and suggestions.
Footnotes
With the exception of the cembrane skeleton in Figure 1, briarane numbering is used for all carbon atoms in this manuscript.
Both Bates and Ito/Iguchi pursued a C1–C2 disconnection, but utilized the C14 stereocenter to control the formation of the C1 stereocenter.
Electronic Supplementary Information (ESI) available: NMR spectra, final coordinates and energies of calculated structures, and complete reference 54. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Berrue F, Kerr RG. Nat Prod Rep. 2009;26:681. doi: 10.1039/b821918b. [DOI] [PubMed] [Google Scholar]
- 2.Reviews: Sung PJ, Sheu JH, Xu JP. Heterocycles. 2002;57:535.Sung PJ, Chang PC, Fang LS, Sheu JH, Chen WC, Chen YP, Lin MR. Heterocycles. 2005;65:195.Sung PJ, Sheu JH, Wang WH, Fang LS, Chung HM, Pai CH, Su YD, Tsai WT, Chen BY, Lin MR, Li GY. Heterocycles. 2008;75:2627.Sung PJ, Su JH, Wang WH, Sheu JH, Fang LS, Wu YC, Chen YH, Chung HM, Su YD, Chang YC. Heterocycles. 2011;83:1241.
- 3.Keifer PA, Rinehart KL, Hooper IR. J Org Chem. 1986;51:4450. [Google Scholar]
- 4.Ravi BN, Marwood JF, Wells RJ. Aust J Chem. 1980;33:2307. [Google Scholar]
- 5.Guerriero A, D’Ambrosio M, Pietra F. Helv Chim Acta. 1987;70:984. [Google Scholar]
- 6.For example: Figueroa J, Vera B, Rodriguez AD. Helv Chim Acta. 2014;97:712.Ortega MJ, Zubia E, Sánchez MC, Carballo JL. J Nat Prod. 2008;71:1637. doi: 10.1021/np8002639.Marrero J, Benitez J, Rodriguez AD, Zhao H, Raptis RG. J Nat Prod. 2008;71:381. doi: 10.1021/np0705561.Sánchez MC, Ortega MJ, Zubia E, Carbello JL. J Nat Prod. 2006;69:1749. doi: 10.1021/np060388x.
- 7.Burks JE, Van der Helm D, Chang CY, Ciereszko LS. Acta Crystallogr, Sect B. 1977;B33:704. [Google Scholar]
- 8.(a) Cardellina JH, II, James TR, Jr, Chen MHM, Clardy J. J Org Chem. 1984;49:3398. [Google Scholar]; (b) Sheu JH, Sung PJ, Huang LH, Lee SF, Wu T, Chang BY, Duh CY, Fang LS, Soong K, Lee TJ. J Nat Prod. 1996;59:935. doi: 10.1021/np960218s. [DOI] [PubMed] [Google Scholar]
- 9.Sung P-J, Su J-H, Wang G-H, Lin S-F, Duh C-Y, Sheu J-H. J Nat Prod. 1999;62:457. doi: 10.1021/np980446h. [DOI] [PubMed] [Google Scholar]
- 10.Gupta P, Sharma U, Schulz TC, Sherrer ES, McLean AB, Robins AJ, West LM. Org Lett. 2011;13:3920. doi: 10.1021/ol201443k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Lin X, Yang B, Liu J, Zhou X, Peng Y. Cytotoxic Briarane-Type Diterpenoids. In: Kim S-K, editor. Marine Pharmacognosy. CRC press; Boca Raton, FL: 2013. pp. 53–63. [Google Scholar]
- 12.Early work toward specific fragments: Roe MB, Whittaker M, Procter G. Tetrahedron Lett. 1995;36:8103.Balasubramaniam RP, Moss DK, Wyatt JK, Spence JD, Gee A, Nantz MH. Tetrahedron. 1997;53:7429.
- 13.Iwasaki J, Ito H, Nakamura M, Iguchi K. Tetrahedron Lett. 2006;47:1483. [Google Scholar]
- 14.Bates RW, Pinsa A, Kan X. Tetrahedron. 2010;66:6340. [Google Scholar]
- 15.Crimmins MT, Knight JD, Williams PS, Zhang Y. Org Lett. 2014;16:2458. doi: 10.1021/ol5008422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.A portion of this work was previously communicated. See: Moon NG, Harned AM. Org Lett. 2015;17:2218. doi: 10.1021/acs.orglett.5b00816.
- 17.Wang H, Houk KN. Chem Sci. 2014;5:462. doi: 10.1039/C3SC52538D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon N. PhD Dissertation. University of Minnesota; 2015. [Google Scholar]
- 19.Nilsson A, Ronlán A, Parker VD. Tetrahedron Lett. 1975:1107. [Google Scholar]
- 20.Grecian S, Wrobleski AD, Aubé J. Org Lett. 2005;7:3167. doi: 10.1021/ol050715c. [DOI] [PubMed] [Google Scholar]
- 21.Zweifel G, Backlund SJ. J Am Chem Soc. 1977;99:3184. [Google Scholar]
- 22.Taber DF, Gerstenhaber DA, Berry JF. J Org Chem. 2011;76:7614. doi: 10.1021/jo2013753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wipf P, Kim Y. J Am Chem Soc. 1994;116:11678. [Google Scholar]
- 24.Stern AJ, Rohde JJ, Swenton JS. J Org Chem. 1989;54:4413. [Google Scholar]
- 25.Schwartz J, Carr DB, Hansen RT, Dayrit FM. J Org Chem. 1980;45:3053. [Google Scholar]
- 26.(a) Solomon M, Jamison WCL, McCormick M, Liotta D, Cherry DA, Mills JE, Shah RD, Rodgers JD, Maryanoff CA. J Am Chem Soc. 1988;110:3702. [Google Scholar]; (b) Swiss KA, Liotta DC, Maryanoff CA. J Am Chem Soc. 1990;112:9393. [Google Scholar]
- 27.A Reaxys search returned a single report of a bimolecular aldol reaction of a cyclic ketone with substituents in the α- and β-positions: Stohrer I, Hoffmann HMR. Helv Chim Acta. 1993;76:2194.
- 28.Lessard J, Tan PVM, Martino R, Saunders JK. Can J Chem. 1977;55:1015. [Google Scholar]
- 29.A values for alkyne (0.18), methyl (~1.7), ester (~1.2): Hirsch JA. Topics in Stereochemistry. 1967;1:199.
- 30.Cowden CJ, Paterson I. Org React. 2004;51:1. [Google Scholar]
- 31.Hamana H, Sasakura K, Sugasawa T. Chem Lett. 1984;13:1729. [Google Scholar]
- 32.Yamago S, Machii D, Nakamura E. J Org Chem. 1991;56:2098. [Google Scholar]
- 33.Volp KA, Johnson DM, Harned AM. Org Lett. 2011;13:4486. doi: 10.1021/ol201211f. [DOI] [PubMed] [Google Scholar]
- 34.Volp KA, Harned AM. J Org Chem. 2013;78:7554. doi: 10.1021/jo4011238. [DOI] [PubMed] [Google Scholar]
- 35.Lopez SA, Pourati M, Gais H-J, Houk KN. J Org Chem. 2014;79:8304. doi: 10.1021/jo501557z. [DOI] [PubMed] [Google Scholar]
- 36.(a) Génisson Y, Tyler PC, Young RN. J Am Chem Soc. 1994;116:759. doi: 10.1021/ja0112341. [DOI] [PubMed] [Google Scholar]; (b) McKillop A, McLaren L, Taylor RJK. J Chem Soc, Perkin Trans 1. 1994:2047. [Google Scholar]; (c) Génisson Y, Tyler PC, Ball RG, Young RN. J Am Chem Soc. 2001;123:11381. doi: 10.1021/ja0112341. [DOI] [PubMed] [Google Scholar]; (d) Liu L, Gao Y, Che C, Wu N, Wang DZ, Li CC, Yang Z. Chem Commun. 2009:662. doi: 10.1039/b817376a. [DOI] [PubMed] [Google Scholar]
- 37.Dohi T, Uchiyama T, Yamashita D, Washimi N, Kita Y. Tetrahedron Lett. 2011;52:2212. [Google Scholar]
- 38.Ahmar S, Fillion E. Org Lett. 2014;16:5748. doi: 10.1021/ol502811j. [DOI] [PubMed] [Google Scholar]
- 39.Discussion of the different factors that contribute to the stereoselective addition of the acetylide can be found in reference 16.
- 40.Brocksom TJ, Coelho F, Deprés J-P, Greene AE, Freire de Lima ME, Hamelin O, Hartmann B, Kanazawa AM, Wang Y. J Am Chem Soc. 2002;124:125313. doi: 10.1021/ja0208456. [DOI] [PubMed] [Google Scholar]
- 41.It is not clear how associated the 18-crown-6 is with ketoester enolate. Only a catalytic amount of the crown is used in the reaction. It is possible that it just serves as a phase transfer catalyst that brings in K2CO3 into solution. After deprotonation of the ketoester, one of the potassium ions can be associated with the enolate (as modeled), the other can then be associated with the crown as the bicarbonate salt.
- 42.An attempt was made to model conformer 22b with an 18-crown-6 ligand on the potassium. This single complex took over a month to optimize at the B3LYPD3/6-31+G(d) level. Unfortunately, attempts at performing a separate frequency calculation were unsuccessful.
- 43.(a) Reed AE, Curtis LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]; (b) Reed AE, Weinhold F. J Chem Phys. 1985;83:1736. [Google Scholar]; (c) Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735. [Google Scholar]
- 44.For a concise description of NBO analysis, see: Alabugin IV, Manoharan M, Zeidan TA. J Am Chem Soc. 2003;125:14014. doi: 10.1021/ja037304g.
- 45.Gawley RE, Aubé J. Principles of Asymmetric Synthesis. 2. Elsevier; Amsterdam: 2012. pp. 21–23. and references therein. [Google Scholar]
- 46.Harned AM. Chem Commun. 2015;51:2076. doi: 10.1039/c4cc07967a. [DOI] [PubMed] [Google Scholar]
- 47.Computational results with CH3I are consistent with the calculations presented here with CH3Cl (see ESI).
- 48.van der Waals radii: K = 2.75 Å, Cl = 173 Å, Csp = 178, H = 120, O = 152 See: Bondi A. J Phys Chem. 1964;68:441.
- 49.(a) Desiraju GR. Acc Chem Res. 1996;29:441. doi: 10.1021/ar950135n. [DOI] [PubMed] [Google Scholar]; (b) Steiner T. Chem Commun. 1997:727. [Google Scholar]
- 50.For discussion of non-classical hydrogen bonds in stereoselective reactions, see: Ajitha MJ, Huang KW. Synthesis. 2016;48:3449.Nacereddine AK, Sobhi C, Djerourou A, Ríos-Gutiérrez M, Domingo LR. RSC Adv. 2015;5:99299.Kanomata K, Toda Y, Shibata Y, Yamanaka M, Tsuzuki S, Gridnev ID, Tarada M. Chem Sci. 2014;5:3515.Johnston RC, Cohen DT, Eichman CC, Scheidt KA, Cheong PHY. Chem Sci. 2014;5:1974. doi: 10.1039/C4SC00317A.Johnston RC, Cheong PHY. Org Biomol Chem. 2013;11:5057. doi: 10.1039/c3ob40828k.
- 51.McAdam CJ, Cameron SA, Hanton LR, Manning AR, Moratti SC, Simpson J. CrystEngComm. 2012;14:4369. and references therein. [Google Scholar]
- 52.Steiner T, Tamm M. J Organomet Chem. 1998;570:235. [Google Scholar]
- 53.Müller TE, Mingos MP, Williams DJ. J Chem Soc, Chem Commun. 1994:1787. and references therein. [Google Scholar]
- 54.Frisch MJ, et al. Gaussian 09, Revision D.01. Gaussian, Inc; Wallingford, CT: 2013. [Google Scholar]
- 55.(a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 56.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 57.Hehre WJ, Radom L, Schleyer PvR, Pople JA. Ab Initio Molecular Orbital Theory. Wiley; New York: 1986. [Google Scholar]
- 58.Wheeler SE, Houk KN. J Chem Theory Comput. 2010;6:395. doi: 10.1021/ct900639j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 60.(a) Peng C, Schlegel HB. Israel J Chem. 1993;33:449. [Google Scholar]; (b) Peng C, Ayala PY, Schlegel HB, Frisch MJ. J Comp Chem. 1996;17:49. [Google Scholar]
- 61.Sunasee R, Clive DLJ. Chem Commun. 2010;46:701. doi: 10.1039/b920884d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.