

Abstract
Obesity and related metabolic disturbances are closely associated with pathologies that represent a significant burden to global health. Epidemiological and molecular evidence links obesity and metabolic status with inflammation and increased risk of cancer. Here, using a mouse model of intestinal neoplasia and strains that are susceptible or resistant to diet-induced obesity, it is demonstrated that high-fat diet-induced inflammation, rather than obesity or metabolic status, is associated with increased intestinal neoplasia. The complement fragment C5a acts as the trigger for inflammation and intestinal tumorigenesis. High-fat diet induces complement activation and generation of C5a, which in turn induces the production of pro-inflammatory cytokines and expression of proto-oncogenes. Pharmacological and genetic targeting of the C5a receptor reduced both inflammation and intestinal polyposis, suggesting the use of complement inhibitors for preventing diet-induced neoplasia.
Introduction
Obesity is an increasingly important risk factor for many cancers (1, 2). While the mechanisms underlying the association between obesity and cancer are not fully understood, hormonal changes and chronic inflammation support a favorable environment for tumor progression (1, 2). Alterations in adipose tissue that occurs in response to excess nutrient intake lead to an imbalance between adipokines that are associated with insulin resistance and a pro-inflammatory environment that characterizes metabolic syndrome. Whereas leptin potentiates tumor growth through induction of cell proliferation, angiogenesis and inflammation, adiponectin has anti-inflammatory properties and is negatively correlated with body mass index (BMI) (3). In colorectal cancer, imbalances in insulin and adipokine metabolism are associated with oxidative stress and increased levels of pro-inflammatory cytokines (3, 4).
Supporting a role for diet-induced obesity (DIO) in promoting inflammation and cancer, levels of IL-6 and tumor burden are both reduced following azoxymethane treatment of obese Lepob/ob mice fed a bean-based diet versus a standard diet (5). Furthermore, medications used to treat hyperlipidemia and hypertension prevent colorectal carcinogenesis by attenuating chronic inflammation (6). Similarly, pre-clinical and clinical studies indicate that non-steroidal anti-inflammatory drugs have anti-tumorigenic activity by inhibiting cyclooxygenases (7). The mechanisms and pathways underlying these effects remain to be identified.
Although a link between inflammation and cancer is appreciated, it is unclear whether increased tumorigenesis results from DIO or instead from independent effects of diet on metabolism and tumorigenesis. Increasing evidence points towards a crucial role of diet in determining tumor growth. Mice fed a high-fat diet (HFD) with reduced vitamin D and calcium show increased weight gain and tumorigenesis compared to counterparts fed a low-fat diet (LFD) (8). Apart from vitamin and nutrient deficiencies, both high-fat and sucrose-rich diets are associated with increased tumorigenesis (9, 10). Mediterranean diets are associated with reduced cancer risk (11). Further, dietary derivatives such as vitamins, β-carotene, resveratrol and the omega-3/6 fatty acid ratio are directly implicated in maintaining immune homeostasis in the intestine and with protection from colorectal cancer (12, 13). Given the complex relations between diet and obesity as well as between obesity and cancer, identifying the individual contributions of diet and obesity-related metabolic disturbances to increased cancer risk is challenging. Such elucidation could contribute to development of effective ways to manage cancer.
To explore the mechanistic relationships among diet, DIO and tumorigenesis, we focused on the Min allele of the Apc tumor suppressor gene, an established mouse model of intestinal neoplasia (14). These mice spontaneously develop intestinal polyps in a manner resembling human familial adenomatous polyposis, a condition that carries a 100% risk of developing colorectal cancer (15). Mutations in Apc are observed in 80% of sporadic colorectal cancer cases (15). In parallel, the previously characterized C57BL/6J-ChrA/J chromosome substitution strains (CSSs) were used to investigate the influence of DIO (16). CSSs are inbred strains that carry a single chromosome from a donor strain (A/J) replacing the corresponding chromosome of the host strain (C57BL/6J [B6]) (16). When exposed to a HFD, strains such as CSS-Chr2A/J (A2) and CSS-Chr9A/J (A9) are susceptible to DIO, while others such as CSS-Chr7A/J (A7) and CSS-Chr17A/J (A17) are resistant (17). Using a series of crosses, we combined the ApcMin mutation with each of these four CSSs to create CSS.ApcMin/+ and CSS.Apc+/+ test and control strains that all share a common B6 inbred genetic background. We hypothesized that if DIO is important for increased intestinal neoplasia, then the DIO-susceptible (CSSs-2 and -9) but not DIO-resistant (CSSs-7 and −17) strains would show increased polyp numbers upon HFD exposure. By contrast, if increased neoplasia results directly from HFD exposure, independent of obesity, increased polyp numbers would be observed in all four CSSs fed a HFD. Thus, with B6.ApcMin/+ or wild-type B6.Apc+/+ mice, alone or in combination with the selected CSSs with contrasting genetic susceptibility to DIO (17), we were able to test the independent effects of diet and DIO on intestinal tumor development.
Here, we show that the chemical composition of dietary fat differentially affects obesity and insulin metabolism. Notably, HFD-induced inflammation from specific dietary fats, rather than obesity or metabolic status, is associated with increased intestinal polyposis. Mechanistically, specific dietary fats activate complement signaling and generate complement fragment C5a, which acts as a key trigger for inflammation and intestinal tumorigenesis. Importantly, C5a-induced tumorigenesis occurs independently of obesity or metabolic status. Pharmacological and genetic targeting of C5a receptor (C5aR) prevented diet-induced local and systemic inflammation and significantly reduced polyp burden
Materials and Methods
Mice
C57BL/6J (B6, Jackson Laboratory Repository number JR000664) and C57BL/6J-ApcMin/+/J (B6.ApcMin/+, JR002020) mice and the CSSs C57BL/6J-Chr2A/J/NaJ (A2, JR004380), C57BL/6J-Chr7A/J/NaJ (A7, JR004385), C57BL/6J-Chr9A/J/NaJ (A9, JR004387), and C57BL/6J-Chr17A/J/NaJ (A17, JR004395) were purchased from The Jackson Laboratory (Bar Harbor, ME). CSSs were chosen according to their resistance (A7 and A17) or susceptibility (A2 and A9) to significant diet-induced weight gain (17). Importantly, at the time of selection, these CSSs were not known to carry genetic modifiers that affected development of intestinal polyps in B6.ApcMin/+ mice. CSSs were backcrossed onto a B6.ApcMin/+ background, and the resulting CSS.ApcMin/+ strains (A2.ApcMin/+, A7.ApcMin/+, A9.ApcMin/+, and A17.ApcMin/+) or the corresponding wild-type controls (A2.Apc+/+, A7.Apc+/+, A9.Apc+/+, and A17.Apc+/+) were maintained on a 12-h light/dark cycle at the Wolstein Research Facility (CWRU). Complement deficient mice, C57BL/6J-C3−/− (B6.C3−/−) and C57BL/6J-C5aR−/− (B6.C5aR−/−), have been previously described (18, 19). These strains were backcrossed for 10 generations onto a C57BL/6J background at the University of Pennsylvania. All experiments used true littermates to assure exposure to similar microbiota composition among test and control mice. Procedures were conducted in accordance with approved Institutional Animal Care and Use Committee (IACUC) standards at Case Western Reserve University.
Diets
Diets were obtained from Research Diets (New Brunswick, NJ). Their composition is detailed in Supplementary Table 1. These diets differed in the source (coconut, corn, or olive oil) and amounts (high or low) of fat. High fat diets (HFD) contained 58% kcal/g from hydrogenated coconut, corn, or olive oil (HFDCoco, HFDCorn, or HFDOlive, respectively), while low-fat (LF) diets contained 10.5% kcal/g from the same oils (LFDCoco, LFDCorn, or LFDOlive, respectively). The HFDCoco diet is rich (99.1%) in saturated fatty acids. HFDCorn is rich in omega-6 polyunsaturated fatty acids (61.5%), similar to Western diets that contain a 25:1 ratio of ω-6:ω-3 fatty acids. HFOlive diet is rich in the monounsaturated fatty acids (71.9%) (20).
Study design
From birth to 30 days of age, mice were fed autoclaved Purina 5010 LabDiet (Richmond, IN), and autoclaved water ad libitum. At 30 days, males were randomized to the HF or LF groups and fed ad libitum until sacrifice after 3 (33 days of age), 30 (60 days of age) or 60 (90 days of age) days on the diet (Fig. 1A). Mice were fasted for 12–14 h and anesthetized with isoflurane prior to blood collection. Whole blood, plasma, and serum samples were collected from the retro-orbital sinus into tubes with or without EDTA. Body weight was measured every other day, and body length was measured at the final time point to calculate body mass index (BMI). At the final time point, mice were euthanized by cervical dislocation, and the epididymal fat pad mass (EFPM) was used as a measure of adiposity. The small and large intestines were immediately removed, flushed with cold PBS, and cut longitudinally. Polyps were counted, and cross-sectional diameter measured in the small intestine and colon using a Leica MZ10F Modular Stereomicroscope. Polyp size and number were used to calculate total polyp mass for each mouse. Intestinal samples were immediately collected for RNA and protein analysis, frozen in liquid nitrogen, and stored at −80°C. Numbers of mice used in each experiment are also indicated individually with data points in each graph. Numbers for molecular analysis are 3–5 samples.
Figure 1. Diet and genetic background determine obesity and insulin resistance.

(A) Study design for diet studies. Male mice were maintained on a stock 5010 (grey) diet until 30 days of age (d.o.a.) before being randomly assigned to a HFD (purple) or LFD (blue). Mice remained on a diet study for 3 days (33 d.o.a.), 30 days (60 d.o.a.) or 60 days (90 d.o.a.). Wild-type B6.Apc+/+, A2.Apc+/+, A9.Apc+/+, A7.Apc+/+, and A17.Apc+/+ strains and the equivalent ApcMin/+ (B6.ApcMin/+, A2.ApcMin/+, A9.ApcMin/+, A7.ApcMin/+, and A17.ApcMin/+) strains were fed the LFDCoco or HFDCoco for 60 days. (B) Body weight curves from B6.Apc+/+ and B6.ApcMin/+. Final body weight (C) and, HOMA-IR (D) were measured at Day 60 of treatment. (E–F) The total number of polyps (E) and polyp mass (F) were measured in the intestine of each mouse at Day 60 of treatment. Values represent means ± s.e.m. (n=5–54 males/group). *P<0.05, **P<0.01, ***P<0.001 in relation to the corresponding LFD-fed group. #####P<0.001 in relation to B6.ApcMin/+ fed HFD.
Metabolic and cytokine analysis
Fasting insulin levels were measured with Mercodia Ultrasensitive Mouse Insulin ELISA (Uppsala, Sweden). Fasting glucose was measured with an OneTouch Ultra glucometer (Life Scan, Inc., Milpitas, CA). Homeostatic model assessment of insulin resistance (HOMA-IR) (21, 22) was calculated using fasting insulin and glucose levels [HOMA-IR = fasting insulin (pmol/L) x fasting blood glucose (mmol/L) /22.5]. Leptin, adiponectin, and complement C5a were measured in plasma with ELISA kits from EMD Millipore (leptin and adiponectin; Temecula, CA) and R&D Systems (C5a; Minneapolis, MN).
Quantitative RT-PCR and protein analysis
Size-matched polyp and normal tissues were collected from the small intestine of B6.ApcMin/+ mice and B6.Apc+/+ mice, respectively. Tissues were frozen in Qiagen RLT buffer (Valencia, CA) and RNA extracted with Qiagen RNeasy Mini Kits. Quantitative RT-PCR was done with SYBR Green (Quanta BioSciences, Gaithersburg, MD) to measure the expression levels of F4/80, IL-6, IL-1b, TNF and Myc in adipose or intestinal tissues (see Supplementary Table 2 for primer sequences). Tissues for Western blot analysis were immediately submerged in RIPA buffer supplemented with phosphatase and protease inhibitors, frozen in liquid nitrogen, and stored at −80°C. All antibodies (AKT, pAKT, IKK, pIKK, NFκB, p65 NFκB, STAT3, pSTAT3) were purchased from Cell Signaling Technology (Beverly, MA) and used according to manufacturer’s instructions. Homogenates from intestinal samples were used to measure intestinal inflammation using R&D Systems (Minneapolis, MN) IL-1β, IL-10, TNF-α, VEGF, and IL-23 ELISAs and the Milliplex Map Mouse Cytokine/Chemokine Panel from EMD Millipore (Temecula, CA).
Immunohistochemistry of intestinal tissue
Tissues from the small intestine (ileum) were fixed overnight at 4°C in 4% paraformaldehyde, pH 7.2 and embedded in paraffin. Cross sections (5 microns) were de-paraffinized in xylene and rehydrated in serial dilutions of ethanol and 1xPBS. Tissues were blocked in 5% goat serum, 3% BSA, 0.3% Triton X100 in 1x PBS without antigen retrieval. Sections were incubated overnight at 4°C with a 1:100 dilution of a rabbit polyclonal anti-C3c antibody (ab15980, Abcam, Cambridge MA) in blocking solution. Sections were washed in 1xPBS and, for secondary detection, incubated with a 1:500 dilution of goat anti-rabbit AlexaFluor 555 (A-21429, Lifetechnologies, Grand Island NY) in blocking solution at room temperature for 1.5 hours. After washes in 1xPBS, coverslips were mounted and nuclei counterstained in Vectashield hardset mounting medium with DAPI (H-1500, Vector Laboratories, Burlingame CA). Images were taken with a Zeiss Axioplan2 and an AxioCam digital camera. Images were processed and layered with Photoshop CS5.
Pharmacological inhibition of C5aR
C5aR was inhibited with the Ac-Phe-[Orn-Pro-(D-Cha)-Trp-Arg] peptide (PMX-53) (23). An inactive peptide containing the same amino acids in a scrambled order served as control. The treatment with PMX-53 or control peptide was initiated concomitantly with the specific LFD or HFD (i.e., 30 days of age). Drugs were diluted in PBS, and subcutaneous injections were administered every other day at 2.0 mg/kg.
Fluorescence-activated cell sorting (FACS) of intestinal immune cells
Lamina propria lymphocytes were isolated using a Percoll gradient. Antibodies for CD11b, CD11c, CD45, and GR1 were purchased from Biolegend (San Diego, CA) and used according to manufacturer’s instructions. Alternatively, cells were stained with the respective isotypes. All cell preparations showed at least 95% viability as measured with DAPI staining (Life Technologies, Grand Island, NY). Samples were analyzed using the BD LSR II flow cytometer (BD Biosciences) and FCS Express software (v4.0; De Novo Software).
Statistical Analysis
Student’s t-tests and one-way analysis of variance (ANOVA) were used to determine statistical significance, which was accepted at the p<0.05 level after Bonferroni correction for multiple hypothesis testing.
Results
Diet and genetic background determine obesity and insulin resistance
To investigate the mechanisms of DIO-induced inflammation and cancer, we first assessed the effect of diet and genetic background on obesity and glucose homeostasis. Previously characterized CSSs were used to distinguish between diet- versus obesity-induced effects (16, 17). Wild-type B6.Apc+/+ and CSS.Apc+/+ mice were fed a calorically equivalent diet high or low in hydrogenated coconut oil, HFDCoco or LFDCoco (Supplementary Table 1). After 60 days on HFD versus LFD, B6.Apc+/+, A2.Apc+/+, and A9.Apc+/+ wild-type mice showed increased final body weight (FBW), BMI and epididymal fat pad mass (EFPM) as well as elevated levels of fasting glucose and insulin with a correspondingly higher insulin resistance index [HOMA-IR] (Fig. 1A–D; Supplementary Table 3). Conversely, A7.Apc+/+ and A17.Apc+/+ mice were resistant to DIO and maintained baseline metabolic parameters (Fig. 1C, D; Supplementary Table 3). These results demonstrate the importance of diet and genetic background on development of obesity as well as glucose homeostasis, and validate the use of CSSs that differ in DIO susceptibility as a model for distinguishing the independent contributions of diet and obesity to intestinal tumorigenesis.
Diet, but not obesity or metabolic status, promotes intestinal tumorigenesis
To explore the association between DIO and tumorigenesis, we used mice heterozygous for a mutant form of the Apc tumor suppressor gene (B6.ApcMin/+) that were fed HFDCoco or LFDCoco. In contrast to B6.Apc+/+ mice on HFDCoco, which were ~10% heavier than animals fed the equivalent LFD after 60 days, B6.ApcMin/+ mice on HFDCoco gained weight during the initial 40 days, but then lost weight progressively and were moribund at the 60-day time-point (90 days of age) (Fig. 1B, C), consistent with the development of multiple intestinal polyps and cachexia (24).
Total intestinal polyp number and mass were significantly increased in all strains fed the HFDCoco but not the LFDCoco after 60 days (Fig. 1E, F; Supplementary Table 3), regardless of obesity or metabolic status (Fig. 1C, D; Supplementary Table 3). While B6.ApcMin/+ mice fed LFDCoco developed an average of 14 polyps, counts increased 6.6-fold in response to HFDCoco. Polyp numbers ranged from 6.6 to 27.4 in CSS.ApcMin/+ strains fed LFDCoco and were 3.3- to 6.6-fold higher in HFDCoco-fed mice, independent of weight gain or glucose and insulin levels (Fig. 1E; Supplementary Table 3). Similarly, polyp mass increased 8.4- to 25.4-fold in strains fed HFDCoco compared to LFDCoco (Fig. 1F; Supplementary Table 3), indicating that dietary fat, rather than obesity and associated metabolic changes, promotes intestinal neoplasia in genetically susceptible B6.ApcMin/+ mice.
Although increased polyp number and mass were observed in obesity-susceptible A2.ApcMin/+ mice fed HFDCoco compared to LFDCoco, these parameters were significantly reduced compared to other strains (Fig. 1E, F). These data suggest presence of a factor on chromosome 2 that modifies intestinal tumorigenesis. Furthermore, an enhanced inflammatory state was observed in the ApcMin/+ mice fed the HFD when compared with their LFD counterparts, as evidenced by increased neutrophil counts but were significantly lower in A2.ApcMin/+ (Supplementary Fig. 1).
We next investigated whether the impact of dietary energy content and chemical composition on intestinal neoplasia with diets containing distinct sources of fat (coconut, corn, or olive oil) (Supplementary Table 1). As observed in B6.Apc+/+ mice exposed to HFDCoco, B6.Apc+/+ mice fed HFDCorn were insulin resistant (Fig. 2B; Supplementary Table 4). However, only HFDCoco- but not HFDCorn-fed mice displayed increases in FBW, EFPM and BMI compared to LFD (Fig. 2A; Supplementary Table 4), suggesting that a diet rich in corn oil induces insulin resistance in the absence of obesity. Mice fed HFDOlive showed significant weight gain without corresponding metabolic changes compared with LFDOlive, as indicated by increased FBW, BMI, and EFPM, but normal levels of fasting glucose and insulin (Fig. 2A, B, Supplementary Table 4), in line with previous findings (20). Thus, chemical composition of dietary fat differentially affects obesity and insulin resistance.
Figure 2. Diet, but not obesity or metabolic status, promotes intestinal tumorigenesis.

B6.ApcMin/+ mice were fed the LFD or HFD with coconut (purple), corn (green), or olive (blue) oil as a source of fat for 60 days. The final body weight (A), HOMA-IR (B), total number of polyps (C) and polyp mass (D) were assessed after Day 60 of diet treatment. Values represent means ± s.e.m. (n=7–54 males/group). *P<0.05, **P<0.01, ***P<0.001 in relation to equivalent LFD-fed group. ###P<0.001 in relation to HFDCoco- or HFDCorn-fed groups.
Importantly, the various diets also differentially affected intestinal tumorigenesis. Total intestinal polyp number and mass were significantly increased in mice fed HFDCoco and HFDCorn compared to corresponding LFD-fed animals (Fig. 2C, D). In contrast, no significant differences in total polyp number or mass were observed in B6.ApcMin/+ fed HFDOlive versus LFDOlive (Fig. 2C, D), demonstrating that although a diet rich in olive oil resulted in DIO, it prevented insulin resistance and intestinal neoplasia, suggesting that tumorigenesis may be associated with specific dietary fats rather than obesity.
Similar degrees of tumorigenesis were observed after shorter exposure to a HFD (30 versus 60 days), independent of fat source (Fig. 2C, D; Supplementary Fig. 2). This dietary effect occurred after a 30-day exposure to dietary fat, prior to significant increases in FBW, BMI or EFPM (Supplementary Fig. 2; Supplementary Table 4), indicating that conditions that favor polyp development are established prior to onset of obesity or insulin resistance.
Intestinal neoplasia is associated with diet-induced inflammation
To identify mechanisms linking dietary fat with intestinal tumorigenesis, we focused on HFDCoco effects over a 30 day period, before the onset of DIO, metabolic changes or tumor-induced cachexia. Circulating levels of the pro-inflammatory cytokines IL-6, IL-1β and monocyte chemotactic protein (MCP-1) and the adipose tissue-derived hormone, leptin, were elevated in mice fed HFDCoco compared to LFDCoco (Fig. 3A–C, E, cf. ref 1). Levels of anti-inflammatory cytokine IL-10 were not affected by diet or strain (Fig. 3D). In contrast, the level of adiponectin, an adipokine that is involved in fatty acid oxidation and has anti-inflammatory properties, was reduced in HFDCoco-fed mice (Fig. 3F). mRNA expression levels of IL-6 and the macrophage marker F4/80 were significantly elevated in adipose tissue from B6.ApcMin/+ in response to HFDCoco (Fig. 3G, H). Expression of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α, was also increased in intestinal tissue of both B6.Apc+/+ and B6.ApcMin/+ mice fed HFD, while c-Myc was significantly elevated in response to HFDCoco only in B6.ApcMin/+ (Fig. 3I–L), supporting the concept that HFDCoco triggers a pro-inflammatory environment that promotes polyp development.
Figure 3. Intestinal neoplasia is associated with diet-induced inflammation.

B6.Apc+/+ and B6.ApcMin/+ mice were fed the LFDCoco or HFDCoco for 30 days. Levels of anti- and pro-inflammatory mediators implicated in metabolic syndrome and cancer were measured in the blood (A–F), adipose tissue (G, H), and intestinal tissue (I–L) at day 30 of treatment. Values represent means ± s.e.m. (n=5–7 males/group). *P<0.05, **P<0.01, ***P<0.001 in relation to the same strain fed the LFDCoco. ##P<0.01 in relation to B6.Apc+/+ fed HFDCoco.
To examine the influence of diet on early tumor environment, mice were exposed to HFD or LFD for 3 days. Prior to any changes in FBW or HOMA-IR (Fig. 4A, B) after HFDCoco exposure, increased intestinal polyp numbers were observed (Fig. 4C, D). Circulating levels of adiponectin were decreased in the HFD-fed mice when compared with the LFD counterparts (Fig. 4F). Remarkably, the short HFD exposure significantly increased IL-6, IL-23, TNF-α and VEGF but not IL-10 in the intestines of mice fed HFDCoco compared to LFDCoco (Fig. 4H–L), suggesting that HFDCoco rapidly triggers a pro-inflammatory and pro-angiogenic environment that promotes polyp development.
Figure 4. Inflammation and intestinal neoplasia are triggered soon after exposure to HFD- the 3 days study.

B6.Apc+/+ and B6.ApcMin/+ mice were fed the LFDCoco or HFDCoco for 3 days. Final body weight (A) and HOMA-IR (B) were measured at Day 3 of treatment. Intestinal polyp number (C) and mass (D) were analyzed. Circulating levels of C5a (E) and adiponectin (F) were measured in plasma. Levels of anti- and pro-inflammatory mediators implicated in inflammation and cancer- C5a (G), IL-6 (H), VEGF (I), IL-23 (J), TNF-α (K), and IL-10 (L) were measured in the intestine of B6.Apc+/+ or B6.ApcMin/+ at Day 3 of treatment. Values represent means ± s.e.m. (A–F, n=6–9 males/group; G–L, n=3–5). *P<0.05, **P<0.01, ***P<0.001 in relation to the corresponding LFD-fed group.
Source of dietary fat determines local pro-inflammatory environment and intestinal polyposis
In light of our observation that HFDOlive did not promote polyposis in genetically susceptible mice despite DIO (Fig. 2), we evaluated intestinal inflammation in mice fed HFDCoco, HFDCorn or HFDOlive. After a 30-day exposure of wild-type B6.Apc+/+ mice to HFD or the corresponding LFD, levels of VEGF, TNF-α, IL-23 and IL-1β were significantly elevated in mice fed HFDCoco and HFDCorn compared with the LFD (Fig. 5A–D). Although no increase in these inflammatory factors was observed in mice fed HFDOlive, the anti-inflammatory cytokine, IL-10, was significantly elevated in HFDOlive-fed mice (Fig. 5E).
Figure 5. Source of dietary fat determines local pro-inflammatory environment and intestinal polyposis.

B6.Apc+/+ and B6.ApcMin/+ mice were fed diets supplemented with coconut (purple), corn (green) or olive (blue) oil for 30 days. Levels of anti- and pro-inflammatory mediators implicated in inflammation and cancer such as VEGF (A), TNF-α (B), IL-23 (C), IL-1β (D) and IL-10 (E) were measured in intestinal tissue at day 30 of treatment. Values represent means ± s.e.m. (n=3–5 males/group). *P<0.05, **P<0.01 in relation to the same strain fed the LFDCoco. #P<0.05, ##P<0.01 in relation to the same strain fed the corresponding LFDCoco or HFDCoco.
CSS A2.ApcMin/+, which showed reduced tumorigenesis regardless of dietary exposure or obesity status, was reassessed to explore the link between diet and inflammation. Notably, chromosome 2 in the A/J strain as well as in CSS A2.ApcMin/+ carries a dysfunctional complement component C5 gene (16). C5 is cleaved in response to complement activation, with the subsequent generation of the C5a fragment, an established pro-inflammatory mediator with a detrimental role in cancer (25–29). This cleavage product signals through the G-coupled receptor, C5aR (30). To test whether dietary fat modulates complement activation, plasma C5a levels were measured in B6.Apc+/+, CSS.Apc+/+, and the corresponding ApcMin/+ strains fed HFDCoco or LFDCoco for 60 days. All strains, except A2.Apc+/+ and A2.ApcMin/+, showed a significant increase in plasma C5a in response to HFDCoco, regardless of obesity status or obesity-associated metabolic changes (Supplementary Fig. 3).
Next, circulating levels of plasma C5a levels were measured in response to the different sources of dietary fat. After 30 days of diet exposure, C5a levels were significantly elevated in both B6.Apc+/+ and B6.ApcMin/+ strains fed HFDCoco or HFDCorn but not HFDOlive when compared with the corresponding LFD-fed mice (Fig. 6A). After a short 3-day exposure to HFDCoco, C5a levels were significantly increased in plasma and the intestine (Fig. 4E, G).
Figure 6. Complement C5a mediates diet-induced inflammation and intestinal neoplasia.

B6.Apc+/+ and B6.ApcMin/+ mice were fed the LFD or HFD with coconut (purple), corn (green), or olive oil (blue) as a source of fat for 30 days. Circulating levels of the complement activation fragment C5a were measured in the plasma at Day 30 of treatment (A). (B–F) B6.ApcMin/+ (+/+; Min/+) and the complement-deficient C5aR+/−; ApcMin/+ (C5aR+/−; Min/+), and C5aR−/−; ApcMin/+ (C5aR−/−; Min/+) strains were fed HFDCoco or LFDCoco for 30 days. The total number of polyps (B) and polyp mass (C) were measured in the intestine of each mouse at Day 30 of treatment. (n=5–20/group). Intestinal inflammation was measured in mice genetically deficient in C5aR (D–F). Protein levels of VEGF (D), TNFα (E), and IL-23 (F) were measured in the intestine (n=3–5/group) B6.Apc+/+ and B6.ApcMin/+ mice were fed HFDCoco for 30 days, and concomitantly treated with the C5aR antagonist peptide (PMX53) or a scrambled inactive peptide (control) (2 mg/kg s.c.) every other day (B, C). (D, E) Intestinal inflammation was measured in males treated with PMX53 or control inhibitor (n=3–5). Total polyp number (B) and polyp mass (C) were measured. Protein levels of VEGF (G), TNF-α (H), and IL-23 (I) and expression of IL-1β (J) and Myc (K) were measured in the intestine of males treated with PMX53 or the control inhibitor (n=3–6/group). (L) The percentage of viable CD11b+/GR-1+ neutrophils was measured with flow cytometry in the intestinal lamina propria of B6.Apc+/+ and B6.ApcMin/+ males treated with PMX53 or the control peptide (n=4/group). Levels of phosphorylated AKT (pS473), IKK (pS176) and NFκB (pS536) were assessed with Western blotting (M) in polyp tissue of B6.ApcMin/+ males or normal intestinal tissue of B6.Apc+/+ males treated with PMX53 or the control peptide. Levels of AKT and NFκB were measured as a control for the protein amount loaded. Results are representative of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001 compared to same strain fed corresponding LFD. #P<0.05, ##P<0.01, ###P<0.001 compared to B6.Apc+/+ or B6.ApcMin/+ fed the same diet. $P<0.05, $$P<0.01, $$$P<0.001 compared to same strain treated with the control inhibitor.
Additionally, using an antibody that recognizes complement C3 fragment C3c, and the C3c epitope of native C3 and C3b, increased deposition of complement C3 in the normal small intestine was observed in response to HFDCoco when compared to LFDCoco (Supplementary Fig. 4A–D). Specifically, antibody staining accumulated within the lamina propria and along the epithelial basement membrane in the underlying connective tissue. These results are consistent with previous observations that activated complement C3 is deposited along the basement membrane in the inflamed intestinal mucosa of ulcerative colitis (UC) and Crohn’s disease (CD) patients and that complement C3 is expressed by subepithelial myofibroblasts (SEMFs), which are positioned subadjacent to IECs along basement membrane, in response to inflammation (31, 32). Curiously, complement C3 also accumulated within polyps of B6.ApcMin/+ mice fed HFDCoco for 30 days (Supplementary Fig. 4E–F). Complement C3 localized primarily to the stroma of polyp lesions but was expressed at greatly diminished levels or was absent in the stroma surrounding normal epithelial cells in deep crypts under polyp lesions and positioned the greatest distance from the intestinal lumen. These results indicate that interactions with specific dietary fats trigger C3 activation and C5a generation shortly after HFD exposure.
Complement C5a mediates diet-induced inflammation and intestinal neoplasia
The link between HFD-induced complement activation and intestinal tumorigenesis was confirmed with a genetically-engineered knockout mutation in the C3 complement component (C3) gene that was backcrossed onto the B6.ApcMin/+ background. Absent (homozygotes) or reduced (heterozygotes) C3 levels, which is normally essential for complement activation (33), resulted in reduced polyp number and mass in the intestine of HFDCoco-fed mice (Supplementary Fig. 5). Similarly, diminished tumorigenesis was observed in C5aR−/− mice on a B6.ApcMin/+ background fed HFDCoco (Fig. 6B, C), supporting a role for the C3-C5a-C5aR axis in diet-induced intestinal neoplasia. In addition, pharmacological targeting of C5aR with the antagonist peptide PMX-53 (23), but not an inactive control peptide, resulted in a significant reduction in polyp number and mass resulting from HFD exposure (Fig. 6B, C; Supplementary Table 5). Complement deficiency did not influence body weight or glucose and insulin levels in mice fed the HFDCoco for 30 days (Supplementary Table 5). Further, genetic or pharmacological inhibition of C5aR did not significantly reduce polyp number or mass in males fed LFDCoco (Supplementary Table 5), supporting the notion that C5a specifically mediates HFD-induced inflammation.
Mechanistically, genetic and pharmacologic inhibition of C5aR signaling reduced HFD-induced inflammation as indicated by reduced levels of intestinal VEGF, TNF-α, and IL-23 (Fig. 6D–I). In addition, abrogation of C5aR signaling with PMX-53 inhibited AKT-NFκB signaling, which has been implicated in inflammation and cancer (34). Whereas increased levels of phosphorylated AKT, IKK, and NFκB were observed in polyp tissue from B6.ApcMin/+ mice fed HFDCoco as compared to normal B6.Apc+/+ tissue, activation was attenuated in mice treated with PMX-53 but not the control peptide (Fig. 6M). Furthermore, pharmacological targeting of C5aR resulted in reduced mRNA expression levels of IL-1β and c-Myc in polyp tissue (Fig. 6J, K), and prevented infiltration of inflammatory neutrophils to the lamina propria (Fig. 6L), which has been implicated as a key inflammatory trigger in DIO response (35).
Discussion
Here we show that complement C5a-induced inflammation promotes intestinal tumorigenesis. Complement signaling modulates various pathophysiological processes and has been previously implicated in tumorigenesis (33). Recent findings indicate that complement activation in the tumor microenvironment leads to tumor growth and metastasis. In vivo models using breast, lung and colon cancer cell lines showed that C5aR-mediated signaling creates a microenvironment that promotes growth through recruitment of myeloid-derived suppressor cells (MDSCs) and inhibition of anti-tumor immune responses mediated by CD8+ and CD4+ T cells (25, 27–29, 36). In addition, C5a-induced inflammation is associated with promotion of angiogenesis and tumor progression. In the absence of C3 or C5aR-mediated signals, mice genetically susceptible to ovarian cancer develop small, poorly vascularized tumors. While MDSCs do not seem to play a role in this model, complement activation is associated with increased tumorigenesis as indicated by elevated regulatory T cell numbers and increased VEGF-induced angiogenesis (37). Complement activation also promotes colitis-associated carcinogenesis via induction of pro-inflammatory IL-1β and IL-17 by bone marrow-derived neutrophils (38). In addition to creating a pro-angiogenic and pro-tumorigenic microenvironment, C3a and C5a derived from tumor cells have an autocrine effect on tumor proliferation via activation of the PI3K/AKT signaling pathway (26). Such findings agree with our observations that the C5aR antagonist PMX-53 prevents activation of AKT/IKK/NFκB following HFDCoco exposure (Fig. 6M). Additionally, deficiency of PTX3 has been associated with increased susceptibility to mesenchymal and epithelial carcinogenesis due to poor regulation of complement activation and consequent inflammation (39). Further, development of hepatocellular carcinoma mediated by HFD-induced inflammation involved c-Myc and NFκB signaling pathways in genetically C5-sufficient (C57BL/6) but not C5-deficient (A/J) mice (40), pointing to complement activation as a link between DIO and cancer. Finally, recent reports suggest that C3aR- and C5aR-mediated signaling modulates neutrophil and CD8+T cell function with consequent increased tumorigenesis (41–43).
Obesity has become a major public health concern because of its association with medical conditions such as hypertension, diabetes, cardiovascular disease, and certain cancers (44). Although mutations in genes such as leptin, leptin receptor and melanocortin-4 receptor affect satiety control, glucose homeostasis and energy metabolism, common obesity is considered a multifactorial disorder controlled by multiple genes combined with environment factors such as diet, physical activity, and intestinal microbiota (45, 46). Given the complexity involved in clinical studies to evaluate obesity-related diseases in humans, mouse strains are essential for studying the physiology of DIO and metabolic syndrome (47, 48). CSSs in particular are a valuable paradigm for disentangling the complex relations among diet, obesity and metabolic status (Fig. 1C, D) (16, 17). These CSSs together with cancer promoting genetic models such as ApcMin/+ (49) are a genetically-defined, reproducible experimental model system that can be used to explore mechanisms underlying interactions among diet, metabolism, and inflammation on tumorigenesis.
Extensive epidemiological research has recently established an association between obesity and increased risk of cancer (40, 50, 51). Obesity is considered a leading environmental risk for colorectal cancer and obesity-induced inflammation has been correlated to various stages of cancer progression including cellular transformation, promotion, survival, invasion, angiogenesis and metastasis (51). Obesity results in the accumulation of large adipocytes that upon necrosis recruit macrophages that secrete pro-inflammatory cytokines and are implicated in the process of insulin resistance. In addition to the influence of diet on tumorigenesis in the GI tract, high fat diets promote tumors with earlier appearance, greater frequency, accelerated growth, larger size, and in some cases more frequent metastasis (50). For example, HFD has been shown to lead to hepatocellular cancer in C57BL/6J mice (40) and feeding a Western type diet, enriched in fat and cholesterol, accelerates tumor progression in mice with an autochthonous model of prostate cancer (52). Further demonstration of diet on tumorigenesis, outside the intestine, is provided by demonstration that a high fat, lard based diet, fed to obesity-resistant BALB/c mice promoted faster tumor growth and increased metastasis in mice orthotopically inoculated with syngeneic mammary carcinoma cells (53).
In this study, specific HFDs induced up-regulation of the pro-inflammatory cytokines MCP-1 IL-1β, IL-6, IL-23, TNF-α, and VEGF in adipose tissue, intestine and blood, which correlated with increased tumorigenesis (Fig. 3–6). The tumor microenvironment favors a collaboration between malignant and immune cells as well as pro-inflammatory mediators secreted by tumor-infiltrating lymphocytes that promote cell proliferation and angiogenesis (54, 55). Increased levels of pro-inflammatory cytokines and angiogenic factors are found in serum and adipose tissue of colorectal cancer patients and are correlated with poor survival (54). Interestingly, lean mice fed HFD presented increased inflammation and tumorigenesis, indicating that the HFD itself and not obesity or the related metabolic status determines the local environment that favors tumor growth. It has recently been shown that HFD promotes tumorigenesis in the small intestine of genetically susceptible K-rasG12Dint mice, independent of obesity, as a consequence of a shift in gut microbiota composition and a decrease in Paneth-cell-mediated antimicrobial host defense (10). Dietary interventions can significantly influence gut microflora, thereby affecting disease outcomes. In this study, HFDs composed of specific dietary fats induced complement activation with consequent release of the pro-inflammatory C5a fragment as soon as after 3 days of diet exposure, suggesting that complement activation may occur prior to the microbiota shift and may influence the composition of intestinal bacteria given the antimicrobial properties of the complement component system (30, 33). Indeed, inhibition of C5aR-mediated signaling has been previously shown to alter the composition and diversity of skin microbiota along with the pro-inflammatory profile (56).
Adipose tissue is a relevant source of complement proteins including C3, Factor B and properdin, that are required for activation of the alternative pathway. Local complement activation and consequent production of C5a regulate infiltration of pro-inflammatory macrophages into adipose tissue and promote insulin resistance (57). This study showed that specific HFDs induced complement activation, adding to the current understanding on the local role of complement in adipose tissue. To our knowledge, only one in vitro but no in vivo study has demonstrated complement activation by a specific dietary component (gluten) (58). Clearly, further investigation is needed to dissect the precise fatty acids responsible for complement activation. Nevertheless, such observations open new avenues for research on diet-induced inflammatory diseases and places complement as a key target for therapeutic intervention in such conditions.
Apart from complement, fatty acids are involved in immunological processes via direct activation of immune cells and release of pro-inflammatory mediators (59). Whereas saturated fatty acids contribute to promoting inflammation, unsaturated fatty acids show both pro- and anti-inflammatory properties and the balance between omega-3/-6 is considered critical for maintenance of healthy levels of fat (12). Reduced levels of complement activation, inflammatory cytokines and tumorigenesis were observed in mice fed HFDOlive despite similar levels of obesity compared to those fed HFDCoco (Fig. 2, 5, 6A). Olive oil, which is rich in the unsaturated oleic acid, has antioxidant properties and together with other components such as phenolic compounds induce anti-proliferative and anti-inflammatory responses (20).
Together, we found that certain HFDs activate complement, regardless of DIO, and generate C5a, which in turn promotes a pro-inflammatory environment by triggering signaling pathways that control expression of proto-oncogenes and release of inflammatory mediators, consequently favoring formation of intestinal polyps in genetically susceptible mice (Fig. 7). Importantly, three observations were made that indicate dissociation between DIO and high dietary fat in tumor development: increased intestinal tumorigenesis in both DIO-susceptible and -resistant CSS mice (Fig. 1C–F), increased polyp numbers in lean mice after only 3 days of HFD exposure (Fig. 4A–D), and lack of tumor progression in obese HFDOlive-fed mice (Fig. 2). As such, these results show that HFD-induced complement activation and C5a generation promote inflammation and intestinal tumorigenesis prior to the onset of obesity. Given that HFD-induced complement activation is an upstream event and is crucial for the establishment of inflammation, and given the beneficial effects of anti-inflammatory therapies in cancer (7), these data point to complement-targeted therapeutics (60) as a potentially powerful means of preventing diet-induced neoplasia.
Figure 7. Molecular mechanisms determining HFD-induced intestinal polyp progression.
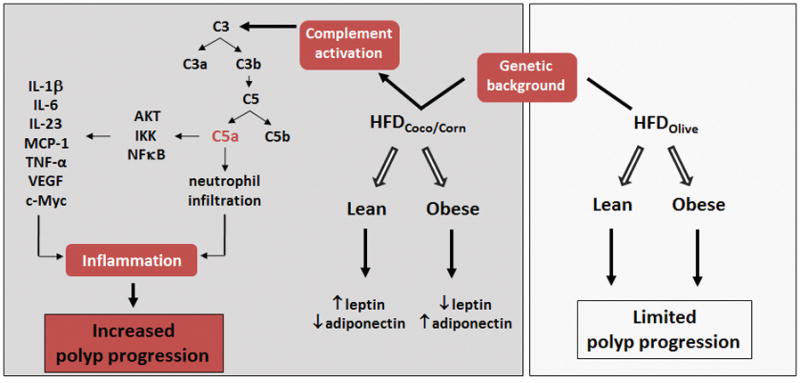
Diets high in specific dietary fats induce an inflammatory environment in the intestine that promotes intestinal tumorigenesis in genetically susceptible APCmin/+mice, independent of obesity and related metabolic conditions. HFD containing coconut or corn oil as a source of saturated fat induce complement activation with subsequent release of C5a. C5a in turn induces the infiltration of inflammatory neutrophils to the intestinal lamina propria and activate the AKT/IKK/NFκB signaling pathway in intestinal cells with consequent production of pro-inflammatory mediators such as IL-1β, IL-6, TNF-α and the proto-oncogene c-myc. Conversely, mice fed a HFD containing olive oil as a source of fat gained significant body weight, but did not promote a pro-inflammatory environment or development of polyps in the intestine.
Supplementary Material
Implications.
This study characterizes the relations between diet and metabolic conditions on risk for a common cancer and identifies complement activation as a novel target for cancer prevention.
Acknowledgments
We thank David A. DeSantis and Carmen Fiuza-Luces for technical assistance. This work was supported by the Transdisciplinary Research in Energy Balance and Cancer Grant #U54 CA116867 to N.A.B. and J.H.N., AI030040 and AI068730 to J.D.L. and P40 RR012305 to J.H.N. and N.A.B.
Footnotes
Competing financial interests: S.K.D., N.A.B., J.D.L. and J.H.N. are co-inventors in a patent application titled “A method to treat polyp formation”.
References
- 1.Renehan AG, Zwahlen M, Egger M. Adiposity and cancer risk: new mechanistic insights from epidemiology. Nat Rev Cancer. 2015;15:484–98. doi: 10.1038/nrc3967. [DOI] [PubMed] [Google Scholar]
- 2.Iyengar NM, Hudis CA, Dannenberg AJ. Obesity and cancer: local and systemic mechanisms. Annu Rev Med. 2015;66:297–309. doi: 10.1146/annurev-med-050913-022228. [DOI] [PubMed] [Google Scholar]
- 3.Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–7. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 4.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 5.Mentor-Marcel RA, Bobe G, Barrett KG, Young MR, Albert PS, Bennink MR, et al. Inflammation-associated serum and colon markers as indicators of dietary attenuation of colon carcinogenesis in ob/ob mice. Cancer Prev Res (Phila) 2009;2:60–9. doi: 10.1158/1940-6207.CAPR-08-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubota M, Shimizu M, Sakai H, Yasuda Y, Ohno T, Kochi T, et al. Renin-angiotensin system inhibitors suppress azoxymethane-induced colonic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Biochem Biophys Res Commun. 2011;410:108–13. doi: 10.1016/j.bbrc.2011.05.115. [DOI] [PubMed] [Google Scholar]
- 7.Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–40. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 8.Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30:88–92. doi: 10.1093/carcin/bgn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Y, Pan Y, Rhea PR, Tan L, Gagea M, Cohen L, et al. A Sucrose-Enriched Diet Promotes Tumorigenesis in Mammary Gland in Part through the 12-Lipoxygenase Pathway. Cancer Res. 2016;76:24–9. doi: 10.1158/0008-5472.CAN-14-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, Aydin B, et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature. 2014;514:508–12. doi: 10.1038/nature13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Couto E, Boffetta P, Lagiou P, Ferrari P, Buckland G, Overvad K, et al. Mediterranean dietary pattern and cancer risk in the EPIC cohort. Br J Cancer. 2011;104:1493–9. doi: 10.1038/bjc.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veldhoen M, Brucklacher-Waldert V. Dietary influences on intestinal immunity. Nat Rev Immunol. 2012;12:696–708. doi: 10.1038/nri3299. [DOI] [PubMed] [Google Scholar]
- 13.Song M, Garrett WS, Chan AT. Nutrients, foods, and colorectal cancer prevention. Gastroenterology. 2015;148:1244–60. e16. doi: 10.1053/j.gastro.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 15.Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66:601–13. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 16.Singer JB, Hill AE, Burrage LC, Olszens KR, Song J, Justice M, et al. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;304:445–8. doi: 10.1126/science.1093139. [DOI] [PubMed] [Google Scholar]
- 17.Burrage LC, Baskin-Hill AE, Sinasac DS, Singer JB, Croniger CM, Kirby A, et al. Genetic resistance to diet-induced obesity in chromosome substitution strains of mice. Mamm Genome. 2010;21:115–29. doi: 10.1007/s00335-010-9247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92:11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hopken UE, Lu B, Gerard NP, Gerard C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996;383:86–9. doi: 10.1038/383086a0. [DOI] [PubMed] [Google Scholar]
- 20.Lucas L, Russell A, Keast R. Molecular mechanisms of inflammation. Anti-inflammatory benefits of virgin olive oil and the phenolic compound oleocanthal. Curr Pharm Des. 2011;17:754–68. doi: 10.2174/138161211795428911. [DOI] [PubMed] [Google Scholar]
- 21.Berglund ED, Li CY, Ayala JE, McGuinness OP, Wasserman DH. Regulation of endogenous glucose production in glucose transporter 4 over-expressing mice. PLoS One. 2012;7:e52355. doi: 10.1371/journal.pone.0052355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 23.Paczkowski NJ, Finch AM, Whitmore JB, Short AJ, Wong AK, Monk PN, et al. Pharmacological characterization of antagonists of the C5a receptor. Br J Pharmacol. 1999;128:1461–6. doi: 10.1038/sj.bjp.0702938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA. Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim Biophys Acta. 2011;1812:1601–6. doi: 10.1016/j.bbadis.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. 2012;189:4674–83. doi: 10.4049/jimmunol.1201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho MS, Vasquez HG, Rupaimoole R, Pradeep S, Wu S, Zand B, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6:1085–95. doi: 10.1016/j.celrep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454–65. doi: 10.1158/0008-5472.CAN-14-0157. [DOI] [PubMed] [Google Scholar]
- 28.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–35. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piao C, Cai L, Qiu S, Jia L, Song W, Du J. Complement 5a Enhances Hepatic Metastases of Colon Cancer via Monocyte Chemoattractant Protein-1-mediated Inflammatory Cell Infiltration. J Biol Chem. 2015;290:10667–76. doi: 10.1074/jbc.M114.612622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klos A, Wende E, Wareham KJ, Monk PN. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev. 2013;65:500–43. doi: 10.1124/pr.111.005223. [DOI] [PubMed] [Google Scholar]
- 31.Ueki T, Mizuno M, Uesu T, Kiso T, Nasu J, Inaba T, et al. Distribution of activated complement, C3b, and its degraded fragments, iC3b/C3dg, in the colonic mucosa of ulcerative colitis (UC) Clin Exp Immunol. 1996;104:286–92. doi: 10.1046/j.1365-2249.1996.17721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol. 2010;160:386–93. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 35.Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–12. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Downs-Canner S, Magge D, Ravindranathan R, O’Malley ME, Francis L, Liu Z, et al. Complement Inhibition: A Novel Form of Immunotherapy for Colon Cancer. Ann Surg Oncol. 2015 doi: 10.1245/s10434-015-4778-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nunez-Cruz S, Gimotty PA, Guerra MW, Connolly DC, Wu YQ, DeAngelis RA, et al. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia. 2012;14:994–1004. doi: 10.1593/neo.121262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ning C, Li YY, Wang Y, Han GC, Wang RX, Xiao H, et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1beta/IL-17A axis. Mucosal Immunol. 2015;8:1275–84. doi: 10.1038/mi.2015.18. [DOI] [PubMed] [Google Scholar]
- 39.Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160:700–14. doi: 10.1016/j.cell.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Hill-Baskin AE, Markiewski MM, Buchner DA, Shao H, DeSantis D, Hsiao G, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum Mol Genet. 2009;18:2975–88. doi: 10.1093/hmg/ddp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. 2016;7:11037. doi: 10.1038/ncomms11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, et al. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J Immunol. 2016;196:4783–92. doi: 10.4049/jimmunol.1600210. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Sun SN, Liu Q, Yu YY, Guo J, Wang K, et al. Autocrine Complement Inhibits IL10-Dependent T-Cell Mediated Antitumor Immunity to Promote Tumor Progression. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-15-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levi JSLM, Laurent RS, Lang A, Rayburn J. F as in Fat: how obesity threatens America’s future 2012. Trust for America’s Health Rep. 2012 http://healthyamericansorg/report/100/
- 45.van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell. 2015;161:119–32. doi: 10.1016/j.cell.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 46.Zhang C, Yin A, Li H, Wang R, Wu G, Shen J, et al. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine. 2015;2:966–82. doi: 10.1016/j.ebiom.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinasac DS, Riordan JD, Spiezio SH, Yandell BS, Croniger CM, Nadeau JH. Genetic control of obesity, glucose homeostasis, dyslipidemia and fatty liver in a mouse model of diet-induced metabolic syndrome. Int J Obes (Lond) 2015 doi: 10.1038/ijo.2015.184. [DOI] [PubMed] [Google Scholar]
- 48.Spiezio SH, Amon LM, McMillen TS, Vick CM, Houston BA, Caldwell M, et al. Genetic determinants of atherosclerosis, obesity, and energy balance in consomic mice. Mamm Genome. 2014;25:549–63. doi: 10.1007/s00335-014-9530-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Zhang Q. Application of the Apc(Min/+) mouse model for studying inflammation-associated intestinal tumor. Biomed Pharmacother. 2015;71:216–21. doi: 10.1016/j.biopha.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 50.Berger NA. Obesity and cancer pathogenesis. Ann N Y Acad Sci. 2014;1311:57–76. doi: 10.1111/nyas.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guffey CR, Fan D, Singh UP, Murphy EA. Linking obesity to colorectal cancer: recent insights into plausible biological mechanisms. Curr Opin Clin Nutr Metab Care. 2013;16:595–600. doi: 10.1097/MCO.0b013e328362d10b. [DOI] [PubMed] [Google Scholar]
- 52.Llaverias G, Danilo C, Wang Y, Witkiewicz AK, Daumer K, Lisanti MP, et al. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. Am J Pathol. 2010;177:3180–91. doi: 10.2353/ajpath.2010.100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim EJ, Choi MR, Park H, Kim M, Hong JE, Lee JY, et al. Dietary fat increases solid tumor growth and metastasis of 4T1 murine mammary carcinoma cells and mortality in obesity-resistant BALB/c mice. Breast Cancer Res. 2011;13:R78. doi: 10.1186/bcr2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi: 10.1155/2014/149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci U S A. 2015;112:E2487–96. doi: 10.1073/pnas.1422445112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A. 2013;110:15061–6. doi: 10.1073/pnas.1307855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D, et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol. 2013;191:4367–74. doi: 10.4049/jimmunol.1300038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Unsworth DJ, Wurzner R, Brown DL, Lachmann PJ. Extracts of wheat gluten activate complement via the alternative pathway. Clin Exp Immunol. 1993;94:539–43. [PMC free article] [PubMed] [Google Scholar]
- 59.Doerner SK, Berger NA. Dietary Fats as Mediators of Obesity, Inflammation and Cancer. In: Dannenberg AJ, Berger NA, editors. Obesity, Inflammation and Cancer. Springer; 2013. pp. 99–132. [Google Scholar]
- 60.Reis ES, Mastellos DC, Yancopoulou D, Risitano AM, Ricklin D, Lambris JD. Applying complement therapeutics to rare diseases. Clin Immunol. 2015;161:225–40. doi: 10.1016/j.clim.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.