

ABSTRACT
Differentially methylated or hydroxymethylated regions (DMRs) in mammalian DNA are often associated with tissue-specific gene expression but the functional relationships are still being unraveled. To elucidate these relationships, we studied 16 human genes containing myogenic DMRs by analyzing profiles of their epigenetics and transcription and quantitatively assaying 5-hydroxymethylcytosine (5hmC) and 5-methylcytosine (5mC) at specific sites in these genes in skeletal muscle (SkM), myoblasts, heart, brain, and diverse other samples. Although most human promoters have little or no methylation regardless of expression, more than half of the genes that we chose to study—owing to their myogenic DMRs—overlapped tissue-specific alternative or cryptic promoters displaying corresponding tissue-specific differences in histone modifications. The 5mC levels in myoblast DMRs were significantly associated with 5hmC levels in SkM at the same site. Hypermethylated myogenic DMRs within CDH15, a muscle- and cerebellum-specific cell adhesion gene, and PITX3, a homeobox gene, were used for transfection in reporter gene constructs. These intragenic DMRs had bidirectional tissue-specific promoter activity that was silenced by in vivo-like methylation. The CDH15 DMR, which was previously associated with an imprinted maternal germline DMR in mice, had especially strong promoter activity in myogenic host cells. These findings are consistent with the controversial hypothesis that intragenic DNA methylation can facilitate transcription and is not just a passive consequence of it. Our results support varied roles for tissue-specific 5mC- or 5hmC-enrichment in suppressing inappropriate gene expression from cryptic or alternative promoters and in increasing the plasticity of gene expression required for development and rapid responses to tissue stress or damage.
KEYWORDS: DNA methylation, 5-hydroxymethylcytosine, cryptic promoters, alternative promoters, H3K36me3, skeletal muscle, brain, heart, myoblasts, transcription factors
Introduction
Although there is growing recognition of the importance of 5-hydroxymethylcytosine (5hmC) in DNA in the normal functioning of the mammalian genome,1-4 most studies of differential DNA methylation associated with development or disease use methods that cannot distinguish genomic 5hmC from 5-methylcytosine (5mC). Genomic 5hmC originates from 5mC residues in CpG sequences by ten eleven translocation (TET) enzyme-catalyzed oxidation.5 These 5-methylpyrimidine dioxygenases (TET1, TET2, and TET3) can also catalyze the conversion of 5hmC to 5-formylcystosine and 5-carboxylcytosine as additional intermediates in an active demethylation pathway but these bases are present in 10–100-fold lower amounts than 5hmC.5 Global levels of 5hmC in mammalian genomes are always lower than 5mC levels and show even more tissue specificity.6-8 The percentage of modified C that is hydroxymethylated is several fold to more than tenfold higher in brain than in other human tissues and is higher in undifferentiated embryonic stem cells (ESC) than in non-embryonic cell lines.1,6,7 Many studies provide evidence for changes in the distribution of genomic 5hmC contributing to differentiation and disease but how much of this is due to associations of 5hmC enrichment with enhancers, gene bodies, exons, promoter regions, and terminal gene regions is unclear.2,9-11 Also uncertain is the extent to which DNA sequence context and cell type influences the effects of DNA hydroxymethylation on gene function.12 The functional significance of 5hmC in DNA is based on its being an intermediate in passive (DNA replication-dependent) or active DNA demethylation pathways as well as a stable component of the genome.5,9
In addition to a surge of recent interest in genomic 5hmC, there has been increased attention to the roles of genomic 5mC in differentiation and disease other than its most studied function in establishing or maintaining promoter silencing. Enrichment in 5mC (or 5hmC) has been also implicated in facilitating transcription (from intragenic regions, especially exons) and regulating co-transcriptional splicing of pre-mRNAs.13 However, the functional relationships between gene-body methylation and expression are complex and still unclear.14,15 Complicating studies of DNA hypermethylation is the question of whether such differentially methylated regions (DMRs) are also enriched in 5hmC (although usually with less 5hmC than 5mC11,16). Distinguishing 5hmC and 5mC is important because genomic cytosine methylation and hydroxymethylation can have opposite effects chemically and biologically,17 e.g., at enhancers.11,18-20 Not only are studies that distinguish genomic 5mC and 5hmC relevant to development and disease but also to physiology. This is illustrated by recent findings implicating hypoxia, which can regulate muscle regeneration21 as well as tumor growth, in decreasing TET enzyme activity and global genomic 5hmC levels and increasing local 5mC levels in tumors.22
We previously identified DMRs throughout the human genome that show high specificity for the skeletal muscle (SkM) lineage based upon reduced representation bisulfite sequencing (RRBS) of 33 different types of tissues and cell cultures.23,24 Because RRBS, like most methods of DNA methylation analysis, cannot resolve 5hmC from 5mC, we also determined the amounts of 5mC and 5hmC by a quantitative, enzyme- and real time PCR-based assay (Epimark) at seven CCGG sites in myogenic hypermethylated DMRs located within or near HOX genes, PAX3, or TBX1.23,25 We found little or no 5hmC in myogenic progenitor cells (myoblasts, Mb, and myotubes, Mt) at these sites but elevated levels of 5hmC in SkM in five of them. These results suggested that there might be an association between hypermethylation (5mC) in Mb and Mt and hydroxymethylation in SkM.
Whole-genome, single-base resolution profiles of genomic 5hmC have been compared with methylomes.2,3,11,16,26 However, such studies are usually limited as to the number of types of samples and replicates analyzed due to the high cost of generating hydroxymethylomes with sufficient genome coverage for detecting infrequent 5hmC residues. In addition, it can be difficult to quantify both 5mC and 5hmC at a given CpG in these separately generated profiles. At a modest cost, the Epimark assay allows quantification of both 5hmC and 5mC levels at selected CCGG sites in a single assay on multiple types of samples with biological replicates. Several recent hydroxymethylome10 and methylome27 analyses of brain indicate the importance of studying biological replicates to account for individual-dependent variation in DNA cytosine modification.
In the current study using the Epimark assay, we focused in detail on a small set of genes in 14 different types of human samples to look for more evidence of the roles of DNA methylation (by which we mean just methylation of C residues in DNA) and hydroxymethylation (5hmC) in regulating gene expression. All the genes had myogenic DMRs previously identified by RRBS.23,24 We found that there is a significant association between 5mC enrichment at hypermethylated DMRs in Mb and Mt with 5hmC enrichment in SkM. We used bioinformatics to demonstrate associations between the tissue-specific DNA hypermethylation or hydroxymethylation and local chromatin structure and gene transcription. For DMRs of two of the genes, transfection assays and bioinformatics provided evidence for hypermethylation at cryptic intragenic promoters near the 3′ ends of the genes functioning to promote gene transcription from the canonical promoters.
Results
5-hydroxymethylcytosine levels at CpGs in skeletal muscle DMRs are highest in skeletal muscle, brain, and heart compared with other tissues
Using the Epimark assay,23 we quantified 5hmC and 5mC at 21 CpG sites in 16 human genes from nine types of normal tissue, four types of non-transformed cell cultures, and B-cell lymphoblastoid cell lines (LCLs; Tables S1–S4). Nineteen of the CpG sites that were chosen are located within SkM-lineage DMRs (Fig. 1, pink or blue labels for hypermethylated or hypomethylated, respectively) as previously determined by RRBS comparisons of 33 types of samples.23 The other two CpGs were in genes that had a SkM lineage DMR elsewhere in the gene's vicinity (black labels in Fig. 1). The Epimark assay involves incubating DNA with T4 β-glucosyltransferase, digestion with restriction endonucleases that distinguish between C(5hmC)GG, C(5mC)GG, and CCGG sequences, and quantitative PCR. Results from this assay indicated that the set of SkM, brain, and heart samples clustered together (Fig. 1a) and had significantly more 5hmC at these sites than did the set of kidney, lung, spleen, and placenta samples (P < 0.001, Tukey's HSD), as seen upon analysis of biological replicates of 14 types of samples. For 5mC, analogous hierarchical clustering grouped SkM with brain, but not with heart (Fig. 1b). Eight CpG sites in SkM, eight in brain, and four in heart had high 5hmC levels, with high being defined as >25% of total C. The relative enrichment for 5hmC among these sites does not simply reflect expression of TET1, TET2, and TET3 in the examined tissues, as monitored by RNA-seq (Table S5). Most of the SkM and brain sites with high 5hmC levels had more 5hmC than 5mC (Fig. 2). Caveats in the analysis are that the Epimark assay scores a hemi-hydroxymethylated CpG dyad as hydroxymethylated, that 5hmC can show strand bias,11 and that there was no scoring of allele-specific cytosine modification,28 but these caveats are unlikely to affect any of our main conclusions.
Figure 1.
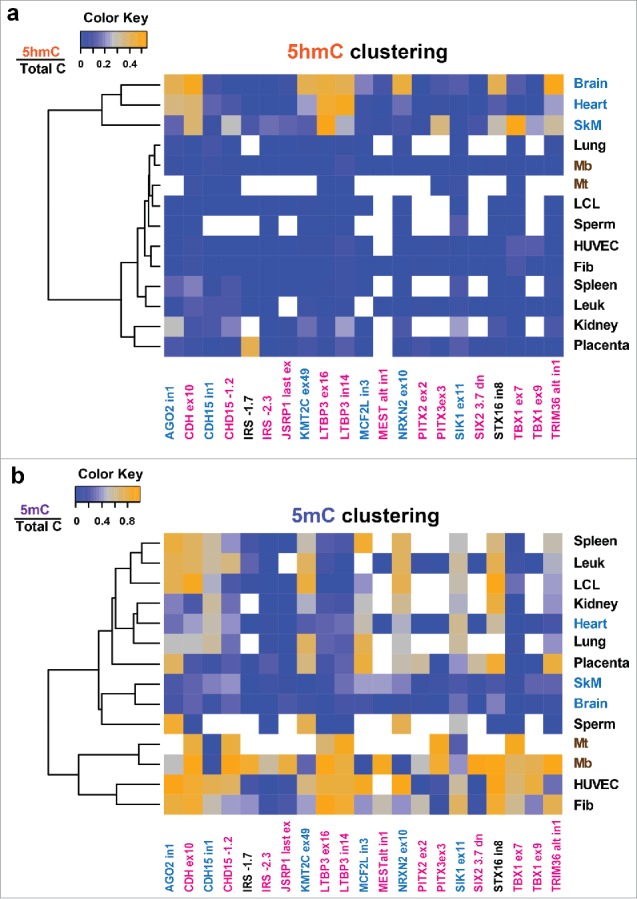
Skeletal muscle, brain, and heart group together upon hierarchical clustering of 5 hmC levels at the analyzed sites in diverse samples. Vectors of aggregate values of 5hmC (a) and 5mC levels (b) for each analyzed tissue at analyzed CpG sites (Tables S1–S4) were clustered as described in Methods. Average values for modified C/total C are shaded on the blue-orange scale with missing values shown in white. The names of the sites that were significantly hypermethylated or hypomethylated in myogenic progenitor cells (Mb and Mt) vs. 16 types of non-muscle cell cultures as previously determined by RRBS23,24 are shown in pink or blue, respectively. No RRBS data were available for the IRS1 −1.7 site; the STX16 in8 site was constitutively methylated in cell cultures but the STX16 gene had a Mb/Mt hypermethylated DMR upstream of the promoter; these sites are shown in black).
Figure 2.
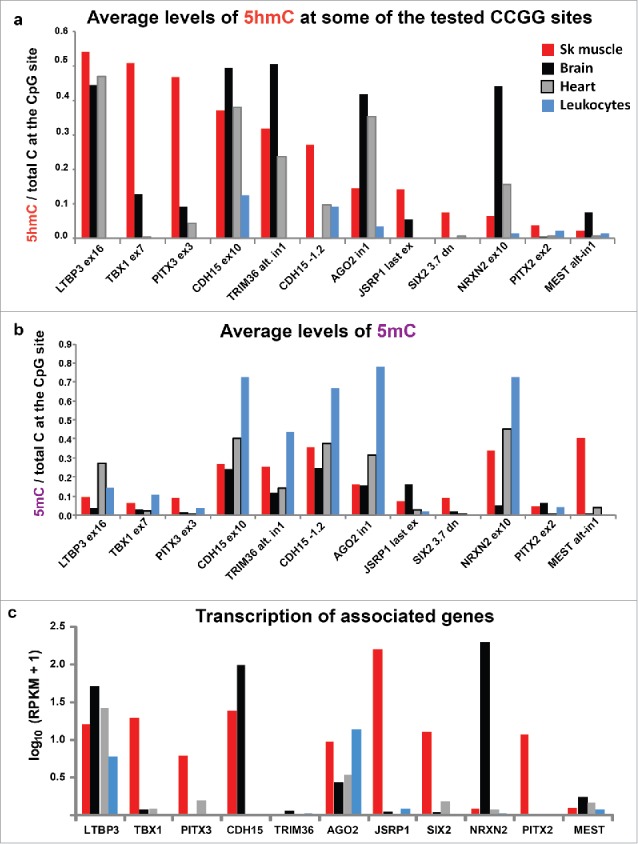
Average levels of 5hmC and 5mC at 12 of the 21 tested CCGG sites and RNA-seq RPKM values for the associated genes. (a) The average levels of 5hmC/total C at 12 of the 21 CCGG sites that were determined by Epimark assays on biological replicates for four of the 14 examined sample types. (b) The average levels of 5mC/total C from these assays. (c) RPKM (reads per kilobase per million mapped reads), median values from 430, 218, 125, and 393 samples of generic SkM, left ventricle, cerebellum, and whole blood samples (data for leukocytes are not available), respectively.30 RPKM values are shown on a log scale for 11 of the examined genes. Tables S3–S5 give the Epimark and RPKM data for all studied sites and samples.
The 5hmC levels of the placenta/kidney set were significantly higher than those of the leukocytes/lung/sperm set (P < 0.01; Tukey's HSD) although lower than those of the brain/SkM/heart set (P < 0.001). Three pairs of studied sites were located close to each other in the genome, TBX1 ex7/ex9, LTBP3 in14/ex16, and IRS1 −2.3 and −1.7 (Table S1). In SkM, brain, and heart, the two LTBP3 sites had similar enrichment in 5hmC to each other. This was also found for the two TBX1 sites (Fig. 1a). The similar results from these pairs of neighboring sites are consistent with findings that the distribution of 5hmC, like that of 5mC, is usually regional, rather than specific, for a given CpG site.16,18 However, we found that in the IRS1 upstream region in placental DNA, a high level of hydroxymethylation was observed at one site (13, 54, and 59% 5hmC from independent samples) but not at another that was only 0.6 kb away (0% 5hmC from the same three samples). This difference between 5hmC levels at neighboring sites in the placental genome might be related to the specific role that IRS1 plays in insulin signaling in placenta.29 The levels of 5hmC at the assayed sites in Mb, Mt, skin fibroblasts, LCLs, and human umbilical vein endothelial cells (HUVECs) were usually negligible (Fig. 1a). However, HUVECs had averages of 11 or 12% 5hmC at the two tested sites in TBX1. In summary, with regard to tissue- and cell-specificity of C modification at the tested sites, samples fell into three main groups as follows: SkM, brain, and heart, which had many high 5hmC levels; kidney and placenta, which had only occasional high 5hmC levels; spleen and leukocytes, lung, sperm, and all studied cell cultures, which displayed low to negligible 5hmC levels.
Enrichment in 5hmC in skeletal muscle correlates with enrichment in 5mC in myoblasts
The 13 sites that had >60% of total C as 5mC in Mb included seven sites with high 5hmC levels in SkM (Tables S3 and S4). Given these findings and the evidence that genomic 5hmC can sometimes be a long-lived intermediate in demethylation of 5mC residues.5 We looked for a relationship between 5mC in Mb and 5hmC in SkM. As predicted, we found a statistically significant correlation between high levels of 5mC in Mb and elevated 5hmC in SkM (Spearman's rho statistic = 0.56; P = 0.008). These results suggest a developmental relationship between DNA methylation in muscle progenitor cells and DNA hydroxymethylation in mature SkM fibers at biologically important regions.
5hmC at sites in some genes are associated with tissue-specific gene expression
By RNA-seq,30 PITX3, CDH15, SIX2, JSRP1, TBX1, and PITX2 were expressed specifically or preferentially in SkM compared with the other tissues, with the exception that CDH15 was expressed at higher levels in cerebellum (but not other parts of brain) than in SkM (Fig. 2c, Table S5). Cerebellum was the source of almost all brain samples in our study. Two other genes (NRXN2 and MCF2L), both of which are thought to encode large numbers of RNA isoforms,31 were preferentially expressed in brain (including cerebellum) compared with SkM and heart. Chromatin state segmentation profiles, which indicate active promoters and actively transcribed chromatin, confirmed the tissue-specific expression of these genes (e.g., Figs. 3–6). The other eight genes showed various expression patterns among the tissues.
Often, considerable 5hmC levels are found in the bodies of active genes in certain tissues,2,10,11 although exceptions have been noted.7 Seventeen of the 21 examined sites were in gene bodies. For the PITX3 ex3, CDH15 −1.2, JSRP1 last ex, TBX1 ex7, TBX1 ex9, LTBP3 ex16, LTBP3 in14, and NRXN2 ex10 sites in SkM and brain (cerebellum), there were correlations between DNA hydroxymethylation and gene expression (Fig. 2a and c; Table S3). For example, the exonic site in brain-associated NRXN2 gene31 had 44% of its C residues as 5hmC and 5% as 5mC in brain, while SkM had 7% 5hmC and 4% 5mC. Surprisingly for a gene strongly associated with neuronal cell signaling, NRXN2 displayed a myogenic promoter in the middle of the gene as indicated by RNA-seq, chromatin state segmentation, and profiling of 5′ caps of RNAs (CAGE; Fig. 5a and b). Upstream of this alternative promoter in NRXN2 were sequences orthologous to mouse Mb binding sites for the MyoD1, a myogenesis-specific transcription factor (TF; Fig. S1a). The Epimark-assayed NRXN2 site is located in this tissue-specific active promoter chromatin, which was seen in aorta as well as Mb and Mt. This alternative promoter is hypomethylated in these samples and in SkM but not in 14 other tissues (Fig. 5 and Fig. S1). A TAB-seq (Tet-assisted bisulfite sequencing) profile of genome-wide 5hmC levels at single-base resolution, which is publicly available for prefrontal cortex but not for the other tissues, revealed high levels of 5hmC throughout much of the NRXN2 gene (Fig. 5e).
Figure 5.
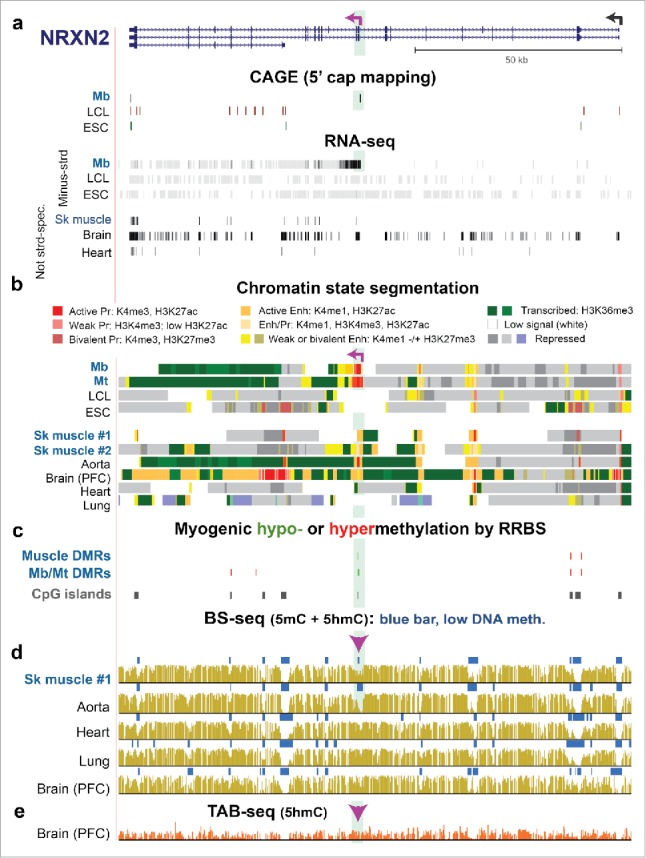
NRXN2, a neuronal gene, displays a Mb/Mt-specific alternative promoter whose DNA hypomethylation persists in SkM despite the loss of promoter activity. (a) RefSeq gene isoforms structures for NRXN2 (chr11:64,371,048–64,493,639) and RNA-seq as in Fig. 3 but also with the ENCODE profile of 5′ cap mapping (CAGE).37 Purple broken arrow on left, TSS for the Mb-associated transcript. (b) Chromatin state segmentation. (c) Significant hyper- or hypomethylated DMRs from 33 RRBS profiles.24 (d) and (e) Bisulfite-seq and TAB-seq. Highlighted green region, Mb/Mt-specific promoter region within NRXN2. Arrowhead, Epimark-tested site with high 5hmC in cerebellum (Fig. 2a).
Most of the genes examined in this study gave 5hmC levels in Epimark assays of brain DNA similar to those seen in the corresponding region from their brain TAB-seq hydroxymethylome profiles (Figs. 3 and 6; Figs. S2–S7). Six of the seven genes lacking appreciable levels of mRNA in cerebellum (TBX1, PITX3, JSRP1, SIX2, PITX2, and MEST) had generally low levels of 5hmC at the Epimark-assayed sites in that tissue. The exception was ubiquitin-protein ligation gene (TRIM36; Fig. S4), which displayed little or no expression in cerebellum, SkM, heart, even though these tissues had averages of 32, 24, and 51% 5hmC, respectively, at the tested TRIM36 site downstream of an alternate transcription start site (TSS). However, TRIM36 is expressed at moderate levels in parts of the brain other than cerebellum (Table S5). Therefore, high levels of 5hmC were seen at TRIM36 in SkM, heart, and cerebellum despite the gene's lack of transcription in those tissues. This contrasts with the moderate-to-high levels of 5hmC at studied sites in PITX3, CDH15, TBX1, JSRP1, LTBP3 and NRXN2 myogenic DMRs in SkM, heart, and/or brain, which positively correlated with expression of these genes.
Figure 3.
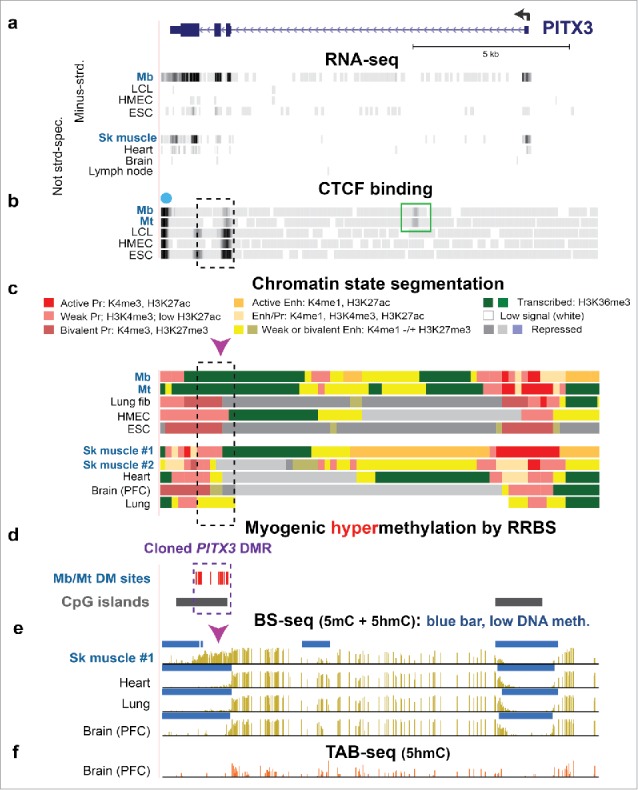
Intragenic Mb/Mt DNA hypermethylation, decreased CTCF binding, and loss of poised promoter chromatin in PITX3 correlated with gene expression in Mb and Mt. (a) RefSeq gene structure for PITX3, a developmental gene, at chr10:103,989,638–104,003,464 (all coordinates for figures are in hg19 and all tracks are aligned) and ENCODE RNA-seq data. The sequence-specific minus-strand RNA-seq profile is shown for cell cultures and the not strand-specific RNA-seq data for tissues.37 (b) CTCF binding from ENCODE data (dot, predicted insulator; green box, preferential Mb/Mt CTCF binding site. (c) Chromatin state segmentation from RoadMap data37 with the indicated color code; Pr, promoter; Enh, enhancer; Enh/Pr, both active promoter-type and enhancer-type histone modification; Repressed, enriched in H3K27me3 (weak, light gray; strong, dark gray) or H3K9me3 (violet). (d) Statistically significant hypermethylated sites as determined by RRBS for comparison of the set of Mb and Mt vs. 16 types of non-muscle cell cultures23 and CGIs from the UCSC Genome Browser.37 (e) Bisulfite-seq profiles37 with blue bars indicating regions with significantly lower methylation compared with most of the given genome.28,72 (f) TAB-seq profile of the distribution of 5hmC in the same prefrontal cortex (PFC) DNA sample from brain used for bisulfite-seq. Mb, myoblasts; LCL, GM12868 lymphoblastoid cell line; HMEC, human mammary epithelial cells; ESC, H1 embryonic stem cells; Sk muscle #1, psoas muscle; Sk muscle #2, unknown type of skeletal muscle; Lung fib, IMR-90, fetal lung fibroblast cell line; heart, left ventricle. Dashed box, cloned DMR sequences; arrowhead, Epimark-assayed CCGG, which had high 5hmC in SkM (Fig. 2a).
Figure 6.
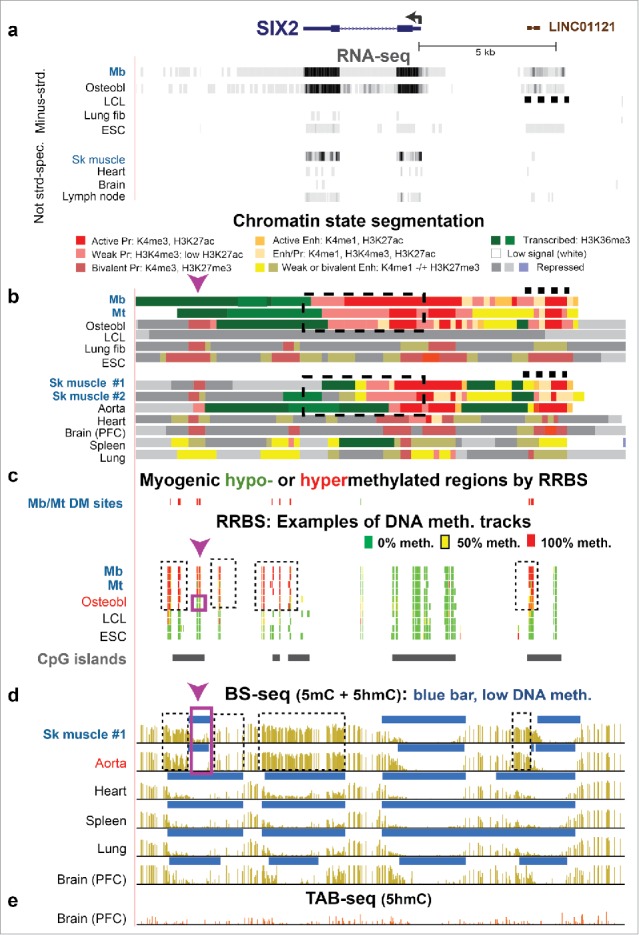
SIX2, a developmental TF gene, exhibits three hypermethylated DMRs in SkM and aorta that correlate with specific gene expression in these tissues. (a) SIX2, the LINC01121 ncRNA gene (dashed line) and RNA-seq for the minus-strand of cell cultures and non-strand-specific RNA-seq for tissues (chr2:45,224,940–45,243,926). (b) Chromatin state segmentation; dashed box, the SIX2 gene body for reference. (c) Significant hyper- or hypo-methylated CpG sites from RRBS profiles.23 (d) and (e) Bisulfite-seq and TAB-seq. Dashed boxes, Mb/Mt- and SkM-hypermethylated region; purple box, subregion in osteoblasts, SkM, and aorta lacking DNA hypermethylation observed in Mb and Mt. Arrowhead, the CCGG site analyzed for by Epimark assay and shown to have the highest 5hmC in SkM but only with an average of 7% of all C as 5hmC (Fig. 2a).
Site-specific 5hmC levels in cancers
Previous studies have found decreased 5hmC in cancer genomes compared with normal tissues.26 We determined 5hmC and 5mC levels at seven sites in DNAs from six ovarian carcinomas, five Wilms' tumors, a glioblastoma, a peritoneal metastasis from the uterus, and a mixed Mullerian cancer, and looked for correlations with our previously determined global DNA 5mC content for the examined DNAs.32 Because we identified CCGG sites with high average 5hmC levels among a set of normal tissues (SkM, brain, and heart), we wanted to test whether such sites might have high 5hmC contents in many cancers. The high 5hmC sites were LTBP3, CDH15, AGO2, NOTCH1, and TBX1, which we compared with low-5hmC sites IRS1 and ZNF556. Among the cancers, we found that the 5hmC levels were both site- and specimen-dependent, as were the 5mC levels (Table S6). Only two cancers (ovarian carcinoma P and the glioblastoma) displayed ≥ 25% 5hmC at any of the studied sites (both at the AGO2 in1 site) but 7 of the other 14 cancers had ≥ 10% 5hmC at one of the seven studied sites. The site in AGO2 (AGO2 in1) had significantly higher levels of 5hmC than the other six sites in the cancers (Tukey HSD, P < 0.01). We found no significant relationship between the total 5mC content of the cancer DNAs, which is often abnormally low in cancer,32 and the 5hmC levels at the seven CpG sites in normal tissues.
Enrichment in 5mC in some intragenic and intergenic DMRs is associated with gene expression
In five of the studied genes, the examined sites (LTBP3 ex16 and ex14, TBX1 ex7 and ex9, CDH15 ex10, PITX3 ex3, JSRP1 last ex and SIX2 dn) in Mb and Mt were 5mC-rich and located in CpG islands (CGIs) overlapping H3K36me3-enriched chromatin (regions of actively transcribed chromatin, txn-chromatin; Tables S3 and S4). One of these genes, JSRP1, had 5-fold higher expression in SkM than the next most highly expressed gene (Table S5). This gene, which encodes a SkM excitation-contraction coupling protein, displayed a DMR that, by RRBS, was significantly hypermethylated in the set of Mb and Mt samples (10 biological replicates) vs. 16 types of non-myogenic cell cultures and in SkM (two biological replicates) vs. 14 types of non-muscle tissue (Mb/Mt/SkM-hypermethylated DMR). The DMR extended from the 3′ half of the gene into the immediately adjacent testis-specific AMH gene (Fig. S2c and d, boxes). The Mb/Mt hypermethylation in this gene region and all the other gene regions examined in this study is likely to be due to 5mC and not 5hmC because little or no 5hmC was observed at all the tested hypermethylated DMR sites in Mb, Mt, and the other tested cell cultures, as described above. The SkM hypermethylation had both 5hmC and 5mC components (Fig. 2a and b).
SIX2 was of particular interest because it has one large expression-linked hypermethylated Mb/Mt DMR (Mb plus Mt samples vs. non-myogenic cell cultures) extending from the last exon to ∼4 kb downstream and another small one at ∼3.9 kb upstream of the TSS (Fig. 6c and d, dashed boxes). This gene encodes a TF involved in various aspects of development, including that of SkM, limb, eye, and kidney31 and has only a single RefSeq or ENSEMBL transcript.31 It is expressed in SkM but at much lower levels than in myogenic progenitor cells (Table S4). SkM has similar hypermethylated DMRs to those of Mb and Mt except that the downstream DMR does not overlap H3K36me3-enriched chromatin and a central 0.8-kb subregion is lacking DNA methylation or hydroxymethylation (Fig. 6b, c, and d, purple box), as verified by Epimark assay (Table S4). Remarkably, aorta and osteoblasts exhibited almost identical DMRs to those of SkM in downstream, upstream, and intragenic regions of SIX2 and also specifically express this gene (Fig. 6a–d; Table S5). The 0.8-kb hole in the hypermethylated DMR downstream of SIX2 in SkM, osteoblasts, and aorta overlapped weak or bivalent promoter chromatin that was not seen in Mb and Mt, where this region was uniquely hypermethylated. Thirteen other tissue types (including heart, bladder, small intestine, and esophagus) and four other cell culture types that lack the downstream, intragenic, and upstream DMRs display little or no expression of this gene (e.g., Fig. 6). Over most of the gene body, Mb, Mt, and osteoblasts had promoter-type chromatin, as previously observed for some active genes.33 The upstream Mb/Mt/SkM DMR, which was also hypermethylated in osteoblasts and aorta, overlaps a long non-coding RNA (lncRNA) gene, LINC01121, which has not been discussed in the literature. LINC01121 exhibited promoter- or enhancer-type chromatin and evidence of expression (Fig. 6a and b, dashed lines) specifically in the SIX2-expressing cells.
Like SIX2 in SkM and aorta, PITX2 in SkM and bladder has expression-linked hypermethylated DMRs upstream, within the gene, and downstream and PITX2's downstream DMR has a central subregion that is mostly unmethylated in SkM but hypermethylated in Mb and Mt (Fig. S5). Other similarities between SIX2 and PITX2 are that they both encode a homeobox TF, are involved in myogenesis,34 and are more highly expressed in Mb and Mt than in SkM (Fig. 6a and b; Table S4). SkM and bladder are the only tissues among those examined that specifically express PITX2 at appreciable levels (Table S5). This gene has three alternative promoter regions, the most distal of which harbors a Mb/Mt hypermethylated DMR, as detected by RRBS. Chromatin state segmentation profiles for Mb, Mt, and SkM and RNA-seq exon analysis for SkM and bladder30 indicate that the hypermethylated distal promoter in these samples is silenced while one or both of the two proximal promoters, which are constitutively unmethylated, are active. Chromatin state profiles also show that the DNA hypermethylation in Mb and Mt overlaps the 3′ end of the most proximal active-promoter region, and that the downstream hypermethylated DMR in SkM does not overlap H3K36me3-enriched chromatin as it does in Mb and Mt.
Mesoderm specific transcript (MEST) gene and MCF2L (a brain-associated G-protein signaling gene), like the previously mentioned NRXN2 (Fig. 5), had myogenic DMRs at tissue-specific alternative promoters (Figs. S5 and S6). In Mb, these genes exhibited hypermethylation-associated promoter silencing (MEST, distal, non-imprinted promoter35) or hypomethylation-linked promoter activation (MCF2L and NRXN2). Surprisingly, in SkM, where these three genes show low or no expression, they retained much of the myogenic DNA hypomethylation or hypermethylation seen in Mb and Mt, where they are highly transcribed.
LTBP3 is a broadly expressed gene encoding a signaling protein that regulates TGFβ proteins and has many functions, including in bone development and in the growth of mature SkM.31,36 In its gene body, RRBS revealed a Mb/Mt hypermethylated DMR, which was also hypermethylated in osteoblasts (Fig. S7c). DNA hypermethylation or high 5hmC levels at the DMR (Mb, Mt, osteoblasts, SkM, heart, and brain) were associated with the absence of promoter-type chromatin that was observed in samples with very little modification of CpGs in this region. In lymphoid lineage samples and in lung and liver, this DMR was mostly unmethylated and overlapped active promoter chromatin. These findings suggest that 5mC or 5hmC enrichment at this DMR located in the gene body of the LTBP3 RefSeq isoforms silences an alternative promoter for a gene isoform that is not in the RefSeq collection but is a provisional isoform in the set of UCSC Genes (Fig. S7a).37 In support of this interpretation, lymphoid cells had CAGE signal or RNA-seq exonic signal corresponding to use of this tissue-specific alternative promoter for transcription initiation. In summary, many of the studied genes (e.g., LTBP3, PITX2, MEST, SIX2, and TBX1) displayed hypermethylated DMRs in their vicinity in SkM lineage samples and sometimes also in a few non-SkM lineage samples that correlated negatively or positively with overlapping or adjacent tissue-specific promoter chromatin.
PITX3 and CDH15 contain intragenic myogenic hypermethylated DMRs associated with gene expression
Epimark assays on the highly tissue-specific CDH15 and PITX3 genes revealed that the assayed sites in their intragenic hypermethylated DMRs had much more 5hmC than 5mC in SkM (Fig. 2a and b, CDH15 ex10 and PITX2 ex3 sites), while in Mb and Mt there was only 5mC at these sites (Tables S3 and S4). CDH15 encodes a muscle- and cerebellum-specific cadherin. PITX3 encodes a homeobox TF that is involved in development of SkM, lens, and the substantia nigra.31,38,39 These genes are associated with SkM regeneration.34,39 They have much sequence conservation between humans and mice, including at these DMRs, which are near their 3′ ends (Figs. S8a and S9a). CDH15 also has a hypomethylated DMR in intron 1, which overlaps enhancer and promoter-type chromatin and myogenic DNaseI hypersensitivity sites (DHS; Figs. 4). In addition, a small myogenic hypermethylated DMR 1.2 kb upstream of the CDH15 TSS correlates with expression of the gene. By Epimark assay the upstream CDH15 −1.2 site was rich in 5mC in Mb and Mt with partial replacement of 5mC by 5hmC in SkM, the only sample with high 5hmC at this CpG (Fig. 2). This site is at the edge of low methylated region (LMR; Fig. 4e, lollipop) and may represent a border where SkM DNA hydroxymethylation is helping to keep the promoter-overlapping LMR from accumulating 5mC.17,40,41 Promoter-upstream enrichment in 5mC or 5hmC has been noted previously to be sometimes positively associated with gene expression.17,42
Figure 4.
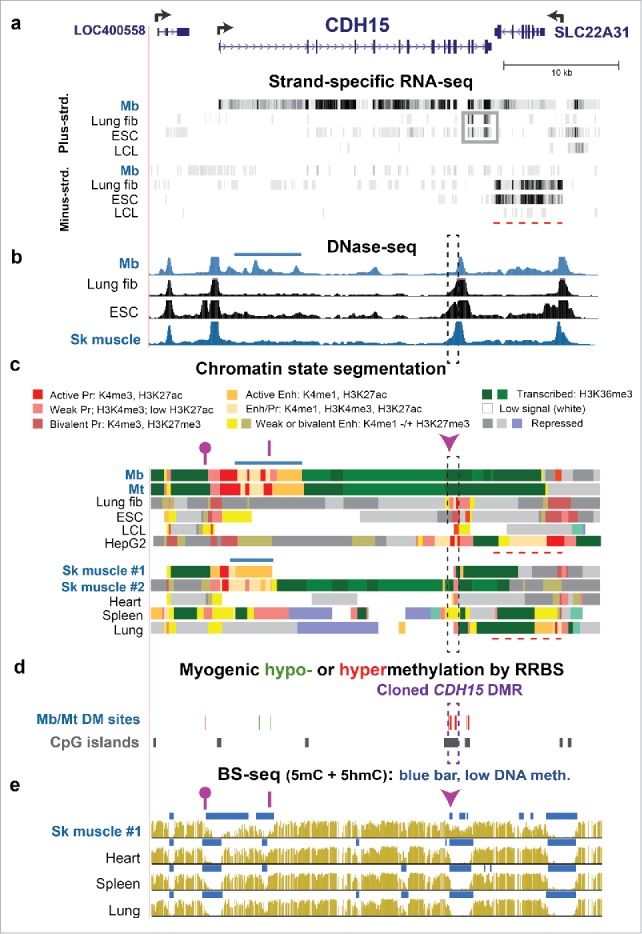
Both hypermethylated and hypomethylated DMRs within the gene body of CDH15 are associated with gene expression. (a) RefSeq structure for CDH15 and adjacent genes (chr16:89,232,229–89,271,355) and RNA-seq data for cell cultures as in Fig. 1. Gray box, an apparent ncRNA seen in lung fibroblasts and ESC; red dashed line, the region of the SLC22A31 transcript. (b) DNase-seq mapping of DNaseI hypersensitive sites. Dashed rectangle CDH15 3′DMR sequence that was cloned and used for reporter gene assays; horizontal blue bar, epigenetic marks at the myogenic enhancer-like region. (c) Chromatin state segmentation. Note 0.2- and 0.4-kb subregions of active promoter chromatin in the cloned region of lung fibroblasts and HepG2 cells, respectively as compared with poised promoter chromatin in the other non-myogenic samples. (d) Significant hyper- or hypomethylated sites from RRBS.23 (e) Bisulfite-seq. Dashed box, cloned DMR sequences. Epimark-tested CCGG sites: arrowhead, site which had high 5 hmC in SkM, heart, and cerebellum; purple line, site with negligible 5 hmC in all tested samples; lollipop, site with high 5 hmC only in SkM (Fig. 2a).
We focused on the PITX3 and CDH15 3′ intragenic DMRs, which are positively associated with expression of these genes in Mb and Mt. The DMRs are located in txn-chromatin in Mb and Mt, where they were very hypermethylated (Figs. 3 and 4, dashed boxes; Figs. S8e and S9c). In non-myogenic cells, these DNA sequences were largely unmethylated and partially overlapped promoter-type chromatin (weak, bivalent, or active promoter chromatin). At the 3′ end of CDH15, fetal lung fibroblasts (IMR-90) and ESC samples displayed a truncated RNA that might have been generated from mixtures of weak, bivalent, and active promoter chromatin at the DMR (Fig. 4a, gray box, and c). The presence of this truncated RNA correlated with transcription of the adjacent gene, SLC22A31 (Fig. 4a, dashed line), which is a little-studied transporter-like gene that is transcribed preferentially in lung.30 These results suggest that the CDH15 DMR can act as a tissue-specific promoter for generation of a transcription-regulatory ncRNA that might promote the transcription of SLC22A31 in lung, but that could downregulate the transcription of CDH15 in the SkM lineage if unmethylated.
The PITX3 and CDH15 intragenic DMRs are methylation-sensitive cryptic promoters
To directly test the cis-acting gene-regulatory activity of the above-described intragenic DMRs, we cloned a 1-kb sequence containing the whole PITX3 DMR and a 0.9-kb sequence containing the half of the CDH15 DMR that did not overlap a constitutive DHS (Figs. 3d and 4d). To examine promoter activity, the cloned sequences were inserted in both directions upstream of a promoter-less, CpG-free luciferase reporter vector. Enhancer activity was examined by inserting the DMRs downstream of the luciferase gene in an analogous CpG-free minimal promoter-containing vector. Upon transfection into mouse Mb (C2C12), the CDH15 DMR and PITX3 DMR constructs for testing promoter activity exhibited >500-fold and ∼7-fold higher luciferase activity, respectively, than did the vector (Vector No-Pr), whereas the DMR had been inserted in the sense or the antisense configuration (CDH15 S and CDH15 AS; Fig. 7a and b). In assays for enhancer activity, the CDH15 DMR gave more luciferase activity than the vector (CDH15 dn vs. Vector Min-Pr; Fig. 7a) but only about one-tenth that of the analogous promoter-test constructs. Parallel transfections into the breast cancer cell line MCF-7 revealed less promoter and enhancer activity for the DMRs than when the host cells were Mb (Fig. 7, note different scales in panels a and b vs. c and d). CpG methylation by M.SssI targeted just to the DMRs essentially silenced their promoter and enhancer activity (Fig. 7).
Figure 7.
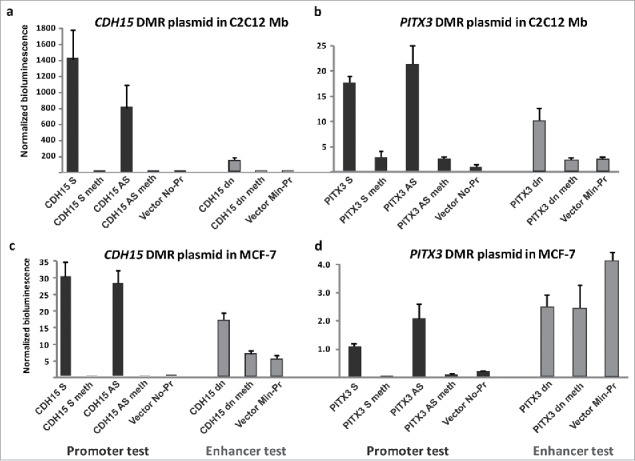
Transient transfection reveals promoter activity in the intragenic PITX3 DMR and exceptionally high promoter activity in the hypermethylated intragenic CDH15 DMR but only when unmethylated. Reference plasmid-normalized reporter gene expression upon transient transfection of promoter- or enhancer-test constructs containing DMRs cloned upstream in the sense or antisense orientation (S or A) or downstream (dn) of the reporter gene. (a) CDH15 DMR and (b) PITX3 DMR transfected into C2C12 Mb. (c) CDH15 DMR and (d) PITX3 DMR transfected into the MCF-7 breast cancer cell line. The data shown are normalized averages from 2–5 independent transfection experiments (including technical replicates) with the standard error indicated. The location of the sequences that were cloned is shown in Figs. 3 and 4 (dashed boxes). Meth, the construct was methylated with M.SssI; if the construct is not designated as “meth,” it was mock-methylated. Note the different scales for bioluminescence in the panels.
Given their strong promoter activity, we examined the cloned regions for promoter motifs. These sequences from PITX3 (exon 2 – exon 4) and CDH15 (exons 10 – 11) DMRs have low-complexity CG-rich repeats that include potential G-quadruplex sequences (PQS), which are overrepresented in promoters.43 One of the G-rich repeats (on the antisense strand) is in the CDH15 DMR and is predicted to be able to form a very stable G-quadruplex when single-stranded (5′-GGGGGTGGGGGGAGCGTGGGGATGGGGGG-3′).44 It is only 65 bp from another strong PQS motif (5′-GGGTGGGGATCCCGAGATCCTGGGCCTGGG-3′). The PITX3 DMR had one strong PQS motif (5′-GGGTC CGGGGTCCGGGGTCCGAGGG-3′; on the antisense strand). Weaker PQS in both the CDH15 and PITX3 DMRs are predicted to overlap a binding site for the TF MAZ, which binds preferentially to PQS (Table S7). With respect to TF binding to these DMRs, SkM-lineage ChIP-seq profiles (Mb and Mt) are available only for CTCF (the CCCTC binding factor, a TF and architectural protein) and EZH2 (a subunit of an H3K27 methyltransferase). These profiles showed that EZH2 bound to the PITX3 DMR in many non-muscle cell types but not to the CDH15 DMR in either myogenic or non-muscle cells. CTCF bound very strongly to the 1-kb PITX3 DMR in many non-myogenic cell types, but weakly in Mb and Mt, and did not bind to the CDH15 DMR in any cell type (Fig. 3b). Two subunits of cohesin, RAD21 and SMC3, which are architectural proteins that often colocalize with CTCF,45 bound to the same subregions of the PITX3 DMR as did CTCF in several tested non-myogenic cell types. Mb and Mt displayed an additional, cell-specific intragenic CTCF site in intron 1 (Fig. 3b, green box). A third CTCF site was seen in all cell types immediately downstream of the gene (Fig. 3b, dot) and is predicted to be an insulator from chromatin state segmentation analysis (data not shown37,46).
From the limited number of available TF ChIP-seq profiles for cell types of interest, we found that many TFs bind to the cloned DMR sequences of CDH15 and PITX3 in non-myogenic cells (Table S7), where these DMRs are mostly unmethylated. ZBTB7A and REST/NRFL (mostly transcription repressors) and ZNF263 (repressor or activator) exhibited moderate-to-strong binding to both DMRs in non-myogenic cell types. BHLHE40 and MXI1 (repressors), ELF1, YY1, MYC, the MYC-heterodimerizing MAX polypeptide, and the MAX-heterodimerizing MXI1 polypeptide (activators or repressors) and MAZ (a transcription initiation and elongation regulator) bind to CDH15 and/or PITX3 DMR sequences in non-myogenic cell cultures. Many of the consensus binding sites identified for these TFs in the studied DMR sequences contain CpGs whose myogenic hypermethylation might inhibit binding in Mb and Mt. In addition, histone modifiers (repression-associated EZH2, SIN3A and HDAC1; activation-associated PHF8 and RBBP5) were found to bind to the CDH15 or PITX3 DMRs in non-myogenic cells. Strong binding of the RNA polymerase II (RNAP II) subunit POLR2A and the TFIID subunit TAF1 was observed at the CDH15 DMR in HepG2, the cell type that displayed active promoter chromatin at this DMR (Fig. 4c). CAGE profiling of HepG2 did not reveal a corresponding 5′-capped RNA; however, 3′ terminal exons of CDH15 were found to be overrepresented in RNA-seq profiles (Fig. S8g). CAGE profiles of Mb and lung fibroblasts did give evidence of a weak transcription start site for a capped RNA near the DMR (Fig. S8h). This result for Mb is surprising because there was >90% 5mC by RRBS and Epimark assay at the DMR in Mb and might not be due to expression in Mb, but rather to expression of large amounts of fibroblasts contaminating the Mb cultures, which is frequent in commercial preparations47 like those used for RNA-seq. For our RRBS and Epimark analyses,23 specially generated, propagated, and characterized Mb and Mt samples were used to avoid this problem (see Methods). In conclusion, these findings are consistent with both CDH15 and PITX3 containing a DMR overlapping an intragenic cryptic promoter that is silenced by 5mC enrichment in Mb and Mt, thereby facilitating expression of the whole gene.
Discussion
Our study of 16 myogenic DMR-containing genes revealed highly tissue-specific DNA methylation and hydroxymethylation, including at alternative or cryptic promoters. We also found a significant correlation between genomic 5mC loss and 5hmC gain with SkM development at the 21 CpG sites quantified for 5hmC and 5mC within or near these genes. The association of high 5mC levels in Mb DNA with enrichment in 5hmC and unmodified C in SkM DNA in these genes, most of which encode TFs or signaling proteins, might reflect a development-linked active demethylation pathway involving 5hmC as an intermediate.5 Consistent with these results, we previously showed that DNA from SkM retains about 30% of the RRBS-determined hypomethylated sites observed in Mb and Mt but only 3% of the Mb- and Mt-hypermethylated sites.23 In DNA epigenetic studies that use DNA methylation analysis methods that do not distinguish 5mC and 5hmC, the deduced cytosine modification may actually reflect the presence of both 5hmC and 5mC. This is especially pertinent in studies of brain,48 but also of other tissues. The importance of addressing whether DNA methylation detected by standard techniques is actually mostly 5mC is illustrated by our study. For example, SkM had 54% of total C as 5hmC and ∼5 times more 5hmC than 5mC at an examined CpG site in a myogenic hypermethylated DMR that doubled as a novel lymphoid-associated promoter within LTBP3, the TGFB-regulatory gene. In contrast, cell cultures (Mb, Mt, LCLs, skin fibroblasts, and HUVEC) almost always had little or no 5hmC at sites examined in this study or in CpG sites analyzed in a small number of genes in our previous studies.23,25,49 The exceptions were intragenic TBX1 sites in HUVEC (Table S3) and a MYOD1 enhancer site in Mb and Mt.47 An explanation for the low 5hmC levels in cell cultures and cancers (Table S6) and for the lower levels of 5hmC in spleen, kidney, and lung relative to brain, SkM, and heart (Fig. 1) is that high cell-turnover50 can contribute to low 5hmC levels.7 Cell division favors the loss of genomic 5hmC because this base is recognized less well than 5mC by the enzyme complexes that maintain DNA methylation upon cell division.51
Our analysis of hypermethylated DMRs in Mb and Mt provides evidence for differential methylation at alternative promoters playing an important role in controlling their genes' expression. Five of the 16 genes we examined displayed DMRs in known alternative promoter regions (TRIM36, MCF2L, TBX1, PITX2, and MEST) and four in novel alternative or cryptic promoter regions (intragenic regions in NRXN2, LTBP3, PITX3, and CDH15) in Mb and Mt relative to many non-myogenic samples. Except for TRIM36, there was an association between low DNA methylation and weak or active promoter-type chromatin at the DMR. DNA methylation may provide more stable and spatially restricted promoter repression than H3K27me3- or H3K9me3-linked promoter silencing, and thereby prevent inadvertent upregulation of these genes by a nearby active transcription-initiation complex.
NRXN2 and MCF2L, neuronal genes implicated in normal cognition and linked to autism,52,53 generated Mb/Mt-associated isoforms from intragenic Mb/Mt-hypomethylated promoters (Fig. 5 and Fig. S6). Hypomethylation at these DMRs persisted in SkM even though this tissue displays little or no detectable expression of the numerous isoforms31 of these genes. Low, but appreciable, levels of 5hmC in the DMRs in SkM are compatible with TET-catalyzed hydroxymethylation helping to maintain DNA hypomethylation by replication-independent DNA demethylation pathways40,41 in this predominantly post-mitotic tissue. We propose that the hypomethylated state of these DMRs/alternative promoters in SkM allows myogenic isoforms of these signaling genes to be poised for activation in response to local physiological changes that necessitate gene upregulation. The DMR/alternative promoter within the MCF2L gene displayed active promoter chromatin in brain, where it was similarly hypomethylated. Therefore, there is probably also a function in the nervous system for this weakly documented gene isoform. Similarly, in the middle of the NRXN2 gene, the highly specific DNA-hypomethylated active promoter chromatin in aorta (but not 14 other tissues) and in Mb and Mt suggests shared functions for a previously undescribed, tissue-specific isoform in these samples. In brain, where documented NRXN2 isoforms are highly expressed, there was much genomic 5hmC throughout NRXN2 as seen in an available TAB-seq profile (Fig. 5e). At the intragenic CpG site that we tested, brain samples had 44% 5hmC and 5% C, which probably reflects the generally observed association of 5hmC with transcriptionally active gene bodies in nerve cells.54 Conversion of gene-body 5mC to 5hmC upon gene activation during nerve development has been correlated with increased intragenic H3K36me354 although gene-body 5mC may also be associated with H3K36me3.55 Intragenic H3K36me3 can increase the efficiency of transcription56 and at exons or exon/intron junctions can help regulate alternative splicing of pre-mRNA.55,57 However, the DNA hypermethylation immediately downstream of the 3′ end of SIX2 and PITX2 in H3K36me3-rich regions in Mb and Mt persisted in SkM while the local H3K36me3 did not (Fig. 6 and Fig. S5). This result indicates that DNA hypermethylation was dissociated from H3K36me3 enrichment at the SkM stage with the DNA hypermethylation possibly serving as a kind of epigenetic memory for these developmental TF genes.
It has been controversial whether gene-body DNA methylation (or hydroxymethylation, from which it is usually not experimentally distinguished), facilitates transcription elongation in certain contexts of DNA sequence, chromatin structure, and cell type48,58 or is just a consequence of H3K36 trimethylation linked to RNAPII movement through the gene body.14,59 To examine this question, we focused on the transcription-associated Mb/Mt-hypermethylated DMRs near the 3′ ends of PITX3 and CDH15, genes involved in myogenesis and neurogenesis.31,60 Because there is little or no alternative splicing of the PITX3 and CDH15 pre-mRNA (Figs. 3 and 430), the effects of myogenic DNA hypermethylation spanning several exons and introns should be on transcription per se. The bidirectional promoter activity of these intragenic CGI/DMRs in transfection assays and the silencing of that promoter activity by in vitro CpG methylation (Fig. 7) are likely to be related to the positive association in vivo between gene expression from the canonical promoter and hypermethylation of these intragenic DMRs. While the cloned DMRs also displayed enhancer activity, their promoter activity was greater and for the CDH15 DMR about 10-fold greater. At their endogenous chromosomal sites in Mb and Mt, where these DMRs are very highly methylated, they are embedded in H3K36me3-rich chromatin and do not display promoter-like or enhancer-like histone modifications. In contrast, in normal non-muscle cell cultures, where these DMRs are predominantly unmethylated, they exhibit mostly poised promoter chromatin. We conclude that these PITX3 and CDH15 DMRs are cryptic promoters.
Intragenic cryptic promoter silencing by Mb/Mt DNA methylation may be more important for expression of CDH15 than for PITX3 expression because the transfection-monitored promoter activity of the cloned PITX3 intragenic DMR was <5% that of the CDH15 intragenic DMR in myogenic or non-myogenic cells. Moreover, only the PITX3 DMR at its endogenous chromosomal location displayed strong CTCF binding in vivo in non-myogenic cells, which suggests an additional mechanism for PITX3 DMR methylation favoring transcription of this gene. Local DNA methylation can decrease CTCF binding (as well as the binding of many other TFs, directly or indirectly61), especially when the binding site has a CpG in it,57 as is the case for the PITX3 DMR (Table S7). Accordingly, there was a large decrease in CTCF binding to the PITX3 DMR in myogenic vs. non-myogenic cell cultures. Although CTCF can act as a part of an insulator,62 it is unlikely that the intragenic PITX3 DMR functions as a methylation-sensitive insulator because CTCF binds constitutively to a nearby gene-downstream site that is more likely to be an insulator (Fig. 3b, dot). However, CTCF can be an architectural protein without functioning as an insulator, as it may do at the PITX3 DMR in conjunction with SMC3, RAD21, and ZNF143, which are CTCF-associated proteins63 that can bind to the unmethylated DMR in non-myogenic cells (Table S7). At the CDH15 DMR, a variety of repressor and activator TFs bind in non-myogenic cells, and some are known to complex with one another.63 One of these is the transcription-pausing protein MAZ, which can bind to both the PITX3 and CDH15 DMRs in non-myogenic cells. These results support the conclusion that the Mb/Mt CpG methylation at these DMRs counteracts the formation of inhibitory chromatin structures that would decrease PITX3 and CDH15 transcription elongation or initiation. In addition, DNA methylation as well as transcription-related H3K36me3 deposition64 might be necessary to suppress the cryptic promoter activity of the PITX3 and CDH15 intragenic DMRs to prevent generation of inhibitory ncRNAs. Such ncRNA transcripts originating from the potentially strong CDH15 cryptic promoter may be needed, particularly at early stages in development. The evidence for this is that Proudhon et al.65 found that mice have orthologous DNA sequences to the human CDH15 cryptic promoter at an imprinted maternal germline DMR, which they found was apparently not imprinted in humans. They inferred that this cryptic promoter directs formation of a paternal-specific ncRNA that they detected in neonatal mouse brain.
A major question raised by our finding is why the intragenic cryptic promoter activity of the CDH15 and PITX3 DMRs is stronger in myogenic than in non-myogenic cells, as we observed in transfection assays. In SkM, these DMRs exhibit weak-promoter chromatin and considerable amounts of unmethylated CpG and 5hmCpG, unlike in Mb and Mt. One SkM-associated function might be to down-modulate expression at the tissue stage vs. the progenitor cell stage. The steady-state levels of CDH15 RNA in SkM are almost 10-fold lower than in Mb, where the 5mC levels at the DMR are >90%. In addition, methylation/hydroxymethylation/demethylation might allow dynamic modulation of CDH15 transcription in SkM in response to physiological changes. Changes in DNA methylation in association with altered gene expression have been implicated in normal muscle physiology and response to muscle damage.66,67 Candidate proteins for mediating such a response include BHLHE40, a repressor, which can bind specifically to a CpG-containing site in this DMR (Table S7) and has been implicated in controlling metabolic pathways in response to exercise in SkM.68 In addition, REST, which is usually a transcription repressor, can bind to the DMRs of both genes, and REST is associated with chromatin plasticity, differential binding to methylated DNA, and recruitment of active TET3.54,69 Decreased local binding of a TF like REST could result in more 5mC at the intragenic DMR/cryptic promoters through unopposed endogenous DNMT activity. This, in turn, could lead to suppression of the proposed transcription down-modulatory activity of these DMRs in normal SkM. Dynamic regulation of CDH15 and PITX3 expression in SkM, including by epigenetic changes at the intragenic cryptic promoter/DMRs, is likely to be needed in order for muscle cells or tissue to rapidly respond to damage, stress, or exercise by upregulating these genes.38,70,71 Hence, we suggest that differential DNA methylation and hydroxymethylation at these DMRs participates in an intricate fine-tuning of tissue-specific gene expression.
Methods
Quantification of 5hmC and 5mC
To quantify 5hmC and 5mC, we used the Epimark assay (New England Biolabs23), which involved incubation of the DNA samples (Table S2) with T4 phage β-glucosyltransferase to glucosylate only 5hmC residues followed by cleavage at CCGG sites by restriction endonucleases (MspI, HpaII, or no digestion). Quantitative PCR (six reactions per sample) was then done to determine the amounts of cleaved and of uncleaved amplicons containing a given CCGG in the reference genome with subtraction of the resulting Ct values, according to the manufacturer's instructions.23 The primers used for PCR had been tested for their quantitative response and specificity by using a series of 2-fold dilutions of HeLa DNA for preliminary PCRs and by checking for single peaks in the melting curves of the products. For the sample analyses, biological replicates (usually three) of SkM, eight other tissues, adult biopsy-derived myogenic progenitor cells (Mb and Mt), and three types of non-myogenic cell cultures were examined. The Mb (∼70% confluent) and Mt (3 – 5 d after serum deprivation) samples used for the Epimark assays as well as for RRBS were derived from adult muscle biopsy tissues and checked by immunocytochemistry to assure that they contained >90% of the nuclei in desmin-positive cells for the Mb cultures and >70% of the nuclei in multinucleated cells for the Mt cultures, as previously described.23 The tissue samples came from non-cancer individuals unless otherwise specified and all samples used for Epimark assay were different from those used for RRBS.
Bioinformatics
Databases with epigenetic and RNA-seq profiles used in the figures are available at the UCSC Genome Browser.37 The bisulfite-seq profiles examined were from the Methylomes from Bisulfite Sequencing Data hub72 at the UCSC Genome Browser with data analysis by Song et al.28 The fifteen types of tissues compared for their bisulfite-seq profiles are illustrated in Fig. 5 and Fig. S2. The chromatin state segmentation (chromHMM, AuxilliaryHMM)72,73 was from a hub for Roadmap Epigenomics Project with the color code for chromatin state segmentation slightly simplified from the original,73 as indicated in the figures. The same psoas SkM sample (SkM #1) had been used for chromatin state segmentation and bisulfite-seq, namely, an unspecified mixture of tissues from a 3 y male and a 34 y male.72 A second SkM sample (SkM #2) had been used just for chromatin state segmentation (one 72 y female; the type of SkM tissue unidentified).72 From the ENCODE project74 we used the following UCSC Genome Browser tracks: DNaseI hypersensitivity profiling, Open Chromatin, DNaseI HS, Duke University;75 RNA-seq (for tissues; not strand-specific), Massachusetts Institute of Technology;76 Transcription Levels by Long RNA-seq for poly(A)+ whole-cell RNA by strand-specific analysis on > 200 nt poly(A)+ RNA (for various cell cultures), Cold Spring Harbor Laboratories and RNA Subcellular CAGE Localization, RIKEN Omics Science Center. For DNaseI-hypersensitivity profiling, the SkM sample was a mixture of psoas muscle from five individuals (male and one female) aged 22–35.75 The RRBS profiles were also from ENCODE and the 33 samples used for RRBS-determined myogenic differential methylation were previously described;23 cell cultures were untransformed cell strains except for the LCLs. Two SkM samples for RRBS (unspecified as to body location; technical duplicates analyzed) were from a 71 y male and 83 y female. For visualizing RNA-seq tracks in the UCSC Genome Browser in figures, the vertical viewing ranges were 0 to 30 for cultured cells and 0 to 2 for tissues, with the exceptions of PITX3 for tissues, 0–1, and CDH15 and SIX2 for cell cultures, 0–50 and 0–100, respectively. For identification of potential MYOD binding sites, orthologous sequences to murine C2C12 Mb and Mt binding sites from MyoD ChIP-seq77 were mapped in the human genome. TF binding sites were from Transcription Factor Binding Sites by ChIP-seq from ENCODE.78 For quantification of RNA-seq from tissues, the GTex database involving large numbers of pooled samples were used;30 the 430 SkM tissues examined had not been classified as to type, age, or gender.
Statistical analysis
Analyses were performed using R version 3.2.4. Aggregate values for 5hmC and 5mC percentages at each site and tissue type were determined using fitted values from logistic two-way ANOVA models. Hierarchical clustering was performed on these aggregate values using Euclidian distance and the complete linkage algorithm. To determine the significance of 5hmC and 5mC percentages associated with tested sites, logistic two-way ANOVA models were fit as a function of site and sample to the full data set and pairwise comparisons with adjustment for multiple testing were performed using the Tukey Honest Significant Difference (HSD). Rank-based correlations between logit-transformed 5mC and 5hmC values were computed using Spearman's rho.
Preparation of constructs and transfection
Cloning into the plasmid vectors pCpGfree-Lucia (no promoter) and pCpGfree-promoter-Lucia (minimal EEF1A1 promoter engineered to be free of CpGs like the rest of the vector; InvivoGen) was by fusion PCR (NEBuilder HiFi Assembly kit, New England Biolabs). Fragments from the CDH15 (chr16:89,258,029–89,258,950, hg19) or the PITX3 DMR (chr10:103,990,812–103,991,824) were obtained by PCR of human DNA using primers containing extensions for fusion PCR cloning. The vectors were first linearized by reverse PCR using primers that overlapped the multicloning region for upstream insertion in the sense or antisense direction into the promoterless vector (promoter test) or for insertion downstream of the minimal promoter/reporter gene's polyadenylation site (enhancer test). Constructs were checked by restriction digestion and by DNA sequencing. Methylation involved treatment with SssI CpG methyltransferase (M.SssI; New England Biolabs) for 4 h under conditions recommended by the manufacturer. The constructs that were not methylated were mock-methylated by analogous incubations except without S-adenosylmethionine or M.SssI. Either 48 or 72 h after transfection in duplicate with 150 or 100 ng of construct and 1.2 ng of the reference plasmid pCMV-CLuc 2 (New England Biolabs) into C2C12 or MCF7 cells (Fast-forward protocol, Qiagen), the Lucia and Cypridina luciferase activities were assayed (Quanti-Luc, InvivoGen and BioLux Cypridina, New England Biolabs, luciferase assay kits).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Drs. Donald Comb, Rich Roberts, William Jack and Clotilde Carlow at New England Biolabs Inc. for research support and encouragement. We also thank Carl Baribault for help with mapping DM sites and DMRs and Melody Badoo and the Tulane Cancer Center for help with the Cufflinks analysis of the ENCODE RNA-seq data.
Funding
This research was supported in part by grants from the National Institutes of Health (NS04885) and the Louisiana Cancer Center to ME and for Cufflinks analysis by COBRE grant NIGMS P20GM103518.
References
- 1.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009; 324:929-30; PMID:19372393; http://dx.doi.org/ 10.1126/science.1169786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al.. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; PMID:23828890; http://dx.doi.org/ 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Green BB, Houseman EA, Johnson KC, Guerin DJ, Armstrong DA, Christensen BC, Marsit CJ. Hydroxymethylation is uniquely distributed within term placenta, and is associated with gene expression. Faseb J 2016; 30:2874-84; PMID:27118675; http://dx.doi.org/ 10.1096/fj.201600310R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, Lahdesmaki H, Rao A. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A 2014; 111:E3306-15; PMID:25071199; http://dx.doi.org/ 10.1073/pnas.1412327111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016; 30:733-50; PMID:27036965; http://dx.doi.org/ 10.1101/gad.276568.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PloS one 2011; 5:e15367; PMID:21203455; http://dx.doi.org/7079182 10.1371/journal.pone.0015367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Gen Res 2012; 22:467-77; PMID:22106369; http://dx.doi.org/7079182 10.1101/gr.126417.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrlich M, Gama-Sosa M, Huang L-H, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res 1982; 10:2709-21; PMID:7079182; http://dx.doi.org/ 10.1093/nar/10.8.2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pang AP, Sugai C, Maunakea AK. High-throughput sequencing offers new insights into 5-hydroxymethylcytosine. Biomol Concepts 2016; 7:169-78; PMID:27356236; http://dx.doi.org/ 10.1515/bmc-2016-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gross JA, Pacis A, Chen GG, Barreiro LB, Ernst C, Turecki G. Characterizing 5-hydroxymethylcytosine in human prefrontal cortex at single base resolution. BMC genomics 2015; 16:672; PMID:26334641; http://dx.doi.org/ 10.1186/s12864-015-1875-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wen L, Li X, Yan L, Tan Y, Li R, Zhao Y, Wang Y, Xie J, Zhang Y, Song C, et al.. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol 2014; 15:R49; PMID:24594098; http://dx.doi.org/ 10.1186/gb-2014-15-3-r49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R, Parlato C, Oliviero S. Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol 2013; 14:R91; PMID:23987249; http://dx.doi.org/ 10.1186/gb-2013-14-8-r91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lev Maor G, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trend Gen: TIG 2015; 31:274-80; PMID:25837375; http://dx.doi.org/ 10.1016/j.tig.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 14.Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R, Bird A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Gen Res 2011; 21:1074-86; PMID:21628449; http://dx.doi.org/22577155 10.1101/gr.118703.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget 2012; 3:462-74; PMID:22577155; http://dx.doi.org/ 10.18632/oncotarget.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012; 336:934-7; PMID:22539555; http://dx.doi.org/ 10.1126/science.1220671 [DOI] [PubMed] [Google Scholar]
- 17.Ehrlich M, Ehrlich KC. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics 2014; 6:563-6; PMID:25531248; http://dx.doi.org/ 10.2217/epi.14.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, Yen CA, Ye Z, Mao SQ, Wang BA, et al.. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Molecular cell 2014; 56:286-97; PMID:25263596; http://dx.doi.org/ 10.1016/j.molcel.2014.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blattler A, Yao L, Witt H, Guo Y, Nicolet CM, Berman BP, Farnham PJ. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol 2014; 15:469; PMID:25239471; http://dx.doi.org/ 10.1186/s13059-014-0469-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrlich KC, Paterson HL, Lacey M, Ehrlich M. DNA hypomethylation in intragenic and intergenic enhancer chromatin of muscle-specific genes usually correlates with their expression. Yale J Biol Med 2016; PMID:28018137;22006022 [PMC free article] [PubMed] [Google Scholar]
- 21.Majmundar AJ, Skuli N, Mesquita RC, Kim MN, Yodh AG, Nguyen-McCarty M, Simon MC. O(2) regulates skeletal muscle progenitor differentiation through phosphatidylinositol 3-kinase/AKT signaling. Mol Cell Biol 2012; 32:36-49; PMID:22006022; http://dx.doi.org/ 10.1128/MCB.05857-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, Ploumakis A, Ghesquiere B, Van Dyck L, Boeckx B, Schoonjans L, et al.. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016; 537:63-8; PMID:27533040; http://dx.doi.org/ 10.1038/nature19081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsumagari K, Baribault C, Terragni J, Varley KE, Gertz J, Pradhan S, Baddoo M, Crain CM, Song L, Crawford GE, et al.. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics 2013; 8:317-32; PMID:23417056; http://dx.doi.org/ 10.4161/epi.23989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lacey MR, Baribault C, Ehrlich M. Modeling, simulation and analysis of methylation profiles from reduced representation bisulfite sequencing experiments. Stat Appl Genet Mol Biol 2013; 12:723-42; PMID:24163200; http://dx.doi.org/ 10.1515/sagmb-2013-0027 [DOI] [PubMed] [Google Scholar]
- 25.Tsumagari K, Baribault C, Terragni J, Chandra S, Renshaw C, Sun Z, Song L, Crawford GE, Pradhan S, Lacey M, et al.. DNA methylation and differentiation: HOX genes in muscle cells. Epigen Chromatin 2013; 6:25; PMID:23916067; http://dx.doi.org/26680004 10.1186/1756-8935-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, Xu Z, Li Z, Liu Y, Ye X, et al.. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res 2016; 26:103-18; PMID:26680004; http://dx.doi.org/ 10.1038/cr.2015.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illingworth RS, Gruenewald-Schneider U, De Sousa D, Webb S, Merusi C, Kerr AR, James KD, Smith C, Walker R, Andrews R, et al.. Inter-individual variability contrasts with regional homogeneity in the human brain DNA methylome. Nucleic Acids Res 2015; 43:732-44; PMID:25572316; http://dx.doi.org/ 10.1093/nar/gku1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Q, Decato B, Hong EE, Zhou M, Fang F, Qu J, Garvin T, Kessler M, Zhou J, Smith AD. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PloS One 2013; 8:e81148; PMID:24324667; http://dx.doi.org/ 10.1371/journal.pone.0081148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Randhawa R, Cohen P. The role of the insulin-like growth factor system in prenatal growth. Mol Genet Metabol 2005; 86:84-90; PMID:16165387; http://dx.doi.org/ 10.1016/j.ymgme.2005.07.028 [DOI] [PubMed] [Google Scholar]
- 30.The GTex Consortium . Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348:648-60; PMID:25954001; http://dx.doi.org/ 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et al.. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics 2016; 54:130.1-1..3; PMID:27322403; http://dx.doi.org/6314264 10.1002/cpbi.5 [DOI] [PubMed] [Google Scholar]
- 32.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 1983; 11:6883-94; PMID:6314264; http://dx.doi.org/ 10.1093/nar/11.19.6883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al.. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014; 158:673-88; PMID:25083876; http://dx.doi.org/ 10.1016/j.cell.2014.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.L'Honore A, Drouin J, Buckingham M, Montarras D. Pitx2 and Pitx3 transcription factors: two key regulators of the redox state in adult skeletal muscle stem cells and muscle regeneration. Free Radic Biol Med 2014; 75 Suppl 1:S37; PMID:26461356; http://dx.doi.org/ 10.1016/j.freeradbiomed.2014.10.781 [DOI] [PubMed] [Google Scholar]
- 35.McMinn J, Wei M, Sadovsky Y, Thaker HM, Tycko B. Imprinting of PEG1/MEST isoform 2 in human placenta. Placenta 2006; 27:119-26; PMID:16338457; http://dx.doi.org/ 10.1016/j.placenta.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 36.Anderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem 2008; 283:7027-35; PMID:18175804; http://dx.doi.org/ 10.1074/jbc.M706678200 [DOI] [PubMed] [Google Scholar]
- 37.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler A, M., Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996-1006; PMID:12045153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.L'Honore A, Commere PH, Ouimette JF, Montarras D, Drouin J, Buckingham M. Redox regulation by Pitx2 and Pitx3 is critical for fetal myogenesis. Dev Cell 2014; 29:392-405; PMID:24871946; http://dx.doi.org/ 10.1016/j.devcel.2014.04.006 [DOI] [PubMed] [Google Scholar]
- 39.Knopp P, Figeac N, Fortier M, Moyle L, Zammit PS. Pitx genes are redeployed in adult myogenesis where they can act to promote myogenic differentiation in muscle satellite cells. Dev Biol 2013; 377:293-304; PMID:23438814; http://dx.doi.org/ 10.1016/j.ydbio.2013.02.011 [DOI] [PubMed] [Google Scholar]
- 40.Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC, Issa JP. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res 2014; 42:6956-71; PMID:24875481; http://dx.doi.org/ 10.1093/nar/gku372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiehle L, Raddatz G, Musch T, Dawlaty MM, Jaenisch R, Lyko F, Breiling A. Tet1 and Tet2 protect DNA methylation canyons against hypermethylation. Mol Cell Biol 2016; 36:452-61; PMID:26598602; http://dx.doi.org/22415013 10.1128/MCB.00587-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hartung T, Zhang L, Kanwar R, Khrebtukova I, Reinhardt M, Wang C, Therneau TM, Banck MS, Schroth GP, Beutler AS. Diametrically opposite methylome-transcriptome relationships in high- and low-CpG promoter genes in postmitotic neural rat tissue. Epigenetics 2012; 7:421-8; PMID:22415013; http://dx.doi.org/ 10.4161/epi.19565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res 2007; 35:406-13; PMID:17169996; http://dx.doi.org/ 10.1093/nar/gkl1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kikin O, D'Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 2006; 34:W676-82; PMID:16845096; http://dx.doi.org/ 10.1093/nar/gkl253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosalia N, Neems D, Kerschner JL, Kosak ST, Harris A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res 2014; 42:9612-22; PMID:25081205; http://dx.doi.org/ 10.1093/nar/gku648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; http://dx.doi.org/ 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandra S, Terragni J, Zhang G, Pradhan S, Haushka S, Johnston D, Baribault C, Lacey M, Ehrlich M. Tissue-specific epigenetics in gene neighborhoods: myogenic transcription factor genes. Hum Mol Genet 2015; 24:4660-73; PMID:26041816; http://dx.doi.org/ 10.1093/hmg/ddv198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al.. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466:253-7; PMID:20613842; http://dx.doi.org/ 10.1038/nature09165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terragni J, Zhang G, Sun Z, Pradhan S, Song L, Crawford GE, Lacey M, Ehrlich M. Notch signaling genes: Myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics 2014; 9:842-50; PMID:24670287; http://dx.doi.org/ 10.4161/epi.28597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson RB, Allan DS, Le Y. Greater organ involution in highly proliferative tissues associated with the early onset and acceleration of ageing in humans. Exp Gerontol 2014; 55:80-91; PMID:24685641; http://dx.doi.org/ 10.1016/j.exger.2014.03.015 [DOI] [PubMed] [Google Scholar]
- 51.Giehr P, Kyriakopoulos C, Ficz G, Wolf V, Walter J. The Influence of hydroxylation on maintaining CpG methylation patterns: A Hidden Markov Model approach. PLoS Comput Biol 2016; 12:e1004905; PMID:27224554; http://dx.doi.org/ 10.1371/journal.pcbi.1004905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dachtler J, Ivorra JL, Rowland TE, Lever C, Rodgers RJ, Clapcote SJ. Heterozygous deletion of alpha-neurexin I or alpha-neurexin II results in behaviors relevant to autism and schizophrenia. Behav Neurosci 2015; 129:765-76; PMID:26595880; http://dx.doi.org/ 10.1037/bne0000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hayashi T, Yoshida T, Ra M, Taguchi R, Mishina M. IL1RAPL1 associated with mental retardation and autism regulates the formation and stabilization of glutamatergic synapses of cortical neurons through RhoA signaling pathway. PloS One 2013; 8:e66254; PMID:23785489; http://dx.doi.org/ 10.1371/journal.pone.0066254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perera A, Eisen D, Wagner M, Laube SK, Kunzel AF, Koch S, Steinbacher J, Schulze E, Splith V, Mittermeier N, et al.. TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell reports 2016; 11:283-94; PMID:25843715; http://dx.doi.org/25510270 10.1016/j.celrep.2015.03.020 [DOI] [PubMed] [Google Scholar]
- 55.Moen EL, Mariani CJ, Zullow H, Jeff-Eke M, Litwin E, Nikitas JN, Godley LA. New themes in the biological functions of 5-methylcytosine and 5-hydroxymethylcytosine. Immunol Rev 2015; 263:36-49; PMID:25510270; http://dx.doi.org/ 10.1111/imr.12242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep 2015; 16:1467-81; PMID:26474904; http://dx.doi.org/ 10.15252/embr.201540945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011; 479:74-9; PMID:21964334; http://dx.doi.org/ 10.1038/nature10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al.. Dynamic DNA methylation across diverse human cell lines and tissues. Gen Res 2013; 23:555-67; PMID:23325432; http://dx.doi.org/25806086 10.1101/gr.147942.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramos MP, Wijetunga NA, McLellan AS, Suzuki M, Greally JM. DNA demethylation by 5-aza-2′-deoxycytidine is imprinted, targeted to euchromatin, and has limited transcriptional consequences. Epigenet Chromatin 2015; 8:11; PMID:25806086; http://dx.doi.org/ 10.1186/s13072-015-0004-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Redies C, Hertel N, Hubner CA. Cadherins and neuropsychiatric disorders. Brain Res 2012; 1470:130-44; PMID:22765916; http://dx.doi.org/ 10.1016/j.brainres.2012.06.020 [DOI] [PubMed] [Google Scholar]
- 61.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, et al.. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013; 152:1146-59; PMID:23434322; http://dx.doi.org/ 10.1016/j.cell.2013.02.004 [DOI] [PubMed] [Google Scholar]
- 62.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell 2009; 137:1194-211; PMID:19563753; http://dx.doi.org/ 10.1016/j.cell.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang K, Li N, Ainsworth RI, Wang W. Systematic identification of protein combinations mediating chromatin looping. Nat Commun 2016; 7:12249; PMID:27461729; http://dx.doi.org/ 10.1038/ncomms12249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucl Acids Res 2013; 41:2881-93; PMID:23325844; http://dx.doi.org/ 10.1093/nar/gks1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Proudhon C, Duffie R, Ajjan S, Cowley M, Iranzo J, Carbajosa G, Saadeh H, Holland ML, Oakey RJ, Rakyan VK, et al.. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell 2012; 47:909-20; PMID:22902559; http://dx.doi.org/ 10.1016/j.molcel.2012.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekstrom TJ, Tegner J, Sundberg CJ. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics 2014; 9:1557-69; PMID:25484259; http://dx.doi.org/ 10.4161/15592294.2014.982445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laker RC, Ryall JG. DNA methylation in skeletal muscle stem cell specification, proliferation, and differentiation. Stem Cells Int 2016; 2016:5725927; PMID:26880971; http://dx.doi.org/ 10.1155/2016/5725927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chung SY, Kao CH, Villarroya F, Chang HY, Chang HC, Hsiao SP, Liou GG, Chen SL. Bhlhe40 represses PGC-1alpha activity on metabolic gene promoters in myogenic cells. Mol Cell Biol 2015; 35:2518-29; PMID:25963661; http://dx.doi.org/ 10.1128/MCB.00387-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005; 121:645-57; PMID:15907476; http://dx.doi.org/ 10.1016/j.cell.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 70.Kodama H, Kumai Y, Nishimoto K, Sanuki T, Yumoto E. Modulation of satellite cells activity and MyoD in rat thyroarytenoid muscle after reinnervation. Laryngoscope 2015; 125:E245-51; PMID:25809587; http://dx.doi.org/ 10.1002/lary.25248 [DOI] [PubMed] [Google Scholar]
- 71.Hsiao SP, Chen SL. Myogenic regulatory factors regulate M-cadherin expression by targeting its proximal promoter elements. Biochem J 2010; 428:223-33; PMID:20334626; http://dx.doi.org/ 10.1042/BJ20100250 [DOI] [PubMed] [Google Scholar]
- 72.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al.. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317-30; PMID:25693563; http://dx.doi.org/ 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chadwick LH. The NIH Roadmap Epigenomics Program data resource. Epigenomics 2012; 4:317-4; PMID:22690667; http://dx.doi.org/21526222 10.2217/epi.12.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Myers RM, Stamatoyannopoulos J, Snyder M, Dunham I, Hardison RC, Bernstein BE, Gingeras TR, Kent WJ, Birney E, Wold B, et al.. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011; 9:e1001046; PMID:21526222; http://dx.doi.org/ 10.1371/journal.pbio.1001046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al.. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Gen Res 2011; 21:1757-67; http://dx.doi.org/ 10.1101/gr.121541.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/ 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al.. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 2010; 18:662-74; PMID:20412780; http://dx.doi.org/ 10.1016/j.devcel.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, et al.. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res 2013; 41:D171-6; PMID:23203885; http://dx.doi.org/ 10.1093/nar/gks1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.