

Abstract
Previous work has provided evidence for E2F-dependent transcription control of both G1/S- and G2/M-regulated genes. Analysis of the G2-regulated cdc2 and cyclin B1 genes reveals the presence of both positive- and negative-acting E2F promoter elements. Additional elements provide both positive (CCAAT and Myb) and negative (CHR) control. Chromatin immunoprecipitation assays identify multiple interactions of E2F proteins that include those previously shown to activate and repress transcription. We find that E2F1, E2F2, and E2F3 bind to the positive-acting E2F site in the cdc2 promoter, whereas E2F4 binds to the negative-acting site. We also find that binding of an activator E2F is dependent on an adjacent CCAAT site that is bound by the NF-Y transcription factor and binding of a repressor E2F is dependent on an adjacent CHR element, suggesting a role for cooperative interactions in determining both activation and repression. Finally, the kinetics of B-Myb interaction with the G2-regulated promoters coincides with the activation of the genes, and RNAi-mediated reduction of B-Myb inhibits expression of cyclin B1 and cdc2. The ability of B-Myb to interact with the cdc2 promoter is dependent on an intact E2F binding site. These results thus point to a role for E2Fs, together with B-Myb, which is an E2F-regulated gene expressed at G1/S, in linking the regulation of genes at G1/S and G2/M.
Keywords: E2F, Myb
Introduction
A variety of experiments have demonstrated the role of E2F proteins in the control of expression of genes important for DNA replication as well as further cell cycle progression (Dyson, 1998; Nevins, 1998). In particular, many studies have detailed the role for E2F activities in controlling gene expression at G1/S, involving the activation of genes encoding DNA replication proteins, enzymes responsible for deoxynucleotide biosynthesis, proteins that assemble to form functional origin complexes, and kinases that are involved in the activation of initiation. In addition to this role for E2F, more recent work has demonstrated that a substantial number of E2F-induced genes are normally regulated at G2 of the cell cycle, encoding proteins known to function in mitosis (Ishida et al, 2001; Polager et al, 2002; Ren et al, 2002). Moreover, previous studies in Drosophila have provided evidence for a connection between E2F activity and the control of mitotic activities (Neufeld et al, 1998).
Although a role for E2F activity in the control of mitotic genes is evident from these past studies, the mechanism for such control remains to be elucidated. It is possible that a particular E2F activity, or modified form of an E2F activity, is uniquely functional at G2. Alternatively, it is also possible that the induction of these genes normally regulated at G2 is a secondary effect of the E2F activities. For instance, the G2-regulated genes could be activated by transcription factors whose expression is controlled at G1/S by E2F activities. In this case, E2F accumulation at G1/S would initiate a cascade of events, initially activating the genes encoding DNA replication activities and then secondarily activating genes encoding mitotic activities. Alternatively, a direct role for E2Fs could extend to the G2-regulated genes, perhaps through a differential repression of transcription, a delayed activation of transcription, or both.
To address these issues, we have analyzed several promoters of genes that are normally regulated at G2 of the cell cycle and which have been shown previously to be subject to E2F control. We find that for the cdc2 promoter and the cyclin B1 promoter, there is a direct role for E2F in both the repression and activation of these promoters. Moreover, we identify specific E2F activities that interact with the sites responsible for this control. Finally, we also identify an important role for the B-Myb transcription factor in the activation of G2-regulated genes. Since B-Myb is itself an E2F-regulated gene expressed at G1/S, these results define a connection in the control of gene expression at G1/S and G2/M through a link with E2F activities.
Results
A role for both positive and negative E2F elements in the control of promoters regulated at G2/M
We have previously identified a group of genes that are subject to E2F control and that are normally regulated at G2/M of the cell cycle (Ishida et al, 2001), a result seen in various other studies (Polager et al, 2002; Ren et al, 2002). An example of this finding is shown by the Northern analysis in Figure 1. Cells were synchronized at G1/S by a hydroxyurea (HU) block and then released into the cell cycle. As seen in Figure 1B, transcripts of the cdc2, cyclin B1, cyclin A2, cdc20, and importin alpha were low in the G1/S cells and then rose as cells progress to the G2/M stage. Each then declines after the peak accumulation and then rises again as cells approach the second G2 (Figure 1B). In contrast, the G1/S-regulated genes cyclin E and PCNA exhibit distinctly different patterns. Each of these declines after the release from G1/S and then reaccumulates as cells re-enter G1/S (Figure 1B).
Figure 1.
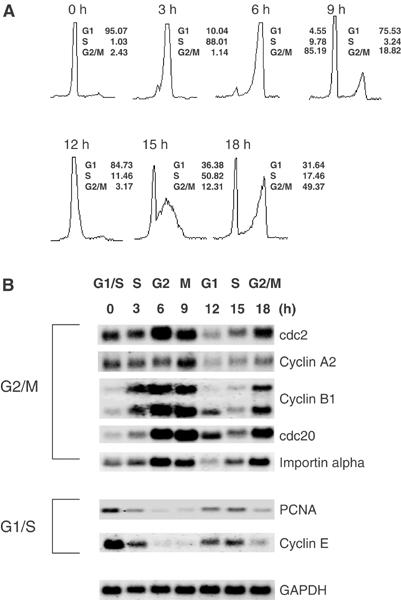
Distinct patterns of G1/S and G2/M cell cycle regulation of E2F-dependent genes. (A) FACS analysis of REF52 cells that were synchronized by HU block, and then released into the cell cycle. (B) Northern analysis of G2/M gene and G1/S gene expression in HU-synchronized REF52 cells. Approximately 2 μg of mRNA was loaded in each lane. Expression of GADPH was evaluated to confirm equal loading.
Although considerable effort has focused on the direct role of E2F in the control of G1/S genes, much less is known of G2/M genes. Previous studies have shown that the cdc2 promoter contains E2F regulatory elements (Dalton, 1992; Tommasi and Pfeifer, 1995). Similarly, our examination of the cell cycle-regulated cyclin B1 promoter sequences (Hwang et al, 1995) has revealed E2F regulatory elements (Figure 2A). Although other work has demonstrated a component of cell cycle-independent expression for cyclin B1 (Hwang et al, 1998), our focus is on the mechanisms underlying cell cycle-dependent expression. We carried out mutagenesis on each putative E2F site within the cdc2 and cyclin B1 promoters, and then assayed transiently transfected luciferase reporter constructs driven by those promoters in cells passing through G2/M stage after HU synchronization. As shown in Figure 2B and C, the luciferase activity driven by the wild-type cdc2 and cyclin B1 promoters quite closely mimics the pattern of accumulation of the endogenous mRNAs as shown in Figure 1. Promoter activity is low at G1/S, rises to a peak level at 7–9 h after release, and then begins to decline. Mutation of a distal E2F element in the human cdc2 promoter (Figure 2B, left panel) and mutation of a proximal E2F element (+1 element) in the human cyclin B1 promoter (Figure 2C, left panel) markedly reduced their activities, indicating that both promoters contain a positive E2F element that is necessary for their expression at G2/M stage. Mutation of a proximal E2F element in human cdc2 promoter (Figure 2B, right panel) and mutation of a distal E2F element (−85 element) in the human cyclin B1 promoter (Figure 2C, right panel) showed increased promoter activities, indicating that both promoters contain a negative E2F element that is necessary for repressing their expression at G0 stage. However, the expression pattern of this mutant promoter is similar to that of wild type with higher magnitude, indicating that the activator must still contribute to the full activation of those genes at G2/M stage. Thus, both positive and negative E2F control contributes to the regulation of the G2/M genes and importantly, this distinction resides in separate E2F elements.
Figure 2.
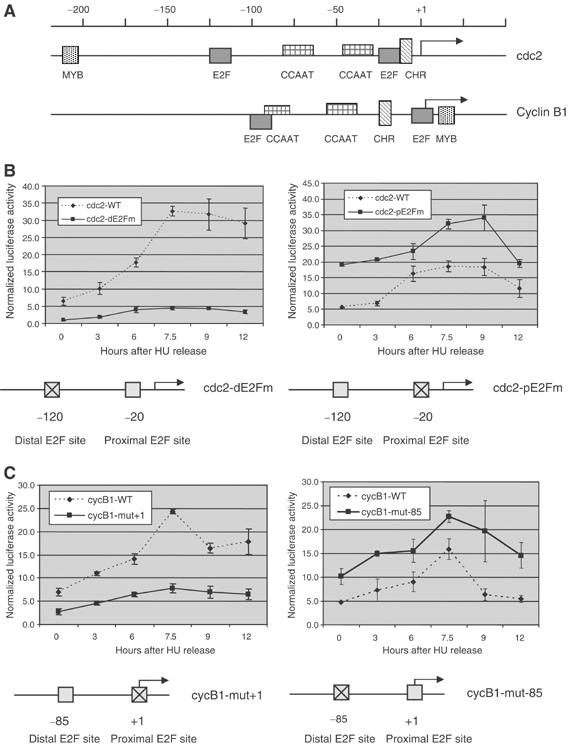
Identification of positive and negative E2F elements controlling activities of G2/M promoters. (A) Schematic of human cdc2 and cyclin B1 promoters indicating E2F binding sites, MYB binding sites, CCAAT elements, and CHR elements. All elements are identified based on previous studies (Tommasi and Pfeifer, 1995; Zwicker et al, 1995a; Yun et al, 1999; Wasner et al, 2003), this study, and sequence alignment between human, mouse, and rat promoters in this study. All elements identified are conserved in all three species. (B) Positive and negative E2F elements are necessary for expression of the cdc2 gene. Luciferase activities assayed from REF52 cells transiently transfected with either wild-type (WT), the distal E2F site mutant (dE2Fm), or the proximal E2F site mutant (pE2Fm) human cdc2 promoter constructs. Left panel: mutation of the positive E2F element (dE2Fm) in human cdc2 promoter abolishes promoter activity. Right panel: mutation of the negative E2F element (pE2Fm) increases promoter activity. (C) Positive and negative E2F elements are necessary for expression of the cyclin B1 gene. Luciferase activities assayed from REF52 cells transiently transfected with either wild-type (WT), the distal E2F site mutant (−85), or the proximal E2F site mutant (+1) human cyclin B1 promoter constructs. Left panel: mutation of the positive E2F element (+1) in human cyclin B1 promoter abolishes promoter activity. Right panel: mutation of the negative E2F element (−85) increases promoter activity.
A role for additional positive- and negative-acting promoter elements
Our recent work has demonstrated a role for cooperative promoter interactions involving E2Fs and other transcription factors, including TFE3 and YY1, as a mechanism to insure specificity of function (Schlisio et al, 2002; Giangrande et al, 2003, 2004). An inspection of cdc2 promoter sequences, together with past work analyzing the effects of mutation of various elements, suggests a role for at least three other sequences—a Myb element upstream of the distal E2F site, CCAAT elements situated between the E2F elements, and a CHR element adjacent to the proximal E2F element (Figure 3A). Previous work has shown Myb proteins to function as positive regulators of several cell cycle genes, including human cdc2 (Ku et al, 1993) and the Drosophila cyclin B1 gene (Okada et al, 2002). Likewise, the CCAAT elements are binding sites for the positive-acting NF-Y transcription factor (Maity and de Crombrugghe, 1998). They have been shown to be important for promoter activity of many cell cycle genes including cdc2, cycA2, and E2F1 (Zwicker et al, 1995a, 1995b; van Ginkel et al, 1997; Zwicker and Muller, 1997; Liu et al, 1998b). A protein(s) binding to the CHR element has not been identified, but the element has been shown to function as a cell cycle regulator in cooperation with E2F control (Liu et al, 1996).
Figure 3.
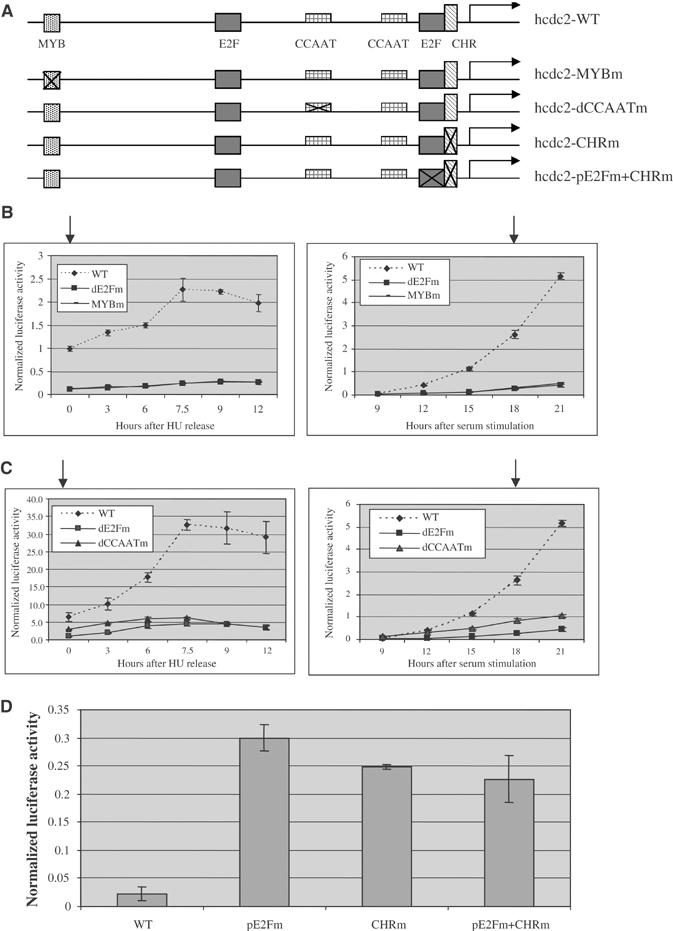
Multiple elements control human cdc2 promoter activity. (A) Schematic of human cdc2 wild-type (WT) and mutant promoters that contain mutations in the MYB binding site (MYBm), the distal CCAAT element (dCCAATm), the CHR element (CHRm), or the proximal E2F site and the CHR element (pE2Fm+CHRm) respectively. The arrows indicate the G1/S transition in the cell cycle. (B) Mutation of the MYB binding site abolishes human cdc2 promoter activity. Left panel: REF52 cells were synchronized by blocking with HU. Right panel: REF52 cells were synchronized by serum starvation. The arrows indicate the G1/S transition. (C) Mutation of the distal CCAAT element dramatically reduces human cdc2 promoter activity. Left panel: REF52 cells were synchronized by blocking with HU. Right panel: REF52 cells were synchronized by serum starvation. The arrows indicate the G1/S transition. (D) Mutation of the CHR element increases human cdc2 promoter activity in quiescence. REF52 cells containing transiently transfected reporter constructs were serum starved for 2 days, and then harvested for luciferase activities.
As shown in Figure 3B, mutation of the Myb element abolished activation of the cdc2 promoter, either after release from an HU block at G1/S or following stimulation of cell growth by serum addition. Likewise, mutation of the distal CCAAT element also abolished promoter activation, equivalent to the mutation of the distal E2F element (Figure 3C). In contrast, mutation of the CHR element led to an increase of promoter activity, similar to the mutation of the proximal E2F site (Figure 3D). This result is the same as previously shown by Zwicker et al (1995b), who referred to the E2F repressive site as a CDE element, which has also been shown to bind E2F4 (Tommasi and Pfeifer, 1995). Taken together, these results suggest a role for multiple elements, both E2F and others, that act in both a positive and negative manner to control the activity of the cdc2 promoter.
Interaction of activator and repressor E2Fs with G2/M-regulated promoters
Given the apparent direct roles for E2F activity in the control of cdc2 and cyclin B1 transcription, both positive and negative, we have investigated the interaction of E2F proteins with the endogenous promoters. Because of availability of human promoter sequence, we made use of T98G cells to examine E2F interaction with these promoters. The expression pattern of these G2/M genes as well as several G1/S genes in the T98G cells was equivalent to that observed in the REF52 cells (Figure 4A, and data not shown). As shown in Figure 4B, both activator and repressor E2Fs were found to interact with the cdc2 and cyclin B1 promoters. Assays for E2F4 revealed an interaction with each of the promoters in the quiescent cell sample, similar to several recent reports (Takahashi et al, 2000; Schlisio et al, 2002). This included both the G2/M (cdc2, cyclin B1, cyclin A2)- and G1/S(PCNA)-regulated genes. This interaction was reduced or absent in the G1/S-blocked cells and remained low as cells progressed through S phase and mitosis (6–9 h time point). Interestingly, the E2F4 interaction then reappeared as the cells came back to the next G1/S (18 h time point).
Figure 4.
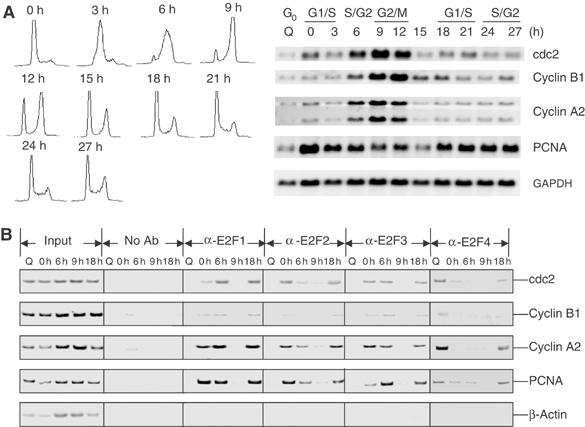
Interaction of E2F proteins with G2-regulated promoters. (A) Analysis of cell cycle gene expression in T98G cells. Left panel: FACS analysis of T98G cells that were synchronized by HU block and released into fresh 10% FBS containing DMEM for the indicated times. Additional sample was prepared from serum-starved cells (Q: quiescence). Right panel: Northern analysis of cdc2, cyclin B1, cyclin A2, and PCNA messages. (B) Both activator and repressor E2Fs bind to endogenous G2-regulated promoters. T98G cells at quiescence (Q), HU block (0 h), or release from HU block (6, 9, and 18 h) were harvested for ChIP analysis using the indicated anti-E2F antibodies. Immunoprecipitated chromatin was analyzed by semiquantitative PCR using primers in the cdc2, cyclin B1, cyclin A2, PCNA, and β-actin promoters. PCNA, an E2F-regulated G1/S gene, is used here as a positive control, while β-actin, a non-E2F-regulated gene, is used as a negative control. Input material represents 0.05% of the total chromatin material used for a given ChIP reaction.
The pattern of interaction of the activator E2Fs was essentially opposite to that for E2F4. In particular, none of these proteins was bound to the promoters in quiescent cells, and then each could be detected on the promoters at G1/S (0 h time point). Each persisted through S phase (6 h sample) and then were absent as cells progressed through G2 and mitosis (9 h time point). Each was then found to interact with the promoters as cells came back to the next G1/S (18 h time point). Taken together, the chromatin immunoprecipitation (ChIP) results provide evidence for interaction of both positive- and negative-acting E2Fs with the G2-regulated promoters. But, the kinetics of these interactions do not suggest a basis for distinguishing control at G1/S versus G2/M.
Activator and repressor E2Fs interact with distinct promoter elements
In light of the clear distinction in the functional role of the individual E2F elements in the promoters, we investigated the relationship between this function and the nature of the E2F proteins that interact with these sites. To do so, we have made use of ChIP assays that specifically measure protein interactions with transfected plasmid sequences rather than interaction with the endogenous promoter sequences (Giangrande et al, 2004). In this way, we can measure the effect of mutation of a particular element on the chromatin interaction profile. We utilized the cdc2 promoter for these assays and measured chromatin interaction in either quiescent REF52 cells that were serum stimulated or G1/S–arrested cells that were released into the cell cycle. The promoter mutants used in the assays, which include alterations in either the proximal or distal E2F elements or both, are shown in Figure 5A.
Figure 5.
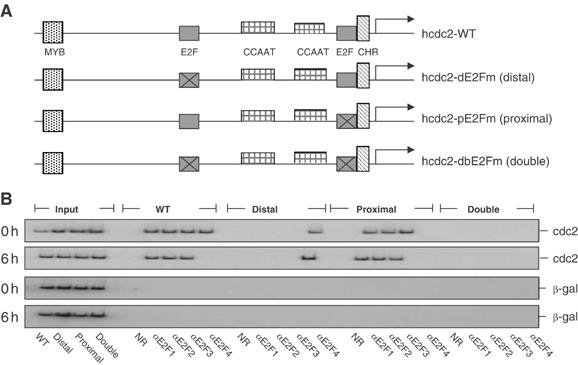
Distinct interaction of E2F proteins with positive- and negative-acting E2F elements. (A) Schematic of wild-type (WT) and mutant human cdc2 promoters that contain mutations in the distal E2F site (dE2Fm), the proximal E2F site (pE2Fm), and both E2F sites (dbE2Fm). (B) Distinct interaction of E2F proteins with positive- and negative-acting E2F elements. REF52 cells containing transiently transfected reporter constructs were harvested at either HU block (0 h) or 6 h after release (6 h). Reporter plasmids containing chromatin material isolated as described in Materials and methods were immunoprecipitated using the indicated anti-E2F antibodies, and then analyzed by semiquantitative PCR using primers specific to transfected plasmids. Control reaction using the purified normal rabbit serum (NR) (Pierce) is shown. Primers specific to cotransfected plasmids harboring either β-gal (shown here) or renilla (data not shown) gene were used as negative controls. Input chromatin represents 0.1% of the total chromatin material in a given ChIP reaction.
Samples of REF52 cells transfected with wild-type and mutant human cdc2 promoter constructs were assayed for chromatin interaction of E2F family members. The results are shown in Figure 5B. As seen in the previous assays of the endogenous cdc2 promoter, E2F4 as well as E2F1–3 was bound to the promoter in the G1/S-arrested cells (0 h) (Figure 5B). As was the case for the endogenous assays, the interaction of E2F4 declined as cells were released into the cell cycle (6 h). Assays of the mutant promoters revealed that the activator E2Fs (E2F1–3) bound to the distal E2F site, while the repressor E2F4 bound to the proximal site. The interaction of E2F4 also coincided with an interaction of either p130 or p107 (data not shown). Strikingly, the E2F4 interaction persisted on the promoter containing the distal site mutation that blocked binding of E2F1–3 to the promoters. These results thus demonstrate that the activator and repressor E2Fs bind to distinct elements within the cdc2 promoter that coincide with function (activator or repressor elements). Moreover, the results suggest that the binding of the activator E2Fs displaces the repressor E2F complexes.
Roles for cooperative interactions involving activator and repressor E2Fs
The fact that the activator E2Fs and the repressor E2F bind to distinct promoter elements implies specificity in site recognition. Yet, the primary sequence suggests little potential for this difference based solely on the E2F interaction. Our recent work has demonstrated a role for cooperative interactions with other transcription factors as a basis for such E2F specificity (Schlisio et al, 2002; Giangrande et al, 2003, 2004). The data presented in Figure 3 demonstrate the role of additional elements in the control of cdc2 promoter activity; to investigate the function of these sequences, particularly in relation to the E2F interactions, we again performed ChIP assays using mutant promoter constructs.
Previous work has shown that the CCAAT element is a binding site for the NF-Y transcription factor (Maity and de Crombrugghe, 1998). As shown in Figure 6A, ChIP assays demonstrate an interaction of two of the NF-Y subunits (NF-YA and NF-YB) with each of the promoters. In contrast to the dynamic nature of the E2F interactions, the interaction of the NF-Y proteins is constant through the cell cycle, consistent with genomic footprint data showing that the CCAAT elements are occupied throughout cell cycle (Tommasi and Pfeifer, 1995). We next used plasmid-based ChIP assays to evaluate the role of the CCAAT element. As shown in Figure 3C, mutation of the distal CCAAT element abolishes promoter activation, equivalent to the mutation of the distal E2F element. The mutation of the CCAAT element also abolished the interaction of NF-YA with the cdc2 promoter, either at 0 or 6 h following release from the HU arrest (Figure 6B). Importantly, this mutation also abolishes the interaction of E2F3 with the cdc2 promoter, suggesting a cooperative binding of E2F3 to the promoter dependent on NF-Y.
Figure 6.
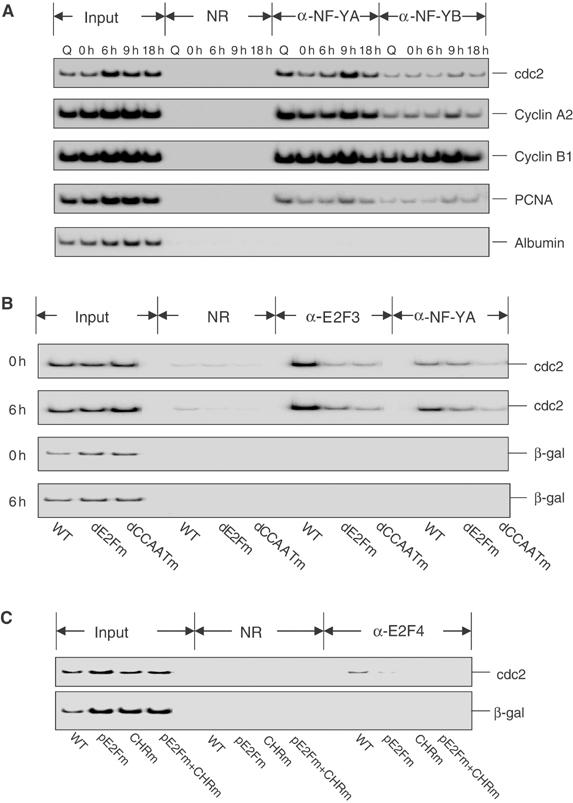
A role for CCAAT and CHR elements in promoting the interaction of activator and repressor E2Fs. (A) NF-Y protein interacts with the endogenous cdc2, cyclin B1, cyclin A2, and PCNA promoters. Albumin promoter with no NF-Y binding was used as a negative control. T98G cells at quiescence (Q), HU block (0 h), or released from HU block (6, 9, and 18 h) were harvested for ChIP analysis using antibodies against NF-Y A and B subunits. (B) The binding of NF-Y to the distal CCAAT element is necessary for the binding of activator E2F3 to the distal E2F site. REF52 cells transiently transfected with wild-type (WT), the distal E2F site mutant (dE2Fm), and the distal CCAAT element mutant (dCCAATm) human cdc2 promoter reporter constructs were harvested at either HU block (0 h) or 6 h after release (6 h). The luciferase activities for these reporter constructs are shown in Figure 3C. The presence of the cdc2 promoter or control sequence (β-gal) from the transfected constructs in immunoprecipitates using anti-E2F3, anti-NF-YA, or control antibodies (NR) was detected by PCR as described in Figure 5. (C) An intact CHR element is necessary for E2F4 binding to the proximal E2F site. REF52 cells transiently transfected with the indicated reporter constructs were made quiescent by serum deprivation for 2 days. The luciferase activities for these constructs are shown in Figure 3D. Plasmid-based ChIP reaction was carried out using anti-E2F4 and control antibody (NR) and the samples were analyzed as described in Figure 5.
Finally, we also examined the effects of mutation of the CHR element, particularly with respect to the E2F4 interaction with the cdc2 promoter. Although the identity of the CHR binding transcription factor(s) has not been established, it does appear that the CHR element is critical for allowing the interaction of E2F4 since the mutation of the CHR element abolished the E2F interaction, equivalent to the effect of mutation of the proximal E2F element (Figure 6C).
A role for B-Myb in the control of cdc2 suggests a mechanism to link G1/S and G2/M transcription control
Finally, given the role for the Myb element as a positive regulator of cdc2 promoter activity, we have investigated the interaction of B-Myb with the cdc2 promoter. Three distinct proteins make up the Myb family: A-Myb, B-Myb, and C-Myb (for reviews, see Weston, 1998; Oh and Reddy, 1999). A-Myb and C-Myb exhibit a more tissue-specific expression pattern, whereas B-Myb is expressed ubiquitously in all cell types and cell lines so far analyzed, and is linked to proliferation. In addition, we have identified that B-Myb as an E2F interacting protein in a yeast two-hybrid selection assay using E2F3 as the bait (data not shown). GST pulldown experiments also showed that B-Myb interacts with the activator E2Fs (E2F1, E2F2, and E2F3) but not with the repressor E2F (E2F4) (data not shown). Also, B-Myb is an E2F-regulated gene expressed at G1/S, and previous work in Drosophila has identified a role for B-Myb in the control of certain mitotic genes including cyclin B1 (Okada et al, 2002). Taken together, these findings suggest a role for B-Myb in E2F-mediated transcription control, which we have now further investigated. As shown in Figure 7A, ChIP assays revealed an interaction of B-Myb with both the cdc2 and cyclin B1 promoters following the stimulation of cell growth. In addition, the kinetics of this interaction were slightly delayed compared to that of the interaction of E2F3 with these promoters. This delay was reflected in a delay in the kinetics of B-Myb protein accumulation relative to E2F3, as seen in Western blot assays (Figure 7B). The delay in B-Myb promoter interaction was also evident in assays of cells released from an HU G1/S arrest (Figure 7C).
Figure 7.
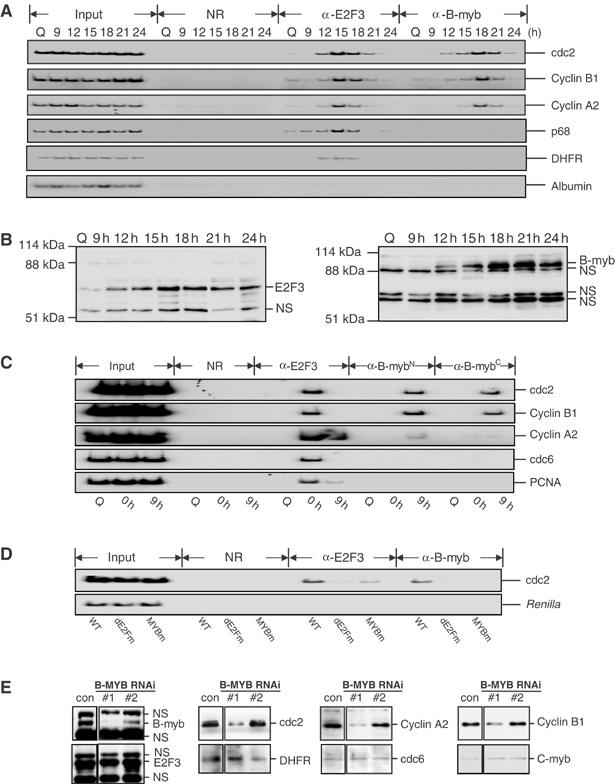
A role for B-Myb in the control of G2/M gene expression. (A) The binding of B-Myb to endogenous G2-regulated promoters (cdc2, CycA2, and cyclin B1) is delayed compared to the binding of the activator E2F (E2F3) to these promoters. T98G cells were released from serum starvation and samples were harvested at quiescence (Q), or 9, 12, 15, 18, 21, and 24 h after serum addition for ChIP analysis using the indicated antibodies. Two E2F-regulated G1/S genes (p68 and DHFR) and a non-E2F-regulated gene (albumin) were used as controls. (B) Relative kinetics of B-Myb and E2F3 protein accumulation. Western analysis of E2F3 and B-Myb using 40 μg of nuclear extract of T98G cells harvested at quiescence (Q) or at the indicated times following serum addition. NS: nonspecific band. (C) The binding of B-Myb to endogenous G2-regulated promoters (cdc2, cyclin B1, and cyclin A2) is prolonged (9 h time point) compared to the binding of activator E2F3 to these promoters. T98G cells were made quiescent (Q), blocked with HU (0 h), released from HU for 6, 9, and 18 h, and then harvested for ChIP analysis using the indicated antibodies (α-B-MybN: N-19; α-B-MybC: H115). Two E2F-regulated G1/S genes (cdc6 and PCNA) were used as controls. (D) Mutation in the MYB binding site abolishes the binding of B-Myb to human cdc2 promoter. REF52 cells transiently transfected with wild-type (WT), the distal E2F mutant (dE2Fm), and the MYB site mutant (MYBm) reporter constructs were made quiescent and then restimulated with serum for 18 h. The luciferase activities for these constructs are shown in Figure 3B. The presence of the cdc2 promoter or control sequence (Renilla) from the transfected constructs in immunoprecipitates using anti-E2F3, anti-B-Myb, or control antibodies (NR) was detected by PCR as described in Figure 5. (E) B-Myb is required for G2/M target gene expression. Western analysis of several G2/M genes (cdc2, cyclin A2, and cyclin B1) and G1/S genes (E2F3, DHFR, and cdc6) and C-Myb using cell extracts from asynchronously growing T98G cells with mock transfected as a control (con), or transfected with an effective B-MYB RNAi duplex 1 (#1) and a noneffective B-MYB RNAi duplex 2 (#2) as another control. Cell lysates were prepared 48 h after siRNA transfection. NS=nonspecific band.
While the interaction of B-Myb was detected with three G2/M-regulated promoters (cdc2, cyclin B1, and cyclin A2), there was no evidence of an interaction with two G1/S-regulated genes (cdc6 and PCNA) (Figure 7C, bottom panel). Based on these results, it would appear that the B-Myb interaction is important for the activation of the G2/M-regulated genes and that this interaction may contribute to the distinct kinetics of activation of these genes during the cell cycle.
We have also measured the interaction of B-Myb, as well as E2F proteins, with a cdc2 promoter bearing mutations in either the Myb or E2F sites. As shown by the assays in Figure 7D, both E2F3 and B-Myb were detected on the wild-type cdc2 promoter at 18 h after growth stimulation, consistent with the observations with the endogenous promoter. Mutation of the Myb binding site eliminated the B-Myb interaction as expected and reduced the E2F3 interaction. Strikingly, mutation of the distal E2F site that functions as a positive control element not only abolished the E2F3 interaction but also abolished the B-Myb interaction. Based on this result, it would appear that the ability of B-Myb to interact with the promoter is dependent on the E2F interaction.
Finally, given these results that implicate B-Myb in the control of cdc2 promoter activity, in relation to E2F function, we have directly investigated a role for B-Myb in the expression of cdc2, using RNAi as a mechanism to reduce the accumulation of B-Myb to determine the effect of loss of B-Myb on expression of various G2/M-regulated genes. Asynchronously growing T98G cells were mock transfected (con), or transfected with two siRNA duplexes designed to target B-Myb mRNA. As shown in Figure 7E, B-Myb duplex 1 (#1) effectively reduced B-Myb protein levels, whereas B-Myb siRNA duplex 2 (#2) had little or no effect on B-Myb levels and thus served as a control. Cell lysates were prepared from 50% of samples 48 h after siRNA transfection and assayed for various proteins encoded by G2/M-regulated genes. As seen in Figure 7E, the reduction of B-Myb protein mediated by B-Myb siRNA duplex 1 coincided with a reduction of cdc2, cycA2, and cycB1 proteins (data from one representative experiment). In contrast, there was no effect on these proteins following the expression of B-Myb siRNA duplex 2. In addition, the reduction of B-Myb levels mediated by duplex 1 had no impact on the accumulated level of either DHFR or cdc6, two G1/S-regulated genes not expected to be regulated by B-Myb. To exclude the possibility that the reduced cdc2, cyclin A2, and cyclin B1 expression is due to the cell cycle arrest prior to the expression of those genes in B-Myb siRNA-treated samples, the cell cycle profiles were analyzed using the other half of samples from the same experiment and showed that there were no marked changes (Supplementary Figure 1). Based on these results, we conclude that B-Myb is important for the expression of several G2/M-regulated genes that are also regulated by E2F proteins, and that this coincides with an ability of B-Myb to interact with these promoters, in an E2F-dependent manner.
Given previous studies showing overexpression of C-Myb can regulate human cdc2 promoter activity in T cells (Ku et al, 1993), and the fact that both C-Myb and B-Myb can recognize the similar MYB binding site in vitro (Mizuguchi et al, 1990; Howe and Watson, 1991), we have examined the possible role of C-Myb in regulating cdc2 expression in vivo in T98G cells. C-Myb is expressed in T98G cells as measured by direct Western (data not shown). Although C-Myb is present in T98G cells, we find no evidence for binding to the endogenous cdc2 promoter (Figure 8A). To exclude the possibility that the lack of C-Myb binding is due to inefficient immunoprecipitation of the protein, we have assayed the ChIP samples by Western. As shown in Figure 8B, while the C-Myb protein was clearly evident in the immunoprecipitates, there was no evidence for an interaction of C-Myb with the promoters assayed. To further evaluate the role of C-Myb in controlling the cdc2 gene, we have utilized an siRNA against C-Myb. As shown in Figure 8D, knocking down C-Myb does not reduce the cdc2, cyclin A2, and cyclin B1 target gene expression. FACS analysis of samples from the same experiment also confirmed that 48 h after the C-Myb siRNA transfection, the cell cycle profile was not substantially altered compared to that of cells transfected with control siRNA (Supplementary Figure 2). Taken together, C-Myb does not appear to play a role in the regulation of the cell cycle expression pattern of the endogenous cdc2 gene in T98G cells. Based on these results, we conclude that B-Myb is important for the expression of several G2/M-regulated genes that are also regulated by E2F protein, and that this coincides with an ability of B-Myb to interact with these promoters in an E2F-dependent manner.
Figure 8.
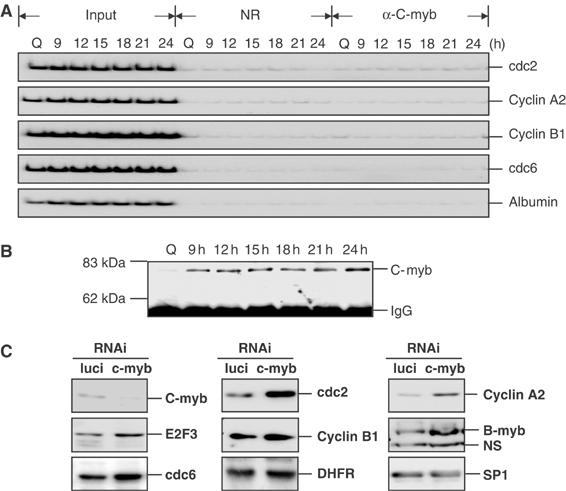
C-Myb is not required for G2/M gene expression. (A) C-Myb does not bind to the endogenous G2-regulated promoters. T98G cells were released from serum starvation and samples were harvested at quiescence (Q), or at 9, 12, 15, 18, 21, and 24 h after serum addition for ChIP analysis using the indicated antibodies. One E2F-regulated G1/S gene (cdc6) and a non-E2F-regulated gene (albumin) were used as controls. (B) Western analysis of the above C-Myb ChIP samples. The mouse monoclonal C-Myb antibody (C-2; Santa Cruz) was used to probe the Western blot. (C) C-Myb is not required for G2/M gene expression. Western analysis of several G2/M genes (cdc2, cyclin A2, and cyclin B1), G1/S genes (E2F3, B-Myb, DHFR, and cdc6), and a noncycling gene SP1 using cell extracts from asynchronously growing T98G cells transfected with either a control luciferase siRNA or a C-Myb siRNA. Cell lysates were prepared 48 h after siRNA transfection. NS=nonspecific band.
Discussion
The role of E2F activity in controlling the expression of genes critical for cell growth extends from repression in quiescent cells to activation of genes at G1/S and now the activation of genes at G2/M. In short, E2F activities play an essential role at each phase of cell growth and cell cycle progression. The mechanism of action of E2Fs at G1/S and in quiescent cells has been described in considerable detail, largely based on a number of studies that have defined essential promoter sequences and then coupled with ChIP assays that directly link specific E2F proteins with regulatory sequences (Takahashi et al, 2000; Rayman et al, 2002). This work has shown a division of function in the E2F family, with E2F4 primarily responsible for quiescent cell repression and E2F1–3 mediating activation at G1/S. The results we present here now extend this understanding to the control of genes expressed in G2, demonstrating a direct role for both positive- and negative-acting E2Fs in the control of mitotic genes.
In addition to the direct role of E2Fs in the control of G2 expression, it was also evident that distinct E2F DNA sequences are required for positive and negative control. Moreover, this coincides with specificity of E2F protein interaction whereby the repressor E2F (E2F4) is seen to bind to the negative site and the activator E2Fs (E2F1–3) bind to the positive site. A similar observation, implicating distinct E2F elements with positive or negative control and binding to distinct E2Fs, has recently been described for the E2F1 promoter (Araki et al, 2003). This is an interesting and important observation in two ways. First, the fact that little or no difference can be discerned in the sequence of the various E2F elements suggests that other aspects of the interaction of the E2F proteins, beyond simple DNA sequence recognition, likely determine the specificity of promoter interaction. Second, the distinction in the sites also might suggest an independent evolution of control by E2Fs whereby positive- and negative-acting regulation could be viewed as independent events. That is, rather than the activator and repressor E2Fs competing for a common site, these results suggest that they instead operate separately through distinct DNA elements. This is emphasized by the fact that the various E2F-regulated promoters do not share an overall conserved structure.
As indicated above, the activator and repressor E2Fs are seen to interact with distinct E2F binding sites in the cdc2 promoter. Although it is conceivable that subtle sequence differences in these sites dictate this specificity, there is little evidence to support such a mechanism. For instance, both E2F1 and E2F4 have been shown to bind to the distal E2F site in the cdc2 promoter, using in vitro binding assays (Tommasi and Pfeifer, 1995; Liu et al, 1998a). Perhaps more compelling are the findings from analysis of an E2F–DNA crystal structure that suggested little possibility for distinct DNA sequence recognition based on amino acid variation within the E2F family (Zheng et al, 1999). In contrast, the specificity of the repressor and activator E2Fs can readily be explained by specificity of interaction with other promoter-specific transcription factors. Our previous work has detailed two examples of such specificity whereby the ability of an E2F to interact with a promoter was dependent on a second transcription factor. These include a specific interaction of E2F3 with TFE3 to activate the DNA pol α p68 promoter (Giangrande et al, 2003, 2004) and E2F2 or E2F3 with RYBP/YY1 to activate cdc6 (Schlisio et al, 2002). We now extend this concept to the G2/M-regulated genes by demonstrating that the ability of each of the activator E2Fs to interact with cdc2 promoter is dependent on the CCAAT element that binds the NF-Y transcription factor. Moreover, we further extend the specificity issue to the repression of transcription by showing that the interaction of E2F4 is dependent on the CHR element, also shown to be critical for repression. As such, the ability of an activator or a repressor E2F to interact with a specific element would be dictated by the presence of the partner protein.
Finally, we note that the role of B-Myb in the control of the G2/M genes provides a mechanism to link G1/S control with the expression of G2/M genes. The B-Myb gene is known to be a target for E2F control at G1/S; thus, by virtue of the fact that G2/M genes are controlled by both E2Fs and B-Myb, a ‘feedforward loop' is established, as proposed from the analysis of yeast cell cycle control (Lee et al, 2002). As noted by Lee and colleagues, a feedforward loop provides the opportunity for temporal control of gene expression networks since the expression of the ultimate target, in this case the G2/M genes, depends on the accumulation of sufficient quantities of the primary (E2Fs) and secondary (B-Myb) regulatory proteins.
The delayed interaction of B-Myb with the cdc2 promoter, relative to the interaction of the E2F proteins, might be explained by previous Northern analyses (Takahashi et al, 2000), as well as the Western assays for B-Myb reported here, that suggest a slight delay in the accumulation of B-Myb relative to the E2F proteins. In addition, other work has implicated phosphorylation by cyclin A/cdk2 in the ability of B-Myb to activate transcription (Saville and Watson, 1998). Since cyclin A/cdk2 kinase activity accumulates after the G1/S transition, this would also impart a delay on the accumulation of functional B-Myb protein. Together, these events could provide a basis for a link between G1/S- and G2-specific transcription and also define a mechanism by which the temporal distinction is achieved.
Materials and methods
Cell culture and FACS analysis
Rat embryonic fibroblast cells (REF52) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) under 5% CO2 at 37°C. Human ganglioblastoma T98G cells (ATCC) were also grown in DMEM containing 10% FBS. Cell samples for FACS analysis were harvested by trypsinization, and then resuspended in 1 ml of nuclear isolation buffer (1 × PBS, 0.5% BSA, 0.1% NP-40) with 10 μl propidium iodide (0.5 mg/ml) and 10 μl RNase A (10 mg/ml). Flow cytometry (FACS) analysis was carried out to confirm the cell cycle synchrony (Duke Cancer Center flow cytometry facility).
Reporter plasmids and mutagenesis
pGL2hcdc2-wt, pGL2hcdc2-pE2Fm, and pGL2hcdc2-dCCAATm were generated by excising the human cdc2 promoters from pCAThcdc2-wt, pCAThcdc2-E2F4m, and pCAThcdc2-dCCAATm (Yun et al, 1999) into the pGL2-basic firefly luciferase reporter plasmid (Promega) respectively with HindIII and SalI. The HindIII site was filled in and then the promoter fragments were ligated to pGL2-basic cleaved with SmaI and XhoI. All the other mutations were generated by site-directed mutagenesis using the GeneEditor kit (Promega) following the manufacturer's instructions. Oligos used to create the distal E2F site mutation, the CHR mutation, the CHR and the proximal E2F site double mutation, and the B-MYB site mutation were ON11 (5′-CCTCTTTCTTTCGTAATCTAGCCACCC-3′), ON41 (5′-TTAGCGCGGTGAGTCGACAACTGCTCGCACTTGG-3′), ON42 (5′-CCTTTAATATTGTGAGTCGACAACTGCTCGCACTTGG CTTC-3′), and ON86 (5′-GCTGACTAGAGCTCGTAGGACGACACTCTCC-3′), respectively. pGL2upcB (Hwang et al, 1995) was kindly provided by Dr RJ Muschel (University of Pennsylvania). Oligos used to create the distal E2F site mutation and the proximal E2F site mutation were as follows: ON28 (5′-TGCGACCGGCAGCAACCAATGGGAAGGGAGTG-3′) and ON34 (5′-TAGGCTGGCTCTTCTCGTTGTGCTGCGGCGGAA-3′).
Luciferase analysis
REF52 cells were transfected using Superfect (Qiagen) following the manufacturer's instruction. The next day, transfected cells were split 1 to 3 into 24-well culture dishes, and starved in 0.1% FBS containing DMEM for 24 h. Cells were then released from starvation and blocked at the G1/S transition by the addition of DMEM containing 10% FBS and 2 mM HU for 18 h. To release from HU, cells were washed twice with DMEM, and then grown in fresh DMEM containing 10% FBS. At desired time points, cells were harvested and activities of firefly, Renilla luciferases, or β-galactosidase were analyzed following the manufacturer's instructions (Promega). pRL-Renilla or pCMV-βgal was used as an internal control for normalization.
RNA preparation and blotting
REF52 cells or T98G cells were brought to quiescence by serum deprivation for 2 days. Cells were then blocked at the G1/S transition by the addition of DMEM containing 10% FBS and 2 mM HU for 18 h, and then released into the cell cycle by the addition of fresh DMEM containing 10% FBS. At desired time points, cells were harvest for total RNA isolation using Trizol Reagent (Invitrogen). Messenger RNA was then purified using the PolyATract mRNA Isolation System IV following the manufacturer's instructions (Promega). Approximately 2 μg of mRNA for each time point was fractionated by electrophoresis in a 1.0% agarose gel under denaturing conditions, transferred to the GeneScreen membrane (NEN Life Science), and probed with 32P-labeled probes as described previously (DeGregori et al, 1995). Mouse cDNA fragments used as probes were made from image clones purchased from Resgenetics Inc.
Chromatin immunoprecipitation
T98G cells were starved in 0.1% FBS containing serum for 2 days, and then grown for 18 h in DMEM containing 10% FBS and 1 mM HU. Cells were then washed twice with DMEM, and released from HU blocking by the addition of 10% FBS medium. At each time point, cells were crosslinked in 1% formaldehyde for 15 min at room temperature. ChIP experiments were carried out as described previously (Takahashi et al, 2000) with the following modifications. Briefly, after washing with Buffer 2, isolated nuclei were lysed in 1 × RIPA buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM DTT) plus a proteinase inhibitor cocktail (1 μg/ml leupeptin, 1 mM AEBSF, 1 μg/ml pepstatin, 1 μg/ml aprotinin) at 4°C for 30 min. The chromatin material was pelleted by centrifugation at 1000 g for 10 min, resuspended in Buffer 3 and then sonicated to generate DNA fragments with an average size of 1 kb. A 2 μg portion of antibody was added to approximately 600 μg of chromatin material and collected with 20 μl of protein A/G agarose beads (Oncogen) that had been preblocked with 1% BSA and 0.1 μg/μl of poly(dIdC) (Roche). Immunoprecipitated chromatin was washed six times with 1 × RIPA buffer. The samples were then de-crosslinked at 65°C in 1 × TE and 0.5% SDS, phenol extracted, ethanol precipitated, and resuspended in 100 μl 1 × TE followed by polymerase chain reaction (PCR) in the presence of [32P]dCTP. PCR products were fractionated on 8% native polyacrylamide gels and visualized by autoradiography. Antibodies used in ChIP experiments were as follows: anti-E2F1 (sc-251), anti-E2F2 (sc-633), anti-E2F3 (sc-879), anti-E2F4 (sc-1082X), anti-NF-YA (CBF-B, sc-10779), anti-NF-YB (CBF-A, sc-13045), anti-B-Myb (a mixture of N19, sc-729 and H115, sc-13028), and anti-C-Myb (a mixture of c-2, SC-8412 and H-144, SC-7874) (Santa Cruz Biotechnology). The sequences of the primers used to amplify promoters were as follows: cdc2 (5′-GCTTGCGCTCGCAC TCAGTTGGCG-3′, 5′-CAGATCCCTGACCTCCAGTCC-3′), cyclin A2 (5′-CTGCTC AGTTTCCTTTGGTTTACC-3′, 5′-CAAAGACGCCCAGAGATGCAG-3′), cyclin B1 (5′-CGATCGCCCTGGAAACGCATTC-3′, 5′-CCAGCAGAAACCAACAGCCGTTC-3′), PCNA (5′-CTTCCTCCAATGTATGCTCTAGG-3′, 5′-AGACAACGACCACTCT GCTACG-3′), p68 (5′-GATGAACCAAGGGCACAACAGGCAG-3′, 5′-GCGTGTGG GCGTCTCTACGCACGC-3′), DHFR (5′-GGCCTCGCCTGCACAAATAG GG-3′, 5′-GGGCAGAAATCAGCAACTGGGC-3′), β-actin (5′-CAAGGCGGCCAACGCCAAA ACTCT-3′, 5′-GCCATAAAAGGCAACTTTCGGAACG-3′), and albumin (5′-TGGGG TTGACAGAAGAGAAAAGC-3′, 5′-TACATTGACAAGGTCTTGTGGAG-3′).
In C-Myb ChIP experiments, immunocomplexes were eluted from protein A/G beads in 1 × TE and 1% SDS buffer by heating at 65°C for 10 min after the final wash. In all, 50% of each sample were fractioned in 8.5% SDS–PAGE gel and subjected to Western blot using the mouse monoclonal anti-C-Myb antibody (SC-8412; Santa Cruz). The other 50% of each sample were de-crosslinked overnight and processed for PCR analysis.
Reporter chromatin immunoprecipitation
REF52 cells at 70% confluence were transfected with reporter plasmids using Superfect following the manufacturer's instructions (Qiagen). Briefly, for a 150 mm tissue culture dish, 30 μg reporter plasmid, 5 μg pRL-Renilla, and 5 μg of pCMV-βgal were mixed with 60 μl Superfect reagent and 700 μl OPTI-DMEM medium. After incubation at room temperature for 20 min, the Superfect-DNA mix was then added to each dish along with 7 ml DMEM containing 10% FBS. After approximately 6 h incubation, cells were washed with 1 × PBS and then recovered overnight in DMEM containing 10% FBS. Transfected cells were split 1 to 3 in DMEM containing 0.1% FBS to render them quiescent. They were either harvested at quiescence, or at different time points after HU blocking as described in ChIP assay (see above). The material used for reporter ChIP is the soluble material extracted with 1 × RIPA buffer after the Buffer 2 wash. The endogenous chromatin material is all in the pellet. Antibodies used in reporter ChIP experiments are described above. The sequences of primers used in this assay were as follows: cdc2 (5′-GCTTGCGCTCGCACTCAGTTGGCG-3′, a pGL2-basic-specific primer GLprimer2) (Promega), Renilla (5′-GGAAACGGATGATAACTGGTC-3′, 5′-TGCCCA TACCAATAAGGTCTGG-3′), and β-gal (5′-ACTGGCAGATGCACGGTTACGATG-3′, 5′-CACATCTGAACTTCAGCCTCCAG-3′).
RNAi transfection and Western blot analysis
T98G cells were plated at 8 × 105 on 60-mm plates a day before transfection with the siRNAs purchased from Dharmacon (Louisville, CO): human B-Myb siRNA duplex #1 (D-010444-01), B-Myb siRNA duplex #2 (D-010444-02), human C-Myb siRNA (D-003910-01), and control luciferase siRNA (P-002099-01-20). Transfections were carried out using Superfect (Qiagen) according to the manufacturer's instructions. Cells were harvested 48 h after transfection. Half of the samples were processed for FACS analysis. The other half of the samples was used to prepare the cell lysates. Approximately 60 μg of proteins was fractioned on either 10 or 15% PAGE gels for Western blotting. The blots were probed with antibodies specific for B-Myb (sc-729), E2F3 (sc-879), cdc2 (sc-54), cyclin A2 (sc-239), cyclin B1 (sc-245), cdc6 (sc-8341), C-Myb (SC-8412) (Santa Cruz Biotechnology), and DHFR (610696, BD Transduction Laboratory).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank Dr Deug Y Shin and Dr RJ Muschel for generously providing us the promoters of human cdc2 and cyclin B1 genes. We also thank Kaye Culler for help with the preparation of the manuscript. JRN is an Investigator of the Howard Hughes Medical Institute. WZ was supported by the Howard Hughes Medical Institute.
References
- Araki K, Nakajima Y, Eto K, Ikeda MA (2003) Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene 22: 7632–7641 [DOI] [PubMed] [Google Scholar]
- Dalton S (1992) Cell cycle regulation of the human cdc2 gene. EMBO J 11: 1797–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Ohtani K, Miron A, Nevins JR (1995) E2F1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity. Genes Dev 9: 2873–2887 [DOI] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245–2262 [DOI] [PubMed] [Google Scholar]
- Giangrande P, Zhu W, Rempel RE, Laakso N, Nevins JR (2004) Combinatorial gene control involving E2F and E box family members. EMBO J 23: 1336–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangrande PH, Hallstrom TC, Tunyaplin C, Calame K, Nevins JR (2003) Identification of the E box factor TFE3 as a functional partner for the E2F3 transcription factor. Mol Cell Biol 23: 3707–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KM, Watson RJ (1991) Nucleotide preferences in sequence-specific recognition of DNA by c-myb protein. Nucl Acids Res 19: 3913–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang A, Maity A, McKenna WG, Muschel RJ (1995) Cell cycle-dependent regulation of the cyclin B1 promoter. J Biol Chem 270: 28419–28424 [DOI] [PubMed] [Google Scholar]
- Hwang A, McKenna WG, Muschel RJ (1998) Cell cycle-dependent usage of transcriptional start sites. A novel mechanism for regulation of cyclin B1. J Biol Chem 273: 31505–31509 [DOI] [PubMed] [Google Scholar]
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR (2001) Role for E2F in the control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 21: 4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku D-H, Wne S-C, Engelhard A, Nicolaides NC, Lipson KE, Marino TA, Calabretta B (1993) c-myb transactivates cdc2 expression via myb binding sites in the 5′-flanking region of the human cdc2 gene. J Biol Chem 268: 2255–2259 [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804 [DOI] [PubMed] [Google Scholar]
- Liu N, Lucibello FC, Engeland K, Muller R (1998a) A new model of cell cycle-regulated transcription: repression of the cyclin A promoter by CDF-1 and anti-repression by E2F. Oncogene 16: 2957–2963 [DOI] [PubMed] [Google Scholar]
- Liu N, Lucibello FC, Zwicker J, Engeland K, Muller R (1996) Cell cycle-regulated repression of B-myb transcription: cooperation of an E2F site with a contiguous corepressor element. Nucleic Acids Res 24: 2905–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Yan H, Dawes JJ, Lu Y, Zhu H (1998b) Transcriptional activation of the p34cdc2 gene by cdc2 promoter binding factor/nuclear factor-Y in fetal rat ventricular myocytes. Circ Res 82: 251–260 [DOI] [PubMed] [Google Scholar]
- Maity SN, de Crombrugghe B (1998) Role of the CCAAT-binding protein CBF/NF-Y in transcription. Trends Biochem Sci 23: 174–178 [DOI] [PubMed] [Google Scholar]
- Mizuguchi G, Nakagoshi H, Nagase T, Nomura N, Date T, Ueno Y, Ishii S (1990) DNA binding activity and transcriptional activator function of the human B-myb protein compared with c-MYB. J Biol Chem 265: 9280–9284 [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AFA, Johnston LA, Edgar BA (1998) Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193 [DOI] [PubMed] [Google Scholar]
- Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and Retinoblastoma families. Cell Growth Differ 9: 585–593 [PubMed] [Google Scholar]
- Oh IH, Reddy EP (1999) The myb gene family in cell growth, differentiation and apoptosis. Oncogene 18: 3017–3033 [DOI] [PubMed] [Google Scholar]
- Okada M, Akimaru H, Hou D-X, Takahashi T, Ishii S (2002) Myb controls G2/M progression by inducing cyclin B expression in the Drosophila eye imaginal disc. EMBO J 21: 675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S, Kalma Y, Berkovich E, Ginsberg D (2002) E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21: 437–446 [DOI] [PubMed] [Google Scholar]
- Rayman JB, Takahashi Y, Indjeian VB, Dannenberg J-H, Catchpole S, Watson RJ, te Riele H, Dynlacht BD (2002) E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev 16: 933–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saville MK, Watson RJ (1998) The cell cycle regulated transcription factor B-Myb is phosphorylated by cyclin A/Cdk2 at sites that enhances its transactivation properties. Oncogene 17: 2679–2689 [DOI] [PubMed] [Google Scholar]
- Schlisio S, Halperin T, Vidal M, Nevins JR (2002) Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J 21: 5775–5786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, Dynlacht BD (2000) Analysis of promoter binding by the E2F and Rb families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 14: 804–816 [PMC free article] [PubMed] [Google Scholar]
- Tommasi S, Pfeifer GP (1995) In vivo structure of the human cdc2 promoter: release of a p130–E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol Cell Biol 15: 6901–6913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ginkel PR, Hsiao KM, Schjerven H, Farnham PJ (1997) E2F-mediated growth regulation requires transcription factor cooperation. J Biol Chem 272: 18367–18374 [DOI] [PubMed] [Google Scholar]
- Wasner M, Haugwitz U, Reinhard W, Tschop K, Spiesbach K, Lorenz J, Mossner J, Engleland K (2003) Three CCAAT-boxes and a single cell cycle genes homology region (CHR) are the major regulating sites for transcription from the human cyclin B2 promoter. Gene 312: 225–237 [DOI] [PubMed] [Google Scholar]
- Weston K (1998) Myb proteins in life, death and differentiation. Curr Opin Genet Dev 8: 76–81 [DOI] [PubMed] [Google Scholar]
- Yun J, Chae HD, Choy HE, Chung J, Yoo HS, Han MH, Shin DY (1999) p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J Biol Chem 274: 29677–29682 [DOI] [PubMed] [Google Scholar]
- Zheng N, Fraenkel E, Pabo CO, Pavletich NP (1999) Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev 13: 666–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker J, Gross C, Lucibello FC, Truss M, Ehlert F, Engeland K, Muller R (1995a) Cell cycle regulation of cdc2C transcription is mediated by the periodic repression of the glutamine-rich activators NF-Y and Sp1. Nucleic Acids Res 23: 3822–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker J, Lucibello FC, Wolfraij LA, Gross C, Truss M, Engeland K, Muller R (1995b) Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J 14: 4514–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker J, Muller R (1997) Cell-cycle regulation of gene expression by transcriptional repression. Trends Genet 13: 3–6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2