

Abstract
Apoptosis-inducing factor (AIF) is a mitochondrial flavoprotein that, after apoptosis induction, translocates to the nucleus where it participates in apoptotic chromatinolysis. Here, we show that human or mouse cells lacking AIF as a result of homologous recombination or small interfering RNA exhibit high lactate production and enhanced dependency on glycolytic ATP generation, due to severe reduction of respiratory chain complex I activity. Although AIF itself is not a part of complex I, AIF-deficient cells exhibit a reduced content of complex I and of its components, pointing to a role of AIF in the biogenesis and/or maintenance of this polyprotein complex. Harlequin mice with reduced AIF expression due to a retroviral insertion into the AIF gene also manifest a reduced oxidative phosphorylation (OXPHOS) in the retina and in the brain, correlating with reduced expression of complex I subunits, retinal degeneration, and neuronal defects. Altogether, these data point to a role of AIF in OXPHOS and emphasize the dual role of AIF in life and death.
Keywords: apoptosis, mitochondria, oxidative phosphorylation, programmed cell death
Introduction
Mitochondria fulfill a dual role in our cells. As the cell's powerhouses, they generate ATP by oxidative phosphorylation (OXPHOS), and constitute a central node for a cornucopia of metabolic pathways. In addition, they play an important role as suicide organelles (Green and Reed, 1998; Kroemer and Reed, 2000; Wang, 2002). Indeed, the fate of a cell driven into apoptosis is sealed when the inner and/or the outer mitochondrial membranes are permeabilized, a process that leads to the release of potentially toxic proteins and ultimately disrupts the vital bioenergetic function of mitochondria. One of the mitochondrial proteins that contributes to the apoptotic process is cytochrome c (Cyt c), a heme protein normally involved in electron shuttling between complexes III and IV of the respiratory chain (Wang, 2002). Upon apoptosis induction, Cyt c is released from the mitochondria and allosterically activates Apaf-1, thereby stimulating the apoptosome caspase activation complex. The proapoptotic function of Cyt c does not depend on its redox activity, since exchange of Fe2+ by Cu2+ in the active center of the heme group (which abolishes electron transfer) does not destroy the capacity of Cyt c to activate caspases in cell-free systems (Budijardjo et al, 1999; Wang, 2002).
Apoptosis-inducing factor (AIF) is another mitochondrial protein that can contribute to apoptosis (Cande et al, 2002; Lipton and Bossy-Wetzel, 2002). AIF is a flavoprotein with NADH oxidase activity normally contained in the mitochondrial intermembrane space (Susin et al, 1999) or loosely associated with the inner mitochondrial membrane (Arnoult et al, 2002). Upon apoptosis induction, AIF translocates from mitochondria to the cytosol and to the nucleus (Susin et al, 1999; Yu et al, 2002). In Caenorhabditis elegans, AIF cooperates with the endonuclease G (Wang et al, 2002), a DNAse that also undergoes a mitochondrio-nuclear translocation process (Li et al, 2001). In mammalian cells, AIF cooperates with cyclophilin A to become an active DNAse (Cande et al, 2004a). AIF carries negatively charged amino acids on the surface (Ye et al, 2002), allowing it to interact with DNA and to contribute to chromatin condensation and chromatinolysis, two hallmarks of apoptosis. Chemical inactivation of the flavine adenine nucleotide (FAD) moiety required for AIF redox activity does not block its apoptogenic function in cell-free systems (Miramar et al, 2001), and mutations that destroy the FAD binding site do not affect the apoptogenic function of AIF in transfection assays (Loeffler et al, 2001). Thus, the apoptogenic activity of AIF does not depend on its NADH oxidase activity. Conversely, mutations of amino-acid residues required for DNA binding (but not for redox activity) abrogate the apoptogenic potential of AIF evaluated on isolated nuclei as well as in intact cells (Ye et al, 2002).
The physiological (nonapoptotic) function of AIF in the mitochondria has been elusive. With the hope to determine this role, AIF-deficient embryonic stem (ES) cells were constructed by homologous recombination of the single AIF gene found in male cells (AIF−/y). Unfortunately, attempts to generate mice from such AIF−/y ES cells failed, precluding the in vivo assessment of the general AIF knockout and restricting the analysis to in vitro cell culture models, in which a partial apoptosis defect was detected (Joza et al, 2001). A hypomorphic mutation of AIF has been reported in the Harlequin mouse strain, which develops neurodegeneration (with ataxia due to cerebellar atrophia as a hallmark) as well as blindness due to retinal degeneration (Klein et al, 2002), two among the most commonly observed symptoms associated with OXPHOS defects in mammals. In Harlequin mice, the expression of AIF is reduced to 10–20% of the normal value due to a retroviral insertion into the first intron of the AIF gene. Increased neuronal cell death in response to oxidative stress was found in such mice (Klein et al, 2002), leading to the speculation that AIF might act as an antioxidant enzyme (Lipton and Bossy-Wetzel, 2002; Klein and Ackerman, 2003).
Driven by these premises and incognita, we decided to evaluate the potential role of AIF in OXPHOS. As shown here, the absence of AIF was found to compromise the composition and function of the respiratory chain, targeting mostly complex I, in both in vitro cell models (HeLa and mouse ES cells) and in vivo in Harlequin mice.
Results
Deficient OXPHOS in AIF-deficient cells
AIF-deficient ES cells (AIF−/y) manifest a reduced in vitro growth (with a duplication interval of 17±2 h as compared to 13±1 h in AIF+/y controls; Figure 1A), yet cause an accelerated acidification of the culture medium (Figure 1B), correlating with enhanced lactate production (Figure 1C), as compared to AIF-expressing controls. This pointed to an altered oxidative metabolism in AIF−/y cells. Complex I-dependent substrate oxidation was assessed by polarographic measurements of oxygen consumption in digitonin-permeabilized cells, upon addition of pyruvate in the presence of malate (which both furnish NADH) and ADP (which can be phosphorylated to ATP). A significant decrease (∼40%) of this complex I-dependent substrate oxidation rate was observed between AIF-deficient ES cells and control cells (Figure 2A and D). In view of the fact that oxidation ratios are accurate indicators of partial respiratory chain deficiencies (Rustin et al, 1991), we compared the complex I-dependent substrate oxidation to succinate oxidation (determined upon addition of the complex II substrate succinate, in the presence of complex I-inhibitor rotenone, ATP, and an uncoupler, m-Cl-CCP), and we consistently detected a marked (∼50%) decrease in complex I activity in AIF−/y mouse ES cells as compared to control ES cells (Figure 2A and E). Thus, complex I was deficient both relative to succinate oxidation (Figure 2A and E) and in absolute terms (Figure 2A and D). Very similar results were found in a human cell line (HeLa) in which the expression of the AIF gene was downregulated by two different small interfering RNA (siRNA) oligonucleotides (data shown for siRNA AIF1; Figure 2B, D, and E).
Figure 1.
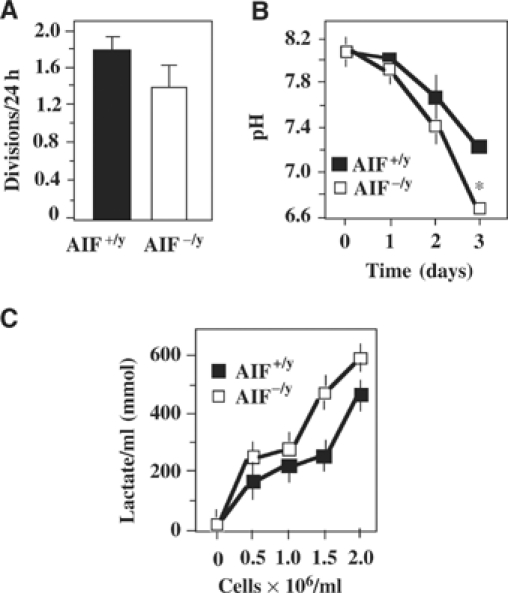
Increased lactate production and decreased complex I activity in AIF-deficient cells. (A) Number of divisions per day of AIF−/y as compared to AIF+/y ES cells. Values are given as mean±s.d. (n=12). (B) Acidification of media by AIF−/y and AIF+/y cells, as determined in nonconfluent cultures (2.5 × 105 cells/ml at day 0). (C) Lactate production by AIF−/y and AIF+/y ES cells, as evaluated at different densities (mean±s.e.m., n=3). Asterisks indicate significant (P<0.01, unpaired Student's t-test) differences between AIF−/y and AIF+/y cells.
Figure 2.
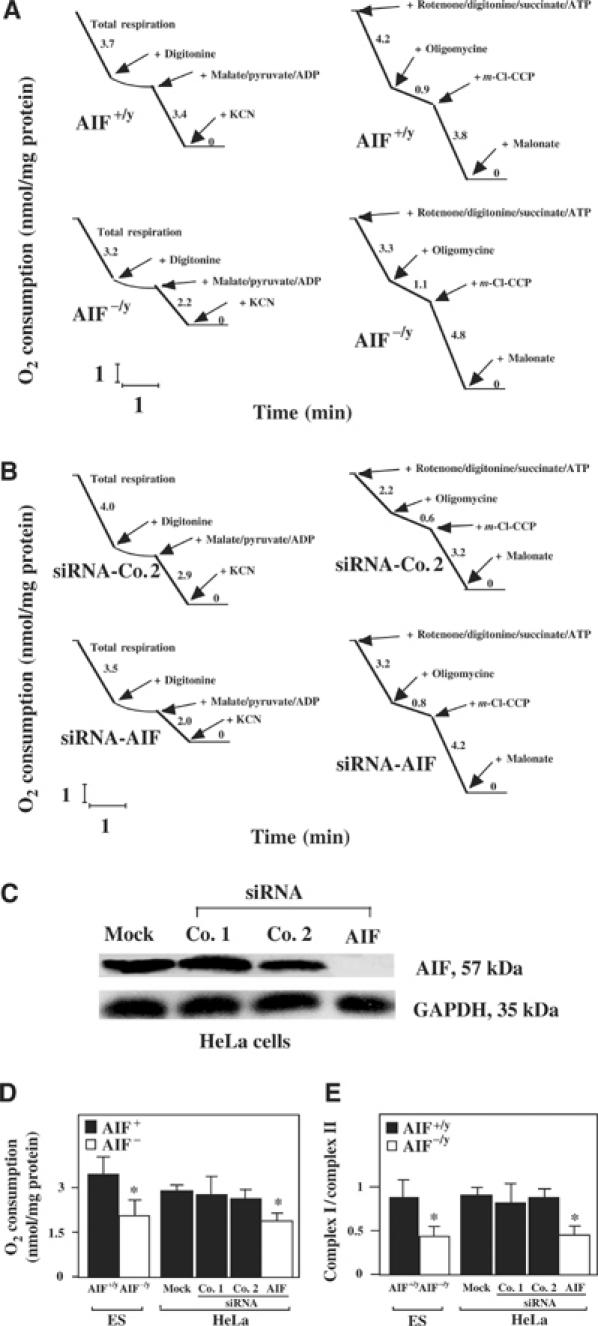
OXPHOS deficiency in AIF-negative cells. (A) AIF−/y or AIF+/y ES cells were introduced into a polarograph to monitor their oxygen consumption, permeabilized with digitonin, followed by addition of the indicated respiratory substrates and inhibitors. The results are representative of three independent experiments. (B) Control HeLa cells or cells manipulated by siRNA to lose AIF expression were analyzed as in (A), yielding similar results in three experiments. (C) Suppression of AIF expression by siRNA. Cells were mock treated or transfected with two different control siRNAs or an AIF-specific siRNA, and the abundance of AIF was determined by immunoblot 72 h later. (D) Reduced absolute O2 consumption after addition of malate, pyruvate, and ADP. (E) Reduced pyruvate plus malate oxidation (as compared to succinate oxidation) after AIF knockout in ES cells or AIF knockdown with siRNA in HeLa cells, as determined by respirometry. Asterisks in (D, E) indicate significant (P<0.01) differences between AIF-deficient cells and their controls.
Noticeably, under the conditions used for succinate oxidation rate determination, oxygen uptake is known to be limited by complex II activity, rather than by complex III or IV (Rustin et al, 1994). The relative inhibition of complex III activity by antimycin was not significantly different (more than 85%) between AIF−/y and AIF+/y cell lines, with residual activity being due to the direct chemical reduction of Cyt c by quinol in solution (Chretien et al, 2004). No indication of significant mitochondrial ubiquinone depletion could be observed, as quinone-dependent activities such as succinate cytochrome c reductase or glycerol 3-phosphate cytochrome c reductase were neither found decreased nor stimulated by exogenous quinone derivatives (not shown). The quantitation of the enzymatic activity of complexes I–V in isolated mitochondria confirmed that complex I (but not complexes II, IV, and V) exhibited a significant (P<0.001) defect (more than 70%) in AIF−/y ES cells, as well as in HeLa cells treated with AIF siRNA. In addition, complex III activity was reduced to about 50% in ES AIF−/y cells but not in AIF siRNA HeLa cells (Figure 3A). Thus, the AIF defect entails an OXPHOS deficiency that mostly affects complex I activity. This complex I defect also affected the NADH-dependent ferricyanide reductase activity, taking place at the NADH oxidation site of complex I, which is not rotenone sensitive. Again, we found a similar 50% decrease for both AIF−/y ES cells and AIF siRNA-treated HeLa cells (Figure 3B), suggesting a general defect of complex I rather than a specific catalytic dysfunction.
Figure 3.
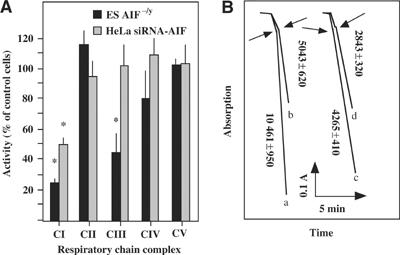
Respiratory chain complex activities in AIF-negative cells. (A) Measurements of isolated respiratory chain complexes. Mitochondrion-enriched fractions from AIF−/y, AIF+y ES cells or control siRNA and AIF siRNA HeLa cells were monitored for the activity of each of the respiratory chain complexes. Data (mean±s.d.) were ratioed to the AIF-positive controls, which were considered as 100% value. This experiment has been performed three times, in triplicate, with similar results. Asterisks denote a significant (P<0.01) AIF deficiency as compared to the control value (100%). (B) Reduced ferricyanide reduction in AIF-deficient ES and HeLa cells. As compared to ES control cells (trace a), AIF−/y ES cells (trace b) show a 50% decreased NADH-ferricyanide reductase activity. A 50% reduction of NADH-ferricyanide reductase activity was also observed in AIF siRNA HeLa cells (trace d) as compared to control HeLa cells (trace c), in three independent determinations. Arrows indicate addition of dodecylmaltoside.
Reduced complex I and III expression in AIF-deficient cells
Blue native (BN) polyacrylamide gel electrophoresis (PAGE) revealed that complex I and, to a less extent, complex III of AIF−/y ES were reduced in their abundance, as compared to AIF+/y controls (∼980 kDa) (Figure 4A). The in-gel activity of complex I was reduced, yet detectable, in AIF-deficient cells (Figure 4A). Two-dimensional gel electrophoresis (BN-PAGE followed by SDS–PAGE) revealed that all subunits of complex I from AIF−/y ES cells were reduced in their expression (Figure 4B). This applies also to complex III subunits, although the reduction is much lower (Figure 4B). Immunoblot experiments revealed that complex I subunits (e.g. the 75, 39, and 30 kDa subunits) migrated within complex I in BN-PAGE (Figure 4B), both in AIF-expressing and AIF-deficient mitochondria, although they were less abundant in the latter. AIF did not comigrate with complex I in BN-PAGE and rather migrated at a higher mobility than complex III (Supplementary Figure 1). Thus, AIF itself is not part of the complex I (nor of a higher order structure comprising complex I). SDS–PAGE followed by immunoblot (Figure 4C) revealed that the AIF knockout or knockdown resulted in the loss of complex I subunits (e.g. the 17, 20, and 39 kDa subunits, also called NDUFB6, NDUFS7, and NDUFA9, as well as the recently identified Grim19 subunit), and this was confirmed for distinct AIF−/y ES cell lines and AIF siRNA heteroduplexes (Supplementary Figure 2). We also found that subunits of complex III were deficient in AIF−/y ES cells, in particular the core subunits (gene numbers UQCRC1 and UQCRC2) and the iron–sulfur subunit (gene number UQCRFS1). A decreased expression of complex I (but not III) subunits was also detected in HeLa cells after suppression of AIF expression (Figure 4C), correlating with the respirometric data. The decreased expression of complex I subunits was a post-transcriptional phenomenon as it was not accompanied by reduced mRNA levels (Figure 4D). Retransfection of AIF−/y ES cells with full-length mouse AIF (which is imported into mitochondria) restored the expression of complex I and III subunits, while transfection with an AIF mutant lacking the mitochondrial targeting sequence (Δ1–100) failed to do so (Figure 4E). Note that the AIF defect did not affect the expression of proteins contained in a variety of different mitochondrial multienzyme complexes, namely complexes IV and V (Figure 4A and B), as well as TOM40 of the TOM complex and the VDAC protein of the permeability transition pore complex (Figure 4C). Moreover, the AIF deficiency did not lead to the disappearance of mitochondrial DNA-encoded respiratory chain subunits such as COX1, 2, and 3 (in complex IV; Figure 4B) and cytochrome b (in complex III; Figure 4B and Supplementary Figure 3) at the protein level, and mitochondrial DNA was normally transcribed in AIF-deficient cells (Figure 4F), thus excluding a gross perturbation of mitochondrial biogenesis and/or a depletion of mitochondrial DNA.
Figure 4.
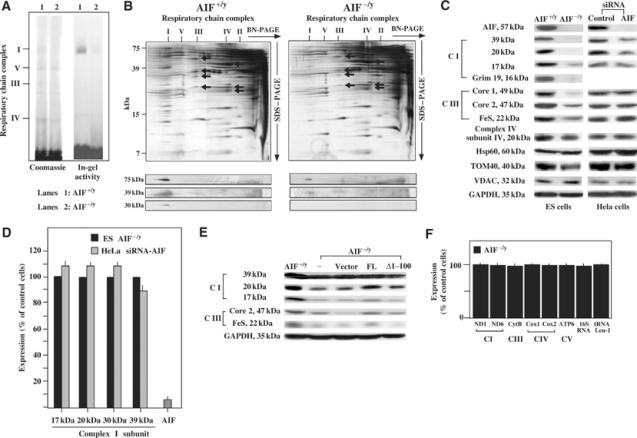
Reduced abundance of complexes I and III. (A) BN-PAGE and in-gel activity of complex I measured on isolated mitochondria from AIF−/y or AIF+/y ES cells. Dodecylmaltoside was used for solubilization and separation of the mitochondrial complexes I, V, III, and IV (I, V, III, IV) by BN-PAGE. Densitometry (normalized to complex V) revealed a relative deficiency in AIF−/y cells, as far as the abundance of complex I (<30% of control AIF+/y value) and its in-gel activity are concerned (14% of control value), a reduced abundance of complex III (54% of control), and no defect in complex IV (84%) and complex V (100%). (B) Two-dimensional resolution of OXPHOS from AIF−/y or AIF+/y ES cells. Silver-stained 2D gels, as well as their immunodecoration with antibodies specific for three complex I subunits are shown. Complex III subunits (black arrows) are core protein II, the mitochondrially encoded cytochrome b, cytochrome c1, and the ‘Rieske' iron–sulfur protein, in the order of descending mass. Complex IV subunits (white arrows) are the mitochondrially encoded subunits COX I, II, and III. (C) SDS–PAGE determination of the subunit composition of complexes I and III using a number of monoclonal antibodies. Whole cell lysates from AIF−/y or AIF+/y ES and control or AIF siRNA (siRNA-AIF1) HeLa cells were subjected to immunoblot detection of the indicated antigens. This experiment has been reproduced five times. Data were confirmed for another ES knock-out cell line and additional siRNA controls (Supplementary Figure 2). (D) Expression of nuclear DNA-encoded complex II subunits in AIF-deficient cells, as determined by RT–PCR, normalized to 18S RNA. The values are given as percentage of the control (AIF+/y ES cells for AIF−/y ES cells and HeLa cells treated with emerin-specific siRNA for HeLa cells treated with AIF-specific siRNA). (E) Expression of AIF into AIF−/y ES restores complex I expression. Cells were transfected (efficiency 12±2%) with full-length (FL) AIF, Δ1–100 AIF (which lacks the mitochondrial targeting sequence), or empty vector, and 48 h later the expression of the indicated complex I and III subunits was monitored. (F) Expression of mitochondrial RNA in AIF-negative cells. Total cellular RNA was extracted from AIF+/y and AIF−/y ES cells, and the levels of the indicated RNA species (ND1, NADH dehydrogenase subunit 1; ND6, NADH dehydrogenase subunit 6; cytB, cytochrome b; cox1, Cyt c oxidase subunit 1; cox2, Cyt c oxidase subunit 2; ATP6, ATP synthase F0 subunit 6; 16S RNA; tRNA Leu-1, tRNA Leucine 1) encoded by mitochondrial DNA were quantified by RT–PCR. The results (mean±s.e.m., n=3) were expressed as percentage of control values (100% in AIF+/y ES cells).
Absence of significant oxidative insult but enhanced glucose dependency of AIF-deficient cells
AIF-deficient and AIF-sufficient ES cells exhibit similar capacities to oxidize the fluorescent indicator dehydrorhodamine 123 (which is nonfluorescent and can be oxidized by reactive oxygen species (ROS) to fluorescent rhodamine 123) and DCF-DA (which measures cellular H2O2 production). AIF-deficient cells contain normal (or slightly elevated) levels of nonoxidized cardiolipin (measured with the fluorescent probe nonylacridine orange (NAO)), normal levels of glutathione (GSH, measured with the fluorochrome monochlorobiman), as well as normal (or slightly elevated) levels of NAD(P)H (measured by assessing its specific autofluorescence; Supplementary Figure 4A). Superoxide dismutase (SOD) activity, which can be considered as an inducible marker for increased superoxide production (Geromel et al, 2002), was found similar in AIF-deficient cells and control cells, in both ES cells and siRNA-treated HeLa cells (Supplementary Figure 4B). Thus, no evidence for oxidative insult in AIF−/y cells could be obtained, suggesting that AIF does not behave as an antioxidant protein in these cells, at least in baseline conditions. Addition of 2-D-deoxyglucose, an inhibitor of glycolysis, did not affect the viability of control ES cells, yet killed a significant fraction of AIF-deficient cells indicative of a high glucose dependency of the latter cells (Figure 5A). Adherent (nonapoptotic) AIF−/y and AIF+/y ES cells exhibited similar base line ATP levels. Upon glucose withdrawal, AIF−/y (but not AIF+/y) manifested a strong decrease in intracellular ATP levels (Figure 5B). Accordingly, glucose withdrawal compromised the growth of AIF-negative cells, yet had no effect on AIF+/y controls (Figure 5C). Fructose, known to readily fuel anaerobic glycolysis, but not galactose or sucrose, which poorly enter glycolysis (Kim et al, 2003; Mazzio and Soliman, 2003), relieved the glucose dependence of AIF−/y cells. Neither glutamine nor respiratory substrates (succinate or pyruvate) could substitute for the need of glucose (Figure 5C).
Figure 5.
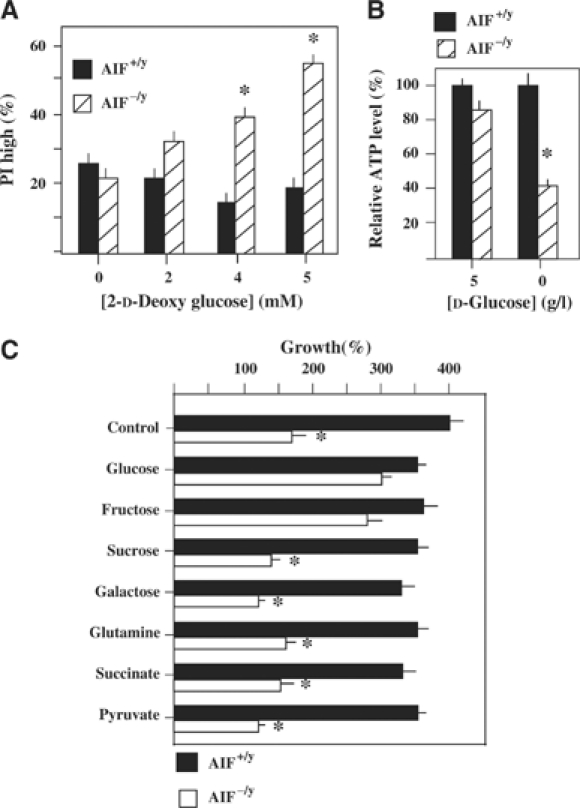
Increased dependence of AIF-deficient cells on glycolysis. (A) Increased death of AIF-deficient cells after inhibition of glycolysis by 2-D-deoxyglucose. Cells (AIF−/y or AIF+/y) were cultured for 72 h in the absence or presence of 6 mM deoxyglucose, followed by staining with propidium iodide (PI) to determine the frequency of dead cells. (B) Reduced ATP production in AIF−/y cells upon glucose withdrawal. Cells were cultured for 36 h in the absence or presence of glucose, and ATP was determined among the adherent fraction of cells. (C) Reduced growth of AIF−/y cells in the absence of glucose. AIF−/y and AIF+/y cells were cultured for 3 days in the presence (5 g/l in C) or absence (Glu−) of glucose, in the presence or absence of the indicated sugars or respiratory chain substrates (all at 5 mM), and the exact number of viable (DAPI−) cells was determined by adding FITC-labeled beads as an internal standard of the FACS analysis. The results are means of three independent determinations (mean±s.d.) and asterisks mark significant (P<0.01) effects of the AIF deficiency.
In response to arsenate, AIF+/y cells exhibited a higher NAD(P)H depletion than AIF−/y ES cells (Cande et al, 2004b), and this effect was abolished by exogenous supplementation of GSH ester or the SOD mimetic Mn(III)tetrakis(4-benzoic acid)porphyrin (MnTBAP) (Figure 6A). These antioxidant agents (as well as tocopherol and the mitochondrion-targeted antioxidant decylubiquinone) failed to cause the re-expression of complex I (as exemplified for the 20 kDa subunit) in AIF−/y ES cells (Figure 6B), although, as an additional control of their antioxidant potential, they did reduce the mitochondrial generation of ROS induced by menadione, both in AIF−/y and AIF+/y cells (Figure 6C). Supplementation with exogenous GSH was also unable to restore the growth of glucose-depleted AIF−/y cells, although it did restore the growth of menadione-treated cells (Figure 6D). Thus, although AIF is involved in NAD(P)H metabolism, this redox function is unlikely to be relevant to the OXPHOS defect that accompanies AIF deficiency.
Figure 6.
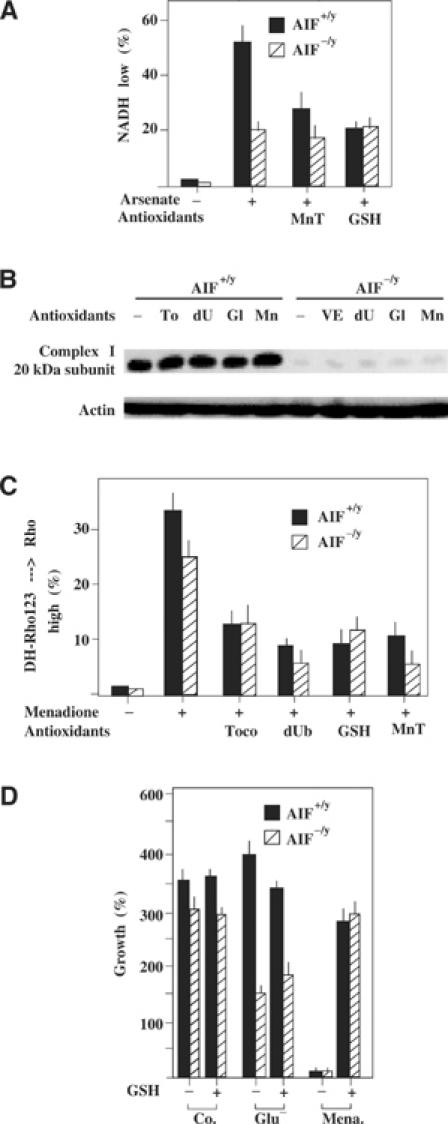
Modulation of the redox consequences of the AIF defect. (A) Enhanced NAD(P)H depletion in AIF+/y as compared to AIF−/y cells in response to arsenate. Cells were treated with 1 mM arsenate for 6 h, in the presence or absence of GSH ester (5 mM) or MnTBAP (50 μM), followed by cytofluorometric determination of cellular NAD(P)H levels. (B, C) Failure of antioxidants to cause re-expression of the 20 kDa complex I subunit. AIF−/y and AIF+/y cells were cultured for 72 h in the presence of tocopherol (200 μM), decylubiquinone (50 μM), GSH ester (10 mM), or MnTBAP (50 μM), all re-added every 12 h, followed by immunoblot (B). Alternatively, cells were treated for 3 h with menadione (100 μM) and the production of ROS was measured by assessing the oxidation of dihydrorhodamine 123 in the cytofluorometer (C). (D) Failure of the antioxidant GSH to rescue AIF−/y cells from glucose withdrawal-induced cell death. AIF−/y and AIF+/y cells were cultured for 48 h in medium without glucose and/or 10 mM GSH ethyl ester, followed by determination of viability as in Figure 5C. As an internal control of the GSH effect, AIF+/y cells were also cultured in the presence of menadione (100 μM). The values are means±s.e.m. of three independent determinations.
AIF deficiency causes a pathogenic OXPHOS deficiency in vivo
Harlequin mice manifest a degenerative disease of the retina and the central nervous system that can be attributed to a reduced (by ∼80%) expression of AIF, as a consequence of a retroviral insertion into the first intron of the AIF gene (Klein et al, 2002). Mitochondria from the retina and brain of male Harlequin mice (genotype AIFHQY) manifested a significant reduction in complex I activity as compared to isogenic controls (Figure 7A and B), a reduced abundance of complex I (as determined by BN-PAGE of brain mitochondria; Figure 7B), reduced expression of complex I subunits both in the brain and in the retina (Figure 7C), but no gross perturbation in other respiratory chain complexes (as determined by respirometry (Figure 7A) and two-dimensional gel electrophoresis of brain mitochondria (Figure 7D)). However, no deficiency of complex I composition or function could be found in the liver and in the heart of AIFHQY mice (Figure 7A and C). In conclusion, it appears that the AIF deficiency characteristic of Harlequin mice compromises complex I in failing organs (neurons and retina), presumably acting in a cell-autonomous fashion.
Figure 7.
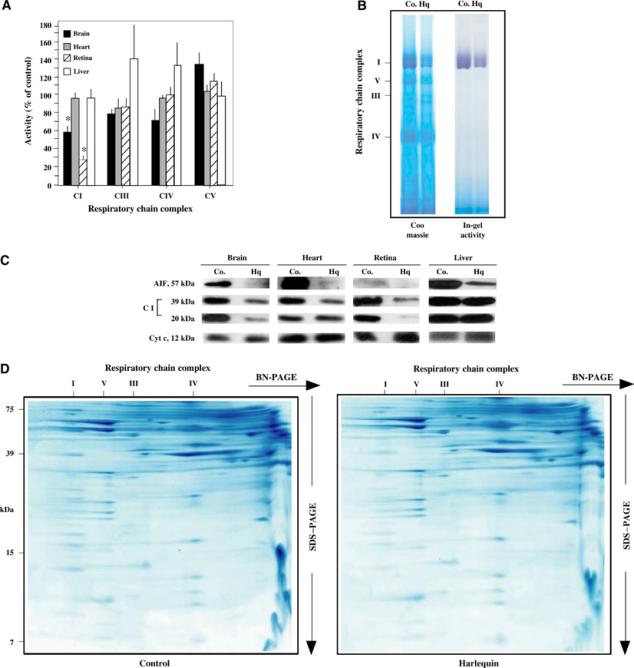
AIF deficiency compromises complex I in vivo. (A) Respiratory chain complex activities in Harlequin mice. Mitochondria from AIFHQY or AIFWTY (Co.) tissues were monitored for the activity of each of the respiratory chain complexes. Data (mean±s.d.) were ratioed to the AIF-positive controls, which were considered as 100% value. Asterisks indicate a significant (P<0.01) respiratory defect, as determined in three independent experiments. (B) BN-PAGE analysis of Harlequin brains. Mitochondria from AIFHQY or AIFWTY brain were analyzed by BN-PAGE, followed by Coomassie staining or an in-gel assay of complex I activity. Densitometric analysis (normalized on complex V) indicated that HQ have 48±4% of complex I, 101±1% of complex III, and 97±3% of complex IV control values. The in-gel activity was reduced in exact proportion to the abundance of complex I. (C) Expression of respiratory chain complex subunits in Harlequin tissues. Mitochondria were subjected to immunoblot detection of AIF and the indicated complex I subunits. Cyt c served as a loading control. (D) Two-dimensional profiles of the respiratory chains from control and Harlequin brains. The analysis was performed as in Figure 4B.
Phylogenetically conserved role of AIF in OXPHOS
Lack of growth on nonfermentable carbon sources is a defining feature of respiratory deficiencies including those in yeast (Saccharomyces cerevisiae). We tested wild-type S. cerevisiae strains and yeast cells in which the AIF ortholog was removed (Δaif1) for their ability to grow on the nonfermentable carbon sources glycerol and lactate. The Δaif1 strain exhibited a significantly longer doubling time than wild-type cells, both on glycerol media (wild type 10.2 h±1.03, Δaif1 23.2 h±2.6, P<0.001) and on lactate media (Figure 8), whereas the doubling time on rich glucose media is equal for both strains (1.3 h; not shown). Thus, the yeast ortholog of AIF is also required for optimal OXHOS.
Figure 8.
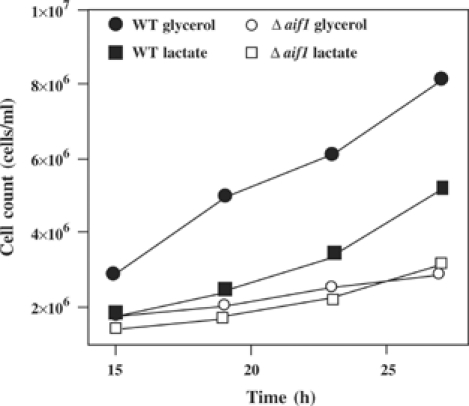
Growth of aif1Δ yeast cells and isogenic controls in lactate or glycerol media. The results are representative of five experiments. No differences were found for growth in rich medium (not shown).
Discussion
The present study reveals a new and unexpected function of the AIF protein, previously only known for its involvement in cell death. The study of both in vitro cell models (siRNA-treated HeLa cells and AIF-KO ES cells) and in vivo AIF-deficient mice (Harlequin mice) establishes that AIF is required for the correct assembly and/or maintenance of the respiratory chain complex I. Presumably, because complex I is also constitutive of a I/III/IV ‘supercomplex', the so-called respirasome (Schägger, 2002), complex I deficiency can be associated with additional, but limited, deficiencies affecting other respiratory chain complexes. How AIF contributes to biogenesis and/or protection of these respiratory complexes is a conundrum, but it appears clear that AIF itself is not part of complex I or of higher-order complexes such as the ‘supercomplexes', within the limits of resolution of BN-PAGE (Supplementary Figure 1). Accordingly, the absence of AIF also compromised OXPHOS in S. cerevisiae (which does not possess a full complex I), indicating a general, pleiotropic role of AIF in the assembly and/or maintenance of respiratory complexes. AIF is an NAD(P)H oxidase (Miramar et al, 2001), and the AIF knockdown or knock-out compromises the utilization of NAD(P)H and the maintenance of GSH in oxidant-stressed cells (Cande et al, 2004b). However, supplementation with exogenous GSH (or addition of the SOD mimetic MnTBAP), which compensates for the AIF effect on NAD(P)H levels (Figure 6A), does not restore defective respiration of AIF-negative cells (Figure 6B and D), indicating that, in principle, defective detoxification of ROS is unlikely to account for the defective assembly of complex I. AIF does not affect the transcription of nuclear-encoded complex I subunits (which is normal in AIF-deficient cells; Figure 4D). Moreover, the lack of AIF does not induce gross perturbation in mitochondria, as indicated by their normal ultrastructure in AIF-KO ES cells (not shown), the normal function and assembly of respiratory chain complexes II, IV, and V (Figures 3A and 4B), and the normal presence of proteins from the permeability transition pore complex (PTPC) and the translocator of the outer mitochondrial membrane (TOM) complex (Figure 4C). AIF-KO cells also produce mitochondrial DNA-encoded subunits of the respiratory chain (Figure 4B) and exhibit normal expression of mitochondrial DNA-encoded mRNA species (Figure 4F), meaning that, in principle, mitochondrial biogenesis is intact. Rather, the AIF-induced defect is likely to concern the intramitochondrial assembly/maintenance of respiratory chain complexes, specifically complex I and to a lower extent complex III. Note that we did not detect the generation of incompletely assembled subcomplexes (Figure 4B). Rather, the abundance of the complete complex I (including all subunits) was reduced in AIF-negative cells (Figure 4A–C), correlating with a reduction in its enzymatic activities (rotenone sensitive in Figures 2 and 3A, and rotenone insensitive in Figure 3B).
Surprisingly, the partial AIF deficiency of AIFHQY mice only affects the brain and retina, yet does not compromise OXPHOS in the heart and liver (Figure 7A and C). This points to a hitherto unexplained organ-specific role of AIF with regard to OXPHOS function. Indeed, it has been observed that loss-of-function mutations of several assembly factors result in tissue-specific OXPHOS deficiencies (Valnot et al, 2000; Jaksch et al, 2001; Shoubridge, 2001; Budde et al, 2003). The molecular mechanisms accounting for this tissue specificity, however, remain entirely elusive. Perhaps due to the intrinsic complexity of assembling respiratory chain complexes (e.g. ∼46 subunits in complex I) (Hirst et al, 2003), there is little knowledge on how exactly assembly factors work (Pecina et al, 2004; Yadava et al, 2004), and AIF is no exception to this rule. It will be necessary to acquire further knowledge on the precise hierarchy through which each of the nascent subunits interact, refold, and self-assemble to generate multipartite building blocks (Vogel et al, 2004) before the detailed mechanisms through which the AIF defect compromises complex I assembly or maintenance can be further explored. We have addressed the question of whether threshold effects may explain the impact of AIF deficiency of different organs of Harlequin mice. While the heart of Harlequin mice (which expresses ∼20% of normal AIF values) (Klein et al, 2002; Figure 7C) has no OXPHOS deficiency (Figure 7A and C), a heart-specific knockout of AIF, leading to the complete elimination of AIF expression from cardiomyocytes, induced a marked reduction of complex I and a dilated cardiomyopathy (Joza et al, submitted). Thus, once below a critical threshold, which may be organ specific, the AIF defect causes an OXPHOS deficiency in many if not all cell types.
Clinically relevant complex I deficiencies can be attributed in about 40% of the cases to mutations in the seven mitochondrion-encoded and seven of the 38 nuclear-encoded complex I subunits (Bénit et al, 2003; Hirst et al, 2003). Based on the speculation that AIF mutations might lead to a complex I deficiency, we have sequenced the AIF gene from 90 patients with confirmed isolated complex I deficiency, yet lacking molecular diagnosis. This analysis failed to identify nonconservative AIF mutations (data not shown). In view of the pleiotropic effect of AIF, in vivo, an inadequate selection of patients can also explain that no mutation could be found in our cohort of isolated complex I-deficient patients. On the other hand, failure to detect structural AIF mutations may reflect embryonic lethality of any major AIF deficiency, presumably resulting from a teratogenic apoptosis defect and/or from a respiratory dysfunction (Joza et al, 2001; Wang et al, 2002). Irrespective of this speculation, the fact that an AIF defect would lead to an OXPHOS dysfunction may explain the pathology of Harlequin mice. Indeed, once below a critical threshold (Villani et al, 1998), OXPHOS deficiencies lead to cell death, in particular in those cells that heavily rely on mitochondrial ATP generation. Thus, in patients, complex I deficiencies have been related to a variety of clinical signs, including neurodegeneration with ataxia and progressive retinal degeneration (Orth and Schapira, 2001; Triepels et al, 2001; DiMauro and Schon, 2003). These are also the clinical and biochemical signs characterizing the Harlequin mouse strain (which underexpress AIF) (Klein et al, 2002), allowing for an effective reinterpretation of the Harlequin phenotype. The discovery of a severe OXPHOS deficiency in the Harlequin mouse provides a simple explanation for the oxidative insult previously reported in this mouse (Klein et al, 2002). Indeed primary defects of the respiratory chain entail tissue-specific oxidative stress in vivo (Larsson and Rustin, 2001). In contrast, genetically engineered mouse models with primary defects in the mitochondrial antioxidant defence, that is, SOD2 or frataxin knockout mice (Melov et al, 1999; Puccio et al, 2001), present a characteristic loss of activity of the oxygen-sensitive iron–sulfur cluster-containing proteins, especially complex II, which was unaffected in the Harlequin mouse. This makes the Harlequin mouse a faithful model of human complex I disorders. The increased H2O2 sensitivity of cerebellar granule cells in AIFHQY mice (Klein et al, 2002) or that of ES cells lacking AIF (data not shown) is likely to be secondary to the complex I defect, because complex I may be involved in the detoxification of exogenous H2O2 (Atorino et al, 2003; Zoccarato et al, 2004).
In conclusion, the present data underscore the notion that the building blocks of the apoptotic machinery have normal functions not related to cell death (Jones et al, 2003). This applies to AIF, a Janus-like molecule with apoptogenic properties, which, in addition, is required for normal OXPHOS. This particular design may prevent the loss of essential parts of the apoptotic executioner (because total loss of AIF leads to cell death in energy-demanding cells) and thus may reduce the probability of oncogenic mutations that invalidate the apoptotic program.
Materials and methods
Cells, culture conditions, transfection, and siRNA
HeLa and ES cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 1 mM sodium pyruvate, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. ES cell culture medium was further supplemented with leukemia inhibitory factor (LIF; 1 μg/l) and 50 μM β-mercaptoethanol. Unless specified, the AIF−/y(1) cell line (Joza et al, 2001) was used. For transfection, HeLa cells were plated in Opti-MEM supplemented with 10% FCS in 12-well plates with a cell density of 4 × 105 cells per well and transfected the next day with 1.2 μg of the siRNA duplex ‘control' (Co. 1 specific for emerin 5′-CCGUGCUCCUGGGGCUGGGdTdT-3′ or Co. 2 specific for murine, not human AIF: 5′-AUGCAGAACUCCAAGCACGdTdT-3′) or ‘AIF' (siRNA-AIF1: 5′-GAUCCUCCCCGAAUACCUCdTdT-3′, siRNA-AIF2: 5′-CUUGUUCCAGCGAUGGCAUdTdT-3′) in 4 μl oligofectamine (Invitrogen). Cells were washed the next day and incubated for another 2–4 days before analysis. ES cells (5 × 106) were transfected by electroporation with 50 μg of mouse AIF cDNA cloned into pcDNA3.1/HisV5 in a Gene Pulser Xcell Electroporation System (Bio-Rad; 250 V, 500 μF) and analyzed after 48 h. Mice carrying the Harlequin mutation were purchased from the Jackson laboratory and analyzed at 10 weeks of age.
Determination of lactate and ATP
Lactate content in culture medium was determined by measuring the oxidized 2,6-dichlorophenol-indophenol (DCPIP), which is reduced by phenazine methosulfate (PMS), which in turn is reduced by NADH produced by lactate dehydrogenase (LDH), which oxidizes at the same time lactate to pyruvate. The diluted culture medium was analyzed in buffer S (10 mM KH2PO4, pH 7.8, 2 mM EDTA, 1 mg/ml bovine serum albumin (BSA), 0.06 mM DCPIP, 0.5 mM PMS, 0.8 mM NAD+, 1.5 mM glutamate, 10 U/ml glutamate-pyruvate-transaminase (GPT), 25 U/ml LDH) and DCPIP oxidation was spectrophotometrically measured at 600 nm. Intracellular ATP levels were measured with the Bioluminescence Assay Kit HS II (Roche). ES cells were trypsinized after 36 h incubation time in culture medium with (5 g/l) or without glucose. The cells were washed once in PBS and resuspended in the kit's dilution buffer.
Cytofluorometry
To assess cell death, ES cells were trypsinized and stained with PI (5 μg/ml) or 4′,6-diamino-2-phenylindole-dihydrochloride (DAPI, 5 μg/ml) and incubated at 37°C for 20 or 5 min, respectively, followed by cytofluorimetric analysis. For the cell count assay, the same amount of FITC-labeled beads (Becton Dickinson) was added to each sample and cells were counted until 1000 beads had passed through the FACS®. The following fluorochromes were employed to assess the redox state of the cells: dihydrorhodamine (50 ng/ml, 30 min incubation) for ROS detection, NAO (500 nM, 20 min incubation) to determine the level of nonoxidized cardiolipin, monochlorobiman (50 μM, 30 min incubation) to determine the level of GSH, and 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, 5 μM, incubation 60 min) to measure peroxides. For all fluorochromes, cells were trypsinized and labeled with the fluorochromes at 37°C followed by cytofluorimetric analysis.
Polarographic and spectrophotometric studies
Polarographic studies of intact cell respiration and of mitochondrial substrate oxidation by digitonin (0.004%)-permeabilized cells (30–50 μg protein) were carried out in a 250 μl chamber equipped with a Clark electrode (Hansatech Instruments Ltd, Norfolk, UK) containing 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, 1 mg/ml BSA, and 10 mM KH2PO4 (pH 7.4) (Rustin et al, 1994). Respiratory chain complex I–V and citrate synthase activities were measured on cells spun down (2500 g × 5 min) through a digitonin (0.01%) and Percoll (5%) solution as described (Chretien et al, 2003). The intactness of the mitochondrial outer membrane of digitonin-treated cells, assessed by their impermeability to exogenous Cyt c, was consistently higher than 95%. Rotenone-sensitive NADH quinone reductase (complex I; EC 1.6.5.3), malonate-sensitive succinate quinone DCPIP reductase (complex II; EC 1.3.99.1), antimycin-sensitive quinol cytochrome c reductase (complex III; EC 1.10.2.2), cyanide-sensitive cytochrome c oxidase (complex IV; EC 1.9.3.1), oligomycin-sensitive ATP hydrolase (complex V; EC 3.6.3.14), and citrate synthase (EC 4.1.3.7) were spectrophotometrically measured using a dual-wavelength spectrophotometer (DW 2000 Aminco/SLM, SLM Instruments, Inc., Urbana, IL) using standard procedures (Rustin et al, 1994). The quinone derivative used to measure complex III was decylubiquinol (Rustin et al, 1994). All measurements were performed at 37°C. NADH-ferricyanide reduction was measured spectrophotometrically at 37°C in a 1 ml cuvette containing 10 mM Tris–HCl (pH 8.0) and 1 mg/ml BSA. Dodecylmaltoside (0.75 mM) was added to disrupt cell membranes (Rustin et al, 1994). Protein levels were determined by the method of Bradford using BSA as a standard. All chemicals were of analytical reagent grade from Sigma Chemical Company. Room temperature difference spectra (either succinate-antimycin minus aerobic, or dithionite reduced minus aerobic) were recorded with a DW-2000 Aminco/SLM spectrophotometer in 1 ml cuvettes containing 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, 1 mg/ml BSA, 10 mM KH2PO4 (pH 7.4), and 520 μg of total cellular protein (Lance and Bonner, 1968).
Biochemical analysis of OXPHOS complexes
Heart muscle, brain tissue, and sedimented ES cells (10–50 mg wet weight each) were homogenized with 500 μl sucrose buffer (250 mM sucrose, 20 mM Na-phosphate (pH 7.2) for tissue specimens, and 83 mM sucrose, 7 mM Na-phosphate (pH 7.2) for ES cells). The homogenates were divided into aliquots corresponding to 5 mg heart muscle or brain tissue, or 10 mg ES cells, centrifuged for 10 min at 10 000 g, and the mitochondria-containing pellets were stored at −80°C. Pellets were resuspended with 20 μl buffer (50 mM NaCl, 50 mM imidazole, 2 mM 6-aminohexanoic acid, 1 mM EDTA, pH 7.0), and the OXPHOS complexes solubilized by adding 5 μl dodecylmaltoside (10%) to heart and brain samples, and 10 μl dodecylmaltoside (10%) to ES cell samples. For the isolation of respiratory chain supercomplexes, the same volumes of digitonin (20%) were used instead of dodecylmaltoside (10%). The mixtures were centrifuged at 100 000 g for 15 min, and the supernatant was supplemented with Coomassie blue-G250 dye (5% stock in 750 mM 6-aminohexanoic acid) to set a detergent/Coomassie-dye ratio of 4 (g/g). This supernatant was loaded onto 5 × 1.5 mm sample wells for BN-PAGE using 4–13% acrylamide gradient gels as described (Schägger, 2003a). The catalytic activity of complex I was determined in BN gels according to Zerbetto et al (1997). For resolution of the subunits of mitochondrial complexes, lanes from the BN gel were cut out and prepared for two-dimensional SDS–PAGE as described (Schägger, 2003a; 2003b).
Immunoblots
Cells were trypsinized, washed once in PBS and lysed (50 mM Tris pH 6.8, 10% glycerol, 2% SDS, 0.005% bromophenol blue, 0.1 mM DTT), and treated at 100°C for 10 min. Samples were resolved in SDS–12% PAGE and transferred to a nitrocellulose membrane. The membranes were blocked for 1 h in PBS/Tween 0.01%/skim milk 5% and then incubated for 1–3 h with different antibodies (AIF 1:1000, Chemicon; complex I subunits, 39 kDa 1:1000, 30, 20, and 17 kDa, all 1:200; complex III subunits core 1, core 2, both 1:1000, FeS 1:200, complex IV subunit IV 1:1000, all Molecular Probes; Hsp60 1:1000, Strenger; VDAC 1:1000, Calbiochem; Grim-19 1:500, Abcam; GAPDH 1:10 000, Chemicon; actin 1:1000, Chemicon; TOM40 1:200, Santa Cruz). A polyclonal rabbit antibody against bovine complex I was used to detect the 75 kDa subunit of complex I (dilution 1:2500). Membranes were washed in PBS/Tween and then incubated for 1 h with appropriate peroxidase-conjugated secondary antibodies (1:5000–1:10 000). The membranes were again washed and then developed by an enhanced chemiluminescence system (Pierce).
Quantitative RT–PCR
cDNAs were synthesized from 1 μg total RNAs with reverse transcriptase MuLV (Applied Biosystems). Then, the TaqMan universal PCR was performed on an ABI PRISM® 7000 Sequence Detection System (Applied Biosystems), following the manufacturer's instructions. The primers and the corresponding Taqman-MGB probes for the mouse and human genes NDUFS7, NDUFA9, NDUFS3, and NDUFB6, and the human gene AIF were purchased from Applied Biosystems as assays-on-demand products (www.Appliedbiosystems.com). For the mouse mitochondrial DNA-encoded genes, the following primers and Taqman probes were purchased from MWG-Biotech AG:
ND1 (forward: 5′-CTCAACCTAGCAGAAACAAACC-3′,
reverse: 5′-GGC CGGCTGCGTATTCTAC-3′,
probe: 5′-CCCCCTTCGACCTGACAGAAGGAGA-3′),
ND6 (forward: 5′-TTGGGAGATTGGTTGATGTAT-3′,
reverse: 5′-TGCCGCTACCCCAATCC-3′,
probe: 5′-ATGATGTTGGAGTTATGTTGGAAGGA-3′),
Cox1 (forward: 5′-TCAGTATCGTATGCTTCAACAAATTTAGA-3′,
reverse: 5′-TGGTTCCTCGAATGTGTGATATG-3′,
probe: 5′-TGACTTCATGGCTGCCCTCC-3′),
Cox2 (forward: 5′-GAGCAGTCCCCTCCCTAGGA-3′,
reverse: 5′-GTCGGTTTGATGTTACTGTTGCTT-3′,
probe: 5′-ATGCCATCCCAGGCCGACTAAAT-3′),
cytochrome b (forward: 5′-AAAGCCACCTTGACCCGATT-3′,
reverse: 5′-GATTCGTAGGGCCGCGATA-3′,
probe: 5′-CGCTTTCCACTTCATCTTACCATT-3′),
ATPase 6 (forward: 5′-TCGTTGTAGCCATCATTATATTTCCT-3′,
reverse: 5′-GAAAGAATGGAGACGGTTGTTGA-3′,
probe: 5′-CAATCCTATTCCCATCCTCAAAACGCCT-3′),
16S RNA (forward: 5′-TGCCTGCCCAGTGACTAAAGT-3′,
reverse: 5′-AACAAGTGATTATGCTACCTTTGCA-3′,
probe: 5′-TAACGGCCGCGGTATCCTGA-3′),
tRNA Leucine 1 (forward: 5′-GGTGGCAGACGGAGGAAA-3′,
reverse: 5′-TATTAGGGAGAGGATTTGAACCT-3′,
probe: 5′-TCGGTAAGACTTAAAACCTTGTTCCC-3′).
Assessment of AIF in yeast
Experiments were carried out in S. cerevisiae BY4741 cells from Euroscarf. Yeast strains BY4741 and Δaif1 (YNR074C) (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) described by Wissing et al (2004) were grown on SC medium containing 0.17% yeast nitrogen base (Difco), 0.5% (NH4)2SO4, 30 mg/l of all amino acids (except 80 mg/l histidine and 200 mg/l leucine), 30 mg/l adenine, 320 mg/l uracil and either 2% glucose, 2% glycerol, 3% ethanol or 2% sodium lactate as carbon source. Yeasts for experiments on nonfermentable media were streaked out on full media, grown for 2 days, and then stored for 3 days at 4°C. By this procedure, the cells remain mainly in G0 and diminished their fermentable carbon sources. Cells were then inoculated to an OD600 of 0.1 on liquid medium containing only nonfermentable carbon sources.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Material
Acknowledgments
We thank Didier Métivier, Brigitte Raynal, and Thierry Ragot (IGR, Villejuif) for their expert help. This work has been supported by a special grant from LNC, and grants from ANRS, FRM, and the European Commission (QLG1-CT-1999-00739 and QLK3-CT-20002-01956) (to GK). NV receives a fellowship from ARC.
References
- Arnoult D, Parone P, Martinou J-C, Antonsson B, Estaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis inducing factor occurs downstream of cytochrome c release in response to several pro-apoptotic stimuli. J Cell Biol 59: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G (2003) Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 163: 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit P, Lebon S, Chol M, Giurgea I, Rötig A, Rustin P (2003) Mitochondrial NADH oxidation deficiency in humans. Curr Genomics 5: 137–146 [Google Scholar]
- Budde SM, van den Heuvel LP, Smeets RJ, Skladal D, Mayr JA, Boelen C, Petruzzella V, Papa S, Smeitink JA (2003) Clinical heterogeneity in patients with mutations in the NDUFS4 gene of mitochondrial complex I. J Inherit Metab Dis 26: 813–815 [DOI] [PubMed] [Google Scholar]
- Budijardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15: 269–290 [DOI] [PubMed] [Google Scholar]
- Cande C, Cecconi F, Dessen P, Kroemer G (2002) Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci 115: 4727–4734 [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G (2004a) AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 23: 1514–1521 [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Metivier D, Tourriere H, Garrido C, Tazi J, Kroemer G (2004b) Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J Cell Sci 117: 4461–4468 [DOI] [PubMed] [Google Scholar]
- Chretien D, Benit P, Chol M, Lebon S, Rotig A, Munnich A, Rustin P (2003) Assay of mitochondrial respiratory chain complex I in human lymphocytes and cultured skin fibroblasts. Biochem Biophys Res Commun 301: 222–224 [DOI] [PubMed] [Google Scholar]
- Chretien D, Slama A, Brière JJ, Munnich A, Rötig A, P R (2004) Revisiting pitfalls, problems and tentative solutions for assaying mitochondrial respiratory chain complex III in human samples. Curr Med Chem 11: 233–239 [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348: 2656–2668 [DOI] [PubMed] [Google Scholar]
- Geromel V, Darin N, Chretien D, Benit P, DeLonlay P, Rotig A, Munnich A, Rustin P (2002) Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol Genet Metab 77: 21–30 [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309–1312 [DOI] [PubMed] [Google Scholar]
- Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE (2003) The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta 1604: 135–150 [DOI] [PubMed] [Google Scholar]
- Jaksch M, Paret C, Stucka R, Horn N, Muller-Hocker J, Horvath R, Trepesch N, Stecker G, Freisinger P, Thirion C, Muller J, Lunkwitz R, Rodel G, Shoubridge EA, Lochmuller H (2001) Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum Mol Genet 10: 3025–3035 [DOI] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES (2003) Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425: 721–727 [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CYJ, Sasaki T, Elia AJ, Cheng H-YM, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong Y-Y, Zuñiga-Pflücker JC, Kroemer G, Penninger JM (2001) Essential role of the mitochondrial apoptosis inducing factor in programmed cell death. Nature 410: 549–554 [DOI] [PubMed] [Google Scholar]
- Kim J-S, He L, Lemasters JJ (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun 304: 463–470 [DOI] [PubMed] [Google Scholar]
- Klein JA, Ackerman SL (2003) Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest 111: 785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419: 367–374 [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6: 513–519 [DOI] [PubMed] [Google Scholar]
- Lance C, Bonner WD (1968) The respiratory chain components of higher plant mitochondria. Plant Physiol 43: 756–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson N, Rustin P. (2001) Animal models for respiratory chain disease. Trends Mol Med 7: 578–581 [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412: 95–99 [DOI] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel E (2002) Dueling activities of AIF in cell death versus survival. DNA binding and redox activity. Cell 111: 147–150 [DOI] [PubMed] [Google Scholar]
- Loeffler M, Daugas E, Susin SA, Zamzami N, Métivier D, Nieminen A-L, Brothers G, Penninger JM, Kroemer G (2001) Dominant cell death induction by extramitochondrially targeted apoptosis inducing factor. FASEB J 15: 758–767 [DOI] [PubMed] [Google Scholar]
- Mazzio E, Soliman KF (2003) The role of glycolysis and gluconeogenesis in the cytoprotection of neuroblastoma cells against 1-methyl 4-phenylpyridinium ion toxicity. Neurotoxicology 24: 137–147 [DOI] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC (1999) Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 96: 846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA (2001) NADH-oxidase activity of mitochondrial apoptosis inducing factor (AIF). J Biol Chem 276: 16391–16398 [DOI] [PubMed] [Google Scholar]
- Orth M, Schapira AH (2001) Mitochondria and degenerative disorders. Am J Med Genet 106: 27–36 [DOI] [PubMed] [Google Scholar]
- Pecina P, Houstkova H, Hansikova H, Zeman J, Houstek J (2004) Genetic defects of cytochrome c oxidase assembly. Physiol Res 53: S213–S223 [PubMed] [Google Scholar]
- Puccio H, Simon D, Cossée M, Criqui-Filippe P, Tiziano F, Melki J, Kahn R, Hindelang C, Matyas R, Rustin P, Koenig M (2001) Mouse models for Friedrich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet 27: 181–186 [DOI] [PubMed] [Google Scholar]
- Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chem Acta 228: 35–51 [DOI] [PubMed] [Google Scholar]
- Rustin P, Chretien D, Bourgeron T, Wucher A, Saudubray JM, Rotig A, Munnich A (1991) Assessment of the mitochondrial respiratory chain. Lancet 338: 60. [DOI] [PubMed] [Google Scholar]
- Schägger H (2002) Respiratory chain supercomplexes of mitochondria and bacteria. Biochim Biophys Acta 1555: 154–159 [DOI] [PubMed] [Google Scholar]
- Schägger H (2003a) Blue native electrophoresis. In Membrane Protein Purification and Crystallization, Hunte C, von Jagow G, Schägger H (eds) pp 105–129. San Diego, CA: Academic Press [Google Scholar]
- Schägger H (2003b) SDS electrophoresis techniques. In Membrane Protein Purification and Crystallization, Hunte C, von Jagow G, Schägger H (eds) pp 85–102. San Diego, CA: Academic Press [Google Scholar]
- Shoubridge EA (2001) Nuclear genetic defects of oxidative phosphorylation. Hum Mol Genet 10: 2277–2284 [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446 [DOI] [PubMed] [Google Scholar]
- Triepels RH, van den Heuvel LP, Trijbels JM, Smeitink JA (2001) Respiratory chain complex I deficiency. Am J Med Genet 106: 37–45 [DOI] [PubMed] [Google Scholar]
- Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, Rotig A (2000) Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase (COX) deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet 67: 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani G, Greco M, Papa S, Attardi G (1998) Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J Biol Chem 273: 31829–31836 [DOI] [PubMed] [Google Scholar]
- Vogel R, Nijtmans L, Ugalde C, van den Heuvel L, Smeitink J (2004) Complex I assembly: a puzzling problem. Curr Opin Neurol 17: 179–186 [DOI] [PubMed] [Google Scholar]
- Wang X (2002) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933 [PubMed] [Google Scholar]
- Wang X, Yang C, Chai J, Shi Y, Xue D (2002) Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298: 1587–1592 [DOI] [PubMed] [Google Scholar]
- Wissing S, Ludovico P, Herker E, Büttner S, Engelhardt SM, Decker T, Link A, Proksch A, Rodrigues F, Corte-Real M, Fröhlich K, Manns J, Candé C, Sigrist SJ, Kroemer G, Madeo F (2004) An AIF orthologue regulates apoptosis in yeast. J Cell Biol 166: 969–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava N, Houchens T, Potluri P, Scheffler IE (2004) Development and characterization of a conditional mitochondrial complex I assembly system. J Biol Chem 279: 12406–12413 [DOI] [PubMed] [Google Scholar]
- Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H (2002) DNA binding as a structural requirement for the apoptogenic action of AIF. Nat Struct Biol 9: 680–684 [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297: 259–263 [DOI] [PubMed] [Google Scholar]
- Zerbetto E, Vergani L, Dabbeni-Sala F (1997) Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis 18: 2059–2064 [DOI] [PubMed] [Google Scholar]
- Zoccarato F, Cavallini L, Alexandre A (2004) Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+. J Biol Chem 279: 4166–4174 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Material