

Abstract
Failure to promptly dispose of undesirable proteins is associated with numerous diseases. In the case of cellular prion protein (PrP), inhibition of the proteasome pathway can generate a highly aggregation-prone, cytotoxic form of PrP implicated in neurodegeneration. However, the predominant mechanisms that result in delivery of PrP, ordinarily targeted to the secretory pathway, to cytosolic proteasomes have been unclear. By accurately measuring the in vivo fidelity of protein translocation into the endoplasmic reticulum (ER), we reveal a slight inefficiency in PrP signal sequence function that generates proteasomally degraded cytosolic PrP. Attenuating this source of cytosolic PrP completely eliminates the dependence on proteasomes for PrP degradation. This allows cells to tolerate both higher expression levels and decreased proteasomal capacity without succumbing to the adverse consequences of misfolded PrP. Thus, the generation of potentially toxic cytosolic PrP is controlled primarily during its initial translocation into the ER. These results suggest that a substantial proportion of the cell's constitutive proteasomal burden may consist of proteins that, like PrP, fail to cotranslationally enter the secretory pathway with high fidelity.
Keywords: cytotoxicity, prion protein, proteasomal degradation, protein aggregation, protein translocation
Introduction
The mammalian prion protein (PrP) is a cell surface glycoprotein whose misfolding is associated with both the transmission and pathogenesis of a variety of neurodegenerative diseases (reviewed by Prusiner, 2001; Aguzzi and Haass, 2003). These diseases, which include Creutzfeldt–Jakob disease (CJD) and Gerstmann–Straussler–Scheinker (GSS) disease in humans, are characterized by a progressive spongiform degeneration of neurons accompanied in many cases by the deposition of PrP-containing aggregates in selected regions of the brain. In most, but not all instances of neurodegeneration associated with PrP, the disease is transmissible. The transmissible agent is predominantly composed of a misfolded conformer of PrP termed PrPSc. While the identification of the transmissible agent and its mechanism of propagation have been the subjects of intense study, relatively little is understood about how changes in PrP folding and/or metabolism can lead to neuronal dysfunction and death.
Increasingly however, it is becoming clear that the accumulation of PrPSc in the brain, while central to transmission of disease, may not directly cause neurodegeneration. For example, extensive deposition of PrPSc in the context of neurons not actively expressing PrP does not lead to neuronal damage and death (Brandner et al, 1996; Mallucci et al, 2003). Conversely, certain naturally occurring or artificially created mutations in PrP can lead directly to neurodegeneration without generating the transmissible PrPSc form (Tateishi and Kitamoto, 1995; Tateishi et al, 1996; Hegde et al, 1998, 1999). Thus, PrPSc accumulation is neither uniformly necessary nor sufficient for neuronal toxicity. These and other findings have led to the idea that other facets of normal PrP metabolism may play critical roles in disease pathogenesis (Hegde and Lingappa, 1999; Chiesa and Harris, 2001; Hegde and Rane, 2003).
Recent studies have begun to implicate events during the initial biogenesis, trafficking, and degradation of PrP at the endoplasmic reticulum (ER) in the development of neurodegeneration (reviewed in Hegde and Rane, 2003). One neurotoxic form of PrP is a transmembrane isoform (termed CtmPrP; Hegde et al, 1998) that is generated during the initial translocation of PrP into the ER (Kim and Hegde, 2002). Although wild-type PrP generates relatively little (<10%) CtmPrP in vivo, several mutations in the potential transmembrane domain significantly increase its generation (Hegde et al, 1998, 1999; Kim et al, 2001; Stewart and Harris, 2001; Kim and Hegde, 2002). Expression of such CtmPrP-favoring mutants in transgenic mice causes neurodegeneration (Hegde et al, 1998, 1999), and at least three naturally occurring human mutations in the transmembrane domain result in increased CtmPrP generation (Hegde et al, 1998; Kim and Hegde, 2002). Thus, CtmPrP appears to play a direct role in at least a subset of PrP-mediated diseases, although the mechanism by which it causes cell death remains unknown.
More recently, it has been demonstrated that inappropriate expression of PrP in the cytosol can be both neurotoxic in mice (Ma et al, 2002) and aggregation-prone in cells (Yedidia et al, 2001; Ma and Lindquist, 2002). However, the pathways by which PrP would appear in the cytosol remain speculative and contentious (Ma and Lindquist, 2001, 2002; Yedidia et al, 2001; Drisaldi et al, 2003; Hegde and Rane, 2003; Roucou et al, 2003). In one model, a proportion of PrP is proposed to misfold in the ER lumen, be recognized by the ER quality control machinery, and subsequently be retrotranslocated to the cytosol for degradation by proteasomes (Ma and Lindquist, 2001, 2002). However, the observation that at least some of the PrP that accumulates upon proteasome inhibition contains an uncleaved signal peptide suggests that another route to the cytosol may involve a failure of PrP translocation into the ER (Drisaldi et al, 2003). In either case, the role of cytosolic PrP in disease pathogenesis remains unclear, ranging from protective (Roucou et al, 2003) to toxic roles (Ma et al, 2002), having been proposed by different investigators.
Clearly, any putative roles for cytosolic PrP in normal physiology (Roucou et al, 2003) or disease pathogenesis (Yedidia et al, 2001; Ma et al, 2002; Ma and Lindquist, 2002) cannot be delineated without the ability to selectively modulate its generation. This requires an understanding of the pathway(s) by which proteins that are normally trafficked through the secretory pathway can reside in the cytosol. Like most secretory and membrane proteins (Rapoport et al, 1996), PrP contains a signal sequence that mediates cotranslational targeting to and translocation across the ER membrane. The general efficiency of signal-mediated segregation to the ER, particularly in vivo is not well studied. However, in vitro studies have recently suggested that certain early steps in protein translocation across the ER may not be uniformly efficient for all substrates (Rutkowski et al, 2001; Kim et al, 2002). In addition, in vivo analysis of the kinetics of signal-mediated targeting (Johnsson and Varshavsky, 1994; Goder et al, 2000) and the steady-state presence of a signal-containing reporter in the cytosol (Levine et al, 2004) suggest that substrates differ in their efficiency of segregation into the ER. Presumably, the nontranslocated fraction of many such proteins would be viewed as undesirable in the cytosol, and need to be disposed by the ubiquitin–proteasome system. The physiological significance or relative contribution of such inefficiencies in ER protein translocation to the total substrate pool handled by cytosolic proteasomes in vivo has not been examined. In this study, we have analyzed this issue for PrP to reveal an unanticipated relationship between protein translocation, protein degradation, and the potential for PrP aggregation in the cytosol.
Results and discussion
Measurement of signal sequence efficiencies in vivo
A transcription factor (TF) sequestration assay (described in detail elsewhere (Levine et al, 2004); see also Supplementary Figure S1A) was used to measure the in vivo efficiency of signal sequence-mediated protein segregation into the ER. Here, any inefficiency of a signal sequence to direct the TF into the ER is detected by activation of a cotransfected luciferase reporter. When TF lacking a signal sequence was tested, a dose-dependent increase in reporter activity is observed over a broad range of expression (Figure 1A), even at TF levels that are below the detection threshold for immunoblotting (Figure 1B). When TF was preceded by the PrP signal sequence (PrP-TF), reporter activity was reduced sharply (∼10–20-fold) despite identical levels of TF expression (Figure 1A and B). Quantitation relative to the standard curve of signal-less TF suggested that, depending on experimental conditions (e.g., growth state and confluency of the cells), between 5 and 15% of total PrP-TF at steady state (∼15% in Figure 1C) was available in the nucleo-cytoplasmic compartment for reporter activation. Even at very low expression levels in cells growing under optimal conditions, at least 5% of PrP-TF was not segregated into the ER.
Figure 1.
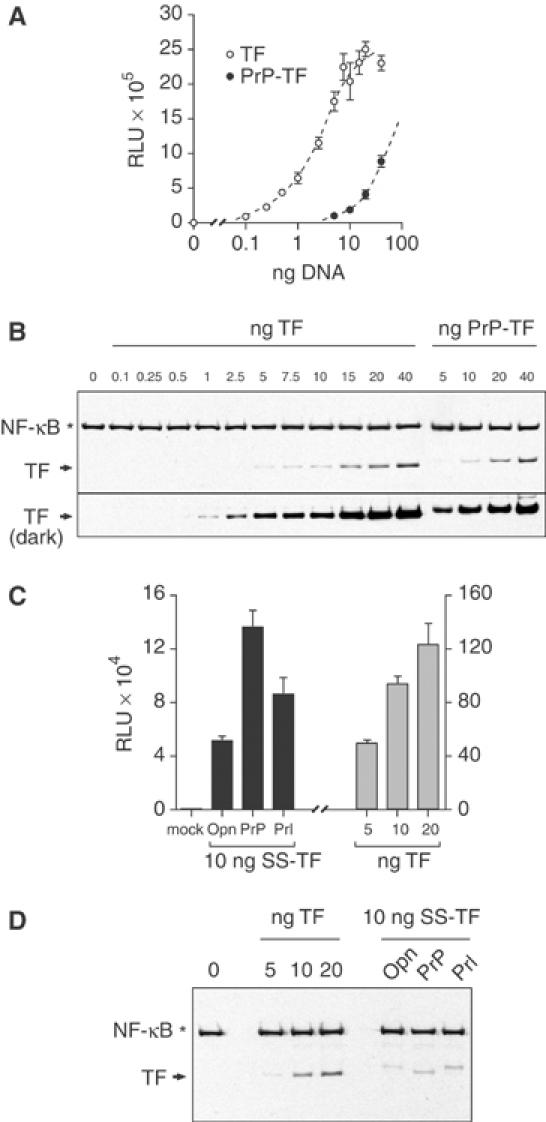
Measurement and modulation of protein translocation in vivo. (A) Varying amounts of plasmid encoding TF or PrP-TF were transfected into MDCK cells and the amount of luciferase reporter activation measured (in relative light units (RLU)). (B) Duplicate samples from panel A were analyzed in parallel by immunoblotting with antibodies against NF-κB. The position of exogenously expressed TF on faint and dark exposures of the blot is indicated with arrows. Endogenous NF-κB (indicated by an asterisk) serves as a loading control. (C) Comparison of translocation efficiencies of the Opn, PrP, and Prl signal sequences fused to TF. Transfection of an unrelated plasmid (mock) resulted in no reporter activation. Varying amounts of TF lacking a signal sequence were analyzed in parallel (note 10-fold difference in y-axis scales). (D) Parallel immunoblots for TF expression of samples from panel C.
When other signal sequences were used in the same assay, the percent of cytoplasmic TF could be substantially increased or decreased relative to PrP-TF (Levine et al, 2004). This is consistent with in vitro observations demonstrating that signal sequences can vary broadly in their functions at early steps in substrate translocation (Rutkowski et al, 2001; Kim et al, 2002). Among the most efficient signal sequences, both in vitro (Kim et al, 2002) and in the TF sequestration assay (Levine et al, 2004; Figure 1C and D and Supplementary Figure S1B), are those from either prolactin (Prl) or osteopontin (Opn). Both of these resulted in ∼50% lower reporter activity relative to PrP-TF at comparable and modest expression levels (Figure 1C and D), indicating that even the highest efficiency signals still permit substantial and readily detectable levels of total TF to exist in a functional state in the nucleo-cytoplasmic compartment.
Consequence of signal sequence inefficiency for PrP metabolism
To explore the relevance of this small but readily detectable inefficiency in the activity of some signals, we turned our attention to native PrP. Modulating PrP translocation by replacement of its signal sequence with the more efficient Opn or Prl signal sequences (termed Opn-PrP and Prl-PrP, respectively) did not lead to obvious changes in several properties of PrP when examined at steady state in unperturbed cultured cells: the glycosylation pattern, resistance to endoglycosidase H digestion, subcellular localization, solubility in nonionic detergents, surface expression, protease sensitivity, and site of signal sequence cleavage were all indistinguishable between these constructs (Figure 2B and Supplementary Figures S2, S3 and S4 and unpublished results). When analyzed by pulse-chase studies, however, small differences could be detected in the metabolism of these three constructs (Figure 2A). Whereas ∼20% of PrP was observed to be unglycosylated after a brief 15 min pulse labeling, substantially less of this population was observed for Opn-PrP and Prl-PrP. By contrast, more than 50% of the synthesized PrP was unglycosylated when the less efficient angiotensinogen (Ang) signal sequence (Kim et al, 2002) was used. The unglycosylated species of PrP for each construct was observed to be largely degraded within the 1-h chase period, a process that was retarded by proteasome inhibitors (Figure 2A). During this same time frame, the glycosylated PrP was trafficked to post-ER compartments, as evidenced by its decreased mobility due to Golgi-specific carbohydrate modifications. Thus, in the absence of proteasome inhibition, the pattern of PrP isoforms within 1 h after a pulse labeling is very similar between the wild-type, Opn-PrP, and Prl-PrP constructs (Figure 2A, top panel), consistent with their identical distributions at steady state. With proteasome inhibition, however, clear differences are observed in the amount of nonglycosylated PrP generated by differences in signal sequence efficiencies (Figure 2A, bottom panel).
Figure 2.
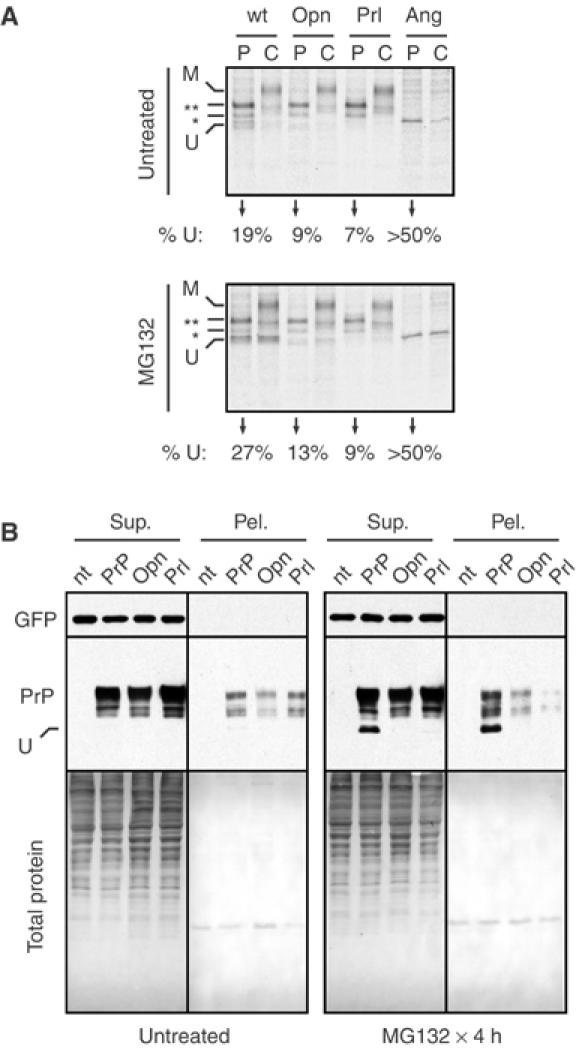
Reduced susceptibility to aggregate formation by modulation of PrP translocation. (A) Biosynthesis and maturation of PrP fused to different signal sequences was assessed by pulse-chase analysis (as described in Materials and methods) of transfected N2a cells in the absence (top panel) or presence (bottom panel) of proteasome inhibition. Samples after a 15 min pulse labeling (‘P' lanes) and following a 1 h chase in unlabeled media (‘C' lanes) are shown. The positions of different species of PrP are indicated: U, unglycosylated, *, singly glycosylated, **, doubly glycosylated, and M, mature. The percent of total PrP synthesized in the unglycosylated form is indicated below each set of lanes. (B) N2a cells expressing PrP with different signal sequences were assessed for aggregate formation by a solubility and sedimentation assay. The total proteins and immunoblots for GFP (a cotransfected control protein) and PrP in the soluble and insoluble fractions are shown (Sup. and Pel., respectively). The position of unglycosylated PrP (U) is indicated. Untreated and MG132-treated cells were analyzed in parallel. Cells not transfected with a PrP construct are indicated (nt).
The consequences of this nontranslocated population were explored further by determining the effects of prolonged proteasome inhibition. A concentration of proteasome inhibitor that only partially impairs the proteasome system (Supplementary Figure S5) was used to mimic the reduced degradative capacity thought to develop during the pathogenesis of various neurodegenerative diseases (Bence et al, 2001; Ciechanover and Brundin, 2003) or even the normal aging process (Gray et al, 2003). With PrP, proteasome impairment with any of three different inhibitors results in the gradual accumulation of nonglycosylated PrP that is resistant to solubilization with mild detergents (Figure 2B and Supplementary Figures S6 and S7A). Remarkably, the generation of this insoluble nonglycosylated form was effectively abolished with Opn-PrP or Prl-PrP (Figure 2B and Supplementary Figure S7A), even upon prolonged inhibition of the proteasome (for up to 8 h; Figure 3A).
Figure 3.
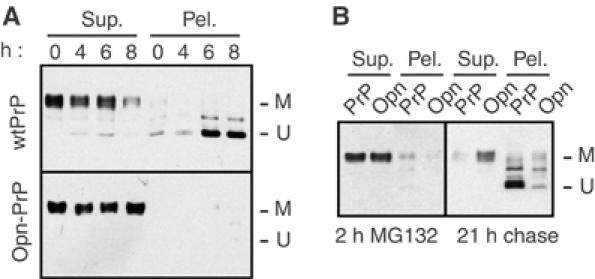
Effect of prolonged proteasome inhibition on the metabolism of PrP and Opn-PrP. (A) N2a cells transfected with either wild-type PrP (upper panel) or Opn-PrP (lower panel) were treated for between 0 and 8 h with 5 μM MG132 before lysis and fractionation into detergent-soluble (Sup.) and insoluble (Pel.) fractions and analysis by immunoblotting. Note that, with wild-type PrP, an insoluble, unglycosylated species of PrP accumulates over 8 h (U); this is not observed with Opn-PrP, which only shows soluble, mature PrP (M). Results identical to those observed with Opn-PrP were also seen for Prl-PrP (unpublished results; see also Figure 2B). (B) Effect of signal sequence on ‘propagation' of cytoplasmic PrP aggregates. Two parallel plates each of N2a cells transfected with either wild-type PrP or Opn-PrP were treated for 2 h with 5 μM MG132. One plate (left panel) was harvested immediately and analyzed for PrP detergent solubility and aggregation. The second set of dishes was rinsed to remove the MG132, and incubated an additional 21 h in normal media (right panel) before harvesting for analysis of PrP detergent solubility and aggregation. After a 2 h ‘pulse' of MG132, note that the immunoblots of PrP and Opn-PrP are largely indistinguishable: both show nearly quantitative solubility and little unglycosylated, insoluble PrP. After a 21 h chase, wild-type PrP was nearly all unglycosylated and insoluble, as has been proposed to occur by a ‘self-propagation' mechanism (Ma and Lindquist, 2002). Even under these conditions, however, relatively little Opn-PrP was found in the unglycosylated, insoluble form.
Furthermore, small amounts of cytosolic PrP generated upon transient treatment with proteasome inhibitors have been reported to accumulate substantially over time by ‘self-propagation' (Ma and Lindquist, 2002). Although the mechanism of ‘propagation' is not yet clear, this treatment protocol allows the detection of very small amounts of cytosolic PrP, which presumably serves as a seed for subsequent accumulation. Under such propagation conditions that cause nearly all of the cellular PrP to become cytosolic, very little Opn-PrP accumulated in the unglycosylated form (Figure 3B). Thus, modulating PrP translocation by using a more efficient signal sequence can substantially reduce or even eliminate the ability of PrP to form nonglycosylated cytosolic aggregates under a variety of conditions.
The consequences of modulating PrP translocation were also examined in live cells expressing wild type and/or Opn-PrP tagged with different color variants (either cyan or yellow) of monomerized green fluorescent protein (mGFP). Co-expression of these two constructs in cultured cells demonstrated them to have the same localization as both each other and nontagged PrP (Figure 4A and Supplementary Figure S2), with the majority of PrP being found on the cell surface and lesser amounts in an intracellular pool colocalizing with the Golgi and endosomal compartments (unpublished results; see also Figure 5A and C). Biochemical analyses confirmed that fluorescent protein-tagged PrP and Opn-PrP were identically glycosylated and resistant to deglycosylation with endoglycosidase H (unpublished results). Upon inhibition of the proteasome in cells expressing PrP-mYFP or Opn-PrP-mYFP, a striking difference was observed (Figure 4B). Numerous PrP-expressing cells began to show a different pattern of localization, with an increasing intracellular population (Figure 4B, left panels). Quantitative single-cell analyses (Figure 4C, closed circles) revealed that the ratio of surface to intracellular localized PrP was lowest for the cells expressing the most PrP. For the lowest-expressing cells, only a modest increase in the intracellular population was observed. Parallel analysis of Opn-PrP-expressing cells revealed them to maintain the normal pattern of subcellular localization, regardless of expression levels (Figure 4B, right panels). Indeed, only a very small difference in the intracellular to surface ratio was observed even in the highest expressing cells (Figure 4C, open circles). Similar results were obtained with indirect immunofluorescence analysis of cells expressing nontagged versions of PrP and Opn-PrP (unpublished results).
Figure 4.
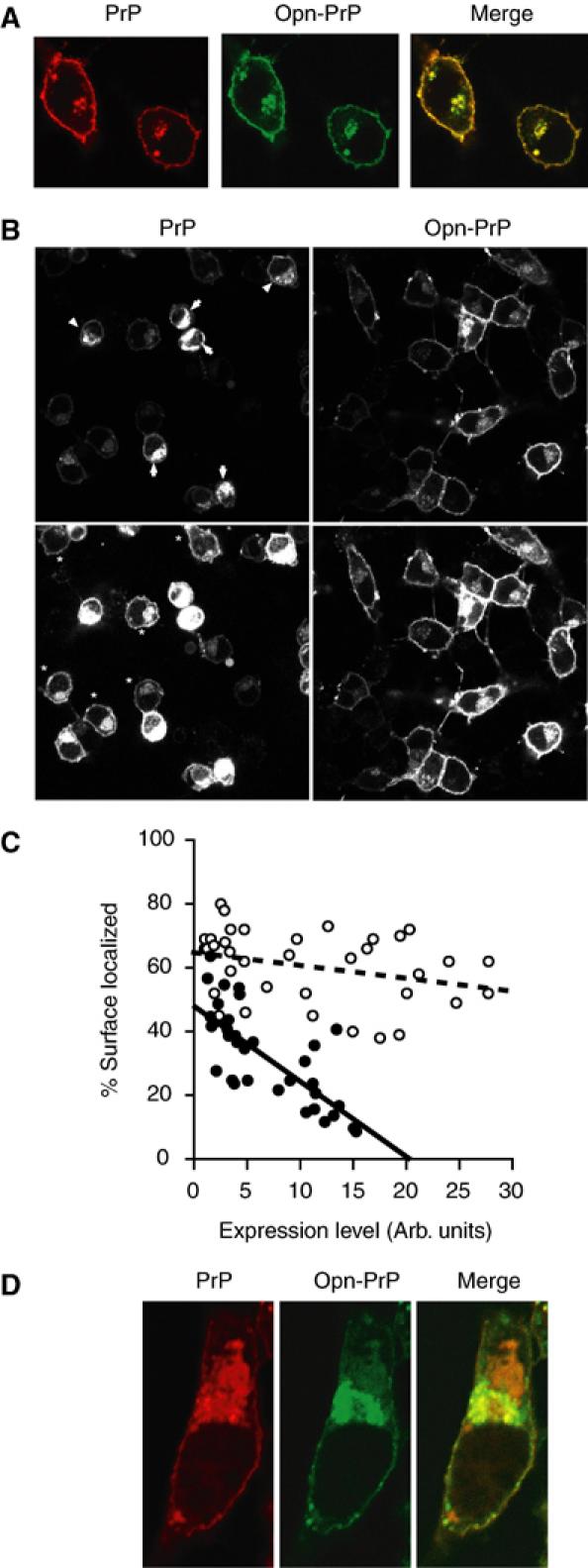
Visualization and modulation of PrP metabolism in single live cells. (A) Expression and colocalization of PrP-mYFP (red) and Opn-PrP-mCFP (green) in N2a cells. Similar results were obtained with PrP-mCFP and Opn-PrP-mYFP (unpublished results). (B) Comparison of localization patterns in N2a cells expressing PrP-mYFP or Opn-PrP-mYFP after proteasome inhibition (with MG132) for 8 h. Two exposures (upper and lower panels) are shown for each representative field to facilitate visualization of cells expressing high (arrows) and low (asterisks) levels of PrP. The PrP-mYFP and Opn-PrP-mYFP cells were visualized using identical imaging conditions to allow direct comparisons between them. (C) Quantitative analysis of subcellular localization relative to expression level for PrP-mYFP (closed circles) and Opn-PrP-mYFP (open circles) after proteasome inhibition as in panel B. (D) Visualization of PrP-mYFP (red) and Opn-PrP-mCFP (green) following proteasome inhibition.
Figure 5.
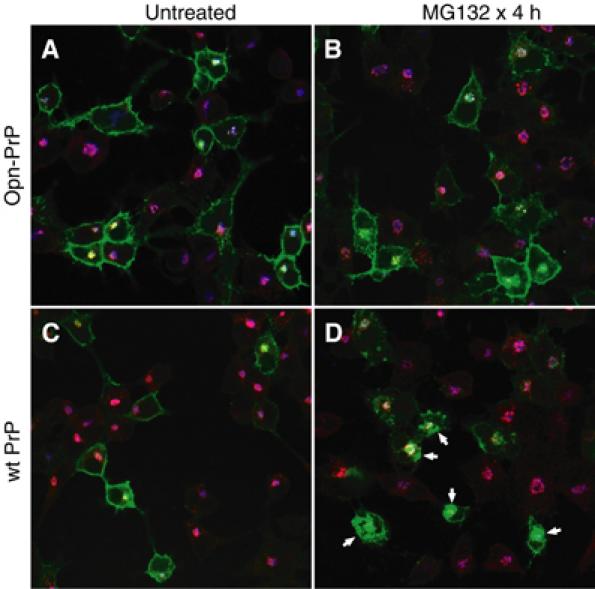
Analysis of PrP and Opn-PrP localization. N2a cells were transfected with Opn-PrP-mYFP (panels A and B) or PrP-mYFP (panels C and D) and either left untreated (panels A and C) or treated with 5 μM MG132 for 4 h. The cells were fixed and processed for double immunofluorescence analysis using antibodies against an endosome marker (EEA1, in red) and a Golgi marker (beta-COP, in blue). Green indicates the position of YFP-tagged PrPs. Note that, in panels A–C, mYFP is localized primarily to the cell surface and perinuclear structures that overlap partially with Golgi and endosome structures. By contrast, panel D shows many cells (indicated with arrows) that have additional accumulations of PrP in other regions of the cytoplasm. Higher magnification images with separated color channels of representative cells from panels B and D are shown in Supplementary Figures S8 and S9, respectively.
When examined in the same cell, a difference in PrP-mYFP and Opn-PrP-mCFP localization upon proteasome inhibition was readily apparent within 2–4 h (Figure 4D). Opn-PrP-mCFP, similar to before proteasome inhibition (compare to Figure 4A), continued to show localization at the cell surface and perinuclear structures. By contrast, PrP-mYFP now clearly showed additional accumulations in the cytoplasm that did not colocalize with Opn-PrP-mCFP (Figure 4D). At longer times after proteasome inhibition, this differential localization was more difficult to see, apparently due to substantial changes in cellular morphology caused by the accumulating PrP-mYFP (unpublished results). That notwithstanding, the ability to observe a difference in the localization for a population of PrP distinct from Opn-PrP within the same cell directly demonstrates that the trafficking pathways diverge for a discrete proportion of these two proteins. Hence, the differential consequences of the PrP and Opn signals on substrate translocation can be directly visualized in live cells during conditions of a compromised proteasome.
Colocalization studies showed that, both before and after proteasome inhibitor treatment, the majority of Opn-PrP-mYFP was at the cell surface and in perinuclear structures consistent with Golgi and endosomes (Figure 5A and B and Supplementary Figure S8). By contrast, a significant proportion of the PrP-mYFP that accumulates after proteasome inhibition is found in cytoplasmic regions that do not colocalize with either Golgi or endosomes (Figure 5D and Supplementary Figure S9). Thus, PrP and Opn-PrP show essentially identical cellular localization patterns before (Figures 4A, 5A and C and Supplementary Figure S2), but not after proteasome inhibition (Figures 4D, 5B and D and Supplementary Figures S8 and S9). We conclude that the additional cytoplasmic accumulations of PrP-mYFP (but not Opn-PrP-mYFP) seen by microscopy are likely to correspond to the unglycosylated PrP that accumulates upon proteasome inhibition in biochemical experiments (e.g., Figures 2 and 3).
Generation of cytosolic PrP independently of expression level
The single-cell analysis of PrP localization (Figure 4C) revealed that, even at relatively low levels of expression, a difference between PrP and Opn-PrP could be observed. This suggests that the basis of cytosolic PrP generation is not likely to be a consequence of saturating the ER targeting and translocation machinery. This conclusion was confirmed in two ways. First, we demonstrated that gross overexpression of Prl-PrP ∼5–10 fold above wild-type PrP still did not generate the aggregated nonglycosylated form of PrP upon proteasome inhibition (Figure 6A and B). Thus, generation of unglycosylated PrP cannot be induced simply as a consequence of overexpression if a sufficiently efficient signal sequence is used for translocation. This indicates that even very high amounts of PrP in the ER lumen are insufficient to result in the generation of unglycosylated PrP by retrotranslocation.
Figure 6.
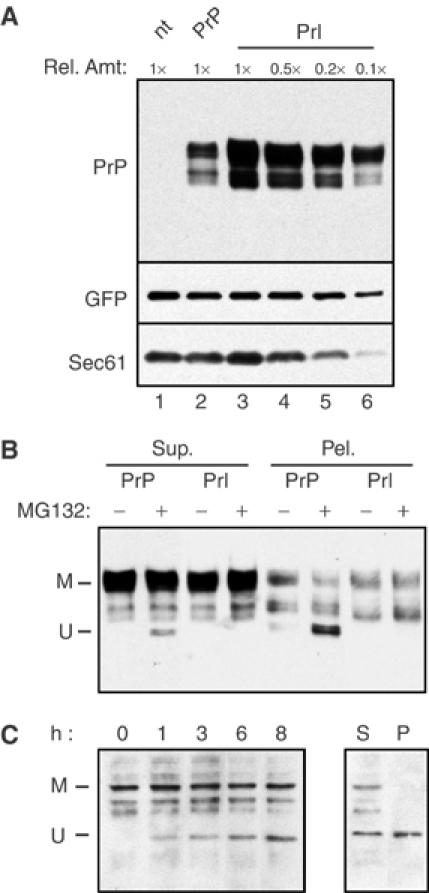
Effect of expression level on cytosolic PrP generation. (A) N2a cells were transfected with either wild-type PrP or a larger amount of Prl-PrP. In both cases, equal amounts of a plasmid expressing GFP were cotransfected. A fixed amount of cell lysate from PrP-expressing cells (1 ×; lane 2) was compared to different amounts of cell lysate from the Prl-PrP-expressing cells (from 0.1 × to 1 ×; lanes 3–6). Cells transfected with just GFP (nt; lane 1) are also shown as a control. Immunoblots were probed with antibodies against PrP, GFP, and an endogenous protein, the beta subunit of Sec61. Note that GFP and Sec61 levels are comparable when equal amounts of nt, PrP, and Prl-PrP cell lysates are compared (lanes 1–3). By contrast, Prl-PrP is expressed at ∼5–10-fold higher levels than PrP, as indicated by the observation that equal intensities are observed when 0.1–0.2 × of Prl-PrP (lanes 5 and 6) is compared to 1 × of PrP (lane 2). (B) Duplicate dishes of cells from lanes 1 and 2 of panel A were either left untreated or treated for 4 h with 5 μM MG132 prior to harvesting and fractionation of detergent-soluble PrP (Sup.) from insoluble PrP (Pel.). One-tenth the amount of Prl-PrP lysate was analyzed on the gel to allow direct comparisons between the samples. Note that, with PrP, unglycosylated, insoluble PrP (U) is generated upon MG132 treatment. However, despite the ∼10 × overexpression of Prl-PrP, this form is not generated. (C) Effect of proteasome inhibition on endogenously expressed PrP. Untransfected N2a cells were treated for between 0 and 8 h with 5 μM MG132 before analysis of total proteins by immunoblotting with the 7D9 anti-PrP antibody (which detects mouse PrP). Note that an unglycosylated species of PrP (U) accumulates over 8 h (left panel). Samples from the 8 h treatment were also analyzed for PrP detergent solubility and aggregation (right panel). Note that a substantial fraction of the unglycosylated PrP is in the detergent insoluble pellet (P), while all of the mature, glycosylated PrP (M) is in the detergent-soluble supernatant (S).
In a second approach, we were able to observe the accumulation of nonglycosylated aggregates of the endogenously expressed PrP in nontransfected N2a cells treated with proteasome inhibitors (Figure 6C). Thus, the progressive generation and accumulation of insoluble, unglycosylated PrP upon proteasome inhibition is not a consequence of exogenous overexpression or the use of a viral promoter, as has been suggested previously (Drisaldi et al, 2003). Many of the conclusions from these previous studies rested on the assumption that cytoplasmic, unglycosylated PrP would necessarily contain an uncleaved signal sequence if it were generated by aborted translocation. However, our previous studies indicate that the step at which some signals are inefficient in initiating translocation involves a post-targeting interaction with the Sec61-translocon (Kim et al, 2002). As the signal peptidase complex is at the translocon and removes signals cotranslationally (Rapoport et al, 1996), the absence of the signal peptide may not necessarily be a reliable indicator of a protein having been completely translocated into the ER lumen. Examples of cytosolic localization after signal sequence cleavage of aborted translocation intermediates have been described previously (Garcia et al, 1988; Ou et al, 1989; Ooi and Weiss, 1992).
Indeed, we have found that, in vitro, cytoplasmic PrP containing a cleaved signal can be generated by chains that were targeted to, but only partially translocated through the translocon before completing synthesis in the cytosol (unpublished results). Consistent with this observation, we often see two unglycosylated species of PrP, separated by approximately the size of the PrP signal peptide, that accumulate to varying degrees in proteasome inhibited cells (e.g., see Supplementary Figure S6). Thus, the use of an anti-signal peptide antibody (Drisaldi et al, 2003; Stewart and Harris, 2003) as a sole marker for cytoplasmic PrP is unreliable, calling into question the interpretation of negative results that failed to detect signal-containing PrP in primary neurons or diseased brain (Stewart and Harris, 2003). Conversely, the conclusion (Ma and Lindquist, 2001), based solely on migration in SDS–PAGE, that PrP has been completely in the ER lumen (and was subsequently retrotranslocated) is potentially premature. Our observation that all forms of unglycosylated PrP (whether they are signal cleaved or not) are abolished by improving translocation into the ER lumen with the Prl or Opn signals indicates that little, if any, of the unglycosylated PrP is generated from chains that retrotranslocated from the lumen (since fully translocated chains are also made in abundance with Prl-PrP and Opn-PrP). Thus, considered together with Figure 1, these results argue that a substantial proportion of the proteasomal burden of PrP is derived from an inherent inefficiency in its signal sequence-mediated translocation into the ER.
Generation of cytosolic PrP in the absence of CtmPrP
As discussed in the Introduction, the CtmPrP transmembrane form, similar to cytosolic PrP, causes neurodegeneration if overexpressed in transgenic mice. In the brains of CtmPrP-expressing mice, the PrP is almost exclusively found in post-ER compartments, suggesting that CtmPrP passes the quality control machinery of the ER and exits to the Golgi (Hegde et al, 1998). Recently, however, the overexpression of a signal sequence and transmembrane domain double mutant of PrP (termed L9R/3AV) that is made predominantly in the CtmPrP form was shown to be degraded by proteasomes (Stewart et al, 2001). Whether the behavior of this mutant reflects the properties of CtmPrP generated by wild-type PrP is not yet clear. However, this observation, although at odds with the findings in brain, suggests that CtmPrP could be a precursor to cytosolic PrP. Such a model would also be consistent with the finding that, in vitro, the generation of CtmPrP depends in part on a slightly inefficient signal sequence (Kim et al, 2001, 2002; Kim and Hegde, 2002).
To test this possibility, we analyzed the behavior of PrP constructs containing mutations in the transmembrane domain that either increase or abolish the ability to generate CtmPrP. Unlike Opn-PrP, however, the PrP(G123P) mutant that cannot make CtmPrP (Hegde et al, 1998) still generated nonglycosylated cytosolic PrP aggregates upon proteasome inhibition (Figure 7B and Supplementary Figure S7B). Furthermore, mutants which increase CtmPrP generation (such as PrP(A117V) and PrP(AV3); see Hegde et al, 1998) also did not influence the generation of nonglycosylated cytosolic PrP aggregates upon proteasome inhibition (Figure 7B and Supplementary Figure S7B). Importantly, we confirmed that, in the N2a cells used for these studies, the PrP(AV3) and PrP(G123P) mutants increase and abolish, respectively, the generation of CtmPrP (Figure 7A and Supplementary Figure S10), exactly as observed in vitro and in mouse brain (Hegde et al, 1998). Thus, while CtmPrP and cytosolic PrP appear to share some common features (such as their potential toxicity, exposure to the cytosol, and dependence on an inefficient signal sequence for generation), they do not appear to have a precursor–product relationship. Instead, cytosolic PrP can be generated largely independently of whether CtmPrP is made or not. This further suggests that the majority of cytosolic PrP is not generated by PrP molecules that are first inserted into the ER, regardless of their topologic form.
Figure 7.
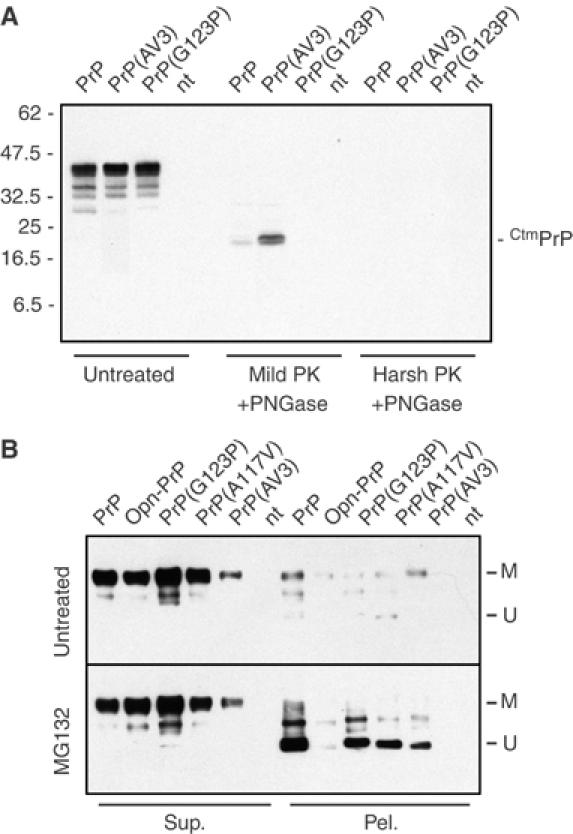
Relationship between CtmPrP and cytosolic PrP generation. (A) Wild-type PrP, PrP(G123P) (a mutant that cannot generate CtmPrP; Hegde et al, 1998) and PrP(AV3) (a mutant that generates increased levels of CtmPrP) were transfected into N2a cells and analyzed for CtmPrP levels as described in Materials and methods. Cell lysates were either left untreated or digested with proteinase K under ‘mild' or ‘harsh' conditions. Protease-digested samples were then deglycosylated with PNGase F prior to SDS–PAGE and immunoblotting with the 3F4 anti-PrP antibody. The position of a diagnostic CtmPrP-specific 18 kDa proteolytic fragment observed after ‘mild' but not ‘harsh' digestion (Hegde et al, 1998, 1999) is indicated to the right of the gel. The positions of molecular weight markers are indicated to the left. Overexposed images of the blot (Figure S10) confirmed the complete absence of a CtmPrP-specific band for the PrP(G123P) mutant. (B) Wild-type PrP, Opn-PrP, or the indicated PrP mutants were transfected into N2a cells and analyzed for the generation of unglycosylated aggregates of PrP. In each case, the cell lysates were separated into detergent-soluble (Sup.) and insoluble (Pel.) fractions before analysis. Samples of untreated cells (top panel) or cells treated with 5 μM MG132 for 8 h (bottom panel) were analyzed in parallel. The positions of mature (M) and unglycosylated PrP (U) are indicated. PrP(A117V), to a lesser extent than PrP(AV3), generates increased levels of CtmPrP relative to wild-type PrP (see Hegde et al, 1998). Similar results were observed with other proteasome inhibitors (Supplementary Figure S7).
Consequences of signal sequence efficiency for susceptibility to cell death
Recent studies have demonstrated that, when inappropriately expressed in the cytosol, PrP is not only susceptible to aggregation, but is selectively toxic to a subset of cells (primarily of neuronal origin), both in culture and in a transgenic mouse model (Ma et al, 2002). We therefore asked whether modulation of PrP translocation into the ER could at least partially alleviate the sensitivity of PrP-expressing cells to the cytotoxic consequences of decreased proteasome activity. Remarkably, as assessed by either Annexin V exposure on the extracellular surface (Figure 8A and B) or overall cell viability (Figure 8C), cells expressing Opn-PrP tolerated partial proteasome inhibition considerably better than cells expressing PrP. In fact, overexpression of Opn-PrP had no detectable adverse consequence beyond that observed in cells expressing either no exogenous proteins or expressing unrelated secretory or cytoplasmic proteins.
Figure 8.
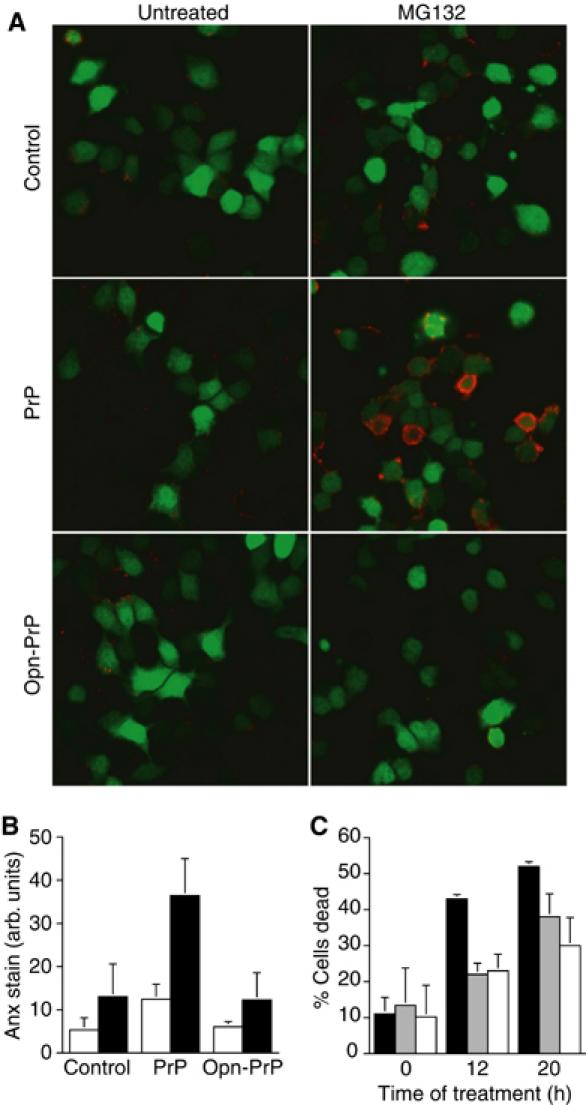
Protection from PrP-mediated cytotoxicity by modulation of protein translocation. (A) N2a cells cotransfected with GFP (a transfection marker, indicated by green cells) and either PrP, Opn-PrP, or an unrelated control plasmid (coding for preprolactin) were analyzed for apoptosis using Annexin V staining (red) before and after treatment with MG132. (B) Quantitative analysis of Annexin V staining before (white bars) and after (black bars) proteasome inhibition (4 h) of N2a cells expressing PrP, Opn-PrP, or an unrelated protein. Equal transfection efficiency and PrP expression levels were confirmed among samples within each experiment (unpublished results; see Materials and methods for details). (C) Quantitative analysis of total cell death (judged by vital dye exclusion) before and after proteasome inhibition for 12 or 20 h of N2a cells expressing PrP (black bars), Opn-PrP (gray bars), or a control protein (white bars).
These results suggest that removing the potential to generate cytosolic PrP by the pathway involving inefficient translocation is sufficient to eliminate the PrP-mediated susceptibility to proteasome inhibition. Thus, overexpression of PrP in the lumen of the ER (e.g., with Opn-PrP) does not increase the susceptibility of cells to proteasome inhibition beyond that observed in control cells. This argues that, at least under nonstressed steady-state conditions, translocation into the ER is the decisive step in avoiding the generation of cytotoxic forms of PrP that would ordinarily be degraded by the proteasome. Although these proteasomally degraded forms of PrP can lead to cell death under some circumstances (e.g., Ma et al, 2002; this study), the roles they play during the pathogenesis of either transmissible or genetic forms of prion disease remain unknown. The ability to eliminate (or at least substantially reduce) the generation of these forms of PrP in vivo should now allow the testing of currently controversial hypotheses proposing their involvement in disease pathogenesis.
Conclusions
The results presented in this study identify inefficiencies in signal sequence-mediated protein segregation to the ER as a major and previously unappreciated pathway for the generation of substrates to be degraded by the proteasome. This inefficiency appears to account for the majority of proteasomally degraded PrP. Importantly, reducing this nontranslocated population by improving signal sequence function reduces the proteasomal burden of cytosolic PrP. This allows cells to normally process PrP in the secretory pathway, while avoiding the generation of an insoluble, aggregated, and potentially cytotoxic population of cytoplasmic PrP even under conditions of substantial overexpression or prolonged diminishment of proteasome activity.
The inefficient translocation pathway for generating cytoplasmic PrP is not mutually exclusive of the previously proposed mechanism involving the cellular quality control machinery and retrotranslocation from the ER lumen (Yedidia et al, 2001; Ma and Lindquist, 2001, 2002; Cohen and Taraboulos, 2003). However, with wild-type PrP in nonperturbed cells, the retrotranslocation pathway does not appear to contribute substantially, since the proteasomally degraded population of PrP could be effectively eliminated by improving its initial import into the ER. Furthermore, the ∼20% of nontranslocated PrP we have observed would appear to sufficiently account for the previous estimates (Yedidia et al, 2001) that ∼10% of total synthesized PrP is metabolized via the proteasome pathway. With mutations or conditions that promote PrP misfolding in the ER lumen, retrotranslocation to the cytosol may be an additional source of proteasomally degraded PrP (Ma and Lindquist, 2001, 2002; Cohen and Taraboulos, 2003). The relative contribution of such putative events to the total amount of proteasome substrate remains to be determined, but can now be evaluated with the identification of PrP variants that largely eliminate inefficient translocation as a source of proteasomal substrate.
Implications for other proteins
The observation of inefficient PrP translocation, together with the finding that the signal sequences of many proteins may be even less efficient than that from PrP (Kim et al, 2002; Levine et al, 2004), has implications for other disease processes that involve proteins trafficked through the secretory pathway. A particularly notable example may be Alzheimer's disease (AD), which involves at least two proteins, APP and ApoE, that contain signal sequences for ER translocation (Selkoe, 2001). How these proteins contribute to cell death during the pathogenesis of AD continues to elude a unifying explanation (Selkoe, 2001; Kawasumi et al, 2002; Caughey and Lansbury, 2003). However, for both APP and ApoE, recent studies have suggested that inappropriate mislocalization to the cytosol can have adverse consequences, some of which resemble the pathological events in AD (Huang et al, 2001; Anandatheerthavarada et al, 2003). Indeed, the observation of ubiquitin-conjugated secretory or membrane proteins in plaques or deposits accumulating during both normal aging and certain neurodegenerative diseases (Gray et al, 2003; Lang-Rollin et al, 2003; Kang et al, 2004) suggests access of the cytoplasmic ubiquitination and degradation machinery to proteins normally trafficked through the secretory pathway. An initial analysis of the signal sequences from APP and ApoE in vitro revealed that they were similar or less efficient than the PrP signal in their function of initiating translocation through the translocon (unpublished results). Thus, it will be of interest to determine whether a substantial population of these proteins transiently traffics through the cytoplasm, and, if so, what role this has in the pathogenesis of disease.
Finally, the nontranslocated population of substrates containing signal sequences may not necessarily be targets for immediate degradation. If a protein can be made in two subcellular locales at varying efficiencies, it may be anticipated that, in at least some instances, both populations have functional roles in the cell. Indeed, proteins with functions in both the ER and the cytoplasm have been proposed (Hegde and Lingappa, 1999), although a mechanism to explain their dual localization remains obscure. Consistent with such an idea, the relative inefficiencies in the signal sequences of many substrates, including PrP, are evolutionarily conserved (Kim et al, 2002). This is particularly puzzling in the case of PrP where, based on the present and previous studies, a cytoplasmic population would appear to be disadvantageous. However, other work suggesting that at least in some cells, a cytoplasmic form of PrP not only exists abundantly (Mironov et al, 2003) but also may be beneficial (Roucou et al, 2003) raises the intriguing idea that PrP has evolved to maintain a balance between multiple forms, all of which have functional roles in subsets of cells at particular times. How the abundances of these different forms are regulated and what their putative functions are remains to be determined. But this may explain why, despite the clear potential for adverse consequences, the inefficient signal sequence of PrP has not evolved to be as efficient as those from Opn or Prl.
Materials and methods
Cells, plasmids, and reagents
MDCK and Neuro 2a (N2a) cells were obtained from the American Tissue Culture Collection (ATCC) and S Priola (NIH), respectively. Fluorescent protein expression plasmids were from Clonetech. The pLuc plasmid (luciferase reporter preceded by the Gal4 DNA-binding element) was from Stratagene. Plasmids for expression of TF- and PrP-containing constructs used the pCDNA vector (Invitrogen) and are described in detail in Supplementary data. Antibodies were obtained from: GFP (JL8; Clonetech); NF-κB (Santa Cruz Biotechnologies); mouse PrP (7D9; Signet Laboratories); hamster PrP (3F4; a gift from S Prusiner); beta-COP (a gift from J Lippincott-Schwartz); EEA1 (BD Transduction Laboratories); Cy3- and Cy5-conjugated secondary antibodies (Jackson ImmunoResearch). Proteasome inhibitors were from Calbiochem.
TF sequestration assay
MDCK cells in 96-well plates were transfected (using Lipofectamine from Invitrogen) with 160 ng pLuc, varying amounts of TF-containing plasmid, and YFP plasmid to make a total of 200 ng DNA per well. After 18–24 h, cells were harvested for parallel luciferase measurements (using a Luciferase Reporter Kit from Roche) and immunoblotting. Between three and six independent replicates were used for luciferase measurements. The assay is characterized extensively elsewhere (Levine et al, 2004) with further details provided in Supplementary data.
Biochemical analyses
Experiments analyzing PrP constructs were performed between 18 and 24 h after transfection (using Lipofectamine 2000) of N2a cells. Pulse-chase experiments were performed in six-well dishes. Pulse labeling was for 15 min with 150 μCi/ml 35S-Translabel (ICN), and chase was for 1 h with unlabeled media. Proteasome inhibition (5 μM MG132) was initiated 2 h prior to pulse labeling, and continued throughout the pulse and chase incubations. Cells were harvested and PrP was immunoprecipitated with the 3F4 antibody as described (Kim et al, 2002; also see Supplementary data). Analysis of PrP solubility and aggregation was as reported previously (Ma and Lindquist, 2001, 2002; Yedidia et al, 2001) and described in detail in Supplementary data. The assay for ‘self-propagation' of cytosolic PrP aggregates was performed as reported previously (Ma and Lindquist, 2002) and described in the legend to Figure 3. Detection of CtmPrP used a previously published assay (Hegde et al, 1998, 1999), and was performed on microsomes isolated from transfected cells as described in Supplementary data. All immunoblots were stained with Ponceau S to verify equal loading of samples (as shown in Figure 2B).
Imaging and quantitative analyses
Fluorescence microscopy utilized an LSM510 confocal microscopy system (Zeiss). Imaging conditions, quantitative analyses of localization, double immunofluorescence, and colocalization studies were essentially as reported previously (Snapp et al, 2003 and references therein), with further details provided in Supplementary data.
Cytotoxicity analysis
Analysis of cell death using Cy5-labeled Annexin V was as recommended by the manufacturer (Molecular Probes). The amount of Annexin staining from three independent experiments was averaged to generate the graph in Figure 8B. Total cell viability (Figure 8C) was determined by trypan blue exclusion. Viable and nonviable cells were tabulated manually using a hemocytometer and the results of multiple independent experiments were averaged.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Methods
Acknowledgments
We thank C Levine and D Mitra for providing reagents for the TF sequestration assay, S Priola for N2a cells, S Prusiner for 3F4 antibody, J Lippincott-Schwartz for the beta-COP antibody, J Bonifacino for the EEA1 antibody, E Snapp for help with microscopy, and H Lorenz, E Cohen, H Bernstein, and members of the Hegde lab for useful discussions. We also appreciate comments on this manuscript from J Bonifacino.
References
- Aguzzi A, Haass C (2003) Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 302: 814–818 [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG (2003) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161: 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996) Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379: 339–343 [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267–298 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Harris DA (2001) Prion diseases: what is the neurotoxic molecule? Neurobiol Dis 8: 743–763 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427–446 [DOI] [PubMed] [Google Scholar]
- Cohen E, Taraboulos A (2003) Scrapie-like prion protein accumulates in aggresomes of cyclosporin A-treated cells. EMBO J 22: 404–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini, Fioriti L, Chiesa R, Harris DA (2003) Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem 278: 21732–21743 [DOI] [PubMed] [Google Scholar]
- Garcia PD, Ou JH, Rutter WJ, Walter P (1988) Targeting of the hepatitis B virus precore protein to the endoplasmic reticulum membrane: after signal peptide cleavage translocation can be aborted and the product released into the cytoplasm. J Cell Biol 106: 1093–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goder V, Crottet P, Spiess M (2000) In vivo kinetics of protein targeting to the endoplasmic reticulum determined by site-specific phosphorylation. EMBO J 19: 6704–6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DA, Tsirigotis M, Woulfe J (2003) Ubiquitin, proteasomes, and the aging brain. Sci Aging Knowledge Environ 2003: RE6. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Lingappa VR (1999) Regulation of protein biogenesis at the endoplasmic reticulum membrane. Trends Cell Biol 9: 132–137 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR (1998) A transmembrane form of the prion protein in neurodegenerative disease. Science 279: 827–834 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Rane NR (2003) Prion protein trafficking and the development of neurodegeneration. Trends Neuro Sci 26: 337–339 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Tremblay P, Groth D, DeArmond SJ, Prusiner SB, Lingappa VR (1999) Transmembrane and genetic prion diseases share a common pathway of neurodegeneration involving transmembrane prion protein. Nature 402: 822–826 [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu XO, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA 98: 8838–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson N, Varshavsky A (1994) Ubiquitin-assisted dissection of protein transport across membranes. EMBO J 13: 2686–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SC, Brown DR, Whiteman M, Li R, Pan T, Perry G, Wisniewski T, Sy MS, Wong BS (2004) Prion protein is ubiquitinated after developing protease resistance in the brains of scrapie-infected mice. J Pathol 203: 603–608 [DOI] [PubMed] [Google Scholar]
- Kawasumi M, Hashimoto Y, Chiba T, Kanekura T, Yamagishi Y, Ishizaka M, Tajima H, Niikura T, Nishimoto I (2002) Molecular mechanisms for neuronal cell death by Alzheimer's amyloid precursor protein-relevant insults. Neurosignals 11: 236–250 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Hegde RS (2002) Cotranslational partitioning of nascent prion protein into multiple populations at the translocation channel. Mol Biol Cell 13: 3775–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Mitra D, Salerno JR, Hegde RS (2002) Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev Cell 2: 207–217 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Rahbar R, Hegde RS (2001) Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J Biol Chem 276: 26132–26140 [DOI] [PubMed] [Google Scholar]
- Lang-Rollin I, Rideout H, Stefanis L (2003) Ubiquitinated inclusions and neuronal cell death. Histol Histopathol 18: 509–517 [DOI] [PubMed] [Google Scholar]
- Levine CG, Mitra D, Sharma A, Smith CL, Hegde RS (2004) The efficiency of compartmentalization into the secretory pathway. Mol Biol Cell, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S (2001) Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA 98: 14955–14960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S (2002) Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298: 1785–1788 [DOI] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S (2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298: 1781–1785 [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302: 871–874 [DOI] [PubMed] [Google Scholar]
- Mironov A Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ (2003) Cytosolic prion protein in neurons. J Neurosci 23: 7183–7193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi CE, Weiss J (1992) Bidirectional movement of a nascent polypeptide across microsomal membranes reveals requirements for vectorial translocation of proteins. Cell 71: 87–96 [DOI] [PubMed] [Google Scholar]
- Ou JH, Yeh CT, Yen TS (1989) Transport of hepatitis B virus precore protein into the nucleus after cleavage of its signal peptide. J Virol 63: 5238–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB (2001) Neurodegenerative diseases and prions. N Engl J Med 344: 1516–1526 [DOI] [PubMed] [Google Scholar]
- Rapoport TA, Jungnickel B, Kutay U (1996) Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu Rev Biochem 65: 271–303 [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC (2003) Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem 278: 40877–40881 [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Lingappa VR, Hegde RS (2001) Substrate-specific regulation of the ribosome-translocon junction by N-terminal signal sequences. Proc Natl Acad Sci USA 98: 7823–7828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766 [DOI] [PubMed] [Google Scholar]
- Snapp EL, Hegde RS, Francolini M, Lombardo F, Colombo S, Pedrazzini E, Borgese N, Lippincott-Schwartz J (2003) Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol 163: 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RS, Drisaldi B, Harris DA (2001) A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic reticulum. Mol Biol Cell 12: 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RS, Harris DA (2001) Most pathogenic mutations do not alter the membrane topology of the prion protein. J Biol Chem 276: 2212–2220 [DOI] [PubMed] [Google Scholar]
- Stewart RS, Harris DA (2003) Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection. J Biol Chem 278: 45960–45968 [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T (1995) Inherited prion diseases and transmission to rodents. Brain Pathol 5: 53–59 [DOI] [PubMed] [Google Scholar]
- Tateishi J, Kitamoto T, Hoque MZ, Furukawa H (1996) Experimental transmission of Creutzfeldt–Jakob disease and related disorders to rodents. Neurology 46: 532–537 [DOI] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A (2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 20: 5383–5391 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Methods