

Abstract
Initiation of transcription in mammalian mitochondria depends on three proteins: mitochondrial RNA polymerase (POLRMT), mitochondrial transcription factor A (TFAM) and mitochondrial transcription factor B2 (TFB2M). We show here that the recombinant mouse and human transcription machineries are unable to initiate transcription in vitro from the heterologous light-strand promoter (LSP) of mitochondrial DNA. This species specificity is dependent on the interaction of TFAM and POLRMT with specific distal and proximal promoter elements. A sequence element localized from position −1 to −2 relative to the transcription start site in LSP functionally interacts with POLRMT. The POLRMT/TFB2M heterodimer is unable to interact with promoter elements and initiate even abortive transcription in the absence of TFAM. TFAM is thus an integral part of the mammalian transcription machinery, and we propose that TFAM induces a structural change of the promoter that is required for POLRMT-dependent promoter recognition.
Keywords: mitochondrion, POLRMT, TFAM, TFB2M, transcription
Introduction
The mitochondrial genome (mtDNA) encodes key components of the oxidative phosphorylation system, and the regulation of mtDNA transcription is therefore of fundamental importance for maintaining metabolic functions in the eukaryotic cell. Mechanistic aspects of mtDNA transcription have mainly been studied in Saccharomyces cerevisiae, but there are significant differences between yeast and mammalian mtDNA. The S. cerevisiae mtDNA (∼86 kb) has a complex organization with abundant noncoding regions and genes interrupted by introns, whereas the smaller mammalian mtDNA of (∼16 kb) contains few noncoding regions and lacks introns (Shadel and Clayton, 1997). Initiation of transcription of S. cerevisiae mtDNA occurs from multiple promoters. In contrast, mammalian mtDNA contains only two major promoters, the light- and heavy-strand promoters (LSP and HSP), which produce near-genomic length transcripts that after RNA processing release individual mRNAs, tRNAs and rRNAs (Ojala et al, 1981; Clayton, 1991). Transcription from LSP is necessary not only for gene expression but also for production of RNA primers required for initiation of mtDNA replication (Shadel and Clayton, 1997).
The basic machinery for transcription of S. cerevisiae mtDNA only consists of two factors: the yeast mitochondrial RNA polymerase, denoted Rpo41 (Masters et al, 1987), and its accessory transcription factor Mtf1, also known as sc-mtTFB (Schinkel et al, 1987; Shadel and Clayton, 1995). In contrast, transcription of mammalian mtDNA promoters is critically dependent on mitochondrial transcription factor A (TFAM), a high-mobility group-box protein (Fisher and Clayton, 1988; Parisi and Clayton, 1991; Shadel and Clayton, 1997). The yeast TFAM homologue, Abf2, does not activate transcription but rather functions as an mtDNA packaging factor (Diffley and Stillman, 1991; Parisi et al, 1993; Dairaghi et al, 1995a).
In yeast, the heterodimeric Rpo41/Mtf1 complex binds to the simple nonanucleotide promoter (consensus ATATAAGTA) and initiates transcription (Mangus et al, 1994). Mtf1 remains associated with Rpo41 during initiation and in the early stages of elongation, but is released when Rpo41 enters into the elongation mode. Rpo41 alone can initiate unspecific transcription from a synthetic poly[d(A-T)] DNA template, whereas the addition of Mtf1 is required for promoter-specific initiation of transcription (Winkley et al, 1985; Schinkel et al, 1988). How Mtf1 contributes to promoter recognition is not understood. There have been suggestions that Mtf1 may be functionally related to sigma factors, which provide bacterial RNA polymerases with the determinants for promoter recognition and DNA melting (Shadel and Clayton, 1995). However, the structural analysis of Mtf1 has revealed homology to ribosomal RNA methyltransferases and does not support a role for Mtf1 as a sigma-like factor (Schubot et al, 2001). Rpo41 displays significant sequence similarity to the monomeric bacteriophage T7 RNA polymerase (T7RNAP), which interacts sequence-specifically with promoters and initiates transcription on its own (Masters et al, 1987). Recent data suggest that Rpo41 has the intrinsic ability to initiate promoter-specific transcription without its specificity factor Mtf1 from negatively supercoiled templates (Matsunaga and Jaehning, 2004).
We have previously reconstituted human mtDNA transcription in a pure in vitro system consisting of a promoter-containing DNA fragment and recombinant TFAM, mitochondrial RNA polymerase (POLRMT) and mitochondrial transcription factor B1 (TFB1M) or B2 (TFB2M) (Falkenberg et al, 2002). Both TFB1M and TFB2M are Mtf1 homologues, but TFB2M is at least two orders of magnitude more active than TFB1M in basal transcription. A human protein denoted mtTFB, which is identical to TFB1M, has been isolated and shown to stimulate transcription in a mitochondrial extract (McCulloch et al, 2002).
Studies of human LSP have revealed that a minimal DNA fragment corresponding to position −28 to +16 relative to the transcription initiation site is able to support transcription initiation in a mitochondrial extract (Chang and Clayton, 1984). TFAM interacts directly with nucleotides (nts) between positions −35 and −17 (Fisher et al, 1987), and the exact distance between the TFAM-binding site and the transcription start site is essential for promoter activity (Dairaghi et al, 1995b). Sequence-specific DNA interactions have so far not been described for TFB1M, TFB2M or POLRMT, and their role in promoter-specific transcription initiation is not understood. We have analyzed the molecular mechanisms of promoter recognition in the mammalian mitochondrion by utilizing the species specificity of the human and mouse mtDNA transcription machineries (Fisher et al, 1989). We conclude that POLRMT, similar to the homologous T7RNAP and yeast Rpo41, recognize promoter elements in a sequence-specific manner. Based on our findings, we propose a model for how promoters are recognized and transcription initiated in mammalian mitochondria.
Results
The mouse and human mitochondrial transcription machineries can only initiate transcription from the species-specific LSP
We decided to use a functional approach to investigate LSP recognition in mammals, by taking advantage of species-specific differences. The human LSP (hLSP) cannot support transcription in a mouse mitochondrial extract, and in a similar way the mouse LSP (mLSP) cannot support transcription in a human mitochondrial extract (Fisher et al, 1989). The exchange of factors between the human and mouse mitochondrial transcription machineries therefore provided us with a strategy to identify the factor/s responsible for interacting directly with promoter DNA sequences.
We first reconstituted a highly purified recombinant in vitro transcription system from mouse (Figure 1A). The mouse system was essentially indistinguishable from our previously reported human in vitro transcription system (Figure 1B and data not shown) (Falkenberg et al, 2002), the only significant difference being that mPOLRMT in isolation was insoluble and therefore could not be dissociated from mTFB2M after purification. We investigated the ability of the human and mouse transcription machineries to initiate in vitro transcription from hLSP and mLSP. Neither the mouse nor the human transcription machinery could initiate transcription from the heterologous promoter (Figure 1C), consistent with previous results from studies with mitochondrial extracts (Fisher et al, 1989). The species specificity of mitochondrial transcription could thus be fully reconstituted with pure components, demonstrating that the specificity was due to differences in the basal transcription machinery components and not caused by additional factors present in mitochondrial extracts.
Figure 1.
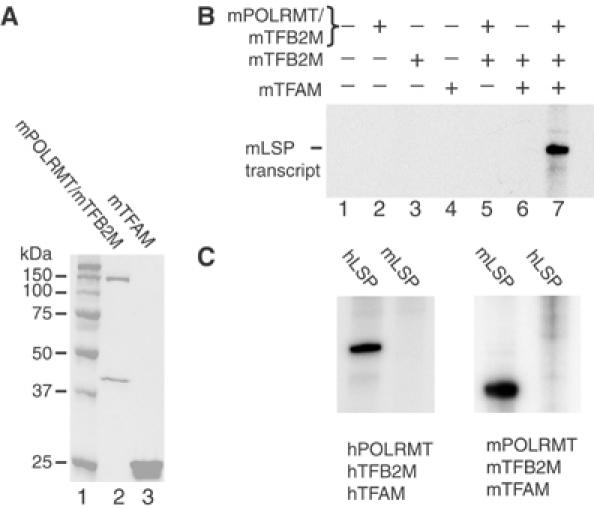
Reconstitution of the mouse mitochondrial transcription system in vitro. (A) SDS–PAGE gel stained with Coomassie blue showing the recombinant mouse proteins used for in vitro transcription reaction. Whereas mTFAM is expressed on its own, mPOLRMT is co-expressed and co-purified with mTFB2M. (B) Transcription from mLSP only occurs when mTFAM (2.5 pmol), mPOLRMT (500 fmol) and mTFB2M (500 fmol) are present simultaneously. (C) The human and mouse transcription machinery cannot initiate transcription from heterologous promoters. Linear templates (85 fmol) containing the mouse or hLSP were used for in vitro run-off transcription assays. The reactions were performed with the following pure recombinant proteins: human or mouse POLRMT (500 fmol), human or mouse TFB2M (500 fmol) and human or mouse TFAM (2.5 pmol).
Species specificity is governed by DNA sequences upstream of the transcription start site
We investigated if the species specificity was due to DNA sequences upstream or downstream of the start site for transcription. We made a series of hybrid promoter constructs with the upstream region of hLSP fused to the downstream region of mLSP and vice versa (Figure 2A). We analyzed how these hybrid DNA constructs interacted with the human and mouse transcription machineries by performing in vitro transcription reactions. We found that the DNA sequences governing species specificity are located upstream of the transcription start site (Figure 2B and C). Interestingly, this region is highly divergent between hLSP and mLSP (Figure 2D). In fact, only nine of 30 base pairs immediately upstream of the transcription start site are conserved between the species.
Figure 2.
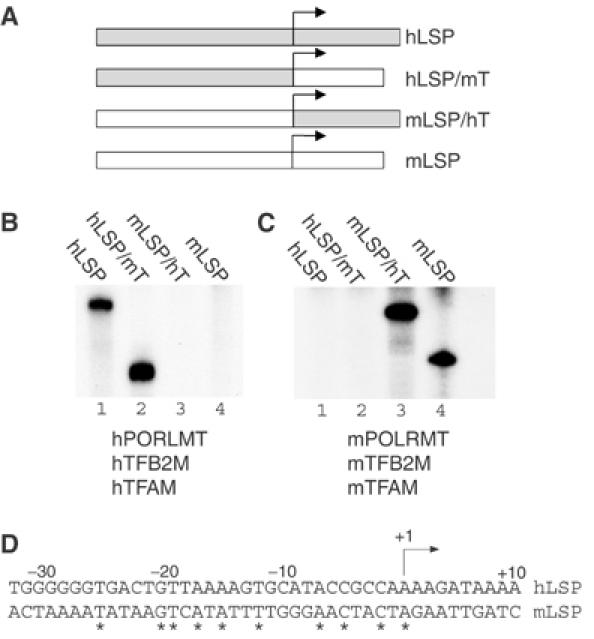
DNA sequences governing species specificity are localized upstream of the transcription initiation site. (A) Schematic representation of the LSP templates harboring a chimeric human LSP-mouse transcript (hLSP/mT) or a mouse LSP-human transcript (mLSP/hT). The transcription initiation site (+1) is indicated with an arrow. (B) The templates were assayed for their ability to support in vitro transcription with the pure recombinant human system. The transcription reaction mixtures contained hPOLRMT (500 fmol), hTFB2M (500 fmol), hTFAM (2.5 pmol) and the indicated mtDNA template (85 fmol). (C) The templates were assayed for their ability to support in vitro transcription with the pure recombinant mouse system. The transcription reaction mixtures were as under (B), but with the corresponding mouse proteins. (D) A sequence comparison between hLSP and mLSP. Conserved nucleotides are indicated with asterisks.
Mapping of essential elements in human LSP
We investigated the primary sequence requirements of hLSP by making a series of promoter constructs with consecutive 2 bp mutations from position −1 to −20 (Figure 3A). The activity of the mutant promoter constructs was investigated in the human in vitro transcription system (Figure 3B). We found that the promoter region spanning position −1 to −4 was especially sensitive to mutations and we denoted this region the proximal promoter element (PPE; Figure 3B, lanes 11 and 12). We also observed a slight decrease in transcription when we mutated the −17 to −20 region (Figure 3B, lanes 3 and 4), and denoted this region the distal promoter element (DPE). It should be noted that although mutations in the PPE and DPE displayed pronounced effects, also mutations of intervening sequences had minor negative effects on the overall levels of transcription. To further verify the importance of PPE, we mutated the corresponding region in mLSP and tested its activity in the mouse in vitro transcription system (Figure 3C). We found that the region −1 to −2 was sensitive to mutations (Figure 3C, lane 2), whereas mutations in region −3 to −4 did not significantly change the levels of transcription relative to the wild-type promoter (Figure 3C, lane 1). We thus conclude that the precise nucleotide sequence of the −1 to −2 region of PPE is essential for full transcriptional activity of both mLSP and hLSP.
Figure 3.
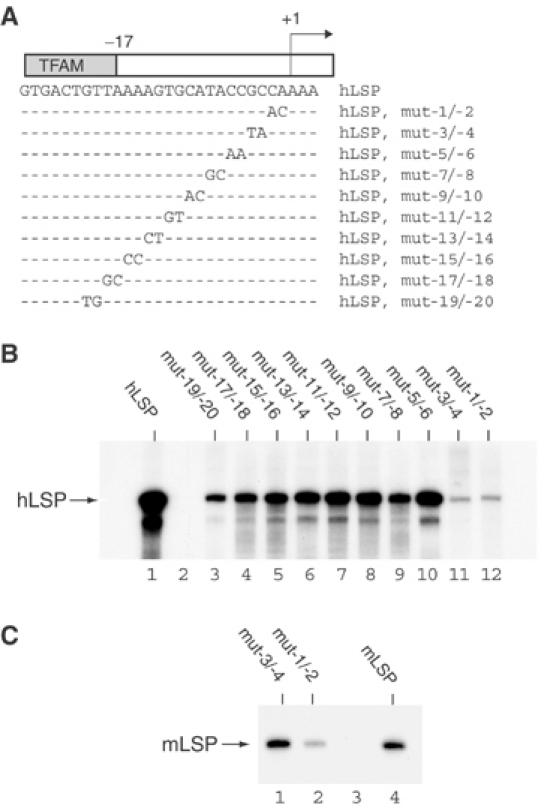
Mutational analysis of the hLSP promoter. (A) A series of LSP promoter constructs in which 2 bp at the time were mutated from position −1 to −20 from the transcription initiation site. The introduced mutations made the following changes in the hLSP sequence: A to C, C to A, T to G and G to T. The transcription initiation site is (+1) indicated with an arrow. Only the hLSP noncoding strand is represented. (B) The ability of the mutant promoter constructs to support transcription was investigated in the complete human in vitro transcription system. The in vitro transcription reaction mixtures contained hTFAM (2.5 pmol), hPOLRMT (500 fmol), hTFB2M (500 fmol) and 85 fmol of human LSP template. (C) Two mLSP promoter constructs were mutated at positions −1/−2 and −3/−4 as described for hLSP under (A). The ability of the mutant mLSP constructs to support transcription was investigated in the complete mouse in vitro transcription system. The in vitro transcription reaction mixtures contained mTFAM (2.5 pmol), mPOLRMT (500 fmol), mTFB2M (500 fmol) and 85 fmol of mLSP template.
TFAM contributes to promoter recognition at mouse LSP
We had previously shown that human TFAM (hTFAM) has a comparatively low affinity for mLSP and therefore is a poor activator of mtDNA transcription in the mouse (Ekstrand et al, 2004). We further investigated the role for hTFAM in species-specific LSP transcription by generating a series of hybrid promoter constructs with the upstream mTFAM-binding DNA sequences of mLSP replaced by the corresponding hLSP sequences (Figure 4A). We used these constructs to perform in vitro transcription reactions with the complete human transcription machinery. As anticipated, the human system could not support transcription from the wild-type mLSP (Figure 4B, lane 1). However, when we introduced the −40 to −11 region from hLSP, we obtained a strong transcription reaction from the hybrid promoter construct (Figure 4B, lane 2). We made the exchanged region containing the hTFAM-binding site gradually smaller and noticed that the activity of the human system dropped dramatically when we changed the −17 to −20 region to the mouse sequence (Figure 4B, lane 5). The transcription was almost abolished when the −21 to −22 region also was mutated (Figure 4B, lane 6). The −17 to −22 region coincides with the 3′ border of the previously characterized hTFAM-binding site (Fisher et al, 1987) and also corresponds perfectly with the DPE. A sequence comparison between hLSP and mLSP revealed that the nucleotides at positions −17, −19 and −20 are conserved between the two promoters, whereas the nucleotide at position −18 is T in hLSP and C in mLSP (Figure 2D). This T to C transition therefore contributes to the inability of hTFAM to stimulate transcription from mLSP. The data presented here and elsewhere (Ekstrand et al, 2004) therefore shows that hTFAM contributes to the observed species specificity at mLSP.
Figure 4.
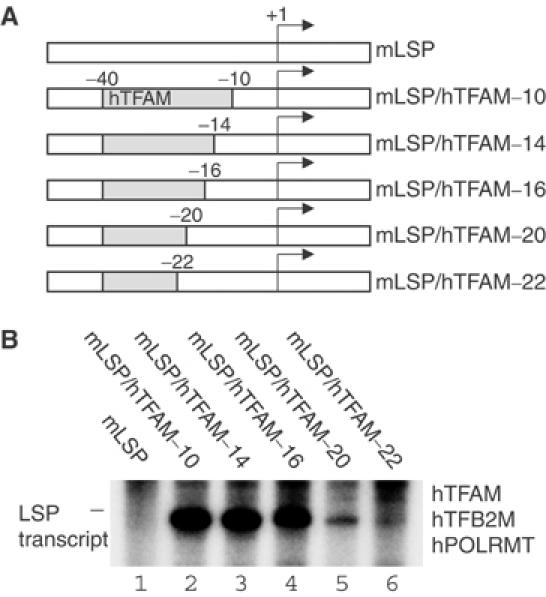
Introduction of the human TFAM-binding sites enables the human transcription machinery to initiate transcription from mouse LSP. (A) Series of hybrid promoter constructs in which the TFAM-binding site of mouse LSP was replaced with corresponding sequences from the human LSP. Shaded boxes correspond to the introduced human sequence. The transcription initiation site (+1) is indicated with an arrow. (B) The ability of the hybrid promoter constructs to support transcription was investigated in the complete human in vitro transcription system. The in vitro transcription reaction mixtures contained hTFAM (2.5 pmol), hPOLRMT (500 fmol), hTFB2M (500 fmol) and 85 fmol of human LSP template.
Mouse POLRMT cannot initiate transcription on human LSP
We next investigated the inability of the mouse mitochondrial transcription machinery to initiate transcription from the human promoter. We first compared the capacity of mTFAM and hTFAM to stimulate hTFB2M/hPOLRMT on wild-type hLSP (Figure 5A). Surprisingly, we found that mTFAM was at least as active as hTFAM in the stimulation of the human transcription machinery. Therefore, in contrast to the situation at mLSP, differences between hTFAM and mTFAM could not explain the observed species specificity at hLSP. Furthermore, mTFB2M could replace hTFB2M, without affecting species-specific promoter recognition (Figure 5B, lane 2). In contrast, mPOLRMT failed to replace hPOLRMT and initiate transcription either in the presence of mTFB2M or hTFB2M (Figure 5B, lanes 3 and 4). Our analysis therefore demonstrated that it was mPOLRMT, and not mTFAM or mTFB2M, which was responsible for the inability of the mouse transcription machinery to initiate transcription at hLSP.
Figure 5.
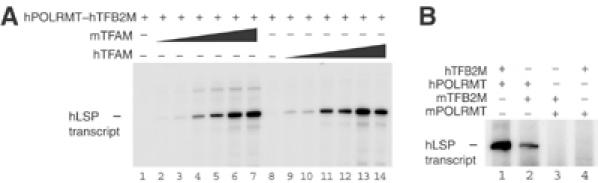
(A) Mouse TFAM can efficiently support transcription on hLSP. Template containing the LSP (85 fmol) of human mtDNA was used for in vitro run-off transcription assays. The reactions were performed with the following pure recombinant proteins: hPOLRMT (300 fmol), hTFB2M (300 fmol) and hTFAM or mTFAM in increasing amounts (0, 0.025, 0.1, 0.25, 1.0, 2.5, 10 pmol). (B) Mouse POLRMT cannot initiate transcription at hLSP. In vitro transcription from hLSP was performed with the proteins indicated (500 fmol) in the presence of (2.5 pmol) of hTFAM.
mPOLRMT recognizes the PPE
We next wanted to define the promoter region recognized by mPOLRMT and therefore made a series of hybrid promoter constructs, in which we gradually replaced the DNA sequences in hLSP with the corresponding sequences from mLSP (Figure 6A). The mPOLRMT could not initiate transcription from either the wild-type hLSP or the hybrid promoter containing the −11 to −40 mTFAM-binding region (Figure 6B, lanes 2 and 3). We thus concluded that an essential region for mPOLRMT function was located in the −1 to −10 region of hLSP.
Figure 6.
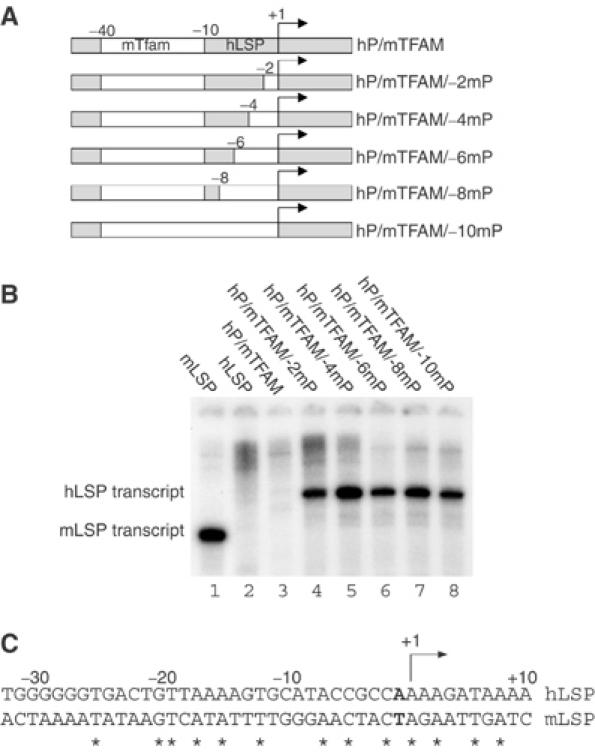
Promoter recognition by mPOLRMT is critically dependent on the PPE. (A) A series of hybrid promoter constructs in which the −1 to −10 hLSP region was gradually (2 bp) replaced with the corresponding sequences from mLSP. (B) Hybrid promoter constructs were used for in vitro run-off transcription assays. The reactions were performed with the following pure recombinant proteins: mPOLRMT (500 fmol), mTFB2M (500 fmol), mTFAM (2.5 pmol) and 85 fmol of template. (C) Sequence comparison between hLSP and mLSP. Conserved nucleotides are marked with asterisks and the A to T transversion at position −1 is in bold font.
We gradually altered the −1 to −10 region of hLSP into the nucleotide sequence present in mLSP (Figure 6A and B). mPOLRMT could efficiently initiate transcription in the presence of hTFAM and hTFB2M from a template containing the −1 to −2 region of mLSP (Figure 6B, lane 4). No further stimulation was observed when we replaced the entire −1 to −10 human region with the mouse sequence (Figure 6B, lanes 5–8). We thus concluded that the activity of the mPOLRMT is dependent on functional interactions with the −1 to −2 region, corresponding to the PPE identified in our mutational analysis of hLSP. A sequence comparison between hLSP and mLSP revealed that the base at position −2 is conserved between the two promoters, whereas the base at position −1 is A in hLSP and T in mLSP (Figure 6C). This A to T transversion is therefore responsible for the inability of mPOLRMT to recognize the human PPE. It is noteworthy that the base at position +1 is identical in mLSP and hLSP; therefore, the transversion at position −1 interrupts a conserved stretch of three nucleotides at the precise site of transcription initiation (Figure 6C).
We next used DNase I footprinting to directly investigate interactions between individual components of the human transcription machinery and hLSP (Figure 7). As expected, hTFAM protected a region between −15 and −38 (Figure 7A, lane 4). In contrast, neither hPOLRMT nor hTFB2M in isolation generated a footprint on hLSP (Figure 7A, lanes 2 and 3).
Figure 7.
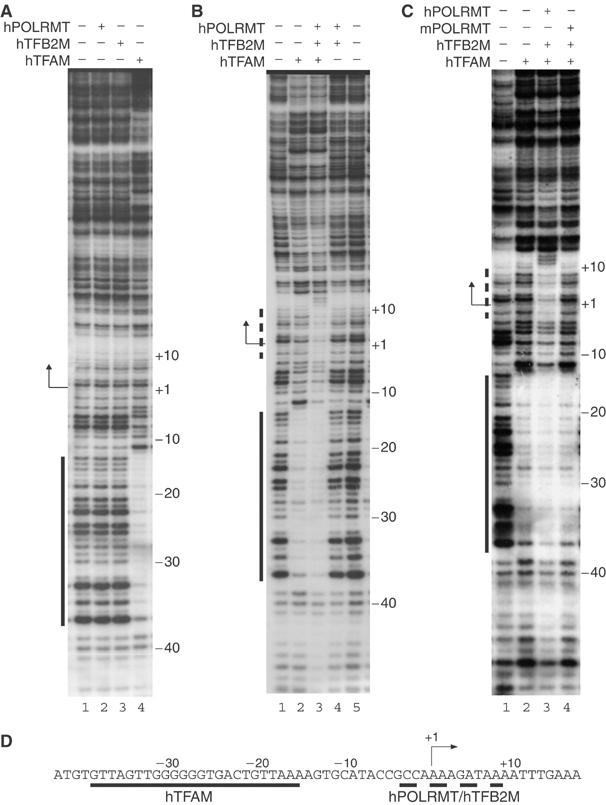
Promoter recognition by hPOLRMT/hTFB2M is strictly dependent on hTFAM (A). DNase I footprinting reveals that neither hPOLRMT nor hTFB2M (lanes 2 and 3) in isolation interacts with hLSP. The binding site for hTFAM (lane 4) is indicated with a solid line. (B) The hPOLRMT/hTFB2M heterodimer interacts with the transcription start site in the presence (lane 3), but not in the absence (lane 4), of hTFAM. The region protected by hPOLRMT/hTFB2M is indicated with a dashed line. (C) The hPOLRMT/hTFB2M complex (lane 3), but not the mPOLRMT/hTFB2M complex (lane 4), interacts with the hLSP transcription start site in the presence of hTFAM. (D) A schematic representation of protein interactions with hLSP. Human TFAM protects the −15 to −38 region (solid line) and the hPOLRMT/hTFB2M heterodimer protects the +10 to −4 region (dashed line).
In yeast, the Rpo41/Mtf1 heterodimer binds sequence specifically to promoter DNA (Mangus et al, 1994) and we therefore speculated that the hPOLRMT/hTFB2M heterodimer might be required for hLSP binding. However, the simultaneous presence of hPOLRMT and hTFB2M also failed to generate a footprint (Figure 7B, lane 4). We finally added all three human transcription factors, hTFAM, hPOLRMT, and hTFB2M, at the same time. We now observed two distinct regions of protection, one corresponding to the hTFAM-binding site and a second region covering position −4 to −1 (PPE) and position +1 to +10 (Figure 7B, lane 3). The data thus indicated that the hPOLRMT/hTFB2M heterodimer interacts with the region surrounding the initiation site for transcription and that this interaction is strictly dependent on the presence of hTFAM. Interestingly, we completely abolished binding to the transcription start site when we replaced hPOLRMT with mPOLRMT (Figure 7C, lane 4). The footprinting experiments therefore supported our previous conclusion that mammalian POLRMT interacts sequence specifically with DNA sequences at the start site for transcription.
TFAM is absolutely required for promoter recognition and transcription initiation
Our footprinting analysis revealed an interesting difference between the yeast and human mitochondrial transcription machinery. In yeast, the TFAM orthologue Abf2 is not required for promoter recognition or transcription initiation. In contrast, our footprinting analysis suggested that hTFAM was absolutely essential for hPOLRMT/hTFB2M-dependent promoter recognition. To further investigate the role of hTFAM, we analyzed the ability of hPOLRMT/hPOLRMT to initiate abortive transcription in vitro. Transcription from hLSP generates two major products, the full-length transcript and a short abortive transcript of 4-nt (Figure 8). The production of both abortive and full-length transcripts was absolutely dependent on the simultaneous presence of hTFAM and the hPOLRMT/hTFB2M heterodimer. Identical data were obtained with the complete mouse transcription system on mLSP (data not shown). The abortive transcription assay therefore supported our conclusion that mammalian TFAM is not simply an activator of a specific phase in transcription initiation, for example, promoter escape, but an integral component of the transcription machinery, essential for promoter recognition and transcription initiation.
Figure 8.
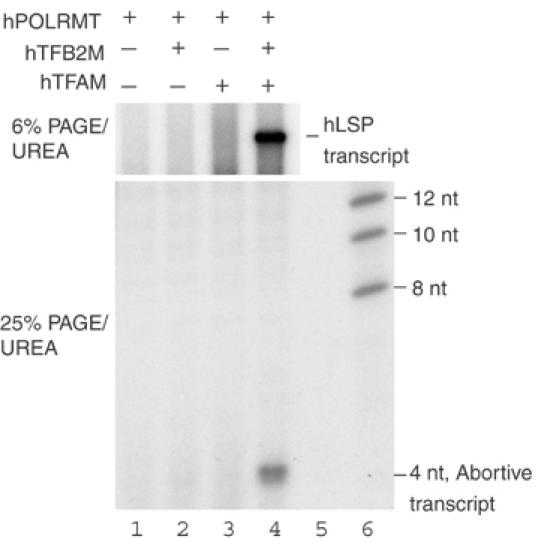
hTFAM and hTFB2M are required for abortive transcription initiation. hTFAM, hPOLRMT and hTFB2M were added as indicated. Transcription reactions were analyzed in parallel on 6% and 25% denaturing polyacrylamide gels.
Discussion
We show here that the recombinant mouse and human transcription machineries are unable to initiate transcription in vitro from the heterologous LSP of mtDNA. We use a factor-swapping approach and find that mTFB2M and mTFAM can function together with hPOLRMT and initiate transcription at hLSP. In contrast, mPOLRMT is unable to initiate transcription at hLSP, even in the presence of hTFB2M and hTFAM. Further analysis demonstrates that the inability of mPOLRMT to initiate transcription from hLSP is dependent on the precise DNA sequence of the PPE, since a single base pair change at position −1 can reactivate the mouse polymerase. We can therefore conclude that the mPOLRMT interacts sequence specifically with mitochondrial promoters.
We also demonstrate that hPOLRMT-dependent promoter recognition and transcription initiation are strictly dependent on both hTFAM and hTFB2M. hPOLRMT is unable to initiate abortive transcription in the absence of these two auxiliary transcription factors, Moreover, DNase I footprinting experiments indicate that the hPOLRMT/hTFB2M heterodimer requires hTFAM binding in order to recognize and bind mitochondrial promoters. This observation is of particular interest for our understanding of TFAM, since its yeast homologue Abf2 is not needed for transcription initiation (Shadel and Clayton, 1997). Our analysis suggests that mammalian TFAM has not just evolved to stimulate transcription, but has become an integral and essential component of the transcription machinery. We can therefore conclude that the mechanisms of mitochondrial transcription initiation are substantially different between yeast and mammalian cells.
The TFAM protein contains two tandem HMG box domains separated by a 27 amino-acid residue linker region and followed by a 25-residue carboxy-terminal (C-terminal) tail. Mutational analysis of TFAM has revealed that the tail region is important for specific DNA recognition and essential for transcriptional activation (Dairaghi et al, 1995a). We demonstrate here that the DPE located between positions −17 and −20 is critically important for hTFAM function, and that a single T to C transition at position −18 is at least partially responsible for the much lower activity of hTFAM on mLSP. It is interesting to note that DPE is perfectly conserved between human and rat LSP (position −16 to −19 in the rat sequence), which may explain why hTFAM can stimulate transcription in isolated rat mitochondria (Garstka et al, 2003).
TFAM can bind, unwind and bend DNA without sequence specificity, similar to other proteins of the HMG domain family (Fisher et al, 1992). It is therefore likely that TFAM binding introduces specific structural alterations in mtDNA, for example, unwinding of the promoter region, which can facilitate promoter recognition by the TFB2M/POLRMT complex. The sequence-specific binding of a TFAM tetramer (Antoshechkin et al, 1997) upstream of HSP and LSP may allow the protein to introduce these structural alterations at a precise position in the promoter region and perhaps partially unwind the start site for transcription. This model may explain why the exact distance between the TFAM-binding site and the start site for LSP transcription is of critical importance (Dairaghi et al, 1995b).
The mitochondrial RNA polymerase belongs to the family of T7-like RNA polymerases (Masters et al, 1987). Promoter recognition by T7RNAP is achieved by the insertion of a ‘specificity loop' (amino acids 739–770) into the DNA major groove (−8 to −12 bp) and of a flexible ‘surface loop' (amino acids 93–110) into the minor grove of an A+T-rich sequence (−13 to −17 bp) (Cheetham and Steitz, 2000). Our finding that mPOLRMT governs sequence-specific interactions at the PPE suggests that sequence-specific DNA binding may be a general property of mitochondrial RNA polymerases. A recent study in S. cerevisiae lends strong support to our conclusions (Matsunaga and Jaehning, 2004). On linear templates, the yeast mitochondrial RNA polymerase Rpo41 is strictly dependent on the TFB2M homologue Mtf1 for initiation of promoter-specific transcription. In contrast, Rpo41 has the intrinsic ability to initiate from promoters without its specificity factor Mtf1 from negatively supercoiled templates. The sequence element in yeast mitochondrial promoters recognized by Rpo41 is localized between positions +1 and −8 relative to the transcription start site. This DNA element thus corresponds perfectly with the PPE identified by us in the present study.
Human TFB1M is a dual-function protein, which not only supports mitochondrial transcription in vitro, but also acts as an rRNA methyltransferase (Seidel-Rogol et al, 2003). Furthermore, the crystal structure of yeast Mtf1 displays homology to the bacterial ErmC′ rRNA methyltransferase (Schubot et al, 2001). ErmC′ is an RNA-binding protein and it is therefore likely that TFB1M and, maybe also, TFB2M have the capacity to bind RNA and/or single-stranded DNA. One possible role for TFB2M could be to bind newly synthesized RNA and prevent the formation of an RNA/DNA hybrid at the promoter, which could inhibit further rounds of transcription initiation. Alternatively, TFB2M could bind single-stranded DNA and stabilize a partially unwound promoter during transcription initiation. Future efforts in our laboratory will be aimed at understanding the molecular role of TFB2M in transcription initiation.
Materials and methods
Cloning, expression and purification of recombinant proteins
We used PCR to amplify DNA fragments encoding mTFB2M, mPOLRMT (without leader peptide), and mTFAM (without leader peptide) from cDNA. The fragments were cloned into the vector pBacPAK9 (Clontech). We also made plasmid constructs in which a 10xHis tag had been introduced at the N-terminus (mPOLRMT) or a 6xHis tag had been introduced at the C-terminus (mTFAM and mTFB2M). We used the plasmid constructs to generate Autographa californica nuclear polyhedrosis recombinant viruses as described in the BacPAK manual (Clontech). The construction of recombinant baculoviruses for expression of hPOLRMT, hTFB2M and hTFAM has been described previously (Falkenberg et al, 2002). The recombinant proteins were expressed in Spodoptera frugiperda (Sf9) cells and whole-cell protein extracts were generated as described (Falkenberg et al, 2002). To generate mPOLRMT/mTFB2M and mPOLRMT/hTFB2M complexes, we co-infected insect cells with viruses expressing the different protein combinations and purified the heterodimeric complexes to near homogeneity over Ni2+-Agarose FF (Qiagen). We further purified the complexes on a 1 ml HiTrap Heparin column (Amersham Biosciences) equilibrated in buffer B (0.2 M NaCl) and used a linear gradient (10 ml) of buffer B (0.2–1.2 M NaCl) (Falkenberg et al, 2002) to elute the heterodimeric complex. We estimated the purity of the purified mPOLRMT/mTFB2M and mPOLRMT/hTFB2M complexes to be at least 95% by SDS–PAGE with Coomassie blue staining.
In vitro mouse transcription
We cloned DNA fragments containing mouse LSP corresponding to nucleotides 15 942–16 260 into the pCR4 vector with the TOPO cloning kit (Invitrogen) generating pmLSP. After linearization, we used the plasmid construct to measure promoter-specific transcription in a run-off assay. Individual reaction mixtures (25 μl) contained 85 fmol of digested template, 10 mM Tris–HCl (pH 8), 10 mM MgCl2, 1 mM DTT, 100 μg/ml bovine serum albovine, 400 μM ATP, 150 μM CTP and GTP, 10 μM UTP, 0.02 μM α-32P UTP (3000 Ci/mmol), 4 U of Rnasin (Amersham Biosciences) and the indicated concentrations of proteins. We stopped the reactions after 30 min at 32°C by adding 200 μl of stop buffer (10 mM Tris–HCl (pH 8.0), 0.2 M NaCl, 1 mM EDTA and 0.1 mg/ml glycogen). We treated the samples with 0.5% SDS and 100 μg/ml proteinase K for 45 min at 42°C, and precipitated them by adding 0.6 ml of ice-cold ethanol. We dissolved the pellets in 10 μl of gel-loading buffer (98% formamide, 10 mM EDTA (pH 8.0), 0.025% xylene cyanol FF, 0.025% bromophenol blue), heated them at 95°C for 5 min, and analyzed the samples on a 4% denaturing polyacrylamide gel in 1 × TBE buffer. We then dried and exposed the gels.
To monitor abortive transcription, we used the same assay conditions, but replaced α-32P UTP with α-32P ATP and analyzed the samples on a 25% denaturing polyacrylamide gel.
Mutational analysis of human LSP
We used an overlap extension PCR method (Ho et al, 1989) to introduce site-specific mutations within the human LSP. Pairs of mutagenic primers (mut-1/-2: 5′ GT GCA TAC CGC ACA AAG ATA AAA TTT G, 5′ CA AAT TTT ATC TTT GTG CGG TAT GCA C, mut-3/-4: 5′ GT TAA AAG TGC ATA CCT ACA AAA GAT AAA ATT TG, 5′ CA AAT TTT ATC TTT TGT AGG TAT GCA CTT TTA AC, mut-5/-6: 5′ GT TAA AAG TGC ATA AAG CCA AAA GAT AAA ATT TG, 5′ CA AAT TTT ATC TTT TGG CTT TAT GCA CTT TTA AC, mut-7/-8: 5′ GT TAA AAG TGC AGC CCG CCA AAA GAT AAA ATT TG, 5′ CA AAT TTT ATC TTT TGG CGG GCT GCA CTT TTA AC, mut-9/-10: 5′ CT GTT AAA AGT GAC TAC CGC CAA AAG ATA AAA TTT G, 5′ CA AAT TTT ATC TTT TGG CGG TAG TCA CTT TTA ACA G, mut-11/-12: 5′ GA CTG TTA AAA GGT CAT ACC GCC AAA AG, 5′ CT TTT GGC GGT ATG ACC TTT TAA CAG TC, mut-13/-14: 5′ GT GAC TGT TAA ACT TGC ATA CCG CCA AAA G, 5′ CT TTT GGC GGT ATG CAA GTT TAA CAG TCA C, mut-15/-16: 5′ GG TGA CTG TTA CCA GTG CAT ACC GCC, 5′ GG CGG TAT GCA CTG GTA ACA GTC ACC, mut-17/-18: 5′ GG GGT GAC TGT GCA AAG TGC ATA CCG, 5′ CG GTA TGC ACT TTG CAC AGT CAC CCC, mut-19/-20: 5′ GG GGG GTG ACT TGT AAA AGT GCA TAC, 5′ GT ATG CAC TTT TAC AAG TCA CCC CCC) were used to direct the synthesis of two overlapping fragments with the human (1–477) LSP (Falkenberg et al, 2002) as template. The fragments were gel purified and used for overlap extension PCR with 5′ and 3′ specific flanking primers for human LSP. The resulting amplified product was digested with appropriate restriction endonucleases (BamHI and HindIII) and cloned into pUC18. The PCR reactions were carried out with the Expand High Fidelity PCR system (Roche). The resulting mutations were confirmed by sequencing. After BamHI linearization, we used the plasmid constructs to measure promoter-specific transcription in a run-off assay.
We also used the overlap extension PCR method to introduce site-specific mutations in the mLSP. Pairs of mutagenic primers (mut-1/-2: 5′ CTA TCA AAC CCT ATG TCC TGA TCA ATT CTC TTA GTT CCC AAA ATA TG, 5′ CAT ATT TTG GGA ACT AAG AGA ATT GAT CAG GAC ATA GGG TTT GAT AG, mut-3/-4: 5′ CTA TCA AAC CCT ATG TCC TGA TCA ATT CTA GGC GTT CCC AAA ATA TG, 5′ CAT ATT TTG GGA ACG CCT AGA ATT GAT CAG GAC ATA GGG TTT GAT AG) were used to direct the synthesis of two overlapping fragments with the mouse (15 942–16 260) LSP (Ekstrand et al, 2004) as template. The fragments were gel purified and used for overlap extension PCR with 5′- and 3′-specific flanking primers for mouse LSP. The PCR products were cloned into the pCR4 TOPO vector. The mutations were confirmed by sequencing and, after linearization with PstI, the plasmid constructs were used to measure promoter specific transcription in a run-off assay.
Hybrid promoter constructs
For the mLSP/hT construct, we performed PCR with the human (1–477) LSP (Falkenberg et al, 2002) as template and the following primers: 5′ GAT CAC AGG TCT ATC ACC C and 5′ TGG TTC ACG GAA CAT GAT TTT GTA AAA TTT TTA CAA GTA CTA AAA TAT AAG TCA TAT TTT GGG AAC TAC TAA AGA TAA AAT TTG AAA TCT GG. For the hLSP/mT, we performed PCR with the pmLSP as template and the following primers: 5′ CAA CAT AGC CGT CAA GG and 5′ATT AGT AGT ATG GGA GTG GGA GGG GAA AAT AAT GTG TTA GTT GGG GGG TGA CTG TTA AAA GTG CAT ACC GCC AAG AAT TGA TCA GGA CAT AGG GTT TGA TAG T. The PCR products were cloned into the pCR4 TOPO vector as above. The expected hybrid promoter constructs were confirmed by sequencing. After linearization, we used the plasmid constructs to measure promoter-specific transcription in a run-off assay.
Construction of altered LSP templates
For the hP/mTFAM construct, we performed PCR with the human (1–477) LSP (Falkenberg et al, 2002) as template and the following primers: 5′ GAT CAC GGT CTA TCA CCC, and 5′ AGT AGT ATG GGA GTG GGA GGG GAA AAT AAT GTA CAA GTA CTA AAA TAT AAG TCA TAT TTT CAT ACC GCC AAA A. For the mLSP/hTFAM-10 construct, we used pmLSP as template and used the following primers: 5′ CAA CAT AGC CGT CAA GG, and 5′ TGG TTC ACG GAA CAT GAT TTT GTA AAA TTT TTG TTA GTT GGG GGG TGA CTG TTA AAA GTT GGG AAC TAC TAG. The hP/mTFAM construct was used as DNA template in an overlap PCR extension together with mutagenic primers first and then specific flanking DNA primers for the human LSP. The mutagenic primers used were: hP/mTFAM/-2mP construct: 5′ CAA ATT TTA TCT TTA GGC GGT ATG, 5′ CAT ACC GCC TAA AGA TAA AAT TTG; hP/mTFAM/-4mP: 5′ CAA ATT TTA TCT TTA GTA GGT ATG, 5′ CAT ACC TAC TAA AGA TAA AAT TTG; hP/mTFAM/-6mP: 5′ CCA GCC TAA CCA GAT TTC AAA TTT TAT CTT TAG TAG TTA TG, and 5′ CAT AAC TAC TAA AGA TAA AAT TTG AAA TCT GGT TAG GCT GG; hP/mTFAM/-8mP: 5′ CCA GCC TAA CCA GAT TTC AAA TTT TAT CTT TAG TAG TTC TGA AAA TAT GAC, 5′ GTC ATA TTT TCA GAA CTA CTA AAG ATA AAA TTT GAA ATC TGG TTA GGC TGG; hP/mTFAM/-10mP: 5′ CCA GCC TAA CCA GAT TTC AAA TTT TAT CTT TAG TAG TTC CCA AAA TAT GAC, 5′ GTC ATA TTT TGG GAA CTA CTA AAG ATA AAA TTT GAA ATC TGG TTA GGC TGG. The PCR products were cloned into the pCR4 TOPO vector and sequenced.
The mLSP/hTFAM-10 construct was used as DNA template in an overlap PCR extension together with mutagenic primers first and then specific flanking DNA primers for the mouse LSP. The mutagenic primers used were: mLSP/hTFAM-14: 5′ CTA GTA GTT CCC AAA ATT TAA CAG TCA CCC CCC, 5′ GGG GGG TGA CTG TTA AAT TTT GGG AAC TAC TAG; mLSP/hTFAM-16: 5′ CTA GTA GTT CCC AAA ATA TAA CAG TCA CCC CCC, 5′ GGG GGG TGA CTG TTA TAT TTT GGG AAC TAC TAG; mLSP/hTFAM-20: 5′ CTA GTA GTT CCC AAA ATA TGA CAG TCA CCC CCC, 5′ GGG GGG TGA CTG TCA TAT TTT GGG AAC TAC TAG, mLSP/hTFAM-22: 5′ CTA GTA GTT CCC AAA ATA TGA CTT TCA CCC CCC, 5′ GGG GGG TGA AAG TCA TAT TTT GGG AAC TAC TAG. The PCR products were cloned into the pCR4 TOPO vector and sequenced.
DNase I ‘footprinting'
A HindIII–BamHI restriction fragment containing hLSP was derived from the (1–477) LSP plasmid (Falkenberg et al, 2002) and end labeled with α-32P dCTP (3000 Ci/mmol) at the HindIII site. The LSP fragment was mixed on ice with 1 pmol hTFB2M, 1 pmol hPOLRMT, 1 pmol mPOLRMT and 5 pmol hTFAM, as indicated in Figure 7. The reaction mixtures were 30 μl and contained 25 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 1 mM ATP, 100 μg/ml bovine serum albumin and 1 mM DTT. We incubated the mixture for 20 min at 20°C and then added DNase I to a final concentration of 0.05 mg/ml. We stopped the reactions after 2 min by transfer to ice and the addition of sonicated calf thymus DNA (0.5 μg) and EDTA (final concentration 50 mM). The samples were extracted by phenol and precipitated with ethanol and subsequently analyzed on 8% polyacrylamide sequencing gels.
Acknowledgments
This work was supported by grants from the Swedish Research Council (to MF, N-GL and CMG), the Swedish Cancer Society (to CMG), the Swedish Foundation for Strategic Research (to N-GL and CMG), the Swedish National Board for Laboratory Animals (to MF and CMG) and the Emil and Wera Cornell's foundation (to MF).
References
- Antoshechkin I, Bogenhagen DF, Mastrangelo IA (1997) The HMG-box mitochondrial transcription factor xl-mtTFA binds DNA as a tetramer to activate bidirectional transcription. EMBO J 16: 3198–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA (1984) Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell 36: 635–643 [DOI] [PubMed] [Google Scholar]
- Cheetham GM, Steitz TA (2000) Insights into transcription: structure and function of single-subunit DNA-dependent RNA polymerases. Curr Opin Struct Biol 10: 117–123 [DOI] [PubMed] [Google Scholar]
- Clayton DA (1991) Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol 7: 453–478 [DOI] [PubMed] [Google Scholar]
- Dairaghi DJ, Shadel GS, Clayton DA (1995a) Addition of a 29 residue carboxyl-terminal tail converts a simple HMG box-containing protein into a transcriptional activator. J Mol Biol 249: 11–28 [DOI] [PubMed] [Google Scholar]
- Dairaghi DJ, Shadel GS, Clayton DA (1995b) Human mitochondrial transcription factor A and promoter spacing integrity are required for transcription initiation. Biochim Biophys Acta 1271: 127–134 [DOI] [PubMed] [Google Scholar]
- Diffley JF, Stillman B (1991) A close relative of the nuclear, chromosomal high-mobility group protein HMG1 in yeast mitochondria. Proc Natl Acad Sci USA 88: 7864–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13: 935–944 [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31: 289–294 [DOI] [PubMed] [Google Scholar]
- Fisher RP, Clayton DA (1988) Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol 8: 3496–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP, Lisowsky T, Parisi MA, Clayton DA (1992) DNA wrapping and bending by a mitochondrial high mobility group-like transcriptional activator protein. J Biol Chem 267: 3358–3367 [PubMed] [Google Scholar]
- Fisher RP, Parisi MA, Clayton DA (1989) Flexible recognition of rapidly evolving promoter sequences by mitochondrial transcription factor 1. Genes Dev 3: 2202–2217 [DOI] [PubMed] [Google Scholar]
- Fisher RP, Topper JN, Clayton DA (1987) Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell 50: 247–258 [DOI] [PubMed] [Google Scholar]
- Garstka HL, Schmitt WE, Schultz J, Sogl B, Silakowski B, Perez-Martos A, Montoya J, Wiesner RJ (2003) Import of mitochondrial transcription factor A (TFAM) into rat liver mitochondria stimulates transcription of mitochondrial DNA. Nucleic Acids Res 31: 5039–5047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Mangus DA, Jang SH, Jaehning JA (1994) Release of the yeast mitochondrial RNA polymerase specificity factor from transcription complexes. J Biol Chem 269: 26568–26574 [PubMed] [Google Scholar]
- Masters BS, Stohl LL, Clayton DA (1987) Yeast mitochondrial RNA polymerase is homologous to those encoded by bacteriophages T3 and T7. Cell 51: 89–99 [DOI] [PubMed] [Google Scholar]
- Matsunaga M, Jaehning JA (2004) Intrinsic promoter recognition by a ‘Core' RNA polymerase. J Biol Chem (in press) [DOI] [PubMed] [Google Scholar]
- McCulloch V, Seidel-Rogol BL, Shadel GS (2002) A human mitochondrial transcription factor is related to RNA adenine methyltransferases and binds S-adenosylmethionine. Mol Cell Biol 22: 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470–474 [DOI] [PubMed] [Google Scholar]
- Parisi MA, Clayton DA (1991) Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 252: 965–969 [DOI] [PubMed] [Google Scholar]
- Parisi MA, Xu B, Clayton DA (1993) A human mitochondrial transcriptional activator can functionally replace a yeast mitochondrial HMG-box protein both in vivo and in vitro. Mol Cell Biol 13: 1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Groot Koerkamp MJ, Tabak HF (1988) Mitochondrial RNA polymerase of Saccharomyces cerevisiae: composition and mechanism of promoter recognition. EMBO J 7: 3255–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Koerkamp MJ, Touw EP, Tabak HF (1987) Specificity factor of yeast mitochondrial RNA polymerase. Purification and interaction with core RNA polymerase. J Biol Chem 262: 12785–12791 [PubMed] [Google Scholar]
- Schubot FD, Chen CJ, Rose JP, Dailey TA, Dailey HA, Wang BC (2001) Crystal structure of the transcription factor sc-mtTFB offers insights into mitochondrial transcription. Protein Sci 10: 1980–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel-Rogol BL, McCulloch V, Shadel GS (2003) Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat Genet 33: 23–24 [DOI] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA (1995) A Saccharomyces cerevisiae mitochondrial transcription factor, sc-mtTFB, shares features with sigma factors but is functionally distinct. Mol Cell Biol 15: 2101–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, Clayton DA (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66: 409–435 [DOI] [PubMed] [Google Scholar]
- Winkley CS, Keller MJ, Jaehning JA (1985) A multicomponent mitochondrial RNA polymerase from Saccharomyces cerevisiae. J Biol Chem 260: 14214–14223 [PubMed] [Google Scholar]