

Abstract
Genetic studies in Caenorhabditis elegans identified lin-9 to function together with the retinoblastoma homologue lin-35 in vulva differentiation. We have now identified a human homologue of Lin-9 (hLin-9) and provide evidence about its function in the mammalian pRB pathway. hLin-9 binds to pRB and cooperates with pRB in flat cell formation in Saos-2 cells. In addition, hLin-9 synergized with pRB and Cbfal to transactivate an osteoblast-specific reporter gene. In contrast, hLin-9 was not involved in pRB-mediated inhibition of cell cycle progression or repression of E2F-dependent transactivation. Consistent with these data, hLin-9 was able to associate with partially penetrant pRB mutants that do not bind to E2F, but retain the ability to activate transcription and to promote differentiation. hLin-9 can also inhibit oncogenic transformation, dependent on the presence of a functional pRB protein. RNAi-mediated knockdown of Lin-9 can substitute for the loss of pRB in transformation of human primary fibroblasts. These data suggest that hLin-9 has tumor-suppressing activities and that the ability of hLin-9 to inhibit transformation is mediated through its association with pRB.
Keywords: differentiation, hLin-9, oncogenic transformation, retinoblastoma, tumor suppressor
Introduction
pRB, the product of the retinoblastoma tumor suppressor gene, is an important regulator of cell cycle progression, apoptosis, differentiation and senescence (reviewed in Classon and Harlow, 2002). pRB inhibits cellular proliferation and apoptosis mainly through binding to E2F transcription factors (Dyson, 1998; DeGregori, 2002; Cam and Dynlacht, 2003). Complexes of hypophosphorylated pRB and E2F act as transcriptional repressors. Repression is mediated by recruitment of proteins that are involved in regulating the chromatin structure, such as histone deacetylases (HDACs), histone-methyl-transferases and BRG1 (reviewed in Harbour and Dean, 2000). During the G1 to S transition, pRB is inactivated by phosphorylation catalyzed by cyclin-dependent kinases. Consequently, pRB is released and E2F is now able to transactivate target genes that play key roles in cell cycle progression, synthesis of nucleotides, DNA replication and apoptosis (reviewed in Trimarchi and Lees, 2002).
Certain pRB mutants that are associated with low-risk retinoblastoma retain the ability to promote differentiation despite their inability to bind to E2F. This observation indicates that the differentiation-promoting function of pRB is, at least in part, independent of E2F (Sellers et al, 1998). Instead, it has been found that pRB can interact with and activate certain transcription factors that play key roles in differentiation, such as MyoD, Cbfa1 and C/EBPβ (Thomas et al, 2001). The ability of pRB to promote differentiation has also been linked to its ability to inhibit the activity of Ras proteins (Lee et al, 1999; Takahashi et al, 2003).
The relationship between Ras and pRB has been studied genetically in Caenorhabditis elegans. The differentiation of the hermaphrodite vulva in C. elegans is initiated by an evolutionary conserved receptor tyrosine kinase/Ras/MAPK signaling pathway (reviewed in Kornfeld, 1997; Sternberg and Han, 1998). Ras signaling is antagonized by the pRB homologue lin-35 and by other so-called synthetic multivulva (synMuv) genes (Kornfeld, 1997; Lu and Horvitz, 1998; Fay and Han, 2000). Like gain-of-function mutations in the Ras pathway, inactivation of synMuv genes results in a multivulva phenotype as a consequence of expression of vulval cell fates in cells that normally adopt an uninduced fate. Genetically, the synMuv genes can be grouped into three classes, A, B and C, that act redundantly (Fay and Han, 2000; Ceol and Horvitz, 2004).
Recently, a number of class B synMuv genes have been shown to encode worm homologues of the pRB pathway, such as lin-35/pRB, efl-1/E2F, hda-1/HDAC and lin-53/RbAp46 (Lu and Horvitz, 1998; Solari and Ahringer, 2000; Ceol and Horvitz, 2001). Although vulva differentiation has been studied genetically in detail, the precise molecular mechanism of Ras inhibition by synMuv proteins is still unclear. As the pRB complex has been implicated in transcriptional regulation, it has been proposed that synMuv proteins play a role in transcriptional repression of genes that are activated by Ras and that are required for vulva cell fate specifications (Solari and Ahringer, 2000; Ceol and Horvitz, 2001).
Novel class B synMuv genes that function in the same genetic pathway have recently been identified. One such novel synMuv gene is lin-9 (Beitel et al, 2000). A function of lin-9 in the pRB pathway is supported also by cDNA microarray data, which showed that lin-9 is coexpressed with lin-35, hda-1 and lin-53 (Jiang et al, 2001; Kim et al, 2001). Besides its role in vulva development, lin-9 also has other functions. For example, lin-9 is required for male reproductive development (Beitel et al, 2000). In addition, a role for Lin-9 in G1 regulation has been suggested (Boxem and van den Heuvel, 2002). However, the molecular function of this protein is still unknown.
We speculated that mammalian homologues of novel synMuv class B proteins might also play a role in the pRB pathway, in inhibition of Ras signaling and possibly in human cancer. To begin to test these predictions, we have isolated a human homologue of Lin-9 (hLin-9). We found that hLin-9 physically associates with pRB in mammalian cells. Secondly, we show that hLin-9 cooperates with pRB during flat cell formation in Saos-2 cells. Surprisingly, hLin-9 is not involved in the acute, E2F-dependent, cell cycle arrest mediated by pRB or in pRB-dependent transcriptional repression. Instead, hLin-9 enhances the ability of pRB to activate transcription. In addition, we found that hLin-9 can inhibit oncogenic transformation of mouse fibroblasts, depending on the presence of pRB. Furthermore, the knockdown of hLin-9 can substitute for the inactivation of the pRB pathway in transformation of primary human fibroblasts. Taken together, our data suggest that hLin-9 functions together with pRB to promote differentiation and to inhibit transformation of mammalian cells.
Results
Identification of a human homologue of lin-9
We identified several overlapping human ESTs with significant homology to C. elegans lin-9 in the Genbank nucleotide database. 5′ and 3′ RACE reactions with primers designed to anneal in a region of Lin-9 that encodes for an evolutionary conserved domain were used to synthesize a human cDNA of approximately 2.5 kb. This novel cDNA (GenBank accession: AY786184) contains an open reading frame of 1626 nucleotides and encodes for a protein of 542 amino acids with a predicted molecular mass of 62 kDa. Importantly, RACE reactions with additional upstream primers failed to produce longer cDNA products. We conclude that we, most likely, have isolated the complete human lin-9 coding sequence. A highly similar mouse lin-9 cDNA was identified in the mouse EST database. While this work was in progress, a cDNA encoding hLin-9 was reported in Genbank (accession number NM_173083). The predicted product of this gene was termed ‘TUDOR gene similar' (TGS) because part of the protein is weakly similar to a TUDOR domain. TUDOR domains, which are often found in RNA-binding proteins, have been implicated in protein–protein interaction (Maurer-Stroh et al, 2003). However, because the similarity of human Lin-9 to the TUDOR domain is well below threshold and there is no evidence that it contains a functional TUDOR domain, we prefer to name this protein hLin-9. Lin-9-related proteins are also found in other organisms, for example, in Drosophila and Arabidopsis thaliana (White-Cooper et al, 2000; Bhatt et al, 2004). hLin-9 and its C. elegans orthologue share 34% sequence similarity overall (Figure 1). Similarity between hLin-9 and other Lin-9 proteins is highest in two conserved regions of unknown function that were previously termed box 1 and box 2 (see Figure 1B; White-Cooper et al, 2000). A comparison of the predicted hLin-9 protein sequence with C. elegans Lin-9 and with two Drosophila orthologues, always early (Aly) and twilight (Twit), is shown in Figure 1A and B. Northern blots showed that mammalian hLin-9 mRNA is expressed in primary untransformed tissues as well as in transformed cell lines (see Supplementary Figure S1).
Figure 1.

(A) ClustalW alignment of human hLin-9 (GenBank accession: AY786184), C. elegans lin-9 (GenBank accession: CAA77454), Drosophila always early (Aly, GenBank accession: CAB86720) and twilight (Twit, GenBank accession: CAA15930). The long C-terminal extension that is unique to Twit is not shown. (B) Schematic comparison of Lin-9 proteins. Percent identity and similarity of hLin-9 to each protein in two conserved regions, box 1 and box 2, is indicated. In the Pfam protein families database (http://pfam.wustl.edu), part of box 1 is now also called Domain in Rb-related Pathway (DIRP).
hLin-9 is a nuclear, chromatin-associated protein
It has been shown previously that Aly, a Drosophila homologue of Lin-9, is associated with chromatin (White-Cooper et al, 2000). Flag-tagged hLin-9 localized to the nucleus as analyzed by immunostaining (Figure 2A). To analyze localization of endogenous hLin-9, we first generated a polyclonal antiserum to hLin-9. In immunoprecipitation–western blot experiments, this antiserum, but not preimmunserum, recognized a single band of 60 kDa in lysates of human 293 cells and mouse embryonic fibroblasts (MEFs; Figure 2B). Importantly, the endogenous protein detected by the antiserum co-migrated with the protein overproduced from our hLin-9 cDNA (Figure 2B, right panels). Next, nuclear localization of hLin-9 was analyzed by biochemical fractionation. Cell lysates from human 293 cells were first fractionated into cytoplasmic and nuclear fractions. Nuclear pellets were extracted with 1 M NaCl. Under these conditions, hLin-9 was detected in the high-salt nuclear extract, suggesting that hLin-9 is tightly associated with nuclear structures (Figure 2C, left panels). In contrast, when nuclear pellets were first subjected to an extraction with DNase I, hLin-9 was found in the DNase I extracted material (Figure 2C, right panels). In each case, Mi-2 and HDAC1, two known chromatin-associated proteins, were found in the same fraction as hLin-9. Chromatin association of hLin-9 was confirmed by a fractionation protocol that has been used before to demonstrate chromatin association of human cdc6 (Supplementary Figure S2; Mendez and Stillman, 2000). In addition, we found that overexpressed flag-tagged hLin-9 as well as endogenous hLin-9 can be efficiently crosslinked to chromatin (Supplementary Figure S2). Together, these data suggest that hLin-9 is a nuclear, chromatin-associated protein.
Figure 2.
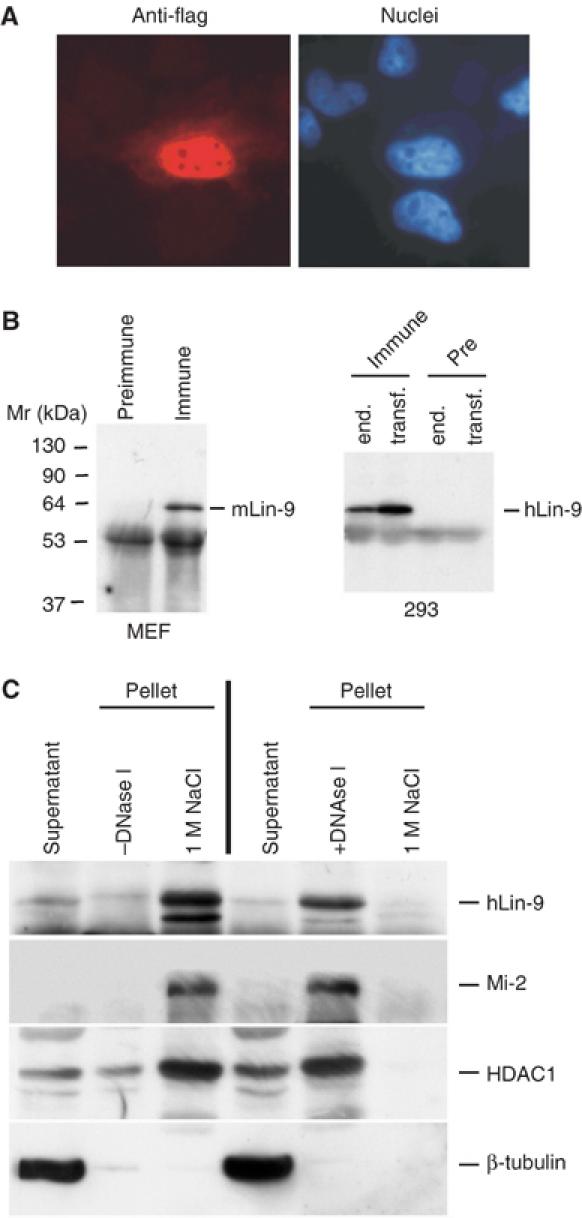
hLin-9 is a nuclear, chromatin-associated protein. (A) Localization of hLin-9 was analyzed by immunostaining of flag-hLin-9 expressing HeLa cells with an anti-flag-antibody (left). Nuclei were counterstained with Hoechst 33285 (right). (B) Characterization of the anti-hLin-9 antiserum. Left: Lysates of primary MEFs were immunoprecipitated with anti-hLin-9 antiserum (Immune) or the corresponding preimmunserum (Pre-Immune). Right: Lysates from 293 cells and from 293 cells transiently overexpressing hLin-9 were immunoprecipitated with anti-hLin-9 antibody or with the preimmunserum. hLin-9 was detected by immunoblotting with anti-hLin-9 antiserum. (C) 293 cells were fractionated into cytoplasmic and nuclear fractions. Nuclei were treated with or without DNase I as indicated and then extracted with 1 M NaCl, followed by a western blot analysis with antibodies to Mi-2, HDAC1 and β-tubulin. hLin-9 was detected by immunoprecipitation followed by immunoblotting with anti-hLin-9 antibody.
Physical interaction of hLin-9 and pRB
In C. elegans, lin-35/pRB and lin-9 genetically function in the same pathway to inhibit vulva differentiation. However, a physical association of these two proteins has not been demonstrated. We analyzed whether hLin-9 and pRB bind to each other. The ability of hLin-9 to associate with pRB was first analyzed in pulldown experiments with recombinant GST-hLin-9. GST-hLin-9 was incubated with extracts of HeLa cells expressing HA-epitope-tagged pRB. Bound pRB was detected by immunoblotting with an anti-HA antibody. In these experiments, HA-pRB specifically bound to GST-hLin-9 but not to GST alone (Figure 3A).
Figure 3.
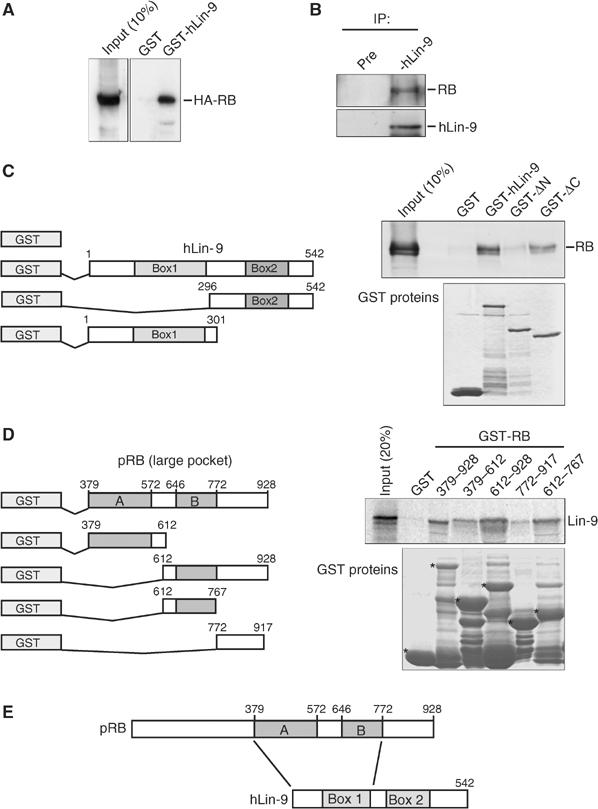
hLin-9 and pRB interact both in vitro and in vivo. (A) Binding of HA-pRB expressed in HeLa cells to GST-hLin-9 and GST. HA-pRB was detected with an anti-HA antibody. (B) Association of endogenous hLin-9 with pRB. hMSC lysates were immunoprecipitated with control preimmunserum or with hLin-9 antiserum and immunoblotted with anti-hLin-9 and anti-pRB antibodies. (C) Mapping of the binding site for pRB on hLin-9. Left: Schematic representation of GST-hLin-9 fusion proteins. Right: Binding of endogenous pRB from nuclear lysates of hMSCs to the indicated GST-hLin-9 fusion proteins. Bound pRB was detected by immunoblotting. Input: 10% of the lysate. The lower panel shows a Coomassie staining of purified GST-fusion proteins. (D) Mapping of the hLin-9-binding site on pRB. Left: Schematic representation of the GST-pRB fusion proteins. Right: The indicated GST proteins were incubated with in vitro translated, 35S-labeled hLin-9. Specifically bound hLin-9 was detected by autoradiography. The input represents 20% of the amount used for pulldown assays. The lower panel shows a Coomassie staining of purified GST-fusion proteins. The position of the full-length GST-fusion proteins is indicated by asterisks. (E) Schematic summary of binding data.
To verify that hLin-9 interacts with pRB in vivo, we performed co-immunoprecipitations with lysates of human mesenchymal stem cells (hMSCs). In these experiments, endogenous pRB was co-precipitated with anti-hLin-9 serum (Figure 3B). Importantly, no pRB was detected in immunoprecipitates with preimmunserum. We concluded that endogenous hLin-9 and pRB associate with each other.
Next, we characterized the sequences in hLin-9 that are required for binding to pRB. As box 1 and box 2 of hLin-9 are conserved in all Lin-9 proteins in different organisms (see Figure 1B), we generated two deletion constructs of hLin-9 that remove either box 1 (GST-hLin-9ΔN) or box 2 (GST-hLin-9ΔC) (Figure 3C). Levels of GST-fusion proteins were comparable, as confirmed by Coomassie staining. Lysates from untransfected hMSCs were incubated with GST or with GST-hLin-9 fusion proteins and bound pRB was detected with an anti-pRB antibody. pRB bound to GST-hLin-9 but not by GST alone, confirming the association of these proteins. Intriguingly, deletion of the amino-terminus of hLin-9 completely abolished binding of pRB. In contrast, pRB bound to GST-hLin-9ΔC, suggesting that hLin-9 interacts with pRB through the amino-terminal half of the protein that includes box 1. As box 1 is highly conserved between Lin-9 proteins, it is possible that it functions as a pRB interaction domain in other species as well. Next, we mapped the binding site for hLin-9 on pRB with fragments of pRB fused to GST and in vitro-translated hLin-9 (Figure 3D). It is evident from these GST pulldowns that hLin-9 binds to the pocket domain of pRB. Strongest binding of hLin-9 was detected with the B pocket, but an additional weaker interaction with the A pocket could also be detected. This suggests that hLin-9 binds to at least two sites in the pRB pocket domain. The regions that are responsible for interaction of hLin-9 and pRB are summarized in Figure 3E.
hLin-9 cooperates with pRB in flat cell induction in Saos-2 cells
In C. elegans, lin-9 functions genetically in the same pathway with the pRB homologue lin-35. To address whether hLin-9 plays a role in pathways controlled by the mammalian retinoblastoma protein, we asked whether hLin-9 has any effect on the ability of pRB to induce flat cells in pRB-negative Saos-2 cells. It has been shown previously that reintroduction of pRB into Saos-2 cells results in morphological changes referred to as flat cells that resemble a senescence-like state (Hinds et al, 1992; Qin et al, 1992). Saos-2 flat cells also express markers of bone differentiation (Sellers et al, 1998). As expected, expression of pRB in Saos-2 cells, together with a small amount of a plasmid with a neomycin resistance marker, resulted in a significant lower number of neomycin-resistant colonies after selection for 10–14 days when compared to control transfections with empty expression vector (Figure 4A). Microscopic examination confirmed that pRB expression resulted in the formation of flat cells (Figure 4B). Expression of hLin-9 on its own had no effect on Saos-2 cell growth and morphology (Figure 4A and B). However, coexpression of hLin-9 with pRB further reduced the number of resistant colonies and enhanced the ability of pRB to form flat cells (Figure 4A and B, right panels). Consequently, when pRB was co-transfected together with hLin-9, almost all cells in the culture turned into flat cells (Figure 4B). Importantly, expression levels of pRB were not affected by coexpression of hLin-9 (Figure 4C), suggesting that the ability of hLin-9 to promote flat cell formation by pRB is not an indirect result of increased pRB expression.
Figure 4.
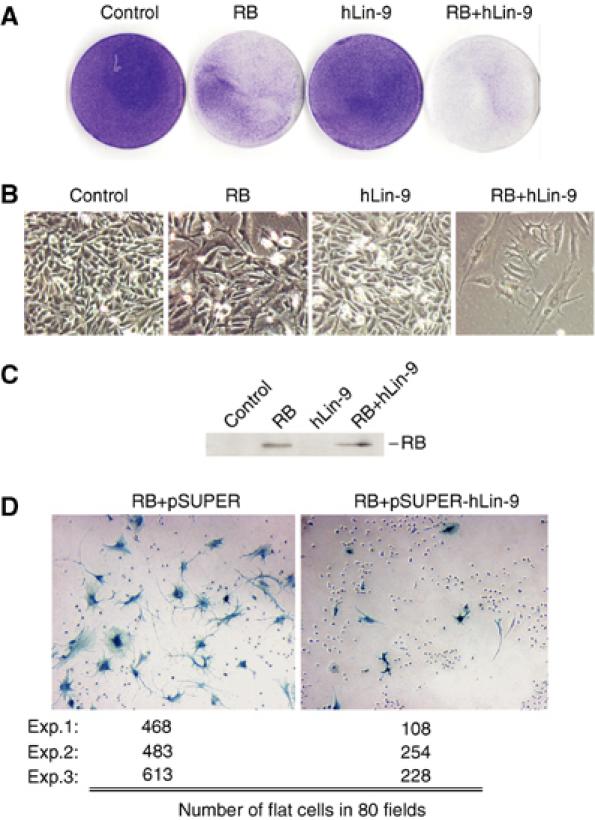
hLin-9 and pRB cooperate in flat cell induction in Saos-2 cells. (A, B) Saos-2 cells were transfected with the indicated expression plasmids. After 2 weeks of selection, dishes were stained with crystal violet to visualize the cells. (A) The whole dishes and (B) photomicrographs (× 20). Results shown are representative of four independent experiments. (C) Immunoblotting of pRB in transfected cells. (D) Saos-2 cells were transfected with pRB, together with empty pSUPER or pSUPER-hLin-9. After 2 weeks of selection, flat cells were detected by staining for senescence-associated β-gal. The number of flat cells in 80 fields in three independent experiments was determined.
To test whether hLin-9 is required for pRB-dependent flat cell formation, we used a pSUPER plasmid-based RNAi strategy to knockdown endogenous hLin-9. Transfection of pSUPER-hLin-9 into Saos-2 cells resulted in a reduction of endogenous hLin-9 of about 80% (Figure 5B and data not shown). pSUPER-hLin-9 was co-transfected together with an pRB expression plasmid into Saos-2 cells. Flat cells were detected by their senescence-associated β-galactosidase (SA-β-gal) activity. Positive cells were manually counted. As summarized in Figure 4D, depletion of hLin-9 significantly suppressed formation of flat cells by pRB in three different experiments. These data demonstrate that hLin-9 is required for efficient flat cell formation by pRB. Taken together, our results suggest that mammalian hLin-9 and pRB cooperate during flat cell formation in Saos-2 cells.
Figure 5.
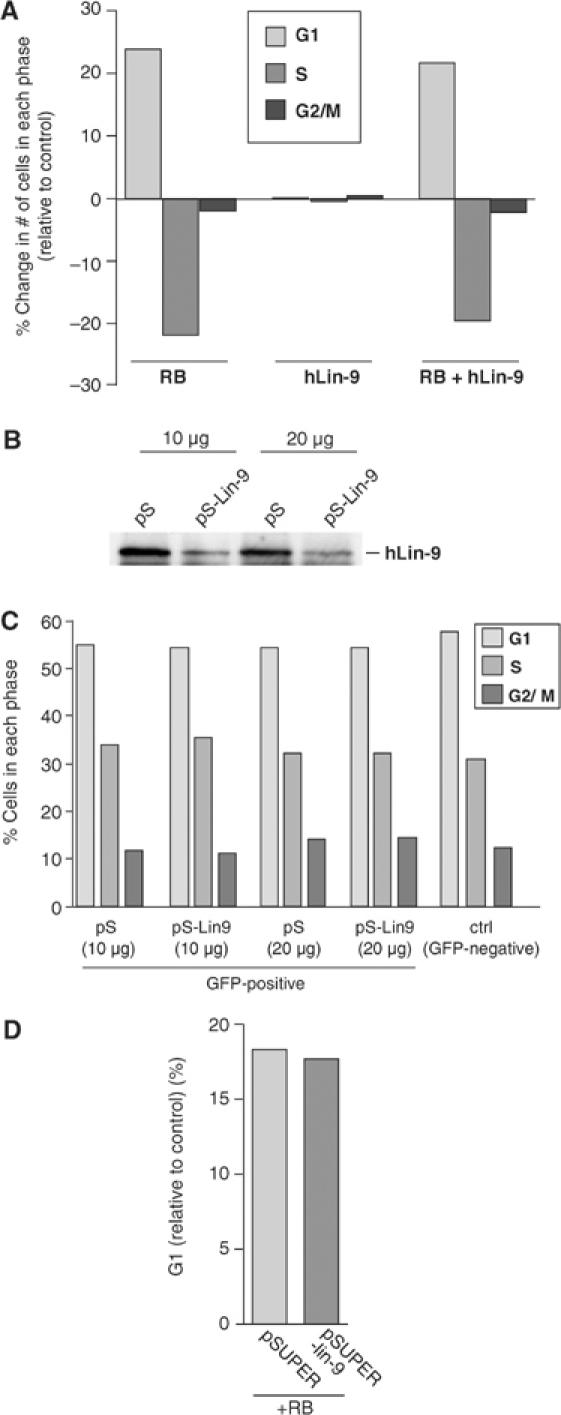
hLin-9 does not inhibit cell cycle progression. (A) The DNA content of Saos-2 cells, transfected with the indicated expression plasmids and a GFP plasmid, was determined 48 h after transfection by FACS. The absolute changes in the number of cells in each phase of the cell cycle compared to cells transfected with GFP alone are shown. (B) Knockdown of endogenous hLin-9 by plasmid-based RNAi. HeLa cells were transiently transfected with 10 or 20 μg empty pSUPER (pS) or pSUPER-hLin-9 (pS-Lin-9). Lysates were immunoprecipitated with an anti-hLin-9 antibody, and immunoblotted with an anti-hLin-9 antiserum. (C) DNA content was analyzed by FACS. Numbers of cells in each phase of the cell cycle are shown. (D) HeLa cells were transfected with pRB, together with empty pSUPER or pSUPER-hLin-9, and the change in the numbers of cells in G1 was analyzed by FACS.
hLin-9 is not involved in pRB-mediated cell cycle arrest
In C. elegans, several class B synMuv genes, including lin-9, function as negative regulators of G1 (Boxem and van den Heuvel, 2002). We therefore wanted to investigate whether hLin-9 cooperates with pRB in inhibition of cell cycle progression. To analyze whether hLin-9 has any effect on cell cycle progression or on the pRB-mediated G1 arrest, we transfected Saos-2 cells with expression constructs for pRB and hLin-9. A GFP expression plasmid was cotransfected for the detection of transfected cells by their green fluorescence. Forty-eight hours after transfection, cell cycle progression was analyzed by FACS. Expression of hLin-9 had no effect on cell cycle distribution (Figure 5A, middle panels). As expected, pRB reduced the number of cells in S-Phase when compared to cells transfected with an empty control vector (Figure 5A, left panels). Coexpression of hLin-9 did not significantly enhance the pRB-induced cell cycle arrest (Figure 5A, right panels). These findings were confirmed independently with several U2-OS cell clones that stably express very high levels of exogenous hLin-9 (Supplementary Figure S3). Consistent with the lack of any observable effect of overexpression of hLin-9 on cell cycle progression, knockdown of endogenous hLin-9 by transient transfection of pSUPER-hLin-9 had no effect on proliferation or on the pRB-mediated G1 arrest in HeLa cells (Figure 5B–D). Taken together, we concluded that hLin-9 is not required for pRB to inhibit cell cycle progression in G1 in mammalian cells. Thus, hLin-9 cooperates with pRB without promoting the acute growth arrest mediated by pRB.
hLin-9 associates with partially penetrant pRB mutants and promotes pRB-dependent transcriptional activation
It has been shown before that the acute inhibition of cell cycle progression by pRB requires binding to E2F and involves repression of transcription. In contrast, the ability of pRB to activate transcription, to form flat cells and to promote differentiation is independent from association with E2F (Sellers et al, 1998; Thomas et al, 2001). This was concluded from the observation that certain tumor-derived mutants of pRB associated with low-risk retinoblastoma retain the ability to promote differentiation despite their inability to associate with E2F. As we found that hLin-9 cooperates with pRB without promoting the pRB-induced acute growth arrest, we wanted to test whether hLin-9 associates with mutants of pRB (e.g. pRB:Δex4 and 661W) that are able to promote differentiation.
To address this possibility, we analyzed binding of hLin-9 to a set of tumor-derived pRB mutants in GST-binding assays (Figure 6A). Lysates of Saos-2 cells expressing HA-tagged versions of pRB mutants were incubated with GST-hLin-9 or with GST as a control. The amount of lysate used for the binding assays was adjusted to account for the lower expression of pRB Δex22, 567L and Δex4 compared to wild-type pRB and 661W (Figure 6A, bottom). Bound pRB was detected with an anti-HA antibody. As expected, wild-type, HA-tagged pRB bound to GST-hLin-9 (Figure 6A, lane 4). In contrast, no binding of the null mutants pRB:Δex22 and 567L to GST-hLin-9 was detectable. Interestingly, however, the two partially penetrant mutants pRB:Δex4 and pRB:661W specifically bound to GST-hLin-9 (Figure 6A, lanes 10 and 12). Thus, hLin-9 associates with pRB mutants that promote differentiation but do not associate with E2F. We therefore hypothesize that hLin-9 is involved in the ability of pRB to promote differentiation and to activate transcription.
Figure 6.
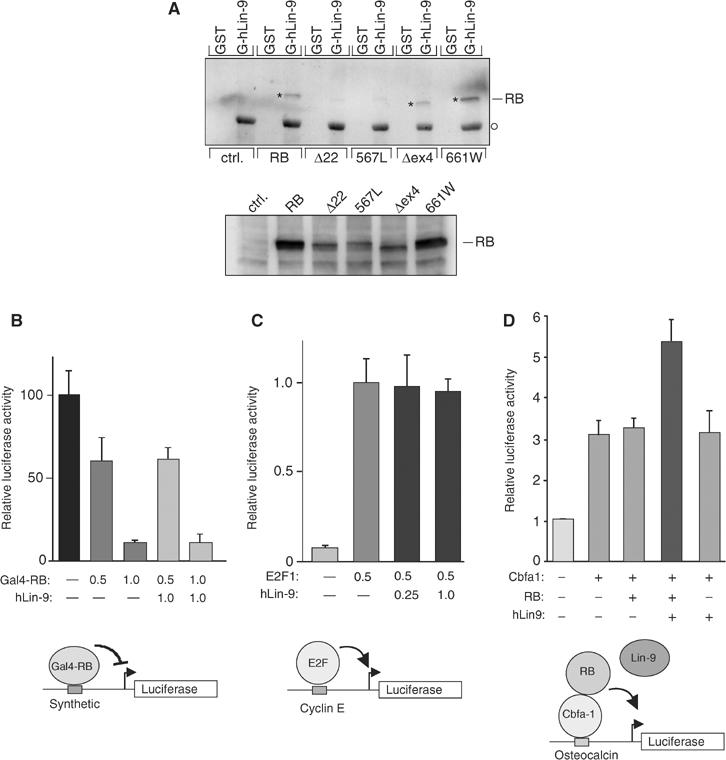
hLin-9 associates with partially penetrant pRB mutants and enhances pRB-mediated transactivation. (A) GST or GST-hLin-9 were incubated with lysates of Saos-2 cells transiently expressing the indicated HA-tagged pRB proteins. Bound pRB was detected with an anti-HA antibody. The lower panel shows the input. To adjust for the lower expression of pRB:Δ22, 567L and Δex4, twice as much lysate was used for the binding assays. The asterisk shows the position of bound pRB. The circle indicates GST-hLin-9 nonspecifically recognized by the secondary antibody. (B) pRB-mediated repression: Saos-2 cells were transfected with the indicated amounts (in μg) of Gal4-pRB and hLin-9 expression plasmids, together with a luciferase reporter plasmid with synthetic Gal4-binding sites in the promoter. (C) E2F-mediated transactivation: Saos-2 cells were transfected with the indicated amounts (in μg) of E2F1 and hLin-9 expression plasmids, together with a cyclin E luciferase reporter plasmid. (D) pRB-mediated transactivation: Saos-2 cells were transfected, as indicated, with 0.1 μg Cbfa1, 0.5 μg pRB and 1.0 μg hLin-9 expression plasmids, together with an osteocalcin-promoter luciferase reporter plasmid. (B–D) An equal amount of a β-gal expression plasmid was cotransfected and luciferase activity was normalized to β-gal activity.
To address this possibility, we measured the effect of hLin-9 on transcriptional activation by the osteogenic transcription factor Cbfa1 in Saos-2 cells. It has been shown before that pRB can activate Cbfa1 (Thomas et al, 2001). In the absence of pRB, hLin-9 had no effect on reporter gene expression when expressed alone or together with Cbfa1. However, in the presence of limiting amounts of pRB, hLin-9 reproducibly activated Cbfa1-dependent transcription about two-fold (Figure 6D). In contrast, hLin-9 had no significant effect on pRB-mediated repression and on E2F1- and E2F4-dependent transactivation (Figure 6B and C and data not shown). Taken together, these data suggest that hLin-9 can cooperate with the retinoblastoma protein in gene activation.
hLin-9 inhibits oncogenic transformation
Our findings support a role for hLin-9 in the pRB tumor suppressor pathway. To test whether hLin-9 itself has tumor-suppressing activities, we generated stable NIH-3T3 cell lines overexpressing high levels of hLin-9 (Figure 7A). Constitutive active, oncogenic RasV12 was introduced into these cell lines by retroviral infection. As expected, expression of RasV12 in normal NIH-3T3 cells and in NIH-3T3 cells transfected with empty vector (NIH-3T3-IRES-neo) resulted in a transformed morphology (Figure 7B, left panels). Interestingly, NIH-3T3 clones constitutively expressing high levels of hLin-9 did not show a transformed morphology after expression of RasV12. In contrast, their morphology was similar to that of cells infected with an empty control retrovirus (Figure 7B, right panels). Thus, hLin-9 inhibited RasV12-induced morphological changes.
Figure 7.
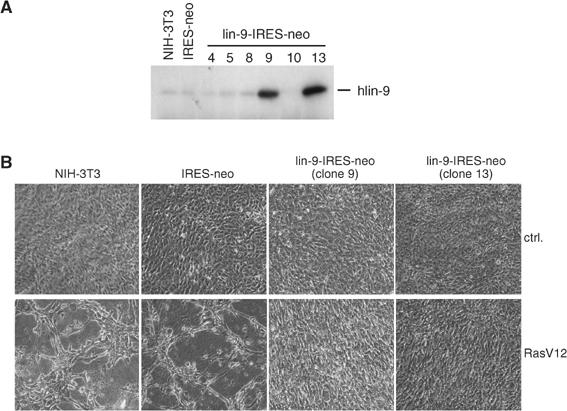
hLin-9 inhibits RasV12-induced morphological changes. (A) Characterization of stable cell lines. Expression of hLin-9 in individual NIH-3T3 clones stably transfected with a hLin-9-IRES-neomycin plasmid. Untransfected NIH-3T3 cells and NIH-3T3 cells transfected with an empty IRES-neo plasmid were analyzed in parallel. (B) The indicated NIH-3T3 cell lines were infected with a RasV12 encoding retrovirus or with empty vector. At 2 days after infection, cells were selected for 7 days and then microscopically examined.
We next asked whether hLin-9 can also antagonize anchorage-independent growth of transformed primary MEFs. MEFs were first infected with a retrovirus encoding hLin-9 and a hygromycin resistance gene (Figure 8A). As a control, an empty retrovirus with a hygromycin resistance gene was used. Next, a second infection with RasV12-, Myc- and Bcl-2-expressing viruses was performed. After selection with hygromycin and puromycin, neoplastic transformation of the MEFs was scored by their ability to grow anchorage independently in soft agar. As expected, cells expressing RasV12, Myc and Bcl-2 readily formed colonies in soft agar within 14 days (Figure 8C and D). Intriguingly, when hLin-9 was coexpressed, only a few colonies grew in soft agar and the size of the few colonies was significantly smaller (Figure 8C). Immunoblot experiments confirmed that the expression of RasV12, Myc or Bcl-2 was comparable in hLin-9-expressing cells and in control-infected MEFs (Figure 8B). We conclude from these results that hLin-9 can antagonize oncogenic transformation of primary cells.
Figure 8.
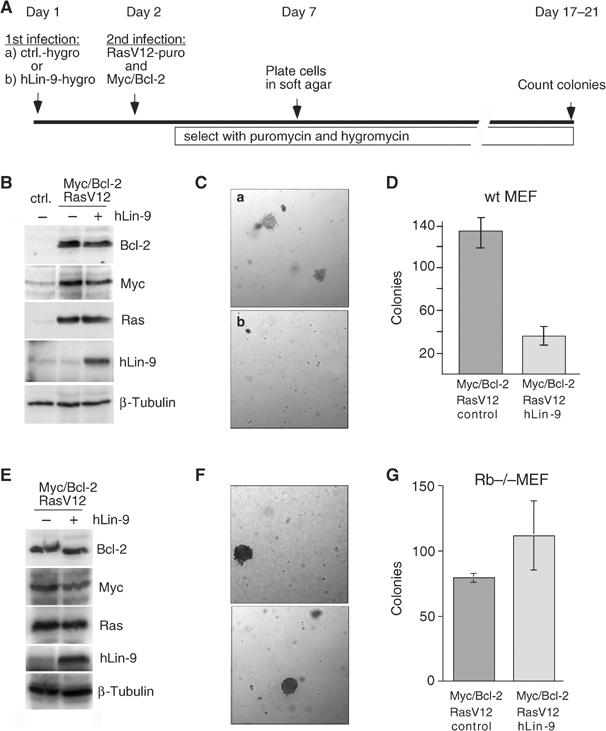
pRB-dependent inhibition of oncogenic transformation of MEFs by hLin-9. (A) Scheme for the infection protocol. (B) Protein expression was analyzed by immunoblotting with antibodies directed against Bcl-2, Myc, RasV12 and hLin-9. As a control, lysates from uninfected MEFs were analyzed in parallel (ctrl). β-Tubulin serves as a loading control. (C) In (a) average size of colonies of MEFs expressing RasV12, Myc and Bcl-2. (b) Size and overall number of colonies when hLin-9 was coexpressed. (D) The number of colonies in soft agar was counted manually. (E–G) Soft agar assays with Rb−/− MEFs as described in (A). (E) Protein expression, (F) photomicrograph of colonies in soft agar and (G) number of colonies.
Next we asked whether the inhibition of oncogenic transformation by hLin-9 is dependent on a functional pRB pathway. To address this question, we repeated the experiment in MEFs derived from Rb null embryos (Figure 8E–G). Intriguingly, in Rb−/− cells, hLin-9 failed to inhibit colony formation by Myc, Ras and Bcl-2 oncogenes, demonstrating that hLin-9 requires pRB to inhibit oncogenic transformation (Figure 8F and G). Inhibition of oncogenic transformation by hLin-9 suggests that hLin-9 could act as a tumor suppressor.
To address more directly the role of hlin-9 in tumor suppression, we analyzed whether the loss of hLin-9 can substitute for the loss of pRB in oncogenic transformation. For these assays, we used immortal primary human BJ-ET fibroblasts (Voorhoeve and Agami, 2003). It has been shown before that expression of RasV12 and SV40 virus small t (st) combined with RNAi-mediated knockdown of p53 and pRB results in oncogenic transformation of these cells, as analyzed by the ability of the cells to form colonies in soft agar (Voorhoeve and Agami, 2003). In this system, the inactivation of pRB can also be substituted by the knockdown of the cdk4 inhibitor p16INK4a, another component of the pRB pathway (Voorhoeve and Agami, 2003). As expected, the expression of RasV12 together with st did not result in transformation (Figure 9). The additional single inhibition of p53, p16INK4a or hLin-9 in these cells was also not sufficient for anchorage-independent growth. As expected, the combined knockdown of p16INK4a and p53 in cells that express RasV12 and st resulted in colony outgrowth. Strikingly, the knockdown of hLin-9 when combined with the knockdown of p53 also resulted in transformation of cells expressing RasV12 and st. In contrast, the loss of hLin-9 did not cooperate with the loss of p16INK4. Thus, the loss of hLin-9 can substitute for the loss of pRB or p16INk4a in oncogenic transformation of primary human cells. These results show that hLin-9 is an integral component in the pRB pathway that controls oncogenic transformation.
Figure 9.
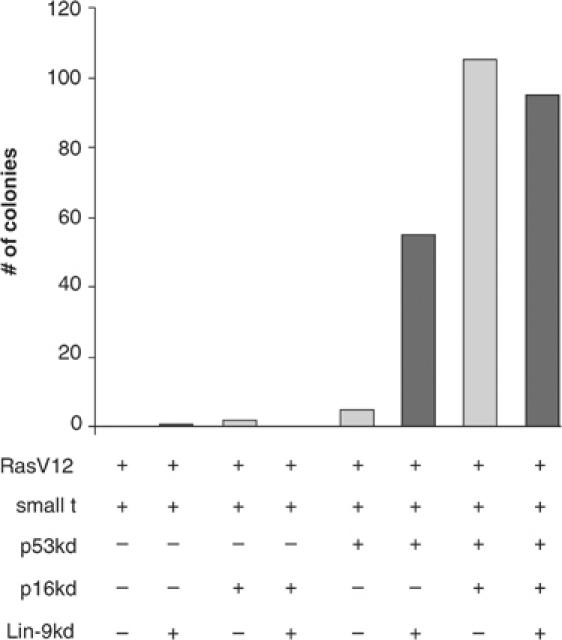
Loss of hLin-9 can substitute for the loss of pRB in cellular transformation of primary human fibroblasts. BJ fibroblasts stably expressing hTERT, st and the ecotrophic retrovirus receptor were infected with the indicated retroviral pSUPER-hygro-based siRNA vectors to knockdown (kd) p53 and p16INK4a and pMSCV-Blast-based siRNA vector to knockdown Lin-9. After selection with hygromycin B and blasticidin, cells were infected with a retrovirus encoding oncogenic RasV12 and plated in soft agar. Visible colonies were counted after 2–3 weeks. The experiment was repeated three times with similar results. One representative experiment is shown.
Discussion
The retinoblastoma protein is a multifunctional tumor suppressor protein that plays important roles in the regulation of cell cycle progression, apoptosis, senescence and differentiation. In C. elegans, a group of genes, the so-called synMuv genes, were shown to function together with lin-35/pRB to antagonize Ras signaling during vulva development (Kornfeld, 1997). One of these synMuv genes that genetically interacts with lin-35/pRB in this pathway is lin-9. However, the molecular function of the product of this gene is unknown. In this study, we now identified a human homologue of lin-9. Our data provide several clues about the function of hLin-9 in the pRB pathway, for example, we report that hLin-9 associates and cooperates with pRB and that it has tumor-suppressing activities.
The genetic studies in C. elegans suggested an interaction of Lin-9 with pRB. In this study, we could now demonstrate that in a mammalian system pRB physically associates with hLin-9 in vivo and in vitro. Binding to pRB requires box 1 of hLin-9, a N-terminal domain that is conserved in several Lin-9 proteins of different species. It is therefore likely that Lin-9 proteins in other organisms bind to pRB homologous as well, and that box 1 functions as the domain that mediates binding to pRB.
That the physical interaction between hLin-9 and pRB is functionally relevant is suggested by our observation that hLin-9 synergizes with pRB in the formation of flat cells in pRB-negative Saos-2 cells. Intriguingly, the ability of hLin-9 to promote the flat cell phenotype is dependent on the presence of a functional pRB protein. In addition, pRB requires endogenous hLin9 to efficiently induce flat cells, as demonstrated by RNAi-mediated knockdown of hLin-9. Although hLin-9 cooperates with pRB in flat cell formation, it appears not to be involved in pRB-mediated cell cycle inhibition. It has been shown before that flat cell formation and inhibition of cell cycle progression are two distinct functions of pRB that can be genetically separated. The induction of the acute G1 arrest depends on the ability of pRB to bind to E2F and to repress transcription. In contrast, the ability of pRB to induce the flat cell phenotype is linked to the ability of pRB to promote differentiation and to activate transcription (Sellers et al, 1998; Thomas et al, 2001). Certain mutants of pRB derived from patients with partially penetrant retinoblastoma retain the ability to promote differentiation despite their inability to associate with E2F (Sellers et al, 1998). Since hLin-9 can bind to these partially penetrant pRB mutants, we propose that hLin-9 has a role in pRB-mediated differentiation. This is also supported by our observation that hLin-9 synergizes with pRB to activate transcription by the bone-specific transcription factor Cbfa1. A role for pRB in bone differentiation and Cbfa1 activation has recently been demonstrated. Although it is not well understood how pRB cooperates with Cbfa1, it was speculated that Cbfa1 recruits pRB to osteogenic promoters and assembles a multiprotein complex that mediates gene activation (Thomas et al, 2001). hLin-9 might be part of such a pRB complex that specifically functions in gene activation during differentiation. A positive role in transcription has been described for the Drosophila homologue of Lin-9, Aly, which is required for the transcriptional activation of terminal differentiation genes in spermatocytes (White-Cooper et al, 2000; Ayyar et al, 2003; Jiang and White-Cooper, 2003). While Aly mainly seems to function in transcriptional activation, the second Lin-9 homologue of Drosophila, Twit, has recently been implicated in transcriptional repression of developmental genes together with the fly orthologue of pRB, RBF (Korenjak et al, 2004). Thus, based on the studies of hLin-9 homologous in other organisms, it is possible that, in addition to activating transcription, human Lin-9 can also repress transcription, depending on the target gene. A systematic analysis of genes regulated by hLin-9 will be necessary to address this question.
As our data implicate hLin-9 in pRB-mediated differentiation, a function that is known to be relevant for pRB-mediated tumor suppression, we investigated whether hLin-9 itself can inhibit transformation. We found that hLin-9 was able to inhibit oncogenic transformation of primary MEFs by a combination of the RasV12, Myc and Bcl-2 oncogenes. Intriguingly, hLin-9 failed to inhibit transformation in Rb−/− cells, indicating that tumor suppression by hLin-9 is dependent on the presence of a functional pRB protein. It is likely that the inhibition of oncogenic transformation by hLin-9 and the ability of hLin-9 to associate with pRB, to promote the flat cell phenotype and to activate transcription, are mechanistically related. Direct experimental evidence that hLin-9 has tumor suppressor activities comes from our observation that the loss of hLin-9 can substitute for the loss of pRB or p16INK4a in transformation of primary human fibroblast (Figure 9). Thus, hLin-9 is an essential component of the pRB tumor suppressor pathway, raising the possibility that the loss or inactivation of hLin-9 could be an important step in neoplastic transformation. So far, we have not analyzed hLin-9 in human tumors. However, it has been reported that 1q42, the chromosomal localization of the human LIN-9 gene, is recurrently lost in breast carcinomas (Pandis et al, 1995). Clearly, further studies are necessary to address whether hLin-9 is involved in the pathogenesis of this or other tumors.
In conclusion, we found that hLin-9 binds to and cooperates with the pRB tumor suppressor protein and inhibits oncogenic transformation in a pRB-dependent manner. The pRB pathway is inactivated in almost all human cancers. Thus, proteins that are involved in pRB-mediated tumor suppression might be attractive targets for cancer therapy.
Materials and methods
Cloning of a human lin-9 cDNA
Human lin-9 was isolated by RACE with human HeLa marathon cDNA (Clontech) as a template. 5′ and 3′ RACE reactions were performed with primers SG151 (5′ CCATCATGAACACCACGTAATCGTGC 3′) and SG150 (5′ CGGCTTATGGGAAAACCACGGAG 3′), respectively. RACE products were subcloned and sequenced. A full-length hLin-9 cDNA was cloned into pCDNA3. Flag- and HA-tagged hLin-9 were generated by PCR.
Plasmids
The osteogenic reporter plasmid (p6OSE2-luc) and expression plasmids for p16INK4a, cbfa1, RasV12, pRB and pRB mutants have been described (Serrano et al, 1997; Sellers et al, 1998; Thomas et al, 2001). pBabe-Myc-Bcl2 was a kind gift of S Adhikary. pSUPER-hLin-9 was generated by inserting oligonucleotides that target nucleotides 1436–1458 of the human hLin-9 coding sequence into pSUPER (Brummelkamp et al, 2002). phLin9-IRES-neo, pBabe-hLin-9-hygro and GST-hLin-9 were generated by inserting the hLin-9 cDNA into pIRES-neo (Clontech), pBabe-hygro and pGEX-4T2, respectively. GST-hLin-9 ΔN (aa 296–542) and ΔC (aa 1–301) were generated by PCR. A retroviral hLin-9 RNAi construct was generated by insertion of the H1 promoter-Lin-9 shRNA cassette from pSUPER-hLin-9 into pMSCV-Blast (Voorhoeve and Agami, 2003). Constructs for retroviral-mediated knockdown of p16INK4a and p53 and for expression of SV40 st (pMSCV-st-GFP) were described previously (Voorhoeve and Agami, 2003).
Cell lines
NIH-3T3 cells were cultivated in 5% BCS in DMEM. U2-OS, Saos-2, HeLa cells, BJ-ET cells and MEFs were cultivated in DMEM with 10% FCS. hMSCs (Cambrex) were maintained in Mesenchymal Stem Cell Growth Medium. NIH-3T3 cell clones stably overexpressing hLin-9 were generated by transfection with pLin9-IRES-neo, selection for 10 days with 800 μg/ml neomycin and isolation of individual clones. Pools stably expressing the empty pIRES-neo vector served as controls.
Antibodies
His-tagged hLin-9 (aa 1–293) was expressed, purified with standard procedures and used to immunize rabbits (Davids Biotechnology). The following antibodies were also used: α-pRB (C-15), α-Bcl-2 (ΔC21), α-HDAC1 (H-51) (Santa Cruz), α-Ras (clone 18, Transduction Laboratories), α-Myc (9B11, Cell Signaling), α-β-tubulin (MAB3408, Chemicon), α-flag (M2, Sigma) and α-HA (MMS-101-P, Babco).
GST pulldowns and immunoprecipitations
GST-fusion proteins were purified from recombinant bacteria by standard procedures. GST proteins bound to glutathione beads were incubated with 600 μg nuclear extracts of hMSCs, transfected HeLa cells or transfected Saos-2 cells overnight at 4°C. After seven washes with TNN (50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, 10 mM Na4P2O7, 2 mM NaVO4, 100 mM NaF, 10 μg/ml PMSF), bound proteins were detected by immunoblotting.
For pulldowns with GST-RB fragments, Lin-9 was in vitro translated and labeled with 35S-methionine in a coupled reticulocyte system (Promega). A volume of 5 μl in vitro translated hLin-9 was incubated with immobilized GST-RB in 200 μl Z-buffer (25 mM HEPES (pH 7.5), 300 mM NaCl, 12.5 mM MgCl2, 20% glycerol, 0.5% NP-40, 2 mM DTT, 150 μg/ml BSA) and incubated for 1 h at room temperature. After five washes in NETN (20 mM Tris (pH 8.0), 300 mM NaCl, 1 mM EDTA, 0.5% NP-40) bound proteins were resolved by SDS–PAGE and detected by autoradiography.
For immunoprecipitations, nuclear extracts were immunopreciptated with α-hLin-9 antibody in a dilution of 1:100. Immunoprecipitates were collected on protein-A–sepharose and washed five times in TNN. Proteins were detected by immunoblotting.
Subcellular fractionation
Nuclear and cytoplasmic cell extracts were prepared as described before (Lindeman et al, 1997). Briefly, cells were resuspended in low-salt buffer (10 mM Hepes (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1 mM DTT, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 0.1 mM phenylmethylsulfonyl fluoride), incubated on ice for 20 min and homogenized using a Dounce homogenizer until disruption of the plasma membrane was confirmed by trypan blue staining. Nuclei were collected by low-speed centrifugation (5 min, 750 g, 4°C) and incubated with or without DNAse I (100 U) for 1 h at room temperature. Chromatin was extracted with 250 mM ammonium sulfate. The insoluble fraction (nuclear matrix) was collected by low-speed centrifugation (5 min, 750 g, 4°C), extracted for 10 min on ice with 1 M NaCl and 5 mM EDTA, and clarified by high-speed centrifugation (10 min, 16 000 g, 4°C). After adjustment to 250 mM NaCl, fractions were subjected to immunoprecipitation with anti-hLin-9 antibody.
Flow cytometry
Cells were transfected with 1 μg CMV-GFP and 10 μg of expression plasmid using calcium phosphate for 48 h. Cells were harvested, fixed in 1% paraformaldehyde in PBS and then in 80% ethanol. DNA was stained with 69 μM propidium iodide in 38 mM sodium citrate and 100 μg/ml RNase for 30 min at 37°C. Samples were analyzed in a Becton Dickinson FACScan. Transfected cells were identified by their green fluorescence.
Reporter assays
1 × 105 Saos-2 cells were plated in six-well culture dishes. After 24 h, 2.5 μg of luciferase reporter plasmid was cotransfected with expression plasmids, as indicated in each experiment. Empty vector was added to maintain a constant amount of DNA. Luciferase assays were performed as described (Lindeman et al, 1997). To normalize for transfection efficiency, 0.5 μg of CMV-β-gal was cotransfected. Error bars represent the standard deviation within a representative experiment. Each experiment has been repeated at least three times.
Flat cell assays
Saos-2 cells were transfected with 20 μg expression plasmids as indicated in each experiment. In all, 4 μg of pCDNA3 was cotransfected. At 48 h after transfection, cells were selected with 800 μg/ml neomycin for 10–14 days and then stained with crystal violet to visualize the cells. In some experiments, SA-β-gal activity was detected as described (Dimri et al, 1995). Flat cells were identified by their strong blue staining and manually counted under the microscope.
Transformation assays
Retroviral supernatants were generated by transient transfection of Phoenix cells with pBABE-based retroviral vectors (pBabe-puro; pBabe-RasV12; pBabe-Myc-Bcl-2; pBabe-hygro and pBabe-hLin9-hygro) as described (Serrano et al, 1997). Supernatants were used to infect low-passage MEFs. Infected MEFs were selected with 1.5 μg/ml puromycin and 100 μg/ml hygromycin for 4 days. Cells (1 × 104, 3 × 104 and 1 × 105) were transferred to 2 ml DMEM containing 0.35% low-gelling agarose and seeded in triplicate into six-well plates containing a 2-ml layer of solidified 0.7% agarose in complete medium. After 2 weeks, the number of foci was determined. Only visible foci (containing at least 10–50 cells) were counted. NIH-3T3 clones were infected with pBABE-puro or pBABE-RasV12-puro and selected with puromycin (3 μg/ml) for 10 days and analyzed by microscopy.
For transformation assays in primary human cells, BJ fibroblasts cells expressing hTERT and the ecotrophic receptor (BJ-ET) were used (Voorhoeve and Agami, 2003). BJ-ET cells were infected with a retrovirus that encodes for st and GFP. Subsequently, cells were infected with the pRS-hygro-based knockdown constructs against p53 and/or p16INK4a, or with empty vector, and selected with 50 μg/ml Hygromycin B (Roche) as described (Voorhoeve and Agami, 2003). Subsequently, cells were infected with the pMSCV-Blast-hLin-9 knockdown virus or with empty virus and selected with 5 μg/ml Blasticidin (Invitrogen). Finally, cells were infected with a retrovirus encoding for RasV12 and plated into soft agar as described above. The number of macroscopically visible foci was scored after 3 weeks. The experiment was performed three times with similar results.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Materials and Methods
Acknowledgments
We like to thank Alexander Brehm for communicating results prior to publication, Martin Eilers, Uta Bauer, Werner Lutz, Caroline Buchard and Jörg Storre for critical reading of the manuscript, all members of the Gaubatz, Eilers and Bauer laboratories for numerous helpful discussions, Martina Seitz for excellent technical support, Denise Hübner and Yvonne Wruck for help with the chromatin isolation and FACS analysis, Jörg Storre, William Sellers, Phil Hinds, Sovana Adhikary and Martin Eilers for reagents. This work was supported by a grant from the VolkswagenStiftung to S Gaubatz.
References
- Ayyar S, Jiang J, Collu A, White-Cooper H, White RA (2003) Drosophila TGIF is essential for developmentally regulated transcription in spermatogenesis. Development 130: 2841–2852 [DOI] [PubMed] [Google Scholar]
- Beitel GJ, Lambie EJ, Horvitz HR (2000) The C. elegans gene lin-9, which acts in an Rb-related pathway, is required for gonadal sheath cell development and encodes a novel protein. Gene 254: 253–263 [DOI] [PubMed] [Google Scholar]
- Bhatt AM, Zhang Q, Harris SA, White-Cooper H, Dickinson H (2004) Gene structure and molecular analysis of Arabidopsis thaliana ALWAYS EARLY homologs. Gene 336: 219–229 [DOI] [PubMed] [Google Scholar]
- Boxem M, van den Heuvel S (2002) C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr Biol 12: 906–911 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Cam H, Dynlacht BD (2003) Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell 3: 311–316 [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR (2001) dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol Cell 7: 461–473 [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR (2004) A new class of C. elegans synMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev Cell 6: 563–576 [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E (2002) The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2: 910–917 [DOI] [PubMed] [Google Scholar]
- DeGregori J (2002) The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim Biophys Acta 1602: 131–150 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12: 2245–2262 [DOI] [PubMed] [Google Scholar]
- Fay DS, Han M (2000) The synthetic multivulval genes of C. elegans: functional redundancy, Ras-antagonism, and cell fate determination. Genesis 26: 279–284 [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 14: 2393–2409 [DOI] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70: 993–1006 [DOI] [PubMed] [Google Scholar]
- Jiang J, White-Cooper H (2003) Transcriptional activation in Drosophila spermatogenesis involves the mutually dependent function of aly and a novel meiotic arrest gene cookie monster. Development 130: 563–573 [DOI] [PubMed] [Google Scholar]
- Jiang M, Ryu J, Kiraly M, Duke K, Reinke V, Kim SK (2001) Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc Natl Acad Sci USA 98: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS (2001) A gene expression map for Caenorhabditis elegans. Science 293: 2087–2092 [DOI] [PubMed] [Google Scholar]
- Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Assland R, White-Cooper H, Dyson N, Brehm A (2004) Regulation of developmental transcription programs by retinoblastoma repressor complexes. Cell 119: 181–193 [DOI] [PubMed] [Google Scholar]
- Kornfeld K (1997) Vulval development in Caenorhabditis elegans. Trends Genet 13: 55–61 [DOI] [PubMed] [Google Scholar]
- Lee KY, Ladha MH, McMahon C, Ewen ME (1999) The retinoblastoma protein is linked to the activation of Ras. Mol Cell Biol 19: 7724–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman JG, Gaubatz S, Livingston DM, Ginsberg D (1997) The subcellular localization of E2F-4 is cell cycle dependent. Proc Natl Acad Sci USA 94: 5095–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Horvitz HR (1998) lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95: 981–991 [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP (2003) The Tudor domain ‘Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28: 69–74 [DOI] [PubMed] [Google Scholar]
- Mendez J, Stillman B (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandis N, Teixeira MR, Gerdes AM, Limon J, Bardi G, Andersen JA, Idvall I, Mandahl N, Mitelman F, Heim S (1995) Chromosome abnormalities in bilateral breast carcinomas. Cytogenetic evaluation of the clonal origin of multiple primary tumors. Cancer 76: 250–258 [DOI] [PubMed] [Google Scholar]
- Qin X-Q, Chittenden T, Livingston DM, Kaelin WG (1992) Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev 6: 953–964 [DOI] [PubMed] [Google Scholar]
- Sellers WR, Novitch BG, Miyake S, Heith A, Otterson GA, Kaye FJ, Lassar AB, Kaelin WG (1998) Stable binding to E2F is not required for the retinoblastoma protein to activate transcription. Genes Dev 12: 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Solari F, Ahringer J (2000) NURD-complex genes antagonise Ras-induced vulval development in Caenorhabditis elegans. Curr Biol 10: 223–226 [DOI] [PubMed] [Google Scholar]
- Sternberg PW, Han M (1998) Genetics of RAS signaling in C. elegans. Trends Genet 14: 466–472 [DOI] [PubMed] [Google Scholar]
- Takahashi C, Bronson RT, Socolovsky M, Contreras B, Lee KY, Jacks T, Noda M, Kucherlapati R, Ewen ME (2003) Rb and N-ras function together to control differentiation in the mouse. Mol Cell Biol 23: 5256–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW (2001) The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell 8: 303–316 [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20 [DOI] [PubMed] [Google Scholar]
- Voorhoeve PM, Agami R (2003) The tumor-suppressive functions of the human INK4A locus. Cancer Cell 4: 311–319 [DOI] [PubMed] [Google Scholar]
- White-Cooper H, Leroy D, MacQueen A, Fuller MT (2000) Transcription of meiotic cell cycle and terminal differentiation genes depends on a conserved chromatin associated protein, whose nuclear localisation is regulated. Development 127: 5463–5473 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Materials and Methods