

Abstract
Three-dimensional cell biology and histology of tissue sections strongly benefit from advanced light microscopy and optimized staining procedures to gather the full three-dimensional information. In particular, the combination of optical clearing with light sheet-based fluorescence microscopy simplifies fast high-quality imaging of thick biological specimens. However, verified in toto immunostaining protocols for large multicellular spheroids or for tissue sections have not been published. We present a method for the verification of immunostaining in three-dimensional spheroids. The analysis relies on three criteria to evaluate the immunostaining quality: quality of the antibody stain specificity, signal intensity achieved by the staining procedure and the correlation of the signal intensity with that of a homogeneously dispersed fluorescent dye. We optimized and investigated variations of five immunostaining protocols for three-dimensional cell biology. Our method is an important contribution to three-dimensional cell biology and the histology of tissues since it allows to evaluate the efficiency of immunostaining protocols for large three-dimensional specimens, and to study the distribution of protein expression and cell types within spheroids and spheroid-specific morphological structures without the need of physical sectioning.
OCIS codes: (170.6900) Three-dimensional microscopy, (180.2520) Fluorescence microscopy
Introduction
Multicellular tumor spheroids (MCTS) resemble characteristics of in vivo tumor tissue. Both develop morphological specialties such as a proliferative, a quiescent and a necrotic zone [1,2], which not only differ in morphology but also in their cellular response to drugs [3,4]. Morphological zones only develop when a spheroid reaches a minimum size. The spheroid diameter and the cultivation time influence the availability of oxygen and nutrients throughout the spheroid, which eventually lead to a reduction of cell survival in the core region [5–7]. The concentric layering in MCTS constitutes a diverse cellular microenvironment that shares similarities to some in vivo tumors. Consequently, it is essential to work with MCTS of an adequate size to study in vivo tumor characteristics.
Providing an insight into the internal morphology of large spheroids has been challenging. The traditional preparation technique for microscopy purposes is physical sectioning of paraffin-embedded or frozen spheroids [8]. The sections are commonly analyzed with conventional fluorescence microscopy [9]. However, following sectioning it is challenging to reconstruct the three-dimensional information. Light sheet-based fluorescence microscopy (LSFM) provides true optical sectioning by moving the sample through a thin cuboid of laser light, which illuminates the focal plane of the detection path. Due to the high imaging speed and good penetration depth with remarkably low photo bleaching, LSFM is well suited for imaging large spheroid [10,11]. The combination with optical clearing methods [12,13] further improves the penetration depth into light scattering and optically dense specimens by rendering the spheroid transparent [14].
An important technique in cancer research is immunofluorescence staining [15]. The frequently used IgG antibodies have a molecular weight of about 150 kDa and hardly penetrate large three-dimensional specimens, thus providing an inhomogeneous stain. Consequently, good stain quality is only obtained for cells on the surface while the stain quality rapidly decreases for cells in deeper regions. A previous study has introduced an in toto immunostaining protocol for small spheroids, which had been generated from colon cancer cell lines with a diameter of 120 to 150 µm [16].
The quality of an immunofluorescence stain is subjectively assessed by a researcher’s opinion. Weiswald et al. commenced to analyze the staining quality by visual inspection of microscopy data and by performing flow cytometry analysis of cells from dissociated spheroids [16]. Especially future developments in three-dimensional cell biology and developing histology without physical sectioning require standardized, applicable and objective quality analysis.
We established a quantitative image analysis pipeline, which assesses specificity, signal intensity, and homogeneity of the stain. With this tool, we systematically evaluated existing immunofluorescence protocols in large MCTS. We tested different fixation and permeabilization techniques and varied antibody incubation temperature and time. We conclude that paraformaldehyde (PFA) fixation in combination with detergent-based permeabilization yields the best stain quality throughout the entire spheroid. Raising the antibody incubation temperature to 37°C significantly improves the penetration of the antibodies. The required time of our staining protocol for large spheroids starting from sample fixation to image acquisition is approximately 1.5 days long. A period that is close to that of immunofluorescence protocols for two-dimensional cell culture, making it an accessible and time-saving application. This allows to perform three-dimensional cell biology at a large scale.
Methods
Cell culture and spheroid preparation
U343 cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco), 2 mM L- glutamine (Roth) and 100 U/ml penicillin/streptomycin (Gibco) at 37°C, with 5% CO2. Spheroids were prepared by the liquid overlay technique [17]. Briefly, 96-well microplates were coated with agarose to form a concave surface. 10,000 cells were seeded in 100 µl culture medium per well. To concentrate cells in the middle of the well, the microplates were centrifuged at 300 g for 4 minutes. Spheroids grew for six days. The murine mammary tumor cell line 4T1 was cultured in RPMI1640 supplemented with 10% FBS and 2 mM L-glutamine. Spheroids seeded from 5,000 cells grew for ten days.
Antibodies
Primary antibodies were α-tubulin (clone 236-10501, Life Technologies), β-catenin (H-102, Santa Cruz) and GM130 (clone 35, BD). Secondary antibodies were anti-mouse Alexa Fluor 568 and anti-rabbit Alexa Fluor 488 (Life Technologies).
Paraffin immunohistochemistry
Following dehydration with ascending ethanol concentrations and incubation in xylol, PFA-fixed spheroids were embedded in paraffin. Sequential sections with a thickness of 4 µm were prepared with a Leica microtome and mounted onto SuperfrostPlus object glasses. Sections were dewaxed by incubation in xylol, followed by a rehydration with descending ethanol concentrations, and incubation in water. Antigen retrieval was performed by incubating the slides in 10 mM citrate buffer pH 6 in a steam cooker. Slides were washed with TBS followed by incubation with the blocking solution (0.5% Triton X-100, 10% FBS in PBS). Primary antibodies (0.5 µg/ml α-tubulin, 2 µg/ml β-catenin, 2.5 µg/ml GM130) were diluted in block solution and incubated on sections over night at 4°C. The secondary antibodies (5 µg/ml) were diluted in block solution and incubated for 90 minutes. Additionally, sections were stained with 1 µg/ml DAPI (Thermo Scientific) and mounted with Mowiol.
Fixation/permeabilization
Four different fixation/permeabilization methods were used in this study and are recapitulated in Table 1.
Table 1. Immunofluorescence protocols applied in this study.
| fixation | permeabilization | block | 1st antibody | 2nd antibody | |
|---|---|---|---|---|---|
| PFA-Triton | 4% PFA for 15 min, RT | 0.3% Triton X-100 for 15 min | 0.1% BSA, 0.2% Triton X-100, 0.05% Tween-20, 10% goat serum for 1 hour, RT | in block sol. o.n., 37°C | in block sol. for 4h or o.n., 37°C |
| PFA-EtOH | 4% PFA for 15 min, RT | dehydration (30%, 50%, 70%, 80%, 90%, 96%, 100% EtOH), rehydration (reverse order) for 2 min each, RT | in block sol. for 4h or o.n., 37°C | ||
| PFA-MetOH | 4% PFA for 15 min, RT | MetOH for 5 min, −20°C | in block sol. for 4h or o.n., 37°C | ||
| MetOH-Ac | MetOH for 5 min, Ac for 1 min, −20°C | - | in block sol. for 4h or o.n., 37°C | ||
| EtOH | 70% EtOH o.n., −20°C | - | in block sol. for 4h or o.n., 37°C | ||
| PFA-Triton 4°C | 4% PFA for 15 min, RT | 0.3% Triton X-100 for 15 min | in block sol. o.n., 4°C | in block sol. o.n., 4°C |
Ac: acetone, EtOH: ethanol, MetOH: methanol, min: minutes, h: hours; o.n.: overnight; RT.: room temperature, sol.: solution.
Antibody staining
After blocking, spheroids were incubated with primary antibodies diluted in blocking solution on a shaker (Eppendorf) at 600 rpm overnight. After washing, spheroids were incubated with 5 µg/ml secondary antibody diluted in blocking solution for the respective time and 600 rpm. Cell nuclei were counterstained with 1 µg/ml DAPI (Thermo Scientific).
Optical clearing and mounting
Spheroids were optically cleared with Murray’s clear [18]. Spheroids were dehydrated at room temperature with increasing concentrations of ethanol in deionized water (30%, 50%, 70%, 90%, 96%, 2x 100%, 2 minutes each). Dehydration reduced spheroid volume. Spheroids were transferred into BABB solution (1:2, benzyl alcohol: benzyl benzoate) with a refractive index of 1.5598 and incubated until transparency. Samples were transferred into rectangular glass capillaries (Hilgenberg), which were previously cleaned with 1% Hellmanex (Hellma Analytics) in deionized water for two hours shaking at 80°C. The capillary was mounted onto the sample holder placed inside the microscope incubation chamber, which was filled with 2,2’-thiodiethanol (TDE, Sigma).
Image acquisition
Images were acquired with the monolithic digital-scanned light sheet-based fluorescence microscope (mDSLM) equipped with a Epiplan-Neofluar 2.5x/NA 0.03 illumination objective and a N-Achroplan 10x/NA 0.3 or N-Achroplan 20x/NA 0.5 detection objective (Carl Zeiss). Three channels were acquired sequentially. The parameters were as follows: channel 00 (α-tubulin/GM130): 561 nm laser, 607/70 bandpass filter, 0.09 mW laser intensity, 100 ms exposure time; channel 01 (β-catenin): 488 nm laser, 525/50 bandpass filter, 0.134 mW laser intensity, 100 ms exposure time; channel 02 (DAPI): 405 nm laser, 447/55 bandpass filter, 0.01 mW laser intensity, 100 ms exposure time.
Image and data analysis
Images were processed with the software FIJI (version 1.48o). First, an average projection was computed for each channel. From these projections, an intensity profile through the center of the spheroid and perpendicular to the orientation of the light sheet was extracted. The analysis of the intensity profiles was performed using OriginLab (version OriginPro 2015G). All intensity profiles were smoothed using adjacent-averaging with a window range of 45 data points.
A signal-to-background (SNB) ratio was measured as the ratio between the mean signal and the standard deviation of the background in the intensity profile. An intensity threshold of 35 was set to obtain the background intensity (values below threshold) and the signal intensity (values above threshold). For each condition, the median ratio value for the samples was calculated and plotted.
To assess the homogeneity of the staining, the intensity profiles of α-tubulin and β-catenin were correlated with the DAPI intensity profile. Linear regression obtained the Pearson’s R to express the degree of similarity of the antibody stain to the DAPI intensity profile.
Results
Overview of the evaluated immunostaining protocols
Existing protocols share common steps necessary for immunolabeling: (1) fixation, (2) permeabilization, (3) blocking, (4a) primary antibody incubation and eventually (4b) secondary antibody incubation. Crucial steps that often require refinement are fixation, permeabilization and the antibody incubation. We thus systematically evaluated fixation conditions using PFA-fixation, ethanol-fixation and methanol-acetone-fixation (Fig. 1). Samples fixed with the crosslinking agent PFA were subjected to either a permeabilization with the detergent Triton X-100, or they were de- and rehydrated with ethanol of different concentrations or by a methanol dehydration to enable antibody penetration into the cells. Fixation with organic solvents does not require a permeabilization step. The blocking was performed with BSA, goat serum, Triton X-100 and Tween-20 [19]. Finally, we tested several antibody incubation conditions. Leaving the antibody concentration constant, we elevated the incubation temperature from the usual 4°C for long-term incubation to 37°C and also tested two incubation times (overnight and four hours) for the secondary antibody (Table 1). We counterstained cell nuclei with DAPI.
Fig. 1.

(A) Schematic illustration of the immunostaining protocols. The workflow shows the different steps of the immunofluorescence staining starting with the collection of the spheroids after six days of formation. The main steps are fixation, permeabilization, blocking, antibody incubation and optical clearing. (B) Single planes at different depths in a typical spheroid data set are shown. The right image shows a 90° rotation around the y-axis of the data set that depicts the location of the single planes. Microscope: mDSLM; illumination objective: Epiplan-Neofluar 2.5x/NA 0.06; detection objective: N-Achroplan 10x/NA 0.3; α-tubulin: 561 nm, 0.09 mW, bandpass filter 607/70; β-catenin: 488 nm, 0.134 mW, bandpass filter 525/50; DAPI: 405 nm, 0.01 mW, bandpass filter 447/55; scale bar: 100 µm. h: hours, min: minutes, RT: room temperature.
Following immunolabeling, we optically cleared the spheroids by dehydration with ascending concentrations of ethanol followed by an incubation in the clearing solution. We observed the cleared spheroids with LSFM. The combination of these methods allowed us to monitor the antibody labeling throughout the whole spheroid. Further, LSFM operates with low laser power and short exposure time. This enabled us to reduce the exposure of the spheroids to laser light, thereby minimizing photo bleaching of the fluorophores. This increases the signal-to-noise ratio and thereby provides a better contrast [20].
The evaluation criteria that assess immunofluorescence quality follow a simple rationale. First, the stain specificity is the most important criterion. In addition to the antibody quality, the staining protocol influences the specificity by affecting the epitope recognition and accessibility. Next, the signal intensity of fluorophores is a criterion, which states the brightness of the signal compared to the background noise. The third aspect is the homogeneity of the stain and is especially important in three-dimensional objects. We used these three criteria (specificity, signal intensity and homogeneity) as a base for the immunofluorescence quality assessment.
Paraformaldehyde fixation preserves staining specificity
Once seeded, U343 cells quickly form a spherical cluster and thereby ensure a high degree of reproducibility. We seeded 10,000 U343 cells and let spheroids form for six days. On average, the resulting spheroid diameter was 600 µm (Fig. 2(A)). We immunolabeled spheroids for β-catenin and α-tubulin and counterstained cell nuclei with the small organic dye DAPI.
Fig. 2.
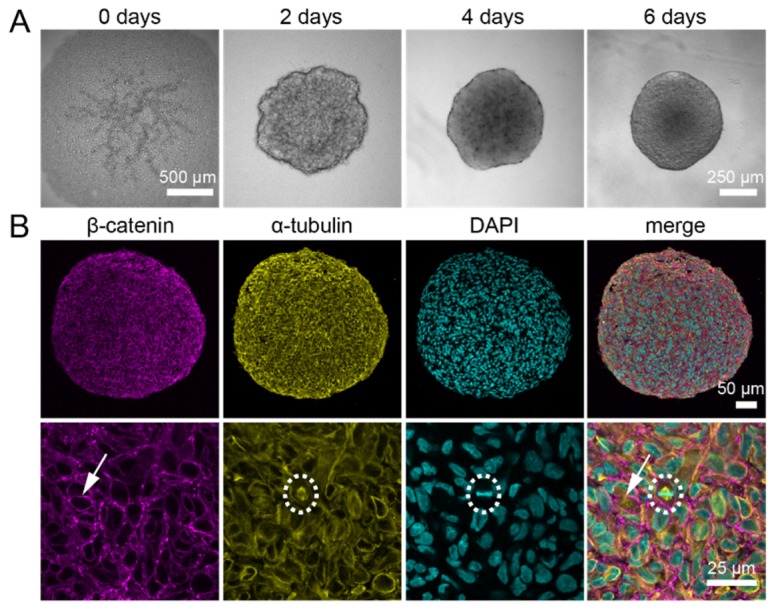
Immunostainings against α-tubulin and β-catenin in histological sections of U343 spheroids show a homogeneous signal throughout the whole spheroid. (A) U343 spheroids were formed for six days from 10,000 seeded cells. (B) Paraffin-embedded spheroids were cut into 4 µm thick sections and stained against α-tubulin and β-catenin. A section from the middle part of a spheroid shows a homogeneous distribution of the signal for α-tubulin as well as β-catenin. Cell nuclei were counterstained with DAPI. β-catenin is located at the plasma membranes of cells (arrow). The α-tubulin antibody specifically labels the microtubules in U343 spheroids, which is best seen in the mitotic spindle during cell division (dashed circle). Microscope: Zeiss LSM780; objective lens (upper panel): Plan-Apochromat 20x/numerical aperture (NA) 0.8; objective lens (lower panel): Plan-Neofluar 40x/NA 1.3.
First, we qualitatively evaluated the antibody specificity. Histological sections showed that both, α-tubulin and β-catenin, were equally expressed throughout the spheroid formed from U343 cells (Fig. 2(B)). At the cellular level, α-tubulin organized into the microtubule cytoskeleton. A specific antibody staining was best observable during mitosis when the microtubules organize to form the spindle (Fig. 2(B), dashed circle). In about 50% of the tested conditions, the spindle apparatus was not detectable in the stained spheroids. This was especially true for staining protocols, which used alcohols at a certain step. Here, specular patterns were visible (Fig. 3, left column). Most portion of β-catenin localizes to the plasma membrane where it connects adherens junctions and the actin cytoskeleton. Hence, β-catenin labeled the cell circumference in spheroids. In all tested protocols, the cell circumference stained with the β-catenin antibody was detectable indicating high specificity of the stain (Fig. 3, middle column).
Fig. 3.
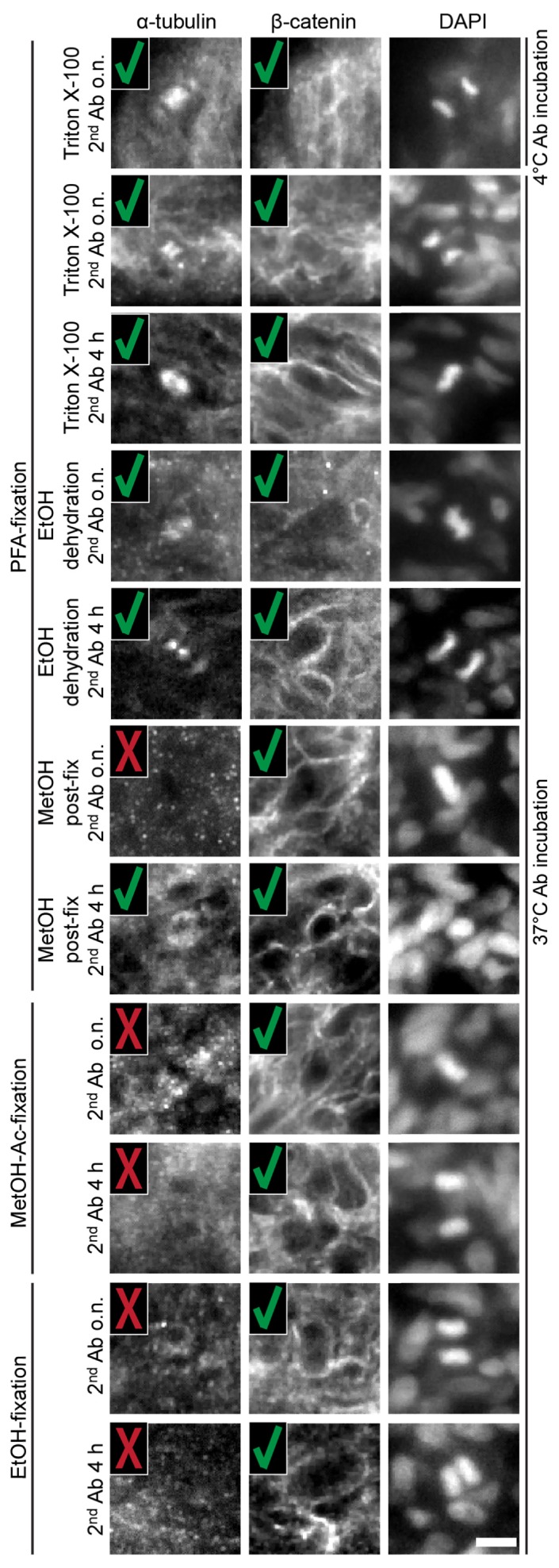
Qualitative evaluation of antibody stain specificity in U343 spheroids at the cellular level. The visibility of the spindle apparatus during mitosis indicates a functioning α-tubulin stain. Since β-catenin attaches to the adherens junctions, the label was characterized by the visibility of the cell circumference. A specific antibody stain is marked with a green check while an unspecific stain is marked with a red cross. Microscope: mDSLM; illumination objective: Epiplan-Neofluar 2.5x/NA 0.06; detection objective: N-Achroplan 20x/NA 0.5; α-tubulin: 561 nm, 0.09 mW, bandpass filter 607/70; β-catenin: 488 nm, 0.15 mW, bandpass filter 525/50; DAPI: 405 nm, 0.01 mW, bandpass filter 447/55; scale bar: 10 µm. Ab: antibody; o.n.: overnight; h: hours.
In summary, the stain specificity is assessed by comparing the obtained staining pattern with the a priori knowledge about the localization and appearance of the antigen of interest. The microtubule component α-tubulin showed diverse staining characteristics when different protocols were applied. Especially protocols without the use of alcohols provided more often a specific antibody stain while alcohol-based protocols may have destructed the microtubule cytoskeleton, thereby only giving a specular staining pattern. Contrary, β-catenin stain always showed the anticipated characteristics when different staining protocols were applied.
Antibody incubation time and temperature influence fluorescence intensity
To quantitatively analyze the staining quality, we developed an image analysis pipeline (Fig. 4). First, average projections of the image z-stacks were generated for the recorded channels. From the average projections, we extracted intensity profiles through the center region of the spheroid perpendicular to the direction of the light sheet.
Fig. 4.

The optically cleared spheroid is mounted onto the sample holder (the edge of the sample holder is indicated by the white dashed line). Following image acquisition, an average projection of the image stack is computed. At the center of the spheroid, the intensity profile along a line orthogonal to the direction of the light sheet (orange line) is measured in each channel (i.e.: α-tubulin, β-catenin and DAPI). The DAPI signal is regarded as the ideal dispersion of the fluorophores in the spheroid. The intensity profiles are smoothed to reduce morphological variations. Finally, the smoothed grey values of the antibodies are plotted against the smoothed DAPI values and a linear regression is calculated. The red circles show the location of the highlighted values in the different representation modes. The output of this analysis is the Pearson’s R value. One and zero indicates that the antibody profile matches, respectively does not match, the DAPI profile.
We then analyzed the obtained intensity profiles and observed a high variability of the profiles (Fig. 5(A)). Thus, we measured the signal-to-background (SNB) ratio for each profile. For each condition, we computed the median of the ratio to summarize the obtained signal intensity.
Fig. 5.

Overview of the signal intensities and the degree of stain homogeneity in spheroids obtained from different immunostaining conditions. (A) Smoothed average signal intensity plots. α-tubulin and β-catenin intensity values of all samples per condition are plotted against the pixel distance along a line through the center of the spheroid. Incubation of the secondary antibody for four hours always results in lower signal intensities compared to samples incubated with the secondary antibody overnight. (B) The ratio between the maximum intensity in an average profile and the background intensity serves as a measure of the intensity yield. Ratios with values above 20 and low variance are desirable whereas values below 10 are considered insufficient. The box-plot shows the median line and the 25 and 75 percentiles. The whiskers represent the min and max values. N [PFA-Triton-4°C] = 5, N [PFA-MetOH o.n.] = 7, N [rest] = 6. Ab: antibody, Ac: acetone, dehyd.: dehydration, EtOH: ethanol, h: hours, MetOH: methanol, o.n.: overnight.
We observed that the different staining protocols resulted in different signal intensities (Fig. 5(B) and Table 2). Antibody incubation at 4°C obtained signal intensities for α-tubulin and β-catenin stain with ratios of 19.40 and 60.63, respectively. When antibodies were incubated at 37°C, the obtained ratio was increased in most cases. An exception was when spheroids were fixed with methanol-acetone and incubated with the secondary antibody for four hours. Here, for both, α-tubulin and β-catenin, we measured a ratio of 15.13 and 33.52, respectively. However, when the secondary incubation period was prolonged, the ratios increased to 22.70 for α-tubulin and 106.41 for the β-catenin stain. For spheroids fixed with methanol-acetone, the resulting fluorescence intensities were for an overnight incubation of the secondary antibodies for α-tubulin 22.69 and for β-catenin 106.41. In contrast, when ethanol was used for fixation, ratios of 36.30 for α-tubulin and 113.56 for β-catenin for a secondary antibody incubation period of four hours, and 109.37 and 260.53 for an overnight incubation were measured. PFA fixation followed by detergent-based permeabilization also resulted in higher ratios (for α-tubulin for four hours and overnight incubation 62.41 and 81.75, and for β-catenin 137.10 and 274.58). Further, ratios of 50.92 for α-tubulin and 119.76 for the β-catenin stain were achieved when methanol was used for permeabilization following PFA fixation and a secondary antibody incubation overnight. Similar values were obtained when ethanol was used to permeabilize the spheroids following PFA fixation.
Table 2. Summary of the immunofluorescence quality analysis. A qualitative as well as the two quantitative characteristics are listed (1) to describe the specificity of the stain, (2) to estimate the signal intensity obtained from the staining protocol described by the SNB ratio and (3) to rate the homogeneity of the antibody stain described by the R-value of the similarity of the intensity profiles to the DAPI stain. Stain specificity: Green indicates that the stain for microtubules shows the spindle apparatus during mitosis and that β-catenin is located at the cell periphery. Signal intensity: Green indicates a ratio above 60, orange indicates ratios between 30 and 60 and white is used for a ratio below 30. Signal homogeneity: Green indicates above 0.75, orange indicates between 0.55 and 0.74 and, white is used for values below 0.54.
| stain specificity | signal intensity | signal homogeneity | |||
|---|---|---|---|---|---|
| PFA-Triton-4°C | o.n. | α-tubulin | yes | 19.40 | 0.568 |
| PFA-Triton-4°C | o.n. | β-catenin | yes | 60.63 | 0.501 |
| PFA-Triton-37°C | 4 hours | α-tubulin | yes | 62.41 | 0.823 |
| PFA-Triton-37°C | o.n. | α-tubulin | yes | 81.75 | 0.797 |
| PFA-Triton-37°C | 4 hours | β-catenin | yes | 137.10 | 0.633 |
| PFA-Triton-37°C | o.n. | β-catenin | yes | 274.58 | 0.433 |
| PFA-EtOH-37°C | 4 hours | α-tubulin | yes | 25.86 | 0.706 |
| PFA-EtOH-37°C | o.n. | α-tubulin | yes | 50.71 | 0.589 |
| PFA-EtOH-37°C | 4 hours | β-catenin | yes | 71.23 | 0.559 |
| PFA-EtOH-37°C | o.n. | β-catenin | yes | 187.75 | 0.488 |
| PFA-MetOH-37°C | 4 hours | α-tubulin | yes | 25.22 | 0.762 |
| PFA-MetOH-37°C | o.n. | α-tubulin | no | 50.92 | 0.857 |
| PFA-MetOH-37°C | 4 hours | β-catenin | yes | 107.27 | 0.435 |
| PFA-MetOH-37°C | o.n. | β-catenin | yes | 119.76 | 0.713 |
| MetOH-Ac-37°C | 4 hours | α-tubulin | no | 15.13 | 0.956 |
| MetOH-Ac-37°C | o.n. | α-tubulin | no | 22.69 | 0.876 |
| MetOH-Ac-37°C | 4 hours | β-catenin | yes | 33.52 | 0.870 |
| MetOH-Ac-37°C | o.n. | β-catenin | yes | 106.41 | 0.709 |
| EtOH-37°C | 4 hours | α-tubulin | no | 36.30 | 0.947 |
| EtOH-37°C | o.n. | α-tubulin | no | 109.37 | 0.884 |
| EtOH-37°C | 4 hours | β-catenin | yes | 113.56 | 0.875 |
| EtOH-37°C | o.n. | β-catenin | yes | 260.54 | 0.846 |
Ac: acetone, EtOH: ethanol, MetOH: methanol, o.n.: overnight.
Alcohol-based fixation improves antibody penetration in large spheroids
Next, we observed that the penetration of antibodies into the spheroid varied between the different immunostaining protocols (Fig. 6(A)). In the following, we measured the dispersion of the antibodies through the spheroids. Therefore, the DAPI intensity profile presented a homogenous distribution of the dye throughout the spheroid. Thus, we measured the degree of similarity between the antibody staining and the DAPI stain by plotting the intensity values of α-tubulin and β-catenin against the intensity values obtained from the DAPI stain. We hypothesized that in case of a homogeneous antibody dispersion, the two variables, the intensities of the antibody stain and the DAPI intensities, followed a linear correlation. Therefore, the homogeneity of the staining was expressed by the Pearson’s R correlation coefficient (Fig. 6(B)).
Fig. 6.
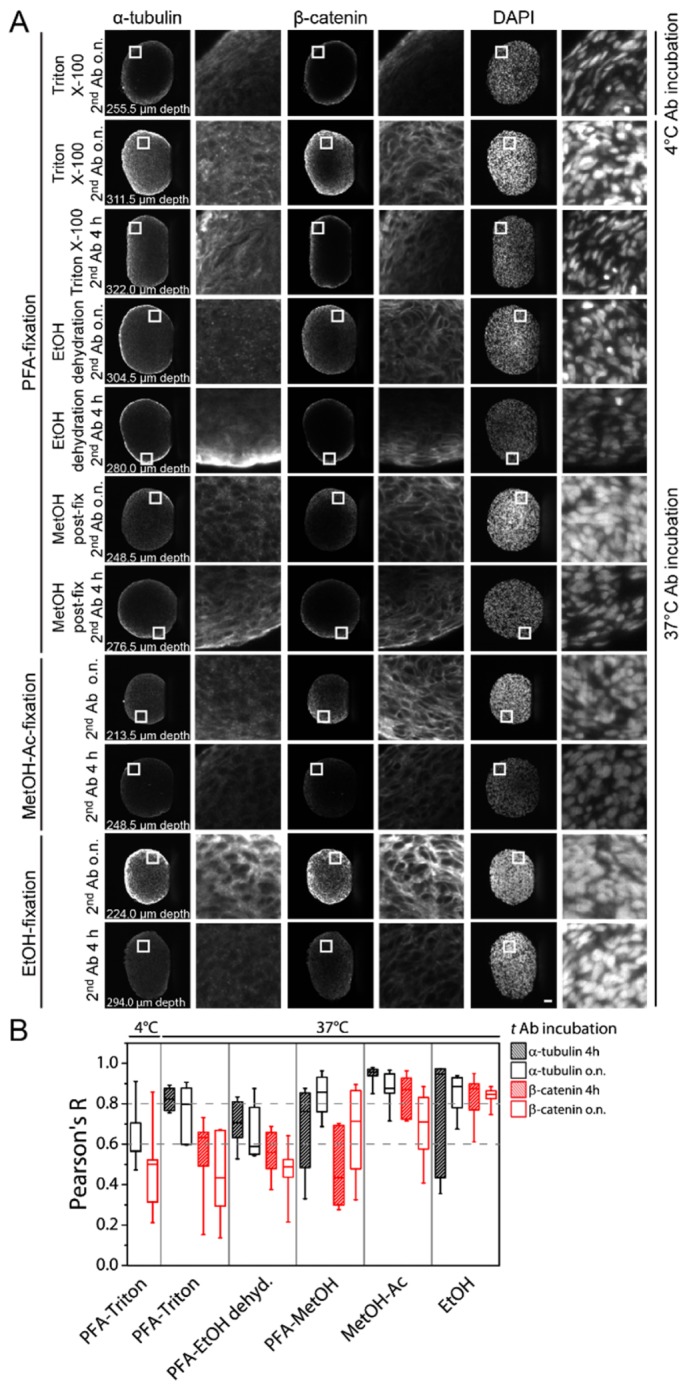
(A) Single planes of immunostained U343 spheroids show the dispersion of the antibody as well as the quality of the stain. Slides were taken from the middle region of the spheroids. The z-depth is indicated for each condition. White squares indicate the magnified areas (8x) in each spheroid. Microscope: mDSLM; illumination objective: Epiplan-Neofluar 2.5x/NA 0.06; detection objective: N-Achroplan 10x/NA 0.3; α-tubulin: 561 nm, 0.09 mW, bandpass filter 607/70; β-catenin: 488 nm, 0.134 mW, bandpass filter 525/50; DAPI: 405 nm, 0.01 mW, bandpass filter 447/55; scale bar: 50 µm. (B) The influence of the fixation/permeabilization and the incubation time of the secondary antibodies is expressed as Pearson’s R. It describes the degree of similarity to the DAPI intensity profile, which represents a homogeneous dispersion of a fluorophore within a spheroid. The box-plot shows the median line and the 25 and 75 percentiles. The whiskers represent the min and max values. Ab: antibody; Ac: acetone, dehyd.: dehydration, EtOH: ethanol, h: hours, MetOH: methanol, o.n.: overnight; h: hours.
First, we saw that when the antibody incubation temperature was set to 4°C, the median R-values were 0.568 for the α-tubulin stain and 0.501 for the β-catenin stain. In all cases regarding the α-tubulin stain, higher R-values were obtained when the antibody incubation temperature was raised. This was also the case for most of the β-catenin stained spheroids. Exceptions were PFA fixation either with Triton X-100 permeabilization or with ethanol de- and rehydration followed by an overnight secondary antibody incubation. Here, the median R-values were 0.433 and 0.488, respectively. When PFA fixation was followed by an ethanol de- and re-hydration to permeabilize the spheroids only poor diffusion of the antibodies into the spheroids was obtained. Here R-values were 0.706 for α-tubulin and 0.559 for β-catenin when the secondary antibody was incubated for four hours, and 0.589 and 0.488 when the antibody incubation was prolonged to overnight. When methanol was used for permeabilization after PFA fixation, R-values for α-tubulin with 0.762 and 0.435 and β-catenin with 0.857 and 0.713 were obtained for secondary antibody incubation of four hours and overnight incubation, respectively. Further, a low correlation coefficient of only 0.435 was obtained when spheroids were subjected to PFA fixation followed by methanol permeabilization and an incubation time of four hours for the secondary antibody. In contrast to this, the most homogenous dispersion of the antibodies was achieved when spheroids were fixed with organic solvents such as ethanol or methanol-acetone. Here, R-values for both antibody labels were between 0.846 and 0.947 for ethanol-fixed samples, and 0.709 and 0.956 for methanol-acetone-fixed samples.
According to the Pearson’s R-value obtained from the linear regression analysis, it appeared that the α-tubulin antibody penetrated in most cases well into the deeper parts of the spheroids while the β-catenin antibody labeled the outer rim of the spheroid very well but only poorly the core region (Fig. 6). Further, the dispersion of the fluorophores into the spheroids was in general more homogeneous using a short incubation time for the secondary antibody.
In summary, we introduced an analysis pipeline to objectively estimate the quality of a fluorescence stain. This rates the stain specificity, the signal intensity and the homogeneity of the fluorophore dispersion. According to this, we tested immunofluorescence staining protocols and concluded that a fixation with PFA is best suitable to specifically label the antigens of interest. Further, a detergent-based permeabilization and a raise of the antibody incubation temperature to 37°C increased fluorescence signal intensity as well as homogeneous dispersion of the fluorophores compared to an alcohol-based permeabilization or incubation temperature at 4°C. This protocol is time-saving, straightforward and inexpensive (Fig. 7(A)). We further applied this protocol with tumor spheroids, generated from the murine mammary tumor cell line 4T1. We stained the cis-Golgi apparatus with an anti GM130 antibody and the cell circumference with an anti β-catenin antibody (Fig. 7(B)).
Fig. 7.

(A) Simplified illustration of the suggested immunofluorescence protocol for large spheroids with time specifications for every step. The overall duration from sample fixation to image acquisition consumes approximately 1.5 days. This time-saving protocol suggests sample fixation with PFA followed by detergent-based permeabilization and block of unspecific binding sites. Antibody incubation temperature at 37°C allows homogeneous diffusion of antibodies. All other steps are performed at room temperature. Following staining, large spheroids are rendered transparent by optical clearing with Murray’s clear. Cleared samples are rapidly investigated with LSFM. (B) Spheroids from the murine mammary tumor cell line 4T1 were immunolabeled according the protocol in (A). Antibodies against β-catenin and GM130, a Golgi marker, were used. A section through the central part of the cleared spheroid shows the staining success. Spheroids were formed for ten days and had an initial diameter of more than 350 µm. Microscope: mDSLM; illumination objective: Epiplan-Neofluar 2.5x/NA 0.06; detection objective: N-Achroplan 20x/NA 0.5; GM130: 561 nm, 0.09 mW, bandpass filter 607/70; β-catenin: 488 nm, 0.15 mW, bandpass filter 525/50; DAPI: 405 nm, 0.01 mW, bandpass filter 447/55; scale bar: 50 µm. RT: room temperature.
Discussion
It is relevant to optimize immunostaining protocols for three-dimensional approaches. Evaluations, which are (1) reproducible and (2) identify specific characteristics for further quantification, require a quantitative quality assessment. Work from other researchers has been done to develop immunofluorescence protocols for in toto protein labeling in MCTS. Fluorescence activated cell sorting (FACS) as well as confocal fluorescence microscopy were used to analyze the staining efficacy [16]. We picked up the idea of an objective estimation of stain quality and developed an image analysis pipeline, which is quick and straightforward. It uses three criteria to estimate the immunolabeling success. First, we rated the stain specificity qualitatively, then we measured the signal intensity (SNB), and finally the dispersion of the fluorophores in the spheroid. The stain specificity investigates the data for the anticipated staining pattern. A highly specific stain is indispensable, but immunofluorescence is sensitive towards the production of artifacts such as speckles or a destruction of the epitope, which results in not interpretable data [21]. The SNB ratio expresses the obtained signal intensity throughout the spheroid. High signal intensities increase contrast and thereby not only increase the image quality but are especially important for downstream applications such as segmentation [22]. This applies also to the stain homogeneity. Antibodies and some dyes form concentration gradients along the spheroid axis resulting in an inhomogeneity of the stain. To estimate the dispersion of fluorophores, a reference is required, which provides information about a homogeneous diffusion. We used the small molecular dye DAPI. Its molecular structure and small size of a few hundred Dalton allows it to diffuse large cellular networks such as spheroids and tissue, thereby providing a homogeneous labeling of cell nuclei.
With our analysis pipeline we tested classical immunostaining procedures with the objective to optimize a protocol for the use in large three-dimensional specimens. We worked with MCTS with an average diameter of about 600 µm, which facilitates layer formation, thereby making them more comparable to in vivo tumors [1,2]. To obtain three-dimensional information about whole mount large spheroids, we used optical clearing and LSFM. We chose Murray’s clear due to its inexpensiveness, reproducibility and simple establishment. Further, it obtains good clearing results, and preserves organic fluorophores [12].
The most significant adaptation of the immunofluorescence staining protocols to three-dimensional whole mount spheroids was to increase the antibody incubation temperature from usually 4°C for long-term incubation to 37°C. The overall signal intensity was increased by more than 100%, and also the dispersion of the antibodies into the inner parts of the spheroids was enhanced by about 44% and 26% of the α-tubulin and β-catenin stain, respectively. Prolongation of the secondary antibody incubation time increased the overall signal intensity but decreased the stain homogeneity throughout the spheroids. We think that a concentration gradient of antibody results in inhomogeneous dispersion of the fluorophores and that a prolonged secondary antibody incubation reinforces this effect by increased binding of antibody at the outer rim of the spheroids. Thus, the incubation time for the secondary antibody was reduced to four hours to decrease to the effect of inhomogeneous staining signal.
We varied other parameter after adaptation of the antibody incubation temperature and time, and found that this affected the immunofluorescence staining in varying aspects. Alcohols as fixatives are frequently used in two-dimensional cell culture with a subsequent cytoskeleton visualization [23]. Further, the use of alcohols is reportedly successful for obtaining a homogeneous antibody staining of thick biological specimens [24]. Fixation with alcohols impaired the visualization of the tubulin network. In most cases, we also measured a reduced signal intensity in the spheroids. The lowest signal intensity was obtained when spheroids were fixed with methanol-acetone. Here, the SNB ratio was about two to four times lower compared to spheroids, which were PFA-fixed and permeabilized with Triton X-100.We suggest that the cytoskeleton in a spheroid is more sensitive towards alcohol-based fixatives making them unfavorable for fixation. In contrast, when alcohols were used to permeabilize spheroids following PFA fixation, the microtubule spindle apparatus remained detectable in most samples. The cytoarchitecture as well as the structure of the cytoskeleton was stabilized by the cross-linking PFA.
Although alcohols such as ethanol provide the most homogeneous staining, they lack stain specificity and also show lower intensity values. The most important sign of quality in immunostaining is the stain specificity, followed by the signal intensity and the stain homogeneity in three-dimensional specimens. According to this, we suggest to subject MCTS to PFA fixation, which maintains cytoarchitecture and epitopes. This in combination with a detergent-based permeabilization such as Triton X-100 also improves towards obtaining high signal intensity and reasonable homogeneity of the staining. The antibody incubation temperature raised to 37°C improves the dispersion of fluorophores and thereby the overall performance of the protocol. With this protocol we achieved specificity to detect α-tubulin and β-catenin as well as good fluorophore dispersion and signal intensity. We recommend an evaluation of the staining quality for any other antibody. This protocol, from fixation to imaging, takes about one and a half working days, due to the fact of the relatively long antibody incubation time. There is a strong perspective to reduce the time to a single working day by the advantages of the heavily developing field of nanobody use [25]. These and other novel approaches [26,27] might also further improve the dispersion of the fluorophores resulting in a perfectly homogeneous stain.
We developed an analysis pipeline that describes three immunofluorescence stain characteristics, which (1) estimate the quality of the stain specificity, (2) quantify the signal intensity and (3) the degree of homogeneity of the antibody stain throughout the spheroid. With this new approach, a quick, reliable and objective validation of the fluorescence staining can be performed. High quality immunostaining is especially required when three-dimensional specimens are stained in one piece. We believe that this is a starting point for performing histology without the need for physical cutting, where tissue biopsies are fixed, stained and imaged in toto.
Funding
This study was supported by the DFG-funded Cluster of Excellence Frankfurt for Macromolecular Complexes (CEF-MC II, EXC115).
References and links
- 1.Bell H. S., Whittle I. R., Walker M., Leaver H. A., Wharton S. B., “The development of necrosis and apoptosis in glioma: experimental findings using spheroid culture systems,” Neuropathol. Appl. Neurobiol. 27(4), 291–304 (2001). 10.1046/j.0305-1846.2001.00319.x [DOI] [PubMed] [Google Scholar]
- 2.Sutherland R. M., “Cell and environment interactions in tumor microregions: the multicell spheroid model,” Science 240(4849), 177–184 (1988). 10.1126/science.2451290 [DOI] [PubMed] [Google Scholar]
- 3.Mellor H. R., Ferguson D. J. P., Callaghan R., “A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs,” Br. J. Cancer 93(3), 302–309 (2005). 10.1038/sj.bjc.6602710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedrich J., Ebner R., Kunz-Schughart L. A., “Experimental anti-tumor therapy in 3-D: spheroids--old hat or new challenge?” Int. J. Radiat. Biol. 83(11-12), 849–871 (2007). 10.1080/09553000701727531 [DOI] [PubMed] [Google Scholar]
- 5.Groebe K., Mueller-Klieser W., “On the relation between size of necrosis and diameter of tumor spheroids,” Int. J. Radiat. Oncol. Biol. Phys. 34(2), 395–401 (1996). 10.1016/0360-3016(95)02065-9 [DOI] [PubMed] [Google Scholar]
- 6.Nichols M. G., Foster T. H., “Oxygen diffusion and reaction kinetics in the photodynamic therapy of multicell tumour spheroids,” Phys. Med. Biol. 39(12), 2161–2181 (1994). 10.1088/0031-9155/39/12/003 [DOI] [PubMed] [Google Scholar]
- 7.Mueller-Klieser W., “Method for the determination of oxygen consumption rates and diffusion coefficients in multicellular spheroids,” Biophys. J. 46(3), 343–348 (1984). 10.1016/S0006-3495(84)84030-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandtzaeg P., “The increasing power of immunohistochemistry and immunocytochemistry,” J. Immunol. Methods 216(1-2), 49–67 (1998). 10.1016/S0022-1759(98)00070-2 [DOI] [PubMed] [Google Scholar]
- 9.Dufau I., Frongia C., Sicard F., Dedieu L., Cordelier P., Ausseil F., Ducommun B., Valette A., “Multicellular tumor spheroid model to evaluate spatio-temporal dynamics effect of chemotherapeutics: application to the gemcitabine/CHK1 inhibitor combination in pancreatic cancer,” BMC Cancer 12(1), 15 (2012). 10.1186/1471-2407-12-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller P. J., Schmidt A. D., Wittbrodt J., Stelzer E. H. K., “Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy,” Science 322(5904), 1065–1069 (2008). 10.1126/science.1162493 [DOI] [PubMed] [Google Scholar]
- 11.Stelzer E. H. K., “Light-sheet fluorescence microscopy for quantitative biology,” Nat. Methods 12(1), 23–26 (2014). 10.1038/nmeth.3219 [DOI] [PubMed] [Google Scholar]
- 12.Dodt H. U., Leischner U., Schierloh A., Jährling N., Mauch C. P., Deininger K., Deussing J. M., Eder M., Zieglgänsberger W., Becker K., “Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain,” Nat. Methods 4(4), 331–336 (2007). 10.1038/nmeth1036 [DOI] [PubMed] [Google Scholar]
- 13.Spalteholz W., Über Das Durchsichtigmachen von Menschlichen Und Tierischen Präparaten (Hirzel, 1911). [Google Scholar]
- 14.Keller P. J., Dodt H. U., “Light sheet microscopy of living or cleared specimens,” Curr. Opin. Neurobiol. 22(1), 138–143 (2012). 10.1016/j.conb.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 15.Coons A. H., Kaplan M. H., “Localization of antigen in tissue cells; improvements in a method for the detection of antigen by means of fluorescent antibody,” J. Exp. Med. 91(1), 1–13 (1949). 10.1084/jem.91.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiswald L. B., Guinebretière J. M., Richon S., Bellet D., Saubaméa B., Dangles-Marie V., “In situ protein expression in tumour spheres: development of an immunostaining protocol for confocal microscopy,” BMC Cancer 10(1), 106 (2010). 10.1186/1471-2407-10-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santini M. T., Rainaldi G., “Three-dimensional spheroid model in tumor biology,” Pathobiology 67(3), 148–157 (1999). 10.1159/000028065 [DOI] [PubMed] [Google Scholar]
- 18.Dent J. A., Polson A. G., Klymkowsky M. W., “A whole-mount immunocytochemical analysis of the expression of the intermediate filament protein vimentin in Xenopus,” Development 105(1), 61–74 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Debnath J., Muthuswamy S. K., Brugge J. S., “Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures,” Methods 30(3), 256–268 (2003). 10.1016/S1046-2023(03)00032-X [DOI] [PubMed] [Google Scholar]
- 20.Stelzer E. H. K., “Contrast, resolution, pixelation, dynamic range and signal-to-noise ratio: fundamental limits to resolution in fluorescence light microscopy,” J. Microsc. 189(1), 15–24 (1998). 10.1046/j.1365-2818.1998.00290.x [DOI] [Google Scholar]
- 21.Schnell U., Dijk F., Sjollema K. A., Giepmans B. N. G., “Immunolabeling artifacts and the need for live-cell imaging,” Nat. Methods 9(2), 152–158 (2012). 10.1038/nmeth.1855 [DOI] [PubMed] [Google Scholar]
- 22.Mathew B., Schmitz A., Muñoz-Descalzo S., Ansari N., Pampaloni F., Stelzer E. H. K., Fischer S. C., “Robust and automated three-dimensional segmentation of densely packed cell nuclei in different biological specimens with Lines-of-Sight decomposition,” BMC Bioinformatics 16(1), 187 (2015). 10.1186/s12859-015-0617-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altieri D. C., Li F., Ambrosini G., Chu E. Y., Plescia J., Tognin S., Marchisio P. C., “Control of apoptosis and mitotic spindle checkpoint by survivin,” Nature 396(6711), 580–584 (1998). 10.1038/25141 [DOI] [PubMed] [Google Scholar]
- 24.Ott S. R., “Confocal microscopy in large insect brains: zinc-formaldehyde fixation improves synapsin immunostaining and preservation of morphology in whole-mounts,” J. Neurosci. Methods 172(2), 220–230 (2008). 10.1016/j.jneumeth.2008.04.031 [DOI] [PubMed] [Google Scholar]
- 25.Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H., “A simple, versatile method for GFP-based super-resolution microscopy via nanobodies,” Nat. Methods 9(6), 582–584 (2012). 10.1038/nmeth.1991 [DOI] [PubMed] [Google Scholar]
- 26.Murray E., Cho J. H., Goodwin D., Ku T., Swaney J., Kim S. Y., Choi H., Park Y. G., Park J. Y., Hubbert A., McCue M., Vassallo S., Bakh N., Frosch M. P., Seung H. S., Wedeen V. J., Chung K., “Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems,” Cell 163(6), 1500–1514 (2015). 10.1016/j.cell.2015.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung K., Deisseroth K., “CLARITY for mapping the nervous system,” Nat. Methods 10(6), 508–513 (2013). 10.1038/nmeth.2481 [DOI] [PubMed] [Google Scholar]