

Abstract
Axin and p53 are tumor suppressors, controlling cell growth, apoptosis, and development. We show that Axin interacts with homeodomain-interacting protein kinase-2 (HIPK2), which is linked to UV-induced p53-dependent apoptosis by interacting with, and phosphorylating Ser 46 of, p53. In addition to association with p53 via HIPK2, Axin contains a separate domain that directly interacts with p53 at their physiological concentrations. Axin stimulates p53-dependent reporter transcription in 293 cells, but not in 293T, H1299, or SaOS-2 cells that are defective in p53 signaling. Axin, but not AxinΔHIPK2, activates HIPK2-mediated p53 phosphorylation at Ser 46, facilitating p53-dependent transcriptional activity and apoptosis. Specific knockdown of Axin by siRNA reduced UV-induced Ser-46 phosphorylation and apoptosis. Kinase-dead HIPK2 reduced Axin-induced p53-dependent transcriptional activity, indicating that Axin stimulates p53 function through HIPK2 kinase activity. Interestingly, HIPK2ΔAxin that lacks its Axin-binding region acts as a dominant-positive form in p53 activation, suggesting that the Axin-binding region of HIPK2 is a putative autoinhibitory domain. These results show that Axin acts as a tumor suppressor by facilitating p53 function through integration of multiple factors.
Keywords: apoptosis, Axin, HIPK2, p53, phosphorylation
Introduction
Axin was initially identified as a negative regulator of axis formation, deficiency of which causes axial duplication as seen in genetically transmitted Fused mice (Zeng et al, 1997). Molecular analysis has revealed that Axin is a master scaffold for the regulation of the cellular levels of β-catenin, a protein believed to be a common denominator of Wnt signaling (Cadigan and Nusse, 1997; Kikuchi, 1999; Peifer and Polakis, 2000). We have previously delineated a signaling pathway in which Axin interacts with MEKK1/4 and activates JNK via MKK4/7 (Zhang et al, 1999; Luo et al, 2003). A peculiar feature of the Axin/JNK pathway is that Axin requires multiple domains/factors for JNK activation. In the course of identifying molecules that may account for the requirement of the C-terminal region of Axin in JNK activation, we carried out a yeast two-hybrid screen and found that Axin interacted with homeodomain-interacting protein kinase-2 (HIPK2) that had been previously shown to phosphorylate p53 at Ser 46 and enhanced p53-dependent transcriptional activity as well as apoptosis (D'Orazi et al, 2002; Hofmann et al, 2002).
The p53 tumor suppressor protein is a potent inhibitor of cell growth, capable of arresting the cell cycle at several points and, under some circumstances, can participate in DNA repair after genotoxic insult, and can activate the apoptotic machinery leading to cell death (Gottlieb and Oren, 1996; Ko and Prives, 1996; Levine, 1997; Sharpless and DePinho, 2002; Attardi and DePinho, 2004). The p53 gene is frequently altered in human cancers (Greenblatt et al, 1994). In addition, apoptosis has recently been suggested to be a major contribution to p53-mediated suppression of tumor formation (Attardi and DePinho, 2004). Besides the well-established roles in tumor suppression, a remarkable recent finding has revealed an unexpected developmental role for p53 in that it promotes mesoderm differentiation (Cordenonsi et al, 2003; Takebayashi-Suzuki et al, 2003). Key TGF-β signaling events depend on functional p53, including the full transcriptional activation of the CDK inhibitor p21waf1. The p53 and Smad proteins physically interact and synergistically activate TGF-β-induced target genes (Cordenonsi et al, 2003). Under normal conditions, p53 is short lived due to degradation upon ubiquitination by Mdm2, a ubiquitin E3 ligase (Michael and Oren, 2003; Vargas et al, 2003). In response to environmental stress, and perhaps developmental cues, it also undergoes a variety of other post-translational modifications including phosphorylation and acetylation (Prives and Manley, 2001; Brooks and Gu, 2003; Xu, 2003). Stress-induced phosphorylation of two critical residues on p53, Ser 15 and Ser 20, results in stabilization and activation of p53 (Chehab et al, 1999; Dumaz and Meek, 1999). In addition to Ser 15 and Ser 20, Ser 46 has been recently shown to be critical for p53 stability regulation (D'Orazi et al, 2002; Hofmann et al, 2002). Phosphorylation of Ser 46 on p53 is mediated by HIPK2, which is activated by ultraviolet radiation (D'Orazi et al, 2002; Hofmann et al, 2002). The HIPK2-mediated phosphorylation of p53 facilitates CBP-mediated acetylation of p53, increasing the transcriptional activity of p53 and enhancing UV-induced apoptosis (D'Orazi et al, 2002; Hofmann et al, 2002). Furthermore, HIPK2 was also shown to downregulate the cellular levels of the transcriptional corepressor CtBP, suggesting that HIPK2 promotes apoptosis by elevating expression of multiple target genes (Zhang et al, 2003).
Here we show that Axin forms a ternary complex with HIPK2 that is bound to p53. In addition, Axin possesses a separate domain that directly interacts with the endogenous p53. Axin stimulates the transcriptional activity of p53 selectively toward some of the p53 target genes, and induces p53-dependent cell death through facilitating HIPK2 phosphorylation of Ser 46. Specific siRNA against Axin reduced UV-induced apoptosis, phosphorylation of p53 at Ser 46, and blocked HIPK2 stimulation of p53-dependent reporter activity. Deletion of the Axin-interacting domain of HIPK2 (HIPK2ΔAxin) failed to diminish Axin-induced p53-dependent transcriptional activities, but rather stimulated p53 transactivation even more dramatically than wild-type HIPK2, suggesting that Axin stimulates HIPK2 by removing an autoinhibitory domain of HIPK2. Taken together, we have found that Axin has an integrating role in the activation of p53-dependent transcription and apoptosis, underscoring its proapoptotic function as a tumor suppressor.
Results
Axin forms a ternary complex with HIPK2 and p53 in vivo and in vitro
In the course of yeast two-hybrid screen using the Axin C-terminal region as bait, we identified multiple clones from a fetal mouse brain cDNA library (Rui et al, 2002), one of which was unexpectedly found to encode HIPK2. HIPK2 was previously shown to interact with p53. To test for the specificity of the yeast two-hybrid interaction in mammalian cells between HIPK2 and Axin, and whether they form a ternary complex with p53, we carried out co-immunoprecipitation of Axin, HIPK2, and p53 in untransfected 293T cells with anti-Axin (for specificity, see Supplementary Figure 1), anti-HIPK2, and the anti-p53 antibody DO-1, respectively (Figure 1A). Western blotting analysis showed that the Axin immunoprecipitate contained p53, and that Axin could be detected in the p53 immunoprecipitate (Figure 1A, left panel). For analysis of HIPK2 interaction with Axin and p53, we carried out immunoprecipitation with the anti-HIPK2 antibody and then detected the copresence of Axin and p53 in its immunoprecipitate (Figure 1A, right panel). Co-immunoprecipitation of Axin and p53 with HIPK2 at their endogenous concentrations was also detected in 293 cells (Supplementary Figure 2). Next, we performed an in vitro GST pulldown assay. GST-fused Axin, but not GST alone, could pull down HA-p53 overexpressed in H1299 cells; likewise, Axin was detected in GST-p53 pulldown, indicating that Axin interacts directly with p53 (Figure 1B). To further examine whether Axin, p53, and HIPK2 could form a ternary complex, we performed a two-step co-immunoprecipitation assay (Figure 1C). HEK 293T cells were transfected with HA-HIPK2 and Myc-Axin. As a control, HIPK2 with no tag was transfected. In the first step of immunoprecipitation, anti-HA was used to pull down HIPK2, and 3 × HA-tag peptide was used to elute the complex. The eluate was then immunoprecipitated with anti-Axin or control IgG, followed by Western blotting to detect p53. As shown in Figure 1C, p53 was present in the final immunoprecipitate but not in the control sample, indicating that Axin, p53, and HIPK2 are in a ternary complex. Since p53 is a nuclear protein and Axin can be visualized as a nuclear protein only after leptomycin B treatment, we carried out immunofluorescent staining of HeLa cells transfected with Axin and p53 or HIPK2 in the presence of leptomycin B. Indeed, Axin is partially colocalized in the nucleus with p53 and HIPK2 (Supplementary Figure 3A and B). We also examined if UV treatment could induce Axin to translocate into the nucleus and colocalize with p53 or HIPK2. As shown in Supplementary Figure 3C and D, some portion of Axin was translocated into the nucleus upon UV treatment, and overlapped with p53 and HIPK2. Finally, to analyze if HIPK2 and p53 could form a complex with monomeric Axin, we transfected 293T cells with HIPK2, and AxinΔC250, which lacks the DIX domain and is defective in dimer formation. As shown in Figure 1D, both HIPK2 and p53 were readily co-immunoprecipitated with AxinΔC250 as well as full-length Axin (Figure 1D). Reciprocally, Axin proteins could be detected in HIPK2 and p53 immunoprecipitates (Figure 1E and F).
Figure 1.
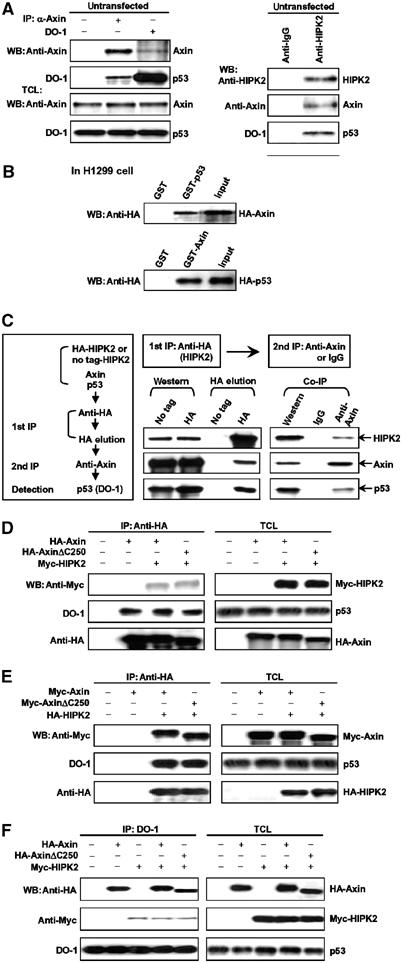
Interaction of Axin with HIPK2 and p53 in vivo and in vitro. (A) Axin and p53 in untransfected 293T cells were separately immunoprecipitated with rabbit anti-Axin that was raised against aa 348–500 of mouse Axin and anti-p53 antibody DO-1, respectively (left panel). The control IgG used was from rabbit. Detection of Axin and p53 in the immunoprecipitates and total cell lysates (TCL) was carried out using anti-Axin and DO-1 antibodies. For analysis of HIPK2 interaction with Axin and p53, we carried out immunoprecipitation with goat anti-HIPK2 antibody or control goat IgG and detected copresence of Axin and p53 in its immunoprecipitate (right panel). (B) GST pulldown assay to evaluate Axin interaction with p53 in vitro. The ability of GST-p53 to retain Axin present in the cell lysate from H1299 cells transfected with pCMV-HA-Axin was analyzed by Western blotting with anti-HA following SDS–PAGE (top panel). Bottom: The ability of GST-Axin to pull down HA-p53 in the cell lysate. In either case, GST alone did not interact with p53 or Axin. Input represents 1/4 of that used for GST pulldown. (C) Two-step co-immunoprecipitation of the complex containing Axin, HIPK2, and p53. The procedures of the two-step co-immunoprecipitation are outlined in the box on the left, according to Harada et al (2003). HEK 293T cells were transfected with plasmids expressing Myc-Axin and HA-HIPK2 (or untagged HIPK2 as control). The first immunoprecipitation was performed using anti-HA and Protein A/G-agarose beads. The complex was eluted with 3 × HA-tag, followed by the second step of co-immunoprecipitation with anti-Axin or control IgG. Protein samples from each step were analyzed by Western blotting separately with anti-Axin, anti-HIPK2, and anti-p53 (DO-1). (D–F) HIPK2 and p53 form a complex on the monomeric Axin. AxinΔC250, which lacks the DIX domain and cannot form homodimer, was used in this experiment. (D) HA-Axin and Myc-HIPK2 were transfected into HEK 293T cells and immunoprecipitation was performed with anti-HA antibody. HIPK2 and endogenous p53 were detected in the Axin immunoprecipitate by Western blotting with anti-Myc and DO-1 (anti-p53) antibody, respectively. Protein levels of Axin, HIPK2, and p53 in the total cell lysates (TCL) were determined by anti-HA, anti-Myc, and DO-1, respectively (on the right). (E) Axin and p53 are copresent in HIPK2 immunoprecipitates. HA-HIPK2 and Myc-Axin were cotransfected into HEK 293T cells and HIPK2 was immunoprecipitated with anti-HA. Co-precipitated endogenous p53 and Myc-Axin were detected with DO-1 and anti-Myc antibody, respectively. (F) Endogenous p53 forms a complex with Axin and HIPK2. Immunoprecipitation was carried out with DO-1. Axin and HIPK2 were detected with anti-HA and anti-Myc, respectively.
Determination of mutual interaction regions in Axin, HIPK2, and p53
To characterize the functional significance of the ternary protein–protein interactions, we first determined their mutual interaction domains. Expression vectors containing different deletions were constructed for Axin, HIPK2, and p53, as indicated schematically (Figures 2 and 3). When Myc-Axin alone was transfected, the anti-HA for HIPK2 did not co-precipitate Myc-Axin (Figure 2A, first lane); however, when cotransfected with HA-HIPK2, the full-length Axin was strongly co-immunoprecipitated with HIPK2. Axin deletion mutants N2, C1, and M1 retained their ability to form a complex with HIPK2, albeit with lower affinity compared to the full-length Axin. However, Axin deletion mutants C2, C3, M2, and M3 lost their ability to interact with HIPK2, indicating that the region around aa 678–753 in Axin is critical for HIPK2 interaction.
Figure 2.
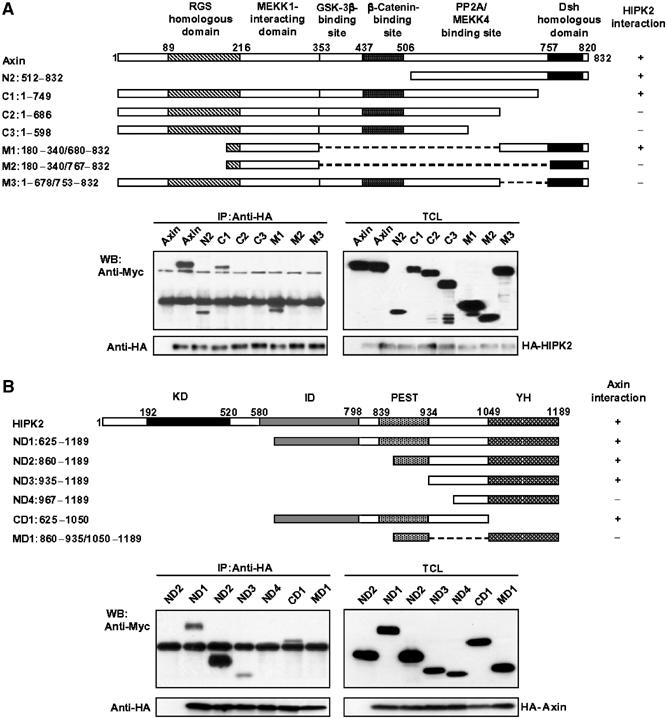
Determination of domains for mutual interaction between Axin and HIPK2. (A) Determination of HIPK2-binding sites on Axin. Schematic diagrams (on the top) depict different Axin deletion constructs used in the domain mapping experiments. The different Axin deletion constructs were cotransfected with the blank vector (first lane) or the full-length HA-HIPK2 into HEK 293T cells. Cells were then subjected to lysis and immunoprecipitation, followed by Western blotting with different antibodies indicated. (B) Identification of HIPK2 sequence critical for Axin binding. On the top are schematic diagrams depicting different HIPK2 deletion mutants used in the domain mapping experiments. HEK 293T cells were transfected with HA-Axin and Myc-tagged HIKP2 deletion constructs. Cell lysates were immunoprecipitated with anti-HA antibody. The immunoprecipitates and cell lysates were then analyzed by Western blotting separately using anti-HA for HA-Axin, and anti-Myc for Myc-HIPK2 deletion constructs.
Figure 3.

Identification of critical regions of Axin and p53 for their complex formation. (A) Endogenous p53 directly interacts with Axin via a domain distinct from the HIPK2-binding site. Schematic diagrams depict different Axin deletion mutants used in the domain mapping experiments. Different HA-tagged Axin deletion constructs were transiently transfected into HEK 293T cells. Cell lysates were immunoprecipitated with anti-p53 antibody DO-1, followed by immunoblotting using anti-HA for Axin deletion mutants and DO-1 for endogenous p53. (B) Determination of Axin-binding sites in p53. Schematic diagrams depict different p53 deletion mutants used in the domain mapping experiments. HEK 293T cells were transfected with HA-Axin and different Myc-tagged p53 deletion constructs as indicated. Cell lysates were immunoprecipitated with anti-HA antibody. The immunoprecipitates and cell lysates were then analyzed by Western blotting separately using anti-HA for HA-Axin and anti-Myc for Myc-p53 proteins.
We then cotransfected different deletion mutants of HIPK2 with the full-length Axin to 293T cells to determine the domain of HIPK2 for Axin interaction. As shown in Figure 2B, all the HIPK2 mutants, with the exception of MD1 and ND4, interacted with Axin, indicating that the region spanning aa 935–1050 of HIPK2 is critical for Axin interaction. However, the weak interaction of ND3 or CD1 with Axin suggests that this region of HIPK2 may not be sufficient for maximal interaction for Axin.
Axin contains an additional domain for direct interaction with p53
We went further to test the ability of different deletion mutants of Axin to form a ternary complex with HIPK2 and p53. Different deletion constructs of Axin were transiently transfected into 293T cells; DO-1 antibody was used to immunoprecipitate the endogenous p53. Unexpectedly, Axin C2, C3, C4, and M7, which all lack the HIPK2 interaction domain, readily co-immunoprecipitated with the endogenous p53 (Figure 3A). We suspected that there existed a separate domain around the MEKK1 interaction region that is responsible for the strong p53 interaction. Indeed, when the region of aa 209–338 in Axin was deleted (Axin-M4), only a minute amount of p53 was detected with the mutant Axin even after prolonged exposure (Figure 3A and Supplementary Figure 4); when both the region of aa 209–338 and the HIPK2-binding region (aa 678–753) were deleted (Axin-M9), the mutant Axin completely lost the ability to interact with p53 (Figure 3A). These results indicate that the region of aa 209–338 in Axin directly interacts with p53, and that the residual amount of p53 detected with Axin-M4 is through the endogenous HIPK2.
Furthermore, we generated different deletion mutants of p53 and carried out complex formation assays to determine which region of p53 is required for direct interaction with Axin. The results showed that the carboxyl half of the DNA-binding domain (DBD) of p53 is responsible for direct interaction with Axin (Figure 3B).
Axin activates p53-dependent transcriptional activity
Recent studies have shown that HIPK2 stimulates p53-dependent transcription of target genes that are important for induction of apoptosis (D'Orazi et al, 2002; Hofmann et al, 2002). To ascertain functional significance of the Axin/HIPK2/p53 ternary complex formation, we first tested if Axin could modulate p53-dependent gene transcription using the PathDetect p53 cis-Reporting System (Stratagene) that carries the p53-specific enhancer element (TGCCTGGACTTGCCTGG)14 derived from the sequence comparison of promoters of p53-inducible genes (el-Deiry et al, 1992; Tokino et al, 1994) and luciferase reporters separately carrying an Mdm2 promoter. Axin activated the p53-dependent gene transcription using the p53-Luc reporter and Mdm2 reporter in a dose-dependent manner in 293 cells (Figure 4A and F). We then transfected into 293 cells with Axin constructs lacking the HIPK2-binding site (Axin-M3), direct p53-binding site (Axin-M4), or both HIPK2- and p53-binding sites (Axin-M9), to assess their contribution to the activation of p53 transcriptional activity (Figure 4B). The Axin-M8 deletion mutant that contains both the p53- and HIPK2-binding sites was also included for comparison. Deletion of the HIPK2-binding site rendered Axin almost completely incapable of activating p53, whereas Axin lacking its p53-binding site retained partial activity presumably contributed by the HIPK2-bound p53, and Axin-M8 activated the reporter as effectively as did the wild-type Axin (Figure 4B). The p53 dominant-negative mutant p53-R175H blocked Axin-mediated p53-dependent reporter activity (Figure 4C). In contrast, in the p53 null H1299 and SaOS-2 cells, the dose-dependent activation of these reporters by Axin was not seen unless the wild-type p53 was cotransfected, indicating that Axin activates the reporter genes through p53 (Figure 4D and E). In agreement with the previous observation, we did not see any activation of the p21 reporter by HIPK2 in H1299 cells (D'Orazi et al, 2002), or Axin in any of the cell lines tested (data not shown). It is also noteworthy here that we could not detect any activation of the reporters by Axin in 293T cells, in which the SV40 large T-antigen binds to p53 in the DBD and blocks its interaction with DNA. The observation with 293T cells therefore also suggests that Axin requires functional p53 to enhance the reporter activity.
Figure 4.

Axin enhances p53-dependent transcriptional activity. PathDetect p53-Luc reporter (A–E) or Mdm2 reporter (F) was transfected together with different Axin or p53 constructs into HEK 293, H1299, and SaOS-2 as indicated. Western blotting of expressed proteins was performed to indicate similar expression levels (insets). In each transfection, an equal amount of GFP expression plasmid was included as internal control and the GFP protein level in each cell lysate was probed with anti-GFP (Molecular Probes) as loading control (l.c.). All transfections were performed in duplicate and the data are means±s.d. of five independent experiments after normalizing luciferase activity from the vector control to 1. (A) Axin can activate p53 transactivation in a dose-dependent manner. Axin (0, 0.5, 1.0, 2.0, and 3.0 μg) and 0.5 μg of the PathDetect p53-Luc reporter (Stratagene) were cotransfected in HEK 293 cells to test the effect of Axin on p53-dependent transcriptional activity. (B) Axin mutants defective in p53 binding (M4), HIPK2 binding (M3), or binding to both p53 and HIPK2 (M9) did not stimulate p53 transactivation except M8, which contains both the p53- and HIPK2-binding sites. (C) The DNA-binding-defective p53 mutant p53-R175H negatively affects Axin-stimulated p53 transcriptional activity. HEK 293 cells were transfected with the p53-Luc reporter in different combinations of an empty vector, Axin, and p53-R175H, as indicated. (D) Axin depends on p53 for stimulation of reporter transcriptional activity in p53−/− H1299 cells. Flag-Axin failed to activate the p53 reporter in H1299 cells; in the presence of wild-type p53 expressed under the control of pCMV5 vector (1 ng each), Axin further stimulates the reporter activity in a dose-dependent manner. (E) HA-Axin activates p53-dependent transcription in p53−/− SaOS-2 cells in a similar manner to that described in panel D. (F) An Mdm2 luciferase reporter was cotransfected with increasing amounts of Axin into HEK 293 cells.
Kinase-dead HIPK2 and HIPK2 siRNA reduced Axin-mediated stimulation of p53 transcription
To evaluate the functional relationship between Axin and HIPK2 in the activation of p53 transcriptional activity, the p53-dependent reporter was cotransfected in different combinations with Axin, HIPK2, kinase-dead HIPK2 (K221R), pSUPER-HIPK2, and pSUPER-Axin (for efficiency of the pSUPER constructs, see Supplementary Figure 5) as indicated in Figure 5. Axin and HIPK2 synergistically stimulated p53-dependent reporter activity (Figure 5A). The HIPK2 mutant K221R retarded the Axin-mediated p53-dependent reporter activation (Figure 5B). Similarly, siRNA against HIPK2, but not the control siRNA, diminished the Axin-mediated p53 transcriptional activation (Figure 5C). Conversely, specific knockdown of Axin by pSUPER-Axin also diminished HIPK2-mediated p53 stimulation (Figure 5D). Intriguingly, AxinΔHIPK2 (M3) defective in binding to HIPK2 could abolish HIPK2 stimulation of p53 transcriptional activity (Figure 5E), possibly due to depletion of p53 and other factors required for HIPK2 normal function. These data indicated that Axin and HIPK2 are mutually dependent in the stimulation of p53 transcriptional activation. However, removal of the Axin-binding domain of HIPK2 (HIPK2ΔAxin) did not lose but rather enhanced its ability to stimulate p53 transactivation of target genes (Figure 5F), indicating that the Axin-binding domain in HIPK2 may exert an autoinhibitory function when not bound by Axin. In line with the data in the transcriptional activity assay, HIPK2ΔAxin is fully active in mediating p53 phosphorylation at Ser 46 in vivo and in vitro (Figure 6E and Supplementary Figure 6).
Figure 5.

Axin and HIPK2 synergistically enhance p53-Luc reporter activity. HEK 293 cells were transfected with expression plasmids in different combinations as indicated. Insets show similar expression levels of relevant transfected proteins. Measurement and data presentation are as described in the legend to Figure 4. (A) Axin and HIPK2 mutually enhance stimulation of p53-Luc transcriptional activity. (B, C) Dominant-negative mutant of HIPK2 (HIPK2-K221R (B)) and siRNA (pSUPER-HIPK2) against HIPK2 (C) diminished Axin activation of p53-Luc reporter gene transcription. It is to be noted that the luciferase activity from cells transfected with HIPK2-K221R alone (bar 3 in panel B) was normalized to 1. RNAi control is expressed from pSUPER that contains an insert with multiple variations to the HIPK2 siRNA. (D) The siRNA against Axin, but not control siRNA, significantly diminished the activation of p53 by HIPK2. (E) AxinΔHIPK2 defective in HIPK2 binding drastically reduced p53-Luc activation by HIPK2. Wild-type (WT) Axin alone was transfected as a positive control for AxinΔHIPK2. (F) HIPK2 and HIPK2ΔAxin defective in Axin binding were separately transfected into HEK 293 cells, and p53 luciferase activity was measured. HIPK2ΔAxin behaved as a dominant positive mutant as it stimulated the reporter activity to a greater extent than the wild-type HIPK2.
Figure 6.

Axin stimulates HIPK2-mediated p53 phosphorylation at Ser 46. (A) Axin stimulates p53 phosphorylation in a dose-dependent manner. Increasing amounts of HA-Axin were transfected in H1299 cells together with Myc-p53, and as an internal control GFP-expressing vector pEGFPN1. Cell lysates were immunoprecipitated with anti-Myc antibody, followed by Western blotting with anti-p53 (FL393), anti-HA (Axin), anti-phospho-Ser 46-p53, and anti-GFP. (B) H1299 cells were transfected with HA-Axin, Flag-HIPK2, Myc-p53, and GFP-expressing vector pEGFPN1 as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody. The immunoprecipitates and total cell lysates were then analyzed by immunoblotting separately using anti-p53 (FL393) for p53, anti-HA for Axin, anti-Flag for HIPK2, anti-phospho-Ser 46 for the level of Ser-46-phosphorylated p53, and anti-GFP that serves as a control for similar transfection efficiency. (C) HIPK2 siRNA diminished Axin-induced p53 phosphorylation. Axin was cotransfected with or without pSUPER-HIPK2 as indicated to assess the effect of siRNA against HIPK2 on p53 phosphorylation at Ser 46. (D) pSUPER-Axin or pSUPER empty vector was cotransfected into MCF-7 cells; after 24 h, cells were treated with ultraviolet at 50 J/m2. Cells were lysed at 10 h post-treatment and p53 was immunoprecipitated by anti-p53 (DO-1), followed by Western blotting with anti-phospho-Ser 46 and anti-p53 (FL393). (E) HIPK2ΔAxin is more potent in p53 phosphorylation. Wild-type HIPK2 or HIPK2ΔAxin defective in Axin binding was cotransfected with Myc-p53 into H1299 cells. P53 was immunoprecipitated with anti-Myc antibody and probed with anti-phospho-Ser 46 to assess the levels of phosphorylated p53. (F) Phospho-Ser-46 p53 is contributed by p53 bound to both Axin and HIPK2. HA-Axin, HA-Axin-M4, HA-Axin-M9, Myc-p53, Myc-p53-S46A, and pEGFPN1 were transfected into H1299 cells as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody. The immunoprecipitates and total cell lysates were then analyzed by immunoblotting separately using anti-p53 (FL393) for p53 and p53-S46A, anti-HA for Axin proteins, anti-phospho-Ser 46-p53, and anti-GFP. (G) HIPK2 mutants lacking both p53-binding and Axin-binding sites failed to phosphorylate p53. Wild-type HIPK2, HIPK2Δp53, or HIPK2Δp53/ΔAxin was cotransfected with Myc-p53 into H1299 cells to determine the ability to phosphorylate p53.
Axin activates HIPK2 phosphorylation of p53 at Ser 46
It has been shown that the nuclear kinase HIPK2 can phosphorylate p53 at Ser 46 (D'Orazi et al, 2002; Hofmann et al, 2002). We examined whether Axin stimulates p53 by increasing HIPK2-mediated p53 phosphorylation. We transfected Myc-p53 together with or without Axin into H1299 cells and carried out immunoblotting analysis with anti-phospho-Ser 46 antibody following immunoprecipitation of p53. As shown in Figure 6A, with increasing levels of Axin, Ser-46 phosphorylation was gradually increased. In the presence of both Axin and HIPK2, p53 Ser-46 phosphorylation was further increased, indicating that Axin and HIPK2 can cooperatively mediate the phosphorylation (Figure 6B). In contrast, Axin had no effect on Ser 15 or Ser 20 phosphorylation (data not shown), in agreement with the previous observations that HIPK2 did not enhance phosphorylation of p53 at these sites (D'Orazi et al, 2002; Hofmann et al, 2002). The requirement of HIPK2 for Axin to stimulate p53 phosphorylation was confirmed by the observation that in the presence of siRNA against HIPK2, Axin failed to increase p53 phosphorylation (Figure 6C).
We then addressed whether Axin played a role in the UV-induced Ser-46 phosphorylation by HIPK2. As expected, UV treatment increased Ser-46 phosphorylation as well as p53 total level in MCF-7 cells (Figure 6D). Specific knockdown of the endogenous Axin by siRNA drastically attenuated UV-induced phosphorylation at Ser 46 in response to UV treatment (Figure 6D).
Based on the transcriptional activity assay, we found that when the Axin-binding domain of HIPK2 was removed, the mutant HIPK2ΔAxin is even more active than its wild type, which pointed to an interesting possibility that the Axin-binding domain is an autoinhibitory domain. We therefore also measured the ability of HIPK2ΔAxin to phosphorylate p53 at Ser 46, showing that HIPK2ΔAxin had slightly higher activity toward p53 phosphorylation (Figure 6E). Consistently, when assayed in vitro using bacterially expressed p53 as substrate, immunoprecipitated HIPK2ΔAxin also exhibited higher kinase activity (Supplementary Figure 6).
As Axin contains two sites for p53 association, we then determined which one is important for Ser-46 phosphorylation. Axin-M4, which can bind to HIPK2 but lacks the N-terminal direct p53-binding site, retained the ability to enhance Ser-46 phosphorylation, in agreement with the previous finding that HIPK2 can bind and phosphorylate its bound p53 (Figure 6F). However, p53 phosphorylation at Ser 46 induced by Axin-M4 was reduced by about half, suggesting that the other p53 pool bound directly to Axin could be phosphorylated by HIPK2. Indeed, when both the HIPK2-binding site and the N-terminal direct p53-binding site in Axin were removed, the double mutant of Axin (M9) completely lost its ability to stimulate p53 phosphorylation. It is to be noted that the previous study with an HIPK2 mutant lacking its C-terminal region (HIPK2Δaa865–1096) that included the Axin-binding domain showed that the mutant failed to phosphorylate p53 (D'Orazi et al, 2002). We reasoned that the discrepancy between our data with HIPK2ΔAxin (Δaa935–1050) and the data obtained with HIPK2Δaa865–1096 is caused by the fact that HIPK2Δaa865–1096 could neither phosphorylate HIPK2-bound p53 nor the Axin-bound p53. For this purpose, we first constructed fine deletion mutants and found that the HIPK2's p53-binding site resides within the PEST domain around aa 839–934, which is N-terminal to the Axin-binding site. As expected, when only the p53-binding site was removed, HIPK2Δp53 retained its activity to phosphorylate p53 (Figure 6G), which was likely derived from the Axin-bound p53, as HIPK2Δp53/ΔAxin defective in binding to Axin and p53 failed to enhance p53 phosphorylation (Figure 6G).
Axin-mediated cell death requires exogenous p53 in H1299 cells
We and others have previously shown that Axin overexpression leads to cell death in hepatocarcinoma cells and in immature thymocytes (Satoh et al, 2000; Hsu et al, 2001). Our current finding that Axin strongly interacts with p53 and its regulator HIPK2 prompted us to determine whether Axin causes apoptosis through functional interaction with HIPK2 as well as p53. Strikingly, in H1299 cells lacking p53, Axin did not induce apoptosis. However, when wild-type p53 was reintroduced into H1299 cells, Axin could further induce apoptosis to a greater extent than in the presence of p53 alone (Figure 7A). Domain requirements of Axin for induction of apoptosis were determined by transiently transfecting Axin, Axin-M3, Axin-M4, Axin-M8, or Axin-M9 into 293 cells (Figure 7B). The results showed that all of the Axin deletion mutants had reduced ability to induce apoptosis in the cells except Axin-M8, which contains both the p53- and HIPK2-binding sites, indicating that each of the p53 interacting domains is required for maximal induction of p53-dependent apoptosis. When pSUPER-p53 (Supplementary Figure 5) was introduced into HEK 293 cells to decrease the endogenous p53 levels, apoptosis rate induced by Axin was significantly reduced (Figure 7C). This effect was not seen with the control siRNA. Consistently, p53-R175H diminished Axin's ability to induce apoptosis in 293 cells (Figure 7D). Next, we asked if pSUPER-Axin could reduce UV-induced apoptosis in U2OS cells as compared to control siRNA (Figure 7E). The results show that knockdown of Axin significantly reduced apoptosis rate after UV treatment. We also performed colony formation assays in HEK 293, U2OS, SaOS-2, and H1299 cells following transient transfection of the empty vector alone, Axin, and Axin-M9. In 293 and U2OS cells, consistent with the data derived from the apoptosis assays, overexpression of Axin resulted in sparse colonies (Figure 7F). The Axin mutant Axin-M9 defective in binding to p53 and HIPK2 did not reduce the number of formed colonies compared to the vector control. P53 dependence of Axin-induced apoptosis was further confirmed by the similar numbers of colonies formed upon transfection of either wild-type Axin or its mutant Axin-M9 in SaOS-2 and H1299 cells.
Figure 7.
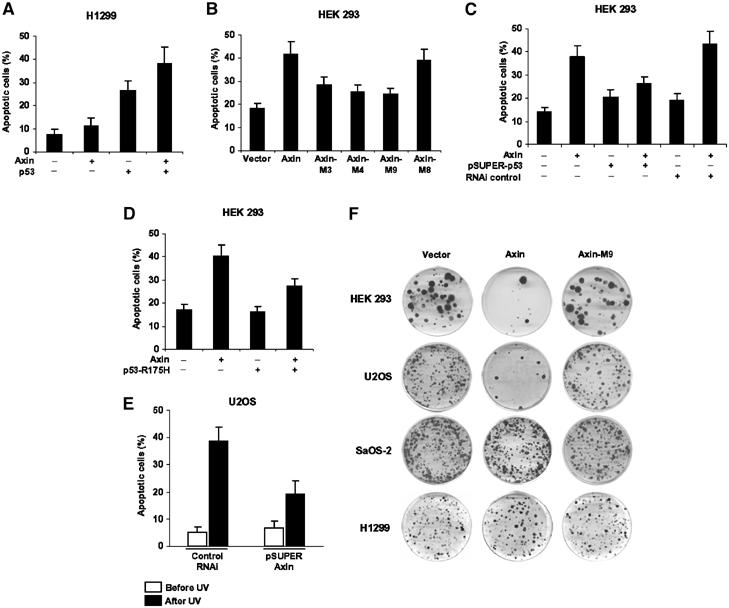
Axin requires functional p53 to induce apoptosis. (A) Axin depends on exogenous p53 for induction of apoptosis in p53-defective H1299 cells. H1299 cells were transfected with different constructs as indicated. Apoptosis was quantified 24 h after transfection by Hoechst staining, and results are means±s.d. (B) Axin requires both HIPK2- and p53-binding sites for maximal induction of apoptosis in 293 cells. (C, D) p53 siRNA and p53-R175H diminished Axin-induced apoptosis in 293 cells, respectively. (E) U2OS cells were transfected with 6 μg of pSUPER-Axin or a control pSUPER. At 24 h post-transfection, cells were left untreated or irradiated with UV (50 J/m2) to induce apoptosis, and analyzed 8 h later. (F) Colony formation assays. HEK 293, U2OS, SaOS-2, and H1299 cells were transfected with empty pCEP4 vector, pCEP4-Axin, or pCEP4-Axin-M9. Transfected cells were selected with medium containing 200 ng/ml hygromycin B and surviving cell colonies were stained with crystal violet. Similar results were obtained from G418-treated cells after transfection with empty pcDNA3.1 vector, pcDNA3.1-Axin, or pcDNA3.1-Axin-M9.
Discussion
In the present work, we provide lines of evidence that Axin, a critical regulator of Wnt signaling, is a scaffold protein for assembly of multimeric protein complex consisting of HIPK2 and p53. We showed that at physiological concentrations, Axin associates with p53 in two ways: one through HIPK2 and the other through direct interaction via a separate domain. Using a combination of deletion mutants and specific siRNAs, we found that Axin mirrors HIPK2-mediated p53 activation in several ways. First of all, Axin stimulates Ser-46 phosphorylation of p53. Second, Axin selectively enhances p53-dependent transcription of target genes, pointing to the possibility that Axin and HIPK2 both preferentially participate in p53-induced apoptosis in tumor suppression. Third, Axin and HIPK2 depend on each other for activation of p53, ultimately enhancing p53-dependent gene transcription and apoptosis. The Axin-mediated enhancement of p53-dependent transcriptional activity and apoptosis is physiologically relevant, as Axin fails to exert these roles in p53-deficient cells including 293T, H1299, and SaOS-2 cells, and specific knockdown of Axin reduced UV-induced apoptosis and Ser-46 phosphorylation of p53.
Mechanism of Axin stimulation of p53 phosphorylation
Axin appears to exert a scaffolding role in mediating HIPK2 phosphorylation of p53. First of all, Axin physically interacts with HIPK2 as well as p53. This brings HIPK2 to the proximity of p53, which is contributed from two pool: one associated with HIPK2 and the other from the direct binding site in Axin. It was previously reported that a deletion mutant of HIPK2, HIPK2Δ865–1096, defective in p53 binding failed to stimulate p53 (D'Orazi et al, 2002). We have fine-mapped the p53-binding domain in HIPK2 to the region of aa 839–934 N-terminal to the Axin-binding site (aa 935–1050; Figure 2B), and constructed a deletion mutant with only aa 839–934 removed (HIPK2Δp53). HIPK2Δp53, which is defective in binding to p53 but retains its ability to bind to Axin, could still stimulate p53 function as assayed for both Ser-46 phosphorylation and transcriptional activity (Figure 6G, and data not shown). It can now be explained that HIPK2Δ865–1096 loses its ability to activate p53 because it cannot get access to either pool of p53 normally bound to the Axin complex. Similarly, HIPK2Δp53 could still stimulate p53 because it can still bind to the endogenous Axin and activate the Axin-bound p53.
The second aspect of Axin function in the stimulation of p53 is to physically occupy the region around aa 935–1050 that might otherwise be an autoinhibitory domain of HIPK2 kinase activity, reminiscent of many kinases in their inactive state. This conjecture is supported by our finding that removal of the Axin-binding region of HIPK2 (mutant HIPK2ΔAxin) is constitutively active in p53 stimulation. More importantly, knockdown of the endogenous Axin by siRNA abolished HIPK2-mediated p53-dependent transcriptional activity or UV-induced p53 phosphorylation at Ser 46. Based on our current findings, together with data obtained by the previous studies, we propose that the kinase domain (KD) of HIPK2 is normally marked by its Axin-binding domain (AD). When bound to Axin, HIPK2 kinase is activated and phosphorylates p53 bound to both HIPK2 and Axin, ultimately enhancing p53-dependent apoptosis (Figure 8).
Figure 8.

A simplified model depicting mechanistic roles of Axin in stimulation of HIPK2 kinase activity. HIPK2 on its own is inactive as its kinase domain (KD) is occupied by the Axin-binding domain (AD), which is a putative autoinhibitory domain. When bound to Axin, the kinase domain (KD) of HIPK2 becomes activated and is accessible to the substrate p53, which is from two pools: one directly associated with Axin and the other bound with HIPK2 itself. Activated HIPK2 phosphorylates Ser 46 of p53.
Axin as a tumor suppressor
Axin has been suggested to be a tumor suppressor for many reasons. In Wnt signaling, Axin plays a central role in the control of the oncogenic protein β-catenin by acting as a scaffold for the APC/GSK-3β/CKIα degradation complex (Liu et al, 2002). Importantly, Axin mutations have been found in primary cancer cells and introduction of wild-type Axin into the cancer cells could lead to cell death (Satoh et al, 2000; Hsu et al, 2001). Moreover, high levels of programmed cell death occurred in the thymus and spleen of transgenic animals overexpressing Axin (Hsu et al, 2001). In the present study, we have provided strong evidence to suggest that Axin stimulates p53, facilitating p53-mediated transcriptional activity and apoptosis. A number of cellular stresses including genotoxic agent, hypoxia, and hyperproliferative signals can activate p53 to stimulate target gene expression. To achieve its role in tumor suppression, activated p53 seems to trigger two separable, albeit not mutually exclusive, pathways: one to induce p21 for cell cycle arrest (el-Deiry et al, 1993) and the other to induce proapoptotic factors such as Bax and Puma (Miyashita and Reed, 1995; Nakano and Vousden, 2001; Attardi and DePinho, 2004). Axin and HIPK2 may preferentially trigger the apoptosis pathway but not the G1 cell cycle arrest pathway. First of all, neither Axin (data not shown) nor HIPK2 could stimulate the promoter activity of p21 (D'Orazi et al, 2002), levels of which correlate with cell cycle arrest. Consistently, even after repeated attempts, no G1 arrest was detected in cells overexpressing Axin. On the contrary, Axin rapidly induces cells to undergo nuclear condensation, leading to apoptotic cell death.
It has been reported that activated p53 or overexpression of p53 downregulates β-catenin in human and mouse cells, which is apparently mediated by ubiquitin–proteasome system (Sadot et al, 2001). This raises another intriguing possibility that Axin may act to decrease cellular levels of β-catenin through stimulation of both abundance and transcriptional activity of p53, in parallel to its scaffolding role for β-catenin degradation by APC/GSK-3β/β-TrCP system. How Axin links its ability to activate p53 to β-catenin downregulation awaits further studies.
In the future, it would be interesting to study whether Fused mice have a higher rate of cancer formation. The Fused mice express, in addition to the normal mature Axin transcript, an aberrant splice form caused by the inserted intracisternal A particle (IAP) transposable element. This aberrant transcript would encode a protein lacking the C-terminal portion that does not contain the HIPK2-binding site as well as the DIX domain. This truncated protein therefore may not have adverse effects on Axin function in the regulation of p53 function and, in particular, it is not present in high abundance (Vasicek et al, 1997).
In sum, we have provided compelling evidence that Axin is an integrating factor for p53 and its regulators such as HIPK2. Considering Axin is a multidomain scaffold binding to a wide array of proteins from many signaling pathways, our finding will expand avenues to understand the functions of p53 as both a tumor suppressor and a developmental regulator.
Materials and methods
Yeast two-hybrid screen
Screening for novel proteins that interact with the C-terminal portion of mouse Axin was carried out with the MATCHMAKER GAL4 Two-hybrid System 3 (Clontech), as previously described (Rui et al, 2002). Briefly, Axin C-terminal 145 aa was inserted into the bait vector pGBKT7 and transformed into the yeast strain AH109, followed by mating with yeast Y187 cells pretransformed mouse brain cDNA library.
Plasmid construction
Various constructs of Axin were generated according to standard molecular techniques, and numbering of amino-acid positions was based on the mouse short alternative form as previously described (Zhang et al, 1999). Full-length cDNA encoding HIPK2 was obtained by fusion of EST clones. Point mutations of HIPK2 and p53 were generated by a PCR-based site-directed mutagenesis method using Pfu polymerase (Stratagene). All the fragments generated by PCR were subjected to sequencing verification. Deletion mutants were created by PCR reaction to obtain DNA fragments followed by fusion to native flanking DNA fragments. The details of the primer sequences used for point mutations and deletion mutations are available upon request.
Cell culture and transient transfection
HEK 293, HEK 293T, SaOS-2 (purchased from ATCC), U2OS, MCF-7, and H1299 cells were maintained in RPMI 1640 medium, with 10% fetal bovine serum, 100 IU penicillin, and 100 μg/ml streptomycin. Transfections were performed in 60-mm dishes using DOSPER liposomal transfection reagent (Roche Molecular Biochemicals) according to the manufacturer's instructions.
GST pulldown assay
The GST-Axin and GST-p53 were expressed in BL21 bacterial cells transformed separately with pGEX bacterial expression plasmids that contain full-length Axin and p53, respectively, and were purified using glutathione-agarose beads (Sigma). Expression was induced with 1 mM IPTG for 4 h at 37°C. Approximately 4 μg of GST fusion protein bound to agarose beads was added to each total lysate from 293T cells, and incubated for 3 h with gentle rotation. The beads were washed three times with cell lysis buffer, and the proteins were eluted with 2 × SDS sample buffer.
Co-immunoprecipitation and Western blotting
Cell lysates were prepared as previously described (Zhang et al, 1999). Antibodies used include anti-HA (F-7), anti-Myc (9E10), anti-HIPK2 (C-15), DO-1 antibodies (Santa Cruz Biotechnology Inc.), and our newly raised anti-Axin antibody. For the analysis of p53-phospho-Ser 46, Myc-p53 was cotransfected into H1299 cells separately with different Axin constructs. After immunoprecipitation with the anti-Myc antibody, Western blot analysis was performed with the anti-phospho-Ser 46 antibody (Cell Signaling Tech.).
Two-step co-immunoprecipitation
Two-step co-immunoprecipitation was performed essentially according to the procedures described by Harada et al (2003). Briefly, six 60-mm dishes of 293T cells were transfected with HA-HIPK2 (3 μg) and Myc-Axin (1 μg). For the control of the first immunoprecipitation, the HIPK2 lacking the HA-tag was used. At 36 h after transfection, the cells were lysed with LSLD buffer, sonicated briefly, and centrifuged. The supernatant was incubated with anti-HA antibody bound to Protein A/G-agarose beads for 2 h at 4°C. The beads were washed with lysis buffer (50 mM HEPES, pH 7.5, 0.2 mM EDTA, 10 μM NaF, 0.5% Nonidet P-40, and protease inhibitor cocktail tablets (Roche Applied Science)) containing 150 mM NaCl three times, and the HA-HIPK2 protein complex was eluted with 300 μl of lysis buffer containing 250 mM NaCl and (HA-tag)3 peptide (250 μg/ml) for 2 h at 4°C. The second immunoprecipitation was performed using 150 μl of eluate from the first immunoprecipitation and 350 μl of lysis buffer containing 464 mM NaCl and 10 μl of purified anti-Axin antibody or control IgG followed by addition of Protein A/G-agarose beads.
Apoptosis assay
HEK 293 cells were plated on glass coverslips in 35-mm culture dishes. Cells were transiently transfected with 0.5 μg of green fluorescent protein (GFP)-expressing vector pEGFPN1 (Clontech), together with 2 μg of empty vector pCMV5, HA-Axin, Axin-M3, Axin-M4, and Axin-M9 using DOSPER according to the manufacturer's instructions. At 36 h post-transfection, cells were rinsed with PBS and then fixed in 4% paraformaldehyde/PBS at room temperature for 30 min. After rinsing with PBS three times, cells were stained with the DNA-binding dye Hoechst 33342 (1 μg/ml) for 15 min at room temperature. Cells were washed in PBS three times and the coverslips were mounted with GelMount (Biomeda Corp., Foster City, CA) and kept at 4°C. Slides were examined under a fluorescence microscope for GFP fluorescence and Hoechst 33342 staining at wavelengths of 488/507 and 360/460 nm, respectively. Cells were counted in five different fields. The percentage of apoptotic cells that were positive was calculated as the ratio of cells positive for both GFP fluorescence and chromatin condensation and cells positive for total GFP.
p53-luciferase reporter gene assay
HEK 293, H1299, and SaOS-2 cells were transfected with 0.5 μg of p53-luciferase reporter (Stratagene) or Mdm2 reporter and 1.5–2 μg of an empty vector or each of the Axin, HIPK2, and p53 constructs indicated using DOSPER liposomal transfection reagent. At 32 h after transfection, cells were lysed and measured for luciferase and β-galactosidase activities (Promega). The ratio of luciferase activity to β-galactosidase activity varied less than 10% among the samples. Data are presented as means plus standard deviation from three separate experiments performed in triplicate.
Colony formation assay
The HEK 293 or U2OS cells were evenly split into 60-mm dishes. After growing to ∼70% confluence, cells were separately transfected with empty pCEP4 vector, pCEP4-Axin, or pCEP4-Axin-M9. After 48 h, the medium was changed to fresh medium containing 200 ng/ml hygromycin B (Invitrogen). After 14–21 days of treatment with hygromycin B, the cells were rinsed with PBS three times and then fixed with 4% paraformaldehyde/PBS, and stained with 1% crystal violet. In addition, we also performed the colony formation assay in H1299 cells and SaOS-2 cells transfected with the same set of Axin sequences constructed in the pcDNA3.1 vector, followed by selection with fresh medium containing 1 mg/ml G418.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Materials and Methods
Acknowledgments
This work was supported by grants from the Hong Kong Research Grants Council (HKUST6141/03M, HKUST6122/04M) and from the HIA Project of HKUST. SCL is a National Young Investigator (NSF30125012). The work was also supported by grants from the Fujian Commission of Science and Technology (2002F002) and the National Science Foundation of China (NSF30370306). We thank Drs V Yu and N Fu (IMCB, Singapore) for providing U2OS cells and plasmids for p53, luciferase reporters containing p21 and Mdm2 promoters, and R Poon for H1299 cells. We also thank our colleagues Y He, M Chen, C Xie, CK Wong, and SC Chan for technical assistance, and Dr Nancy Ip for comments.
References
- Attardi LD, DePinho RA (2004) Conquering the complexity of p53. Nat Genet 36: 7–8 [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W (2003) Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 15: 164–171 [DOI] [PubMed] [Google Scholar]
- Cadigan KM, Nusse R (1997) Wnt signaling: a common theme in animal development. Genes Dev 11: 3286–3305 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 96: 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S (2003) Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 113: 301–314 [DOI] [PubMed] [Google Scholar]
- D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 4: 11–19 [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW (1999) Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 18: 7002–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1: 45–49 [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Gottlieb TM, Oren M (1996) p53 in growth control and neoplasia. Biochim Biophys Acta 1287: 77–102 [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54: 4855–4878 [PubMed] [Google Scholar]
- Harada J, Kokura K, Kanei-Ishii C, Nomura T, Khan MM, Kim Y, Ishii S (2003) Requirement of the co-repressor homeodomain-interacting protein kinase 2 for ski-mediated inhibition of bone morphogenetic protein-induced transcriptional activation. J Biol Chem 278: 38998–39005 [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML (2002) Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 4: 1–10 [DOI] [PubMed] [Google Scholar]
- Hsu W, Shakya R, Costantini F (2001) Impaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic mice. J Cell Biol 155: 1055–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A (1999) Roles of Axin in the Wnt signalling pathway. Cell Signal 11: 777–788 [DOI] [PubMed] [Google Scholar]
- Ko LJ, Prives C (1996) p53: puzzle and paradigm. Genes Dev 10: 1054–1072 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108: 837–847 [DOI] [PubMed] [Google Scholar]
- Luo W, Ng WW, Jin LH, Ye Z, Han J, Lin SC (2003) Axin utilizes distinct regions for competitive MEKK1 and MEKK4 binding and JNK activation. J Biol Chem 278: 37451–37458 [DOI] [PubMed] [Google Scholar]
- Michael D, Oren M (2003) The p53–Mdm2 module and the ubiquitin system. Semin Cancer Biol 13: 49–58 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Peifer M, Polakis P (2000) Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science 287: 1606–1609 [DOI] [PubMed] [Google Scholar]
- Prives C, Manley JL (2001) Why is p53 acetylated? Cell 107: 815–818 [DOI] [PubMed] [Google Scholar]
- Rui HL, Fan E, Zhou HM, Xu Z, Zhang Y, Lin SC (2002) SUMO-1 modification of the C-terminal KVEKVD of Axin is required for JNK activation but has no effect on Wnt signaling. J Biol Chem 277: 42981–42986 [DOI] [PubMed] [Google Scholar]
- Sadot E, Geiger B, Oren M, Ben-Ze'ev A (2001) Down-regulation of beta-catenin by activated p53. Mol Cell Biol 21: 6768–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y (2000) AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 24: 245–250 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA (2002) p53: good cop/bad cop. Cell 110: 9–12 [DOI] [PubMed] [Google Scholar]
- Takebayashi-Suzuki K, Funami J, Tokumori D, Saito A, Watabe T, Miyazono K, Kanda A, Suzuki A (2003) Interplay between the tumor suppressor p53 and TGF-beta signaling shapes embryonic body axes in Xenopus. Development 130: 3929–3939 [DOI] [PubMed] [Google Scholar]
- Tokino T, Thiagalingam S, el-Deiry WS, Waldman T, Kinzler KW, Vogelstein B (1994) p53 tagged sites from human genomic DNA. Hum Mol Genet 3: 1537–1542 [DOI] [PubMed] [Google Scholar]
- Vargas DA, Takahashi S, Ronai Z (2003) Mdm2: a regulator of cell growth and death. Adv Cancer Res 89: 1–34 [DOI] [PubMed] [Google Scholar]
- Vasicek TJ, Zeng L, Guan XJ, Zhang T, Costantini F, Tilghman SM (1997) Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 147: 777–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y (2003) Regulation of p53 responses by post-translational modifications. Cell Death Differ 10: 400–403 [DOI] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL III, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F (1997) The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90: 181–192 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH (2003) Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 115: 177–186 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Neo SY, Wang X, Han J, Lin SC (1999) Axin forms a complex with MEKK1 and activates c-Jun NH(2)-terminal kinase/stress-activated protein kinase through domains distinct from Wnt signaling. J Biol Chem 274: 35247–35254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Materials and Methods