

Abstract
Mitochondrial disorders (MIDs) due to respiratory-chain defects or nonrespiratory chain defects are usually multisystem conditions [mitochondrial multiorgan disorder syndrome (MIMODS)] affecting the central nervous system (CNS), peripheral nervous system, eyes, ears, endocrine organs, heart, kidneys, bone marrow, lungs, arteries, and also the intestinal tract. Frequent gastrointestinal (GI) manifestations of MIDs include poor appetite, gastroesophageal sphincter dysfunction, constipation, dysphagia, vomiting, gastroparesis, GI pseudo-obstruction, diarrhea, or pancreatitis and hepatopathy. Rare GI manifestations of MIDs include dry mouth, paradontosis, tracheoesophageal fistula, stenosis of the duodeno-jejunal junction, atresia or imperforate anus, liver cysts, pancreas lipomatosis, pancreatic cysts, congenital stenosis or obstruction of the GI tract, recurrent bowel perforations with intra-abdominal abscesses, postprandial abdominal pain, diverticulosis, or pneumatosis coli. Diagnosing GI involvement in MIDs is not at variance from diagnosing GI disorders due to other causes. Treatment of mitochondrial GI disease includes noninvasive or invasive measures. Therapy is usually symptomatic. Only for myo-neuro-gastro-intestinal encephalopathy is a causal therapy with autologous stem-cell transplantation available. It is concluded that GI manifestations of MIDs are more widespread than so far anticipated and that they must be recognized as early as possible to initiate appropriate diagnostic work-up and avoid any mitochondrion-toxic treatment.
Keywords: diarrhea, gastrointestinal, mitochondrial DNA, oxidative, respiratory chain, stress, vomiting
Introduction
Syndromic and nonsyndromic mitochondrial disorders (MIDs) are in the majority of cases multisystem conditions [mitochondrial multiorgan disorder syndrome (MIMODS)], manifesting in each organ of the body but with markedly different prevalence and high variability [DiMauro and Hirano, 2009]. The organs most frequently affected are the muscle, the central nervous system (CNS), the endocrine glands, the eyes, the ears, and the heart [Finsterer and Bastovansky, 2015]. More rarely affected are the kidneys, gastrointestinal (GI) tract, bone marrow, arteries, bones, lungs, and the skin [Finsterer and Wakil, 2015]. GI compromise may be due to an intestinal or extra-intestinal cause. Among syndromic MIDs, mitochondrial neurogastrointestinal encephalopathy (MNGIE) is the MID most well-known for its GI involvement [Hirano et al. 1994]. MIDs may also mimic cycling vomiting syndrome where mitochondrial dysfunction may play a pathogenetic role [Gelfand and Gallagher, 2016]. It is estimated that more than 50% of the MIDs present with some form of GI compromise, but GI manifestations of MIDs are often not well appreciated. However, recognition of GI manifestation may facilitate the diagnosis of MIDs. This review aimed at summarizing and discussing current knowledge and recent findings concerning the clinical presentation, diagnosis, and treatment of GI involvement in MIDs and attempted to reinforce to the gastroenterologist that GI manifestations of MIDs should be managed symptomatically.
Methods
Data for this review were identified by searches of MEDLINE, Current Contents, EMBASE, Web of Science, Web of Knowledge, LILACS, SCOPUS, and Google Scholar for references of relevant articles using the search terms ‘appetite’, ‘sicca syndrome’, ‘dry mouth’, ‘dysphagia’, ‘vomiting’, ‘reflux’, ‘pancreatitis’, ‘pancreatic cyst’, ‘liver cysts’, ‘hepatopathy’, ‘steatosis hepatis’, ‘intestinal pseudo-obstruction’, ‘megacolon’, ‘pancreas lipomatosis’, ‘diarrhea’, ‘constipation’, in combination with ‘mtDNA’, ‘respiratory chain’, ‘MID’, ‘MNGIE’, ‘cycling vomiting’, and ‘neuromuscular’. Randomized (blinded or open label) clinical trials, longitudinal studies, case series, and case reports were considered. Abstracts and reports from meetings were not included. Only articles in English, French, Spanish, or German and published between 1966 and 2015 were included. Appropriate papers were studied and discussed for their usefulness to be incorporated in this review.
Figure 1.
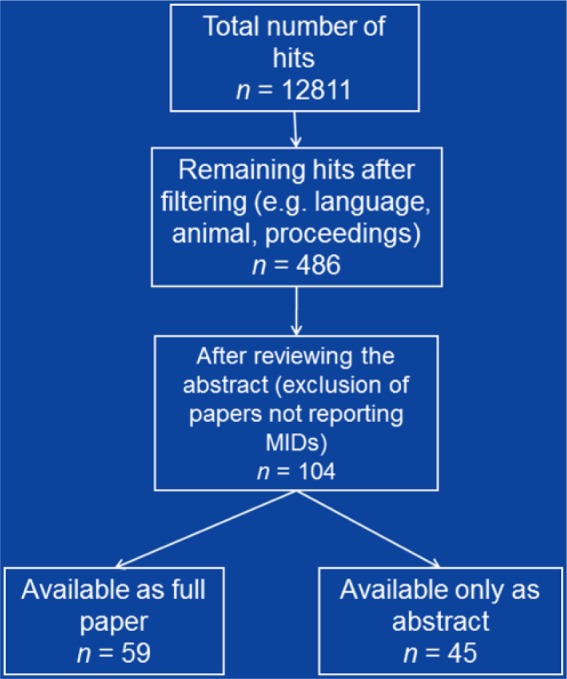
Overview about about the selection process of the included literature.
Results
The number of hits achieved by combining the applied search terms is given in Table 1. The main GI manifestations of MIDs include abnormalities of the GI hollow organs, such as poor appetite, gastroesophageal reflux and achalasia, constipation, dysphagia, vomiting, gastroparesis, diarrhea, and pseudo-obstruction, abnormalities of GI parenchymatous organs, such as aseptic pancreatitis, pancreatic cysts, pancreas lipomatosis, hepatopathy, steatosis hepatis, and liver cysts. The abnormalities are described in more detail in the following sections.
Table 1.
Number of hits achieved by combining the applied search terms in PubMed.
| mtDNA | RC | MID | MNGIE | CV | Neuromuscular | |
|---|---|---|---|---|---|---|
| Appetite | 13 | 26 | 31 | 0 | 0 | 24 |
| Sicca syndrome | 5 | 10 | 37 | 0 | 0 | 33 |
| Dry mouth | 4 | 7 | 20 | 0 | 0 | 75 |
| Dysphagia | 52 | 40 | 87 | 3 | 0 | 837 |
| Vomiting | 90 | 109 | 181 | 12 | 19 | 302 |
| Reflux | 6 | 18 | 14 | 1 | 0 | 242 |
| Pancreatitis | 53 | 66 | 83 | 1 | 0 | 45 |
| Pancreatic cyst | 1 | 4 | 2 | 0 | 0 | 3 |
| Liver cysts | 33 | 20 | 35 | 0 | 0 | 3 |
| Hepatopathy | 1519 | 1423 | 3278 | 10 | 0 | 486 |
| Steatosis | 505 | 833 | 811 | 2 | 0 | 42 |
| Pseudo-obstruction | 62 | 18 | 118 | 59 | 0 | 76 |
| Megacolon | 6 | 5 | 11 | 2 | 0 | 51 |
| Pancreas lipomatosis | 0 | 0 | 1 | 0 | 0 | 0 |
| Diarrhea | 55 | 323 | 100 | 13 | 3 | 117 |
| Constipation | 10 | 2 | 20 | 0 | 1 | 192 |
| Total | 2424 | 2904 | 4829 | 103 | 23 | 2528 |
CV, cycling vomiting; MID, mitochondrial disorder; MNGIE, mitochondrial neurogastrointestinal encephalopathy; mtDNA, mitochondrial DNA; RC, respiratory chain.
Abnormalities of gastrointestinal hollow organs
Poor appetite
Poor appetite is a frequent GI manifestation of MIDs, which may result in weight loss, anorexia, or cachexia. Reduced appetite in children may result in trophic disturbance with poor weight gain [Chapman et al. 2014]. Poor appetite may have a CNS cause, may be due to mood disturbance, or may be due to GI compromise. CNS causes may be leukoencephalopathy, dementia, or cognitive impairment. If CNS abnormalities cause poor appetite this may lead to dysphagia. GI causes of poor appetite include dysmotility, pseudo-obstruction, hepatopathy, diarrhea, vomiting, pancreatitis, postprandial abdominal pain, or constipation. Syndromic MIDs and nonsyndromic MIDs may be associated with poor appetite. Among the syndromic MIDs poor appetite has been described in Kearns–Sayre syndrome (KSS) [Ho et al. 2014] and MNGIE [Benureau et al. 2014] but also in other syndromes. There are also reports about nonsyndromic MIDs in which an eating disorder has been observed [Deutsch et al. 2007]. Loss of appetite was, for example, described in a 7-year-old girl carrying the sporadic mitochondrial DNA (mtDNA) mutation m.14739G>A in the tRNA(Glu) gene [Mayr et al. 2006]. If an MID manifests as autism in children, poor feeding is a frequent complaint. In patients with MNGIE poor appetite may result from bloating or abdominal pain [Chapman et al. 2014]. In mice it has been shown that polg mutations are associated with reduced appetite thereby reducing the diabetic phenotype in these animals [Fox et al. 2011].
Gastroesophageal sphincter dysfunction
Gastroesophageal sphincter dysfunction may manifest as either gastroesophageal reflux or as achalasia. Gastroesophageal reflux is common in MIDs and is pathophysiologically the opposite of achalasia. Gastroesophageal reflux has been reported in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) [van Biervliet et al. 2009], pontocerebellar hypoplasia (PCH) [Sánchez-Albisua et al. 2014], MNGIE [Hirano, 2016], and in nonsyndromic MIDs [Bhardwaj et al. 2012] (Table 2). Achalasia can occur at various locations along the GI tract, for example at the cricopharyngeal sphincter, the gastroesophageal sphincter, or the rectum. The cause of achalasia in MID patients remains unknown, but it can be speculated that affection of the smooth muscle cells by the metabolic defect or affection of the autonomic innervation with reduced parasympathetic tone may lead to sympathetic overinnervation or permanent contraction of smooth muscle cells. Possibly, achalasia results from a failure of smooth muscle fibers to relax which may cause a sphincter to remain closed and fail to open when needed. Achalasia is an exceedingly rare GI manifestation of pediatric MIDs but has been occasionally reported in adults [Chelimsky et al. 2005; Kornblum et al. 2001]. In a 6-year-old female and a 6-month-old male with multiple mtDNA deletions, achalasia was the dominant feature and occurred at 3 and 7 years of age respectively [Chelimsky et al. 2005]. Gastroesophageal reflux was found only in the male starting at age 6 months [Chelimsky et al. 2005]. Cricopharyngeal achalasia has been reported in KSS patients and may lead to dysphagia [Kornblum et al. 2001]. Cricopharyngeal achalasia has been also reported in mtDNA deletion syndromes, such as KSS or chronic progressive external ophthalmoplegia (CPEO) [Kornblum et al. 2001].
Table 2.
GI manifestations in syndromic and nonsyndromic MIDs.
| MID | PAP | DYP | VOM | GED | GPDE | PC | HP | IPO | COS | DAR |
|---|---|---|---|---|---|---|---|---|---|---|
| MELAS | − | − | + | + | − | + | + | + | + | − |
| MERRF | − | + | − | − | − | + | + | + | − | − |
| CPEO | − | + | − | − | − | + | + | − | − | − |
| KSS | − | + | + | + | − | + | − | − | − | − |
| PS | − | + | + | − | − | + | + | − | − | − |
| LHON | − | − | − | − | − | − | − | − | − | − |
| NARP | − | − | − | − | − | − | − | − | − | − |
| LS | − | + | + | − | − | − | + | − | − | + |
| MIDD | − | − | − | − | − | + | − | + | + | + |
| MLASA | − | − | − | − | − | − | − | − | − | − |
| XLASA | − | − | − | − | − | − | − | − | − | − |
| PCH | − | + | − | + | − | − | + | − | − | − |
| ADOA | − | − | − | − | − | − | − | + | + | − |
| ARCO | − | − | − | − | − | − | − | − | − | − |
| ARSAL | − | − | − | − | − | − | − | − | − | − |
| WS | − | + | − | − | − | − | − | − | − | − |
| MTS/DDS | − | + | − | − | − | − | − | − | − | − |
| DYTCA | − | − | − | − | − | − | − | − | − | − |
| LBSL | − | − | − | − | − | − | − | − | − | − |
| MEGDEL | − | − | − | − | − | − | + | − | − | − |
| SANDO (ANS) | − | + | − | − | + | − | − | − | − | − |
| MIRAS (ANS) | − | − | − | − | − | − | + | − | − | − |
| MEMSA (ANS) | − | − | − | − | − | − | − | − | − | − |
| SCAE (ANS) | − | − | − | − | − | − | − | − | − | − |
| MNGIE (MDS) | + | + | + | + | + | − | + | + | + | + |
| IOSCA (MDS) | − | − | − | − | − | − | + | − | − | − |
| AHD (MDS) | − | − | + | − | − | + | + | + | + | − |
| HC-MDS | − | − | − | − | − | − | + | − | − | − |
| EM-MDS | − | − | − | − | − | − | + | + | − | + |
| MP-MDS | − | − | − | − | − | − | + | − | − | − |
| MCHS (MDS) | − | − | + | − | − | − | + | − | − | − |
| Ns MID | + | + | + | + | + | + | + | + | + | + |
ADOA, autosomal dominant optic atrophy; AHD, Alpers–Huttenlocher disease; ANS, ataxia neuropathy spectrum; ARCO, autosomal recessive cardiomyopathy and ophthalmoplegia; ARSAL, autosomal recessive spastic ataxia with leukoencephalopathy; COS, constipation; CPEO, chronic progressive external ophthalmoplegia; DAR, diarrhea; DYP, dysphagia; DYTCA, dystonia ataxia syndrome; EM-MDS, encephalomyopathic MDS; GED, gastro-esophageal sphincter dysfunction; GPDE, gastroparesis and delayed emptying; HC-MDS, hepato-cerebral mitochondrial depletion syndrome; HP, hepatopathy; IOSCA, infantile-onset spinocerebellar ataxia; IPO, intestinal pseudo-obstruction; KSS, Kearns–Sayre syndrome; LBSL, leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation; LHON, Leber’s hereditary optic neuropathy; LS, Leigh syndrome; MCHS, myo-cerebro-hepatopathy spectrum; MDS, mitochondrial depletion syndrome; MELAS, mitochondrial encephalopathy, lactacidosis, and stroke-like episodes; MEGDEL, 3-methylglutaconic aciduria type IV with sensorineural deafness, encephalopathy and Leigh-like syndrome; MEMSA, myoclonus, epilepsy, myopathy, and sensory ataxia; MERRF, myoclonic epilepsy with ragged red fibers; MIDD, maternally inherited diabetes and deafness; MIRAS, mitochondrial recessive ataxia syndrome; MNGIE, mitochondrial neurogastrointestinal encephalopathy; MLASA, myopathy-lactic acidosis-sideroblastic anemia; MP-MDS, myopathic MDS; MTS, Mohr-Tranebjaerg syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; Ns, nonsyndromic MIDs; PAP, poor appetite; PC, pancreatitis; PCH, ponto-cerebellar hypoplasia; PS, Pearson syndrome; SANDO, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; SCAE, spinocerebellar ataxia with epilepsy; VOM, vomiting; WS, Wolfram syndrome; XLASA, X-linked lactic acidosis and sideroblastic anemia.
Constipation
Constipation results from GI dysmotility and is one of the most frequent GI manifestations of MIDs. In single cases it may be of such an intensity that colectomy may be necessary [Prayson and Wang, 1998]. Constipation has been particularly reported in MELAS [Prayson and Wang, 1998], maternally inherited diabetes and deafness (MIDD) [Bergamin et al. 2008], autosomal dominant optic atrophy (ADOA) [Bonneau et al. 2014], Alpers–Huttenlocher disease (AHD) [Crimmins et al. 1993], and nonsyndromic MIDs [Spiegler et al. 2011] (Table 2). GI dysmotility with markedly delayed emptying and megacolon requiring surgery has been also reported in MNGIE [Chapman et al. 2014; Bhardwaj et al. 2012]. Dietary measures are often ineffective and patients require regular administration of laxatives.
Dysphagia
Dysphagia may be due to CNS involvement, involvement of the peripheral nerves, or due to involvement of the smooth muscle cells in the underlying MID. Dysphagia in MIDs is common and may be progressive to such a degree that swallowing becomes impossible [DiMauro and Hirano, 2011]. If dysphagia is associated with tongue atrophy, amyotrophic lateral sclerosis (ALS) must be considered as a differential [Read et al. 2012; Finsterer and Stöllberger, 2014]. Dysphagia may occur in syndromic MIDs as well as nonsyndromic MIDs. Among the syndromic MIDs it has been reported in myoclonic epilepsy with ragged-red fibers (MERRF) syndrome with brain stem stroke-like episodes [Poungvarin and Viriyavejakul, 1991], CPEO [Zaganas et al. 2009; Reyes et al. 2015; Pfeffer et al. 2014], KSS [Gonzalez-Moron et al. 2013; Kapeller et al. 1996], Pearson syndrome (PS) [Lacbawan et al. 2000], Leigh syndrome (LS) [Ma et al. 2011], PCH [Szabó et al. 2008], Wolfram syndrome (WS) [Saiz et al. 1995], Mohr–Tranebjaerg syndrome (MTS) [Merchant et al. 2001], sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) [Tanaka et al. 2013; Gáti et al. 2011], or MNGIE [Hussein, 2013] (Table 2). In a family carrying a mutation in the RNASEH1 gene, CPEO was accompanied by dysphagia, general weakness, and spinocerebellar ataxia [Zaganas et al. 2009].
The RNASEH1 gene encodes the RNAase H1, an endonuclease which acts in the nucleus and in mitochondria where it digests the RNA component of RNA/DNA hybrids. Patients carrying SPG7 mutations may not only present with CPEO but also with general myopathy, spasticity, ataxia, and dysphagia [Reyes et al. 2015]. Dysphagia may be also part of the phenotype in patients carrying PEO1 mutations, which may additionally manifest with CPEO, general myopathy, and respiratory insufficiency [Pfeffer et al. 2014]. In nonsyndromic MIDs, dysphagia was reported in an Italian female with multisystem disease due to a deletion in the cytb gene [Carossa et al. 2014]. In patients carrying POLG1 mutations, dysphagia may be accompanied by CPEO, seizures, neuropathy, ataxia, hepatopathy, or migraine [Woodbridge et al. 2013]. In a 6-year-old boy with a single mtDNA deletion manifesting as stroke-like episodes and Leigh-like neuropathology, dysphagia was one among numerous other phenotypic features [Bene et al. 2003].
Vomiting
Cyclic or episodic vomiting is a frequent GI manifestation of syndromic and nonsyndromic MIDs. Most well-known is MELAS syndrome in which vomiting can be one of the cardinal phenotypic manifestations or may be the presenting manifestation. Recurrent vomiting may be so strong that it is occasionally confused with bulimia [Chapman et al. 2014]. Compared with cyclic vomiting syndrome, a putative MID, vomiting in MELAS, is less prominent. Migraine-like headache, a frequent CNS manifestation in various MIDs [Wang et al. 2004], is often associated with profuse and recurrent vomiting, intractable to standard therapy during the attacks. Other syndromic MIDs in which vomiting has been reported include KSS [Ho et al. 2014], PS [Lacbawan et al. 2000], LS [Lombardo et al. 2014], MNGIE [Chapman et al. 2014; Benureau et al. 2014], myo-cerebro-hepatopathy syndrome (MCHS), a mitochondrial depletion syndrome (MDS) due to POLG1 mutations [Montassir et al. 2015; Prasad et al. 2013], and AHD [Narkewicz et al. 1991] (Table 2). In nonsyndromic MIDs vomiting, together with other GI manifestations, has been reported in 3 of 6 patients with a nonsyndromic MID [Chapman et al. 2014]. In a 16-year-old male with recurrent vomiting the Leigh-like phenotype was due to the primary Leber’s hereditary optic neuropathy (LHON) mutation m.3460G>A in the ND1 gene [La Morgia et al. 2014]. In patients with MNGIE, vomiting may occur only during the night [Chapman et al. 2014]. In case recurrent vomiting lasts for months or even years, chronic malnutrition may ensue.
Gastroparesis
Gastroparesis is rather infrequent among MIDs but has been occasionally reported [Hom and Lavine, 2004; Chitkara et al. 2003]. Gastroparesis may be due to affection of the smooth muscle cells by the metabolic defect or due to reduced parasympathetic innervation leading to sympathetic overstimulation. Gastroparesis results in gastric dysmotility with delayed gastric emptying and dilation of the stomach. Gastroparesis with delayed gastric emptying has been particularly reported in patients with nonsyndromic MIDs due to POLG1 mutations [Bostan et al. 2012]. These patients additionally present with sensory-ataxic neuropathy, CPEO, and dysarthria [Bostan et al. 2012]. Delayed gastric emptying was also reported in MID patients carrying the m.3243A>G mutation [Nohara et al. 2006]. Even children with nonsyndromic MIDs may present with delayed gastric emptying and prolonged intestinal transit time (ITT) [Bhardwaj et al. 2012]. Among the syndromic MIDs, a grossly dilated stomach has been most often reported in MNGIE patients. [Chapman et al. 2014] and gastroparesis in SANDO [Bostan et al. 2012]. Intractable gastroparesis may be also a phenotypic feature of pyruvate-dehydrogenase deficiency [Tatekawa and Komuro, 2010].
Intestinal pseudo-obstruction
Intestinal pseudo-obstruction is rare and characterized by recurrent episodes of bowel obstruction without a mechanical obstructive cause [Faber et al. 1987]. Intestinal pseudo-obstruction is actually ‘the most severe form’ of MIDs affecting GI motility. Intestinal pseudo-obstruction has to be differentiated from mechanical intestinal obstructive syndrome, which can be challenging, since both abnormalities present with similar clinical manifestations [Vîlcea and Vasile, 2004]. Intestinal pseudo-obstruction has been reported in syndromic and nonsyndromic MIDs but also in respiratory chain MIDs and nonrespiratory chain MIDs. Among the respiratory chain MIDs it has been found in MELAS [Primiano et al. 2014], MERRF [Sekino et al. 2012], MIDD [Bergamin et al. 2008], AHD [Bonneau et al. 2014], encephalo-myopathic MDS [Slama et al. 2005], and nonsyndromic MIDs [Amiot et al. 2009] (Table 2). In patients with MELAS syndrome administration of phenytoin may trigger the development of intestinal pseudo-obstruction [Chiyonobu et al. 2008].
Diarrhea
GI dysmotility resulting in diarrhea has a similar frequency as constipation. Diarrhea in MIDs may be due to intestinal overactivity or exocrine pancreas insufficiency. Diarrhea may be also due to small intestinal bacteria overgrowth (SIBO). Diarrhea has been particularly reported in LS [van Coster et al. 1991], MIDD [Narbonne et al. 2004], MNGIE [Oztas et al. 2010], encephalomyopathic MDS [Bornstein et al. 2008], and nonsyndromic MIDs [Bhardwaj et al. 2012; Uncini et al. 1994] (Table 2). In particular, patients with MNGIE may be easily satiated, nauseated, and may have an increased bowel frequency [Chapman et al. 2014]. Other patients with MNGIE may present with intermittent diarrhea [Chapman et al. 2014].
Rare manifestations of gastrointestinal hollow organs
Rare manifestations of GI hollow organs in MIDs include dry mouth (sicca syndrome, xerostomia) [personal communication], paradontosis [Finsterer, 2006], tracheoesophageal fistula [Skinner et al. 2014], stenosis of the duodeno-jejunal junction [Chapman et al. 2014], atresia or imperforate anus [Skinner et al. 2014], congenital stenosis or obstruction of the GI tract with dilation of the pre-stenotic sections [Chapman et al. 2014], recurrent bowel perforations with intra-abdominal abscesses [Dreznik et al. 2014], postprandial abdominal pain in MNGIE [Bhardwaj et al. 2012; Bonneau et al. 2014] or nonsyndromic MIDs [Chapman et al. 2014], diverticulosis [Perez-Atayde et al. 1998], or pneumatosis coli [Chapman et al. 2014]. Whether rare GI manifestations of MIDs are more prevalent among MIDs than in the general population is unknown.
Abnormalities of gastrointestinal parenchymatous organs
Pancreatitis
Pancreatitis with or without diabetes and with or without exocrine pancreas insufficiency has been occasionally described in respiratory chain and nonrespiratory chain MID patients. Pancreatitis is usually of the aseptic type with a good tendency to recover but a high risk to recur. In some patients, pancreatitis may manifest exclusively as amylase or lipase elevation without clinical manifestations or abnormalities on imaging. The cause of pancreatitis in MIDs is unknown, but it can be speculated that the metabolic defect in exocrine pancreas cells promotes a defect in exocrine enzyme secretion, which could result in intracellular protease accumulation and activation. It is also conceivable that the composition of the pancreatic secretions is nonphysiological thus leading to obstruction of the exocrine pathways and consecutively to pancreatitis. MIDs in which pancreatitis has been reported include MELAS [Ishiyama et al. 2013], MERRF [Toyono et al. 2001], CPEO [Ishiyama et al. 2013], KSS [Debray et al. 2006], PS [Crippa et al. 2015], MIDD [Schleiffer et al. 2000], AHD [Montine et al. 1995], and nonsyndromic MIDs [Verny et al. 2008] (Table 2). Interestingly, pancreatitis has not been reported in MNGIE.
Hepatopathy
Involvement of the liver is a frequent phenotypic manifestation of MIDs and occurs as elevation of liver transaminases [Hakonen et al. 2007], as steatosis hepatis, hepatomegaly [Montassir et al. 2015], fibrosis [Montassir et al. 2015], as liver cysts, hepatoma, biliary malignancy, or as acute liver failure [Bijarnia-Mahay et al. 2014]. Liver involvement may occur in up to 20% of MID patients [Cloots et al. 2013]. Liver involvement is more frequent in children compared with adults [Bianchi et al. 2011]. Steatosis or elevation of liver enzymes typically occurs in the absence of hyperlipidemia, intake of liver toxic drugs, a history of alcohol consumption, or antibodies against hepatitis viruses. Involvement of the liver may manifest clinically or may be subclinical [De Kremer et al. 2001]. In some patients, liver involvement becomes evident only after application of liver toxic medication [Krähenbühl et al. 2000]. It is also striking that many MID patients have a history of cholecystolithiasis or choledocholithiasis [personal communication]. Liver involvement occurs in syndromic and nonsyndromic MIDs. The most well-known among the syndromic MIDs are the MDSs, such as AHD or the hepato-cerebral MDS. Liver involvement has been also reported in respiratory chain MIDs such as MELAS [Ohno et al. 2010; Ishikawa et al. 1995; Ban et al. 1992], MERRF [Lombes et al. 1989], CPEO [Stewart et al. 2009], PS with progressive liver failure and steatosis [Shapira et al. 2014], LS [Leshinsky-Silver et al. 2003], PCH [Sans-Fitó et al. 2002], 3-methyl-glutaconic aciduria, deafness, encephalopathy, and Leigh-like (MEDGEL) syndrome [Sarig et al. 2013], mitochondrial recessive ataxia syndrome (MIRAS) [Hakonen et al. 2010], MNGIE (steatosis hepatis) [Finkenstedt et al. 2013], infantile-onset spinocerebellar ataxia (IOSCA) (elevated transaminases) [Hakonen et al. 2007], AHD [Montine et al. 1995; Simonati et al. 2003], hepato-cerebral MDS [Al-Hussaini et al. 2014], encephalomyopathic MDS [Carrozzo et al. 2016], myopathic MDS [Chanprasert et al. 2012], and in nonsyndromic MIDs [Bianchi et al. 2011; De Kremer et al. 2001; Wong et al. 2006; McDonald et al. 2002] (Table 2) but also in nonrespiratory chain MIDs such as hyper-ornithinemia-hyper-ammonemia-homo-citrullinuria (HHH) syndrome [Al-Hassnan et al. 2008]. Hepatopathy seems to be particularly prevalent among patients carrying POLG1 mutations [Scalais et al. 2012]. Autopsy studies, as well as muscle biopsy analyses, have shown that in patients with MIDs the activity of respiratory chain complexes can be reduced [Szymanska-Debinska et al. 2015].
Rare manifestations of gastrointestinal parenchymatous organs
Rare manifestations of GI parenchymatous organs in MIDs include liver cysts, pancreatic cysts [Debray et al. 2006], and pancreatic lipomatosis [personal communication], [Finsterer, 2006]. Whether these GI manifestations of MIDs are more prevalent among MIDs than in the general population is unknown.
Complications of abnormal gastrointestinal function
Complications of abnormal GI function in MIDs include chronic aspiration and consecutive pneumonia in cases of GI stenosis, dysmotility, achalasia, or obstruction, gastroesophageal reflux, malnutrition with polyneuropathy, or bacterial GI infections [Chapman et al. 2014]. In cases of recurrent vomiting, electrolyte disturbances, exsiccosis, hypokalemia, and hypochloremic alkalosis may ensue. Secondary GI infection may be another complication of GI dysmotility.
Treatment
Generally, treatment of GI manifestations in MIDs may be symptomatic or causative, noninvasive or invasive, surgical or nonsurgical. In the vast majority of cases, GI involvement in MIDs can be treated only by symptomatic measures. Only for MNGIE is a causal therapy with bone marrow transplantation available [Halter et al. 2015].
Noninvasive treatment
Mainstays of treating GI abnormalities in MIDs include treatment of constipation, enteral nutrition, including tube feeding, tricyclic antidepressants for chronic irritable bowel syndrome-type pain, and treatment of vomiting and cyclic vomiting. Dietary measures may play a supplementary role. Particularly in patients with hepatic involvement, pancreatitis, dysphagia, constipation, or diarrhea, an appropriate diet can support recovery. Appetite stimulation is also a key among those who swallow normally. In cases where the GI tract shuts down, metabolic stabilization can be achieved by intravenous glucose administration. In cases of dysphagia or low appetite due to mood disorders, application of antidepressants can be helpful. In case of noninfectious diarrhea, application of carbo medicinalis or loperamide may be indicated. More challenging is the treatment of cyclic vomiting syndrome during an episode, which may include lorazepam, ondansetron, or zolmitriptan in cases of abdominal cramping [Finsterer and Hayman, 2015]. In cases of constipation, domperidone (20 mg/day) or laxatives may be helpful. In cases of secondary infection of the GI tract, application of an appropriate antibiotic after identification of the causative agent by a stool culture may be effective. Patients with postprandial abdominal pain may benefit from treatment with antispasmodics or proton-pump inhibitors. If there is pain from reflux esophagitis, application of proton-pump inhibitors, H2-blockers, or sucralfate can be helpful. Children with abdominal pain may profit from amitriptyline, but the mitochondrion-toxic side effects of this compound [e.g. causes coenzyme-Q (CoQ) deficiency, reduces the mitochondrial membrane potential, causes oxidative stress, blocks citrate synthetase] have to be considered [Moreno-Fernández et al. 2012]. Patients with aseptic pancreatitis and exocrine pancreas insufficiency may benefit from the substitution of pancreas enzymes. This may also have a beneficial effect on diarrhea if this is due to exocrine pancreas insufficiency.
Whether the application of CoQ, idebenone, vitamins, co-factors, L-arginine, succinate, or L-carnitine, frequently given in MIDs, also have a beneficial effect on GI manifestations of MIDs is largely unknown, but it can be speculated that they may exert a positive effect also in this indication in some of the patients. The putative beneficial effect of these agents may be also attributable to a placebo effect. However, no randomized-controlled studies are available to solve these issues. One of the most important measures to treat GI manifestations of MIDs is the avoidance of mitochondrion-toxic drugs, such as chloramphenicol, aminoglycosides, linezolide, valproic acid, nucleoside reverse transcriptase inhibitors, dichloroacetate, carbamazepine, phenytoin, phenobarbital, or statins. [Gonzalez-Moron et al. 2013], which is not well known by many of the treating physicians. Valproic acid should not be given to patients with AHD or other MIDs with liver involvement, since it may cause acute liver failure particularly in patients carrying POLG1 mutations.
Invasive treatment
In cases of cachexia or malnourishment or a failure to thrive, implementation of a percutaneous gastrostomy (PEG) or temporary insertion of a nasogastric tube can be inevitable. In cases of congenital stenosis or obstruction the most adequate measure to solve the problem is surgery. In cases of diverticulosis or recurrent diverticulitis, surgical resection of the affected section may be indicated. In cases of severe constipation or dysmotility or megacolon colectomy may be the only option. Stem cell therapy of MNGIE is carried by infusion of bone-marrow derived hematopoietic stem cells as a source of thymidine phosphorylase [Halter et al. 2015]. In cases of severe constipation colectomy or colostomy may be beneficial.
Clinical implications and conclusion
GI manifestations of MIDs are manifold and need to be recognized in order to support the work-up for establishing the diagnosis. Typical MID manifestations are dysphagia, vomiting, GI sphincter dysfunction, reduced contractility or increased contractility of the GI tract, hepatopathy, pancreatitis, and congenital obstruction. GI manifestations of MIDs may be the dominant or nondominant phenotypic feature. GI problems may be the sole manifestation of a MID or one among others. Diagnostic work-up needs to be intensified if a MID is suspected to establish the correct diagnosis and to initiate appropriate symptomatic treatment in due time. For MNGIE a more causal therapy is available, which is more effective the earlier it is initiated. Early diagnosis of a MID is thus important and GI manifestations can contribute to raise the suspicion of a MID and, if part of a MIMODS, strongly support the diagnosis. Neurologists and gastroenterologists must be aware of the fact that GI symptoms and signs may be attributable to a MID particularly if clinical or subclinical manifestations are present also in other systems. Since avoidance of mitochondrion-toxic drugs is one of the key measures to treat MIDs, it is important to exclude a MID as the cause of GI symptoms and signs and not to worsen GI manifestations by application of mitochondrion-toxic drugs. Overall, it is important to note that GI manifestations may be useful in the diagnosis of MIDs (e.g. in patients with motility disorders), but also need to be anticipated so that complications can be managed.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Josef Finsterer, Krankenanstalt Rudolfstiftung, Postfach 20, 1180 Vienna, Austria.
Marlies Frank, First Medical Department, Krankenanstalt Rudolfstiftung, Vienna, Austria.
References
- Al-Hassnan Z., Rashed M., Al-Dirbashi O., Patay Z., Rahbeeni Z., Abu-Amero K. (2008) Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with stroke-like imaging presentation: clinical, biochemical and molecular analysis. J Neurol Sci 264: 187–194. [DOI] [PubMed] [Google Scholar]
- Al-Hussaini A., Faqeih E., El-Hattab A., Alfadhel M., Asery A., Alsaleem B., et al. (2014) Clinical and molecular characteristics of mitochondrial DNA depletion syndrome associated with neonatal cholestasis and liver failure. J Pediatr 164: 553–559. [DOI] [PubMed] [Google Scholar]
- Amiot A., Tchikviladzé M., Joly F., Slama A., Hatem D., Jardel C., et al. (2009) Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction. Gastroenterology 137: 101–109. [DOI] [PubMed] [Google Scholar]
- Ban S., Mori N., Saito K., Mizukami K., Suzuki T., Shiraishi H. (1992) An autopsy case of mitochondrial encephalomyopathy (MELAS) with special reference to extra-neuromuscular abnormalities. Acta Pathol Jpn 42: 818–825. [DOI] [PubMed] [Google Scholar]
- Bene J., Nádasi E., Kosztolányi G., Méhes K., Melegh B. (2003) Congenital cataract as the first symptom of a neuromuscular disease caused by a novel single large-scale mitochondrial DNA deletion. Eur J Hum Genet 11: 375–379. [DOI] [PubMed] [Google Scholar]
- Benureau A., Meyer P., Maillet O., Leboucq N., Legras S., Jeziorski E., et al. (2014) Mitochondrial neurogastrointestinal encephalopathy disease. Arch Pediatr 21: 1370–1374. [DOI] [PubMed] [Google Scholar]
- Bergamin C., Rolim L., Dib S., Moisés R. (2008) Unusual occurrence of intestinal pseudo obstruction in a patient with maternally inherited diabetes and deafness (MIDD) and favorable outcome with coenzyme Q10. Arq Bras Endocrinol Metabol 52: 1345–1349. [DOI] [PubMed] [Google Scholar]
- Bhardwaj J., Wan D., Koenig M., Liu Y., Hashmi S., Rhoads J. (2012) Impaired gastric emptying and small bowel transit in children with mitochondrial disorders. J Pediatr Gastroenterol Nutr 55: 194–199. [DOI] [PubMed] [Google Scholar]
- Bianchi M., Rizza T., Verrigni D., Martinelli D., Tozzi G., Torraco A., et al. (2011) Novel large-range mitochondrial DNA deletions and fatal multisystemic disorder with prominent hepatopathy. Biochem Biophys Res Commun 415: 300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijarnia-Mahay S., Mohan N., Goyal D., Verma I. (2014) Mitochondrial DNA depletion syndrome causing liver failure. Indian Pediatr 51: 666–668. [DOI] [PubMed] [Google Scholar]
- Bonneau D., Colin E., Oca F., Ferré M., Chevrollier A., Guéguen N., et al. (2014) Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 137: e301. doi: 10.1093/brain/awu187 [DOI] [PubMed] [Google Scholar]
- Bornstein B., Area E., Flanigan K., Ganesh J., Jayakar P., Swoboda K., et al. (2008) Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul Disord 18: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan A., Glibert G., Dachy B., Dan B. (2012) Novel mutation in spacer region of POLG associated with ataxia neuropathy spectrum and gastroparesis. Auton Neurosci 170: 70–72. [DOI] [PubMed] [Google Scholar]
- Carossa V., Ghelli A., Tropeano C., Valentino M., Iommarini L., Maresca A., et al. (2014) A novel in-frame 18-bp microdeletion in MT-CYB causes a multisystem disorder with prominent exercise intolerance. Hum Mutat 35: 954–958. [DOI] [PubMed] [Google Scholar]
- Carrozzo R., Verrigni D., Rasmussen M., de Coo R., Amartino H., Bianchi M., et al. (2016) Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients. J Inherit Metab Dis 39: 243–252. [DOI] [PubMed] [Google Scholar]
- Chanprasert S., Wong L., Wang J., Scaglia F. (2012) TK2-related mitochondrial DNA depletion syndrome, myopathic form. In: Pagon R., Adam M., Ardinger H., Wallace S., Amemiya A., Bean L., et al. (eds), GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2015. Available from http://www.ncbi.nlm.nih.gov/books/NBK114628/ (accessed 6 December 2012). [Google Scholar]
- Chapman T., Hadley G., Fratter C., Cullen S., Bax B., Bain M., et al. (2014) Unexplained gastrointestinal symptoms: think mitochondrial disease. Dig Liver Dis 46: 1–8. [DOI] [PubMed] [Google Scholar]
- Chelimsky G., Shanske S., Hirano M., Zinn A., Cohen M., McNeeley K., et al. (2005) Achalasia as the harbinger of a novel mitochondrial disorder in childhood. J Pediatr Gastroenterol Nutr 40: 512–517. [DOI] [PubMed] [Google Scholar]
- Chitkara D., Nurko S., Shoffner J., Buie T., Flores A. (2003) Abnormalities in gastrointestinal motility are associated with diseases of oxidative phosphorylation in children. Am J Gastroenterol 98: 871–877. [DOI] [PubMed] [Google Scholar]
- Chiyonobu T., Noda R., Yoshida M., Fujiki A., Ishii R., Nukina S., et al. (2008) Intestinal pseudo-obstruction in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) associated with phenytoin therapy. Brain Dev 30: 430–433. [DOI] [PubMed] [Google Scholar]
- Cloots K., Verbeek J., Orlent H., Meersseman W., Cassiman D. (2013) Mitochondrial hepatopathy in adults: a case series and review of the literature. Eur J Gastroenterol Hepatol 25: 892–898. [DOI] [PubMed] [Google Scholar]
- Crimmins D., Morris J., Walker G., Sue C., Byrne E., Stevens S., et al. (1993) Mitochondrial encephalomyopathy: variable clinical expression within a single kindred. J Neurol Neurosurg Psychiatry 56: 900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crippa B., Leon E., Calhoun A., Lowichik A., Pasquali M., Longo N. (2015) Biochemical abnormalities in Pearson syndrome. Am J Med Genet A 167A: 621–628. [DOI] [PubMed] [Google Scholar]
- Debray F., Drouin E., Herzog D., Lortie A., Lambert M., Garel L., et al. (2006) Recurrent pancreatitis in mitochondrial cytopathy. Am J Med Genet A 140: 2330–2335. [DOI] [PubMed] [Google Scholar]
- De Kremer R., Paschini-Capra A., Bacman S., Argaraña C., Civallero G., Kelley R., et al. (2001) Barth’s syndrome-like disorder: a new phenotype with a maternally inherited A3243G substitution of mitochondrial DNA (MELAS mutation). Am J Med Genet 99: 83–93. [DOI] [PubMed] [Google Scholar]
- Deutsch S., Rosse R., Yaseen M., Schulman H., Abraham S. (2007) Mitochondrial myopathy complicated by eating disorder: a case report highlighting the potential interaction of genetic, metabolic, and psychodynamic factors. CNS Spectr 12: 289–292. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Hirano M. (2009) Pathogenesis and treatment of mitochondrial disorders. Adv Exp Med Biol 652: 139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S., Hirano M. (2011) Mitochondrial DNA deletion syndromes. In: Pagon R., Adam M., Ardinger H., Wallace S., Amemiya A., Bean L., et al. (eds), GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2015. Available from http://www.ncbi.nlm.nih.gov/books/NBK1203/ (accessed 17 December 2003, updated 3 May 2011). [Google Scholar]
- Dreznik Y., Gutman M., Weiss B., Nevler A. (2014) Mitochondrial neuro-gastrointestinal encephalomyopathy presenting with recurrent bowel perforations and intra-abdominal abscesses. J Gastrointest Surg 18: 2054–2056. [DOI] [PubMed] [Google Scholar]
- Faber J., Fich A., Steinberg A., Steiner I., Granot E., Alon I., et al. (1987) Familial intestinal pseudoobstruction dominated by a progressive neurologic disease at a young age. Gastroenterology 92: 786–790. [DOI] [PubMed] [Google Scholar]
- Finkenstedt A., Schranz M., Bösch S., Karall D., Bürgi S., Ensinger C., et al. (2013) MNGIE syndrome: liver cirrhosis should be ruled out prior to bone marrow transplantation. JIMD Rep 10: 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J. (2006) Overview on visceral manifestations of mitochondrial disorders. Neth J Med 64: 61–71. [PubMed] [Google Scholar]
- Finsterer J., Bastovansky A. (2015) Multiorgan disorder syndrome (MODS) in an octogenarian suggests mitochondrial disorder. Rev Med Chil 143: 1210–1214. [DOI] [PubMed] [Google Scholar]
- Finsterer J., Hayman J. (2015) Cyclic vomiting syndrome in multisystem mitochondrial disorder. Tunis Med 93: 424–426. [PubMed] [Google Scholar]
- Finsterer J., Stöllberger C. (2014) Noncompaction in alleged motor neuron disease suggests myopathy. Int J Cardiol 177: 639–640. [DOI] [PubMed] [Google Scholar]
- Finsterer J., Wakil S. (2015) Abnormalities of skin and cutaneous appendages in neuromuscular disorders. Pediatr Neurol 53: 301–308. [DOI] [PubMed] [Google Scholar]
- Fox R., Kim H., Reddick R., Kujoth G., Prolla T., Tsutsumi S., et al. (2011) Mitochondrial DNA polymerase editing mutation, PolgD257A, reduces the diabetic phenotype of Akita male mice by suppressing appetite. Proc Natl Acad Sci U S A 108: 8779–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gáti I., Danielsson O., Jonasson J., Landtblom A. (2011) Sensory ataxic neuropathy with dysarthria/dysphagia and ophthalmoplegia (SANDO). Two case reports. Acta Myol 30: 188–190. [PMC free article] [PubMed] [Google Scholar]
- Gelfand A., Gallagher R. (2016) Cyclic vomiting syndrome versus inborn errors of metabolism: a review with clinical recommendations. Headache 56: 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Moron D., Bueri J., Kauffman M. (2013) Progressive external ophthalmoplegia (PEO) due to a mutation in the C10orf2 (PEO1) gene mimicking a myasthenic crisis. BMJ Case Rep pii: bcr2013010181. DOI: 10.1136/bcr-2013-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonen A., Isohanni P., Paetau A., Herva R., Suomalainen A., Lönnqvist T. (2007) Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 130: 3032–3040. [DOI] [PubMed] [Google Scholar]
- Hakonen A., Isohanni P., Rantamäki M., Kälviäinen R., Nordin A., Uusimaa J., et al. (2010) Mitochondrial recessive ataxia syndrome (MIRAS) and valproate toxicity. Duodecim 126: 1552–1559. [PubMed] [Google Scholar]
- Halter J., Michael W., Schüpbach M., Mandel H., Casali C., Orchard K., et al. (2015) Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 138: 2847–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M. (2016) Mitochondrial neurogastrointestinal encephalopathy disease. In: Pagon R., Adam M., Ardinger H., Wallace S., Amemiya A., Bean L., et al. (eds), GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2016. Available from http://www.ncbi.nlm.nih.gov/books/NBK1179/ (accessed 22 April 2005, updated 14 January 2016). [PubMed] [Google Scholar]
- Hirano M., Silvestri G., Blake D., Lombes A., Minetti C., Bonilla E., et al. (1994) Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 44: 721–727. [DOI] [PubMed] [Google Scholar]
- Ho J., Pacaud D., Rakic M., Khan A. (2014) Diabetes in pediatric patients with Kearns-Sayre syndrome: clinical presentation of 2 cases and a review of pathophysiology. Can J Diabetes 38: 225–228. [DOI] [PubMed] [Google Scholar]
- Hom X., Lavine J. (2004) Gastrointestinal complications of mitochondrial disease. Mitochondrion 4: 601–607. [DOI] [PubMed] [Google Scholar]
- Hussein E. (2013) Non-myeloablative bone marrow transplant and platelet infusion can transiently improve the clinical outcome of mitochondrial neurogastrointestinal encephalopathy: a case report. Transfus Apher Sci 49: 208–211. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y., Asuwa N., Ishii T., Masuda S., Kiguchi H., Hirai S., et al. (1995) Severe mitochondrial cardiomyopathy and extra-neuromuscular abnormalities in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode (MELAS). Pathol Res Pract 191: 64–69; [DOI] [PubMed] [Google Scholar]
- Ishiyama A., Komaki H., Saito T., Saito Y., Nakagawa E., Sugai K., et al. (2013) Unusual exocrine complication of pancreatitis in mitochondrial disease. Brain Dev 35: 654–659. [DOI] [PubMed] [Google Scholar]
- Kapeller P., Fazekas F., Offenbacher H., Stollberger R., Schmidt R., Berglöff J., et al. (1996) Magnetic resonance imaging and spectroscopy of progressive cerebral involvement in Kearns Sayre Syndrome. J Neurol Sci 135: 126–130. [DOI] [PubMed] [Google Scholar]
- Kornblum C., Broicher R., Walther E., Seibel P., Reichmann H., Klockgether T., et al. (2001) Cricopharyngeal achalasia is a common cause of dysphagia in patients with mtDNA deletions. Neurology 56: 1409–1412. [DOI] [PubMed] [Google Scholar]
- Krähenbühl S., Brandner S., Kleinle S., Liechti S., Straumann D. (2000) Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver 20: 346–348. [DOI] [PubMed] [Google Scholar]
- Lacbawan F., Tifft C., Luban N., Schmandt S., Guerrera M., Weinstein S., et al. (2000) Clinical heterogeneity in mitochondrial DNA deletion disorders: a diagnostic challenge of Pearson syndrome. Am J Med Genet 95: 266–268. [PubMed] [Google Scholar]
- La Morgia C., Caporali L., Gandini F., Olivieri A., Toni F., Nassetti S., et al. (2014) Association of the mtDNA m.4171C>A/MT-ND1 mutation with both optic neuropathy and bilateral brainstem lesions. BMC Neurol 14: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshinsky-Silver E., Levine A., Nissenkorn A., Barash V., Perach M., Buzhaker E., et al. (2003) Neonatal liver failure and Leigh syndrome possibly due to CoQ-responsive OXPHOS deficiency. Mol Genet Metab 79: 288–293. [DOI] [PubMed] [Google Scholar]
- Lombardo B., Ceglia C., Tarsitano M., Pierucci I., Salvatore F., Pastore L. (2014) Identification of a deletion in the NDUFS4 gene using array-comparative genomic hybridization in a patient with suspected mitochondrial respiratory disease. Gene 535: 376–379. [DOI] [PubMed] [Google Scholar]
- Lombes A., Mendell J., Nakase H., Barohn R., Bonilla E., Zeviani M., et al. (1989) Myoclonic epilepsy and ragged-red fibers with cytochrome oxidase deficiency: neuropathology, biochemistry, and molecular genetics. Ann Neurol 26: 20–33. [DOI] [PubMed] [Google Scholar]
- Ma Y., Wu T., Liu Y., Wang Q., Song J., Xiao J., et al. (2011) Leigh syndrome due to mitochondrial respiratory chain complex II deficiency. Zhongguo Dang Dai Er Ke Za Zhi 13: 569–572. [PubMed] [Google Scholar]
- Mayr J., Moslemi A., Förster H., Kamper A., Idriceanu C., Muss W., et al. (2006) A novel sporadic mutation G14739A of the mitochondrial tRNA(Glu) in a girl with exercise intolerance. Neuromuscul Disord 16: 874–877. [DOI] [PubMed] [Google Scholar]
- McDonald D., McMenamin J., Farrell M., Droogan O., Green A. (2002) Familial childhood onset neuropathy and cirrhosis with the 4977bp mitochondrial DNA deletion. Am J Med Genet 111: 191–194. [DOI] [PubMed] [Google Scholar]
- Merchant S., McKenna M., Nadol J., Jr, Kristiansen A., Tropitzsch A., Lindal S., et al. (2001) Temporal bone histopathologic and genetic studies in Mohr-Tranebjaerg syndrome (DFN-1). Otol Neurotol 22: 506-511. [DOI] [PubMed] [Google Scholar]
- Montassir H., Maegaki Y., Murayama K., Yamazaki T., Kohda M., Ohtake A., et al. (2015) Myocerebrohepatopathy spectrum disorder due to POLG mutations: a clinicopathological report. Brain Dev 37: 719–724. [DOI] [PubMed] [Google Scholar]
- Montine T., Powers J., Vogel F., Radtke R. (1995) Alpers’ syndrome presenting with seizures and multiple stroke-like episodes in a 17-year-old male. Clin Neuropathol 14: 322–326. [PubMed] [Google Scholar]
- Moreno-Fernández A., Cordero M., Garrido-Maraver J., Alcocer-Gómez E., Casas-Barquero N., Carmona-López M., et al. (2012) Oral treatment with amitriptyline induces coenzyme Q deficiency and oxidative stress in psychiatric patients. J Psychiatr Res 46: 341–345. [DOI] [PubMed] [Google Scholar]
- Narbonne H., Paquis-Fluckinger V., Valero R., Heyries L., Pellissier J., Vialettes B. (2004) Gastrointestinal tract symptoms in Maternally Inherited Diabetes and Deafness (MIDD). Diabetes Metab 30: 61–66. [DOI] [PubMed] [Google Scholar]
- Narkewicz M., Sokol R., Beckwith B., Sondheimer J., Silverman A. (1991) Liver involvement in Alpers disease. J Pediatr 119: 260–267. [DOI] [PubMed] [Google Scholar]
- Nohara S., Iwase M., Imoto H., Sasaki N., Nakamura U., Uchizono Y., et al. (2006) Gastric emptying in patients with Type 2 diabetes mellitus and diabetes associated with mitochondrial DNA 3243 mutation using 13C-octanoic acid breath test. J Diabetes Complications 20: 295–301. [DOI] [PubMed] [Google Scholar]
- Ohno A., Mori A., Doi R., Yonenaga Y., Asano N., Uemoto S. (2010) Successful left hemihepatectomy and perioperative management of a patient with biliary cystadenocarcinoma, complicated with MELAS syndrome: report of a case. Surg Today 40: 878–882. [DOI] [PubMed] [Google Scholar]
- Oztas E., Ozin Y., Onder F., Onal I., Oguz D., Kocaefe C. (2010) Chronic intestinal pseudo-obstruction and neurological manifestations in early adulthood: considering MNGIE syndrome in differential diagnosis. J Gastrointestin Liver Dis 19: 195–197. [PubMed] [Google Scholar]
- Perez-Atayde A., Fox V., Teitelbaum J., Anthony D., Fadic R., Kalsner L., et al. (1998) Mitochondrial neurogastrointestinal encephalomyopathy: diagnosis by rectal biopsy. Am J Surg Pathol 22: 1141–1147. [DOI] [PubMed] [Google Scholar]
- Pfeffer G., Gorman G., Griffin H., Kurzawa-Akanbi M., Blakely E., Wilson I., et al. (2014) Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 137: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poungvarin N., Viriyavejakul A. (1991) Motor neurone disease in Thailand: the clinical aspects of 77 patients. J Med Assoc Thai 74: 181–186. [PubMed] [Google Scholar]
- Prasad C., Melançon S., Rupar C., Prasad A., Nunez L., Rosenblatt D., et al. (2013) Exome sequencing reveals a homozygous mutation in TWINKLE as the cause of multisystemic failure including renal tubulopathy in three siblings. Mol Genet Metab 108: 190–194. [DOI] [PubMed] [Google Scholar]
- Prayson R., Wang N. (1998) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome: an autopsy report. Arch Pathol Lab Med 122: 978–981. [PubMed] [Google Scholar]
- Primiano G., Plantone D., Forte F., Sauchelli D., Scaldaferri F., Gasbarrini A., et al. (2014) Acute refractory intestinal pseudo-obstruction in MELAS: efficacy of prucalopride. Neurology 82: 1932–1934. [DOI] [PubMed] [Google Scholar]
- Read J., Whittaker R., Miller N., Clark S., Taylor R., McFarland R., et al. (2012) Prevalence and severity of voice and swallowing difficulties in mitochondrial disease. Int J Lang Commun Disord 47: 106–111. [DOI] [PubMed] [Google Scholar]
- Reyes A., Melchionda L., Nasca A., Carrara F., Lamantea E., Zanolini A., et al. (2015) RNASEH1 mutations impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am J Hum Genet 97: 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz A., Vila N., Muñoz J., Martí M., Graus F., Tolosa E., et al. (1995) Wolfram’s syndrome: correlation of clinical signs and neurological images. Neurologia 10: 107–109. [PubMed] [Google Scholar]
- Sánchez-Albisua I., Frölich S., Barth P., Steinlin M., Krägeloh-Mann I. (2014) Natural course of pontocerebellar hypoplasia type 2A. Orphanet J Rare Dis 9: 70 DOI: 10.1186/1750-1172-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans-Fitó A., Campistol-Plana J., Mas-Salguero M., Póo-Argüelles P., Fernández-Alvarez E. (2002) Pontocerebellar hypoplasia type 2 and Reye-like syndrome. J Child Neurol 17: 132–134. [DOI] [PubMed] [Google Scholar]
- Sarig O., Goldsher D., Nousbeck J., Fuchs-Telem D., Cohen-Katsenelson K., Iancu T., et al. (2013) Infantile mitochondrial hepatopathy is a cardinal feature of MEGDEL syndrome (3-methylglutaconic aciduria type IV with sensorineural deafness, encephalopathy and Leigh-like syndrome) caused by novel mutations in SERAC1. Am J Med Genet A 161A: 2204–2215. [DOI] [PubMed] [Google Scholar]
- Scalais E., Francois B., Schlesser P., Stevens R., Nuttin C., Martin J., et al. (2012) Polymerase gamma deficiency (POLG): clinical course in a child with a two stage evolution from infantile myocerebrohepatopathy spectrum to an Alpers syndrome and neuropathological findings of Leigh’s encephalopathy. Eur J Paediatr Neurol 16: 542–548. [DOI] [PubMed] [Google Scholar]
- Schleiffer T., ‘t Hart L., Schürfeld C., Kraatz K., Riemann J. (2000) Maternally inherited diabetes and deafness (MIDD): unusual occult exocrine pancreatic manifestation in an affected German family. Exp Clin Endocrinol Diabetes 108: 81–85. [DOI] [PubMed] [Google Scholar]
- Sekino Y., Inamori M., Yamada E., Ohkubo H., Sakai E., Higurashi T., et al. (2012) Characteristics of intestinal pseudo-obstruction in patients with mitochondrial diseases. World J Gastroenterol 18: 4557–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira A., Konopnicki M., Hammad-Saied M., Shabad E. (2014) Pearson disease in an infant presenting with severe hypoplastic anemia, normal pancreatic function, and progressive liver failure. J Pediatr Hematol Oncol 36: 402–403. [DOI] [PubMed] [Google Scholar]
- Simonati A., Filosto M., Savio C., Tomelleri G., Tonin P., Dalla Bernardina B., et al. (2003) Features of cell death in brain and liver, the target tissues of progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher disease). Acta Neuropathol 106: 57–65. [DOI] [PubMed] [Google Scholar]
- Skinner S., Doonanco K., Boles R., Chan A. (2014) Homozygous TRAP1 sequence variant in a child with Leigh syndrome and normal kidneys. Kidney Int 86: 860. [DOI] [PubMed] [Google Scholar]
- Slama A., Lacroix C., Plante-Bordeneuve V., Pillant H., Joly P., Haut S., et al. (2005) Thymidine phosphorylase gene mutations in patients with mitochondrial neurogastrointestinal encephalomyopathy syndrome. Mol Genet Metab 84: 326–331. [DOI] [PubMed] [Google Scholar]
- Spiegler J., Stefanova I., Hellenbroich Y., Sperner J. (2011) Bowel obstruction in patients with Alpers-Huttenlocher syndrome. Neuropediatrics 42: 194–196. [DOI] [PubMed] [Google Scholar]
- Stewart J., Tennant S., Powell H., Pyle A., Blakely E., He L., et al. (2009) Novel POLG1 mutations associated with neuromuscular and liver phenotypes in adults and children. J Med Genet 46: 209–214. [DOI] [PubMed] [Google Scholar]
- Szabó N., Szabó H., Hortobágyi T., Túri S., Sztriha L. (2008) Pontocerebellar hypoplasia type 1. Pediatr Neurol 39: 286–288. [DOI] [PubMed] [Google Scholar]
- Szymanska-Debinska T., Karkucinska-Wieckowska A., Piekutowska-Abramczuk D., Jurkiewicz E., Iwanicka-Pronicka K., Rokicki D., et al. (2015) Leigh disease due to SCO2 mutations revealed at extended autopsy. J Clin Pathol 68: 397–399. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Tateishi T., Kawamura N., Ohyagi Y., Urata M., Kira J., et al. (2013) A case of sensory ataxic neuropathy, dysarthria, and ophthalmoparesis with multiple mitochondrial DNA deletions. Rinsho Shinkeigaku 53: 205–211. [DOI] [PubMed] [Google Scholar]
- Tatekawa Y., Komuro H. (2010) A technical surgery for refractory gastroparesis in a patient with a mitochondrial disorder. Pediatr Surg Int 26: 65565–65568. [DOI] [PubMed] [Google Scholar]
- Toyono M., Nakano K., Kiuchi M., Imai K., Suzuki H., Shishikura K., et al. (2001) A case of MERRF associated with chronic pancreatitis. Neuromuscul Disord 11: 300–304. [DOI] [PubMed] [Google Scholar]
- Uncini A., Servidei S., Silvestri G., Manfredi G., Sabatelli M., Di Muzio A., et al. (1994) Ophthalmoplegia, demyelinating neuropathy, leukoencephalopathy, myopathy, and gastrointestinal dysfunction with multiple deletions of mitochondrial DNA: a mitochondrial multisystem disorder in search of a name. Muscle Nerve 17: 667–674. [DOI] [PubMed] [Google Scholar]
- Van Biervliet S., Verloo P., Vande Veldel S., Van Winckel M., Smet J., Seneca S., et al. (2009) Abdominal pain and vomiting as first sign of mitochondrial disease. Acta Gastroenterol Belg 72: 365–368. [PubMed] [Google Scholar]
- Van Coster R., Lombres A., De Vivo D., Chi T., Dodson W., Rothman S., et al. (1991) Cytochrome c oxidase-associated Leigh syndrome: phenotypic features and pathogenetic speculations. J Neurol Sci 104: 97–111. [DOI] [PubMed] [Google Scholar]
- Verny C., Amati-Bonneau P., Letournel F., Person B., Dib N., Malinge M., et al. (2008) Mitochondrial DNA A3243G mutation involved in familial diabetes, chronic intestinal pseudo-obstruction and recurrent pancreatitis. Diabetes Metab 34: 620–626. [DOI] [PubMed] [Google Scholar]
- Vîlcea D., Vasile I. (2004) Chronic intestinal pseudoobstruction syndrome in adults. Chirurgia (Bucur) 99: 117–124. [PubMed] [Google Scholar]
- Wang Q., Ito M., Adams K., Li B., Klopstock T., Maslim A., et al. (2004) Mitochondrial DNA control region sequence variation in migraine headache and cyclic vomiting syndrome. Am J Med Genet A 131: 50–58. [DOI] [PubMed] [Google Scholar]
- Wong L., Yim D., Bai R., Kwon H., Vacek M., Zane J., et al. (2006) A novel mutation in the mitochondrial tRNA(Ser(AGY)) gene associated with mitochondrial myopathy, encephalopathy, and complex I deficiency. J Med Genet 43: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbridge P., Liang C., Davis R., Vandebona H., Sue C. (2013) POLG mutations in Australian patients with mitochondrial disease. Intern Med J 43: 150–156. [DOI] [PubMed] [Google Scholar]
- Zaganas I., Latsoudis H., Papadaki E., Vorgia P., Spilioti M., Plaitakis A. (2009) A8344G tRNALys mutation associated with recurrent brain stem stroke-like episodes. J Neurol 256: 271–273. [DOI] [PubMed] [Google Scholar]