

Abstract
As a leading cause of cancer deaths worldwide, colorectal cancer (CRC) results from accumulation of both genetic and epigenetic alterations. Disruption of epigenetic regulation in CRC, particularly aberrant histone methylation mediated by histone methyltransferases (HMTs) and demethylases (HDMs), have drawn increasing interest in recent years. In this paper, we aim to review the roles of histone methylation and associated enzymes in the pathogenesis of CRC, and the development of small-molecule modulators to regulate histone methylation for treating CRC. Multiple levels of evidence suggest that aberrant histone methylations play important roles in CRC. More than 20 histone-methylation enzymes are found to be clinically relevant to CRC, including 17 oncoproteins and 8 tumor suppressors. Inhibitors of EZH2 and DOT1L have demonstrated promising therapeutic effects in preclinical CRC treatment. Potent and selective chemical probes of histone-methylation enzymes are required for validation of their functional roles in carcinogenesis and clinical translations as CRC therapies. With EZH2 inhibitor EPZ-6438 entering into phase I/II trials for advanced solid tumors, histone methylation is emerging as a promising target for CRC.
Keywords: colorectal cancer, drug targets, epigenetic regulation, histone demethylase, histone methyltransferase
Introduction
Over 1.3 million new cases of colorectal cancer (CRC) are recorded each year, with more than 0.6 million deaths worldwide [Torre et al. 2015].
Current management for CRC includes surgery, radiofrequency ablation, radiation therapy, chemotherapies, and targeted therapies. For patients in cancer stage III or IV, chemotherapy or targeted therapies are normally used. Based on biomarker analysis, targeted therapies such as epidermal growth factor receptor (EGFR) monoclonal antibodies, cetuximab and panitumumab, can significantly improve therapeutic effects in patients [Pritchard and Grady, 2011]. However, due to molecular heterogeneity and drug resistance, new therapies are required for patients who do not respond to current treatment approaches.
In-depth understanding of pathogenesis will lead to novel therapies for CRC. It has been widely accepted that CRC results from the sequential accumulation of both genetic [Fearon and Vogelstein, 1990; Kinzler and Vogelstein, 1996] and epigenetic changes [Grady and Carethers, 2008; Wong et al. 2007] that induce the transformation of normal glandular epithelium into invasive adenocarcinomas. Both genetic and epigenetic alterations contribute to the tumor formation by activating oncogenes or inactivating tumor suppressors that regulate CRC-associated signaling pathways. These pathways include wingless-type MMTV integration site family (WNT)-, tumor protein 53 (TP53)-, transforming growth factor (TGF)/bone morphogenetic protein(BMP)/SMAD-, receptor tyrosine kinase (RTK)-, NOTCH-, and phosphoinositide 3 kinase (PI3K)-signaling pathways, which affect functions like proliferation, migration, differentiation, adhesion and cell death [Van Engeland et al. 2011]. They also include microsatellite instability (MSI)-, chromosomal instability (CIN)-, and CpG island methylator phenotype (CIMP)-pathways, which regulate the genomic stability [Al-Sohaily et al. 2012].
In recent years, the importance of epigenetic alterations in CRC has been rapidly realized. Epigenetic alterations affect many components of epigenetic regulation, including DNA methylation, histone modifications, nucleosomal occupancy and remodeling, chromatin looping and noncoding RNAs, and contribute to the development of CRC by affecting cancer-associated pathways [Van Engeland et al. 2011]. DNA methylation is one of the mostly well characterized epigenetic alterations in cancer. By searching ‘DNA methylation and cancer’ in PubMed on 28 March 2016, the author got 17,270 publications. However, taking a close look at the number of publications by year between 2001 and 2015, this topic was found to reach a peak in 2014, and flatten in 2015 (Figure S1a, available online). The same tendency has also been observed in the area of ‘DNA methylation and CRC’.
Like DNA methylation, histone modifications have been frequently linked with CRC. Histone modifications are important epigenetic markers that regulate transcription, repair, replication and recombination of genes by affecting the chromatin structure, recruiting remodeling enzymes or transcription-complex proteins [Bannister and Kouzarides, 2011]. Many modifications have been found within histones, with reference to acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation [Bannister and Kouzarides, 2011]. Among them, acetylation and methylation are mostly investigated since the pioneering studies by Allfrey and colleagues in the early 1960s [Allfrey et al. 1964]. By searching ‘histone acetylation or methylation and cancer’ in PubMed, the number of relevant publications was 1392 and 513, respectively. Unlike DNA methylation, the topics of ‘histone acetylation or methylation and cancer’ have made much faster progress in the past 15 years (Figure S1a, available online). A similar pattern also exists in the area of ‘histone acetylation or methylation and CRC’ (Figure S1b, available online).
In line with these observations, the importance of DNA methylation and histone acetylation in CRC were highlighted by a series of reviews [Bardhan and Liu, 2013; Khare and Verma, 2012; Mottamal et al. 2015; Vaiopoulos et al. 2014; West and Johnstone, 2014]. Several DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi), such as azacitidine, decitabine, vorinostat and romidepsin, have been approved by the US Food and Drug Administration for cancers, including chronic leukemia, and more recently, panabinostat for myeloma.
However, less attention has been paid to histone methylation in CRC, although in recent years, we have witnessed rapid progress in this area, which grows even faster than histone acetylation (Figure S1b, available online). Histone-methylation modulators have entered into phase I/II trials for advanced solid tumors, giving hope to the idea that regulating histone methylation can be developed as a novel therapy for CRC. This review will focus on histone methylation, associated enzymes, and potential modulators’ development for treatment of CRC.
Histone methylation in colorectal cancer
Histone methylation occurs on the side chains of lysine and arginine (Figure 1). Two enzyme families mediate the addition and removal of methyl groups: histone methyltransferases (HMTs) and histone demethylases (HDMs). Distinguished by substrates, HMTs are further divided into protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs). PKMTs catalyze transferring of the methyl group from the cofactor S-adenosylmethionine (SAM) to the ϵ-amino group of the lysine side chain, which can be mono-, di-, and trimethylated [Luo, 2012]. Similarly, PRMTs catalyze the methyl group transferring to the ω-guanidino group of arginine with the same methyl donor, SAM. The arginine side chain can be mono-, and symmetrically or asymmetrically di-methylated. Compared with histone acetyltransferases, HMTs are more substrate-specific, in terms of methylation sites and states [Luo, 2012].
Figure 1.

Histone methylation. Histone methylation is regulated by two families of enzymes: histone methyltransferases (HMTs) and histone demethylases (HDMs). The methylation occurs at side chains of both lysine and arginine, which are catalyzed by protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs), respectively. The methylation states are substrate specific: for lysine, there are mono-, di-, and trimethylation; for arginine, there are mono-methylation and symmetrical/asymmetrical dimethylation.
Unlike histone acetylation, histone methylation does not change electrostatic charge of histones or affect the chromatin structure. Instead, it creates docking sites that can be recognized by structural motifs like Tudor-, malignant brain tumor (MBT)-, PWWP-domains, and chromodomains [Bonasio et al. 2010; Holdermann et al. 2012; Pek et al. 2012; Qin and Min, 2014]. These structural domains normally exist in proteins comprising transcriptional complexes or other molecular machines. Histone lysine methylation is associated with both transcriptional activation and repression. For example, trimethylation of histone 3 lysine 4 (H3K4me3) is a conserved marker for transcription activation, while trimethylation of histone 3 lysine 9 (H3K9me3) and histone 3 lysine 27 (H3K27me3) are signals for gene silencing [Bannister and Kouzarides, 2011; Kouzarides, 2007]. Histone arginine methylation is also involved in transcriptional regulatory mechanism [Di Lorenzo and Bedford, 2011]. For instance, asymmetrical dimethylation of histone 4 arginine 3 (H4R3me2a) is a transcriptional activating marker, while the symmetrical dimethylation of histone 4 arginine 3 (H4R3me2s) is associated with transcriptional repression [Bedford and Clarke, 2009]. Beside gene transcription, histone-methylation markers also recruit proteins associated with DNA repairing and other functions. For instance, trimethylation of histone 3 lysine 36 (H3K36me3) recruits hMutSα, the mismatch recognition protein, via direct interactions between H3K36me3 and the PWWP domain of human mutS homolog 6 (hMSH6) [Li et al. 2013a].
Methylated lysine can be restored by the flavin-dependent enzymes of lysine-specific histone demethylase-1, 2 (LSD-1, 2) [Fang et al. 2010; Shi et al. 2004], or the Jumonji family of 2-oxoglutarate-dependent demethylases [Tsukada et al. 2006]. Initially, converting arginine to citrulline via a deamination reaction was considered an indirect approach to reversal of arginine methylation [Cuthbert et al. 2004]. Recently, Jumonji domain-containing 6 (JMJD6) was reported to directly demethylate histone 3 arginine 2 (H3R2) and histone 4 arginine 3 (H4R3) [Chang et al. 2007].
Histone methylation not only regulates many biological functions, including gene transcription, nucleosomal positioning, DNA replication and repair, but also influences the carcinogenesis of cancers by affecting various cancer pathways [Esteller, 2007; Jones and Baylin, 2007]. Indeed, aberrant histone methylation has been frequently found in CRC tumor samples and cell lines (Table 1).
Table 1.
Aberrant histone-methylation markers in colorectal cancer.
| Histone markers | Alterations in CRC | Effects on CRC | Affected functions | Reference |
|---|---|---|---|---|
| H4K20me3 | Decreased in cell lines and primary tumor tissue | Poor prognosis | Hypomethylation of DNA repetitive sequences | Fraga et al. [2005], Benard et al. [2014] |
| H3K4me3 | Elevated in tumor tissue of patients and cells | Unclear | Interacting with β-catenin and promoting WNT-signaling target genes | Salz et al. [2014] |
| H3K4me1/2/3 | Decreased at MLH1 promoter under hypoxia | Unclear | Silencing MLH1 and resulting in DNA mismatch repair defect | Lu et al. [2014] |
| H3K9me3 | Increased in invasive CRC tissue; increased under hypoxia | Metastasis | Promoting cell motility; repression of APAK | Yokoyama et al. [2013]; Olcina et al. [2016] |
| H3K27me3 | Elevated in tumor tissue of patients; increased in patients with poor prognosis | Poor prognosis | Unclear | Benard et al. [2013, 2014]; |
| H3K79me2 | Elevated in patients with poor prognosis | Poor prognosis | Promoting IL-22 induced cancer stemness | Kryczek et al. [2014] |
CRC, colorectal cancer; WNT, wingless-type MMTV integration site family; APAK, ATM and p53-Associated KZNF Protein; IL, interleukin.
Initially, loss of trimethylation of histone 4 lysine 20 (H4K20me3) was identified as one of the common hallmarks of human cancers [Fraga et al. 2005]. Consistently, high expression of H4K20me3 and H3K9me3, and low nuclear expression of H3K4me3, were associated with good prognosis in early-stage CRC patients [Benard et al. 2014]. As a well known gene activation marker, H3K4me3, was found to be elevated in tumor tissue of CRC patients and several cell lines, resulting in activated expression of WNT-signaling target genes through interaction between SET Domain containing 1A (SETD1A) and of β-catenin [Salz et al. 2014]. Interestingly, H3K4me1/2/3 were all decreased at the MutL Homolog 1 (MLH1) promoter in SW480 cells under hypoxia, leading to silence of MLH1 and DNA mismatch repair defects, a key process in the development of sporadic CRC [Lu et al. 2014]. Meanwhile, well known transcription repression marker, H3K9me3 was increased in invasive tumor tissue of CRC patients, resulting in enhanced cell motility [Yokoyama et al. 2013]. It was also found that the H3K9me3 level was elevated along ATM and p53-Associated KZNF Protein (APAK) loci under hypoxia, leading to repression of APAK and p53-dependent apoptosis [Olcina et al. 2016]. The transcription-repression marker H3K27me3 was found to be increased in tumor tissue of CRC patients with poor prognosis [Benard et al. 2013, 2014]. Additionally, dimethylation of histone 3 lysine 79 (H3K79me2) was elevated in CRC patients with poor prognosis, enhancing IL-22-induced cancer stemness [Kryczek et al. 2014].
Very recently, direct mutations in histone-methylation sites have been found to contribute to abnormal histone-methylation profile, then cancer development. Histone 3 lysine 36-to-methionine (H3K36M) mutation was identified in a CRC sample [Shah et al. 2014]. This mutation has been proved to impair mesenchymal progenitor cell differentiation and promote undifferentiated sarcoma in vivo [Lu et al. 2016], suggesting that H3K36 methylation is an important epigenetic marker for tumor suppression.
Histone-methylation enzymes and colorectal cancer
Histone methylation in CRC is regulated by HMTs and HDMs. Targeting histone-methylation enzymes may restore normal methylation profile, therefore there is a potential to develop the therapeutic reagents. To evaluate the prospects of histone-methylation enzymes as drug targets in CRC, evidence from preclinical studies was collected. Among the 87 histone-methylation enzymes accessed in this study, including 60 HMTs and 27 HDMs, 25 proteins were found to have links with CRC, namely 17 oncoproteins and 8 tumor suppressors (Figure 2; Table 2).
Figure 2.
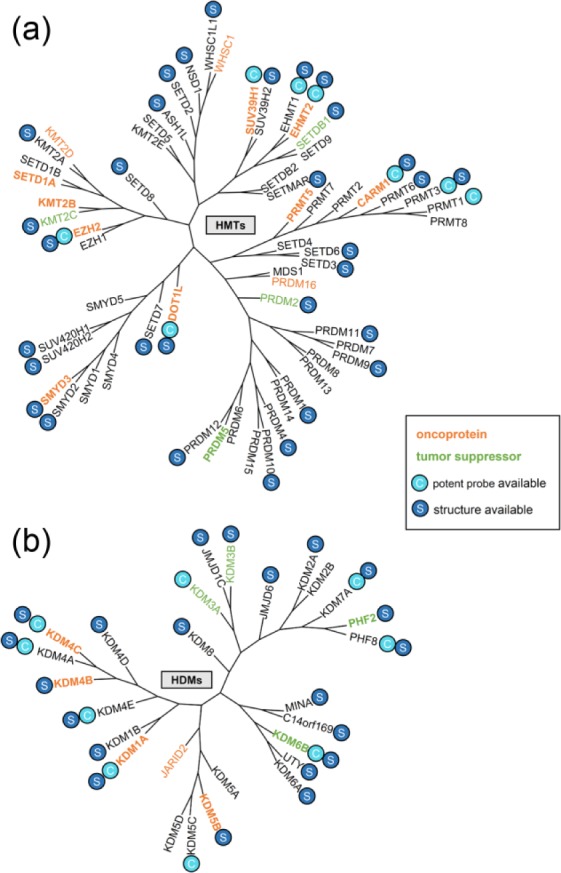
Histone-methylation enzymes in colorectal cancer (CRC). Enzymes of histone methyltransferase (HMT) and histone demethylase (HDM) are plotted in a polygenetic tree based on sequence homology. The gene name of each enzyme is highlighted by different colors according to its role in CRC: orange, oncoproteins and blue, tumor suppressor. Gene name in bold represents a validated target. If the enzyme has potent chemical probes, it is labeled with ‘C’ in a circle; if it has crystal structure(s) which contains the catalytic domain, it is labeled with ‘S’ in a circle.
HMTs, histone methyltransferases; HDMs, histone demethylases.
Table 2.
Histone-methylation enzymes associated with colorectal cancer.
| Family | Enzyme | Synonyms | Substrates | Role in CRC | Target validation | Reference |
|---|---|---|---|---|---|---|
| HMT | KMT2B | MLL4 | H3K4 | Oncoprotein | Knockdown | Ansari et al. [2012] |
| KMT2C | MLL3 | H3K4 | Tumor suppressor | Not yet | Watanabe et al. [2011]; Li et al. [2013b]; Huhn et al. [2014] | |
| KMT2D | MLL2 | H3K4 | Oncoprotein | Not yet | Natarajan et al. [2010] | |
| SETD1A | hSETD1A | H3K4 | Oncoprotein | Knockdown | Salz et al. [2014] | |
| SUV39H1 | KMT1A | H3K9 | Oncoprotein | Knockdown | Kang et al. [2007]; Yokoyama et al. [2013] | |
| EHMT2 | G9a | H3K9 | Oncoprotein | Knockdown; pharmacological inhibition | Zhang et al. [2015b] | |
| PRDM2 | RIZ;RIZ1 | H3K9 | Tumor suppressor | Not yet | Chadwick et al. [2000]; Emterling et al. [2004] | |
| PRDM16 | MEL1 | H3K9 | Oncoprotein | Not yet | Burghel et al. [2013] | |
| SETDB1 | KMT1E | H3K9 | Tumor suppressor | Not yet | Kim et al. [2012a]; Olcina et al. [2016] | |
| EZH2 | KMT6A | H3K27 | Oncoprotein | Knockdown | Fluge et al. [2009]; Wang et al. [2010]; Takawa et al. [2011]; He et al. [2015]; Liu et al. [2015] | |
| DOT1L | KMT4 | H3K79 | Oncoprotein | Pharmacological inhibition | Kryczek et al. [2014] | |
| SMYD3 | KMT3E | H4K5 | Oncoprotein | Knockdown | Xi et al. [2008]; Van Aller et al. [2012]; Peserico et al. [2015] | |
| WHSC1 | MMSET; NSD2 | H4K20 | Oncoprotein | Not yet | Hudlebusch et al. [2011] | |
| PRDM5 | PFM2 | Unknown | Tumor suppressor | Overexpression | Watanabe et al. [2007]; Bond et al. [2015] | |
| CARM1 | PRMT4 | H3R17 H3R26 |
Oncoprotein | Knockdown | Di Lorenzo and Bedford [2011]; Ou et al. [2011] | |
| PRMT5 | SKB1 | H3R8 H4R3 |
Oncoprotein | Knockdown; pharmacological inhibition | Zhang et al. [2015a] | |
| HDM | KDM1A | LSD1 | H3K4 | Oncoprotein | Knockdown | Ding et al. [2013]; Jie et al. [2013]; Jin et al. [2013] |
| KDM5B | JARID1B | H3K4 | Oncoprotein | Knockdown | Ohta et al. [2013] | |
| KDM3A | JMJD1A | H3K9me2 | Tumor suppressor | Not yet | Zuo et al. [2008]; Liu et al. [2013] | |
| KDM3B | JMJD1B | H3K9 | Tumor suppressor | Not yet | Liu et al. [2013] | |
| PHF2 | JHDM1E | H3K9me2 | Tumor suppressor | Not yet | Lee et al. [2015] | |
| KDM4B | JMJD2B | H3K9; H3K36 | Oncoprotein | Knockdown | Liu et al. [2013]; Berry et al. [2014] | |
| KDM4C | JMJD2C | H3K9 | Oncoprotein | Knockdown; pharmacological inhibition | Kim et al. [2014] | |
| KDM6B | JMJD3 | H3K27 | Tumor suppressor | Knockdown | Tokunaga et al. [2016] | |
| JARID2 | JMJ | Unknown | Oncoprotein | Not yet | Tange et al. [2014] |
HMT, histone methyltransferases; HDM, histone demethylase.
Histone-methylation enzymes as oncoproteins in colorectal cancer
H3K4 methylation-associated enzymes
KMT2B/MLL4, KMT2D/MLL2, and SETD1A are all H3K4 methyltransferases [Denissov et al. 2014; Nguyen et al. 2008; Nightingale et al. 2007] that promote the development of CRC. Knockdown of KMT2B by antisense suppressed tumor growth in CRC xenograft-implanted nude mouse. Further experiments in cancer cells revealed that KMT2B regulated expression of several critical cell-cycle regulatory genes; while knockdown of KMT2B affected cell-cycle progression and induced apoptosis [Ansari et al. 2012]. Further, KMT2D was found to be significantly elevated in tumor tissue compared with adjacent benign mucosa in CRC patients’ samples and cell lines [Natarajan et al. 2010]. KMT2D could be a potential oncoprotein in CRC, but such a role remains to be validated by knockdown or pharmacological inhibition experiments. Levels of SETD1A and H3K4me3 were elevated in human CRC cells and patient samples; while depletion of SETD1A inhibited CRC cell growth and affected about 50% WNT target genes [Salz et al. 2014].
Interestingly, H3K4 demethylases, KDM1A/LSD1 and KDM5B/JARID1B [Secombe and Eisenman, 2007] also promote the development of CRC, suggesting that the local H3K4 methylation profile might be better associated with CRC. Overexpression of KDM1A was found in colon cancer specimens, and associated with advanced Tumor-Node-Metastasis (TNM) stages and metastasis [Ding et al. 2013; Jie et al. 2013]. Depletion of KDM1A in human CRC cell line HCT116 resulted in reduced cell proliferation both in vitro and in vivo [Jin et al. 2013]. KDM5B is involved in CRC maintenance, and depletion of KDM5B led to loss of epithelial differentiation and suppression of CRC cell growth [Ohta et al. 2013].
H3K9 methylation-associated enzymes
SUV39H1 and PRDM16 are two H3K9 methyltransferases [Pinheiro et al. 2012; Rea et al. 2000] found to be associated with CRC. Increased level of SUV39H1 mRNA was found in 25% of 219 CRC cases [Kang et al. 2007]. SUV39H1-mediated H3K9me3 was specifically increased in invasive regions of CRC tissue [Yokoyama et al. 2013]. CRC cell migration was activated by overexpression of wild-type SUV39H1 and reduced by knockdown of SUV39H1 [Kang et al. 2007], indicating that SUV39H1 is an oncoprotein in CRC. PRDM16 was one of the gained focal-minimal common-region genes identified in 53 microsatellite-stable sporadic CRC cases [Burghel et al. 2013]. It is a potential oncoprotein, but such a role remains to be established. EHMT2/G9a is responsible for dimethylation of H3K9 (H3K9me2) [Tachibana et al. 2002]. Very recently, EHMT2 was found to be much higher expressed in CRC tumor tissue than peritumoral counterparts. Knockdown of EHMT2 by antisense inhibited proliferation and induced DNA damage of CRC cells [Zhang et al. 2015b]. These data suggest that EHMT2 is an oncoprotein in CRC.
KDM4B and KDM4C are both demethylases of H3K9 [Berry and Janknecht, 2013]. High expression of KDM4B was correlated with lymph node status, Duke’s classification and tumor invasion of CRC patients [Liu et al. 2013]. Consistent with this finding, KDM4B was upregulated in colon and rectal adenocarcinomas, which stimulated β-catenin and colon cancer cell growth; downregulation of KDM4B by shRNA resulted in β-catenin/TCF4 target genes [Berry et al. 2014], indicating that KDM4B is an oncoprotein in CRC. Overexpression of KDM4C was found in colon cancer cell lines, while the downregulation of KDM4C led to reduced growth and clonogenic capacity of colon cancer cells [Kim et al. 2014], suggesting that KDM4C is also an oncoprotein in CRC.
H3K27 methylation-associated enzymes
EZH2 methylates H3K27 [Kuzmichev et al. 2004]. This PKMT belongs to the polycomb group genes involved in the tumor-suppressor gene silencing. Overexpression of EZH2 was found in tumor tissue compared with adjacent nonneoplastic tissue in CRC patients [Fluge et al. 2009; Wang et al. 2010], which was further validated by two independent studies [Liu et al. 2015; Takawa et al. 2011]. EZH2 was responsible for the methylation-dependent resilencing of RUNX3 after the removal of demethylating agents [Kodach et al. 2010]. EZH2 was regulated by the ERK and AKT pathways, which resulted in silencing integrin alpha2 and enhancing the epithelial–mesenchymal transition associated with metastasis [Ferraro et al. 2013, 2014]. The vitamin D receptor (VDR) has also been identified as an EZH2 target; and the downregulation of VDR contributes to the EZH2-induced CRC cell invasion [Lin et al. 2013]. Further study revealed that HAND1 [Tan et al. 2014] and CLDN23 [Maryan et al. 2015] are also silenced by EZH2 in CRC tissue. EZH2 knockdown by siRNA led to the inhibited proliferation and migration of SW620 cells and apoptosis [He et al. 2015]. These results suggested that EZH2 is deeply involved in the carcinogenesis of CRC as an oncoprotein.
Others
DOT1L is the only non-SET-domain-containing PKMT that methylates H3K79 [Steger et al. 2008]. High expression of DOT1L in CRC tissue is a predictor for poor prognosis, and it was found that IL-22-dependent colon cancer stemness is regulated by DOT1L via H3K79 methylation. When using treatment with selective DOT1L inhibitor, EPZ004777, primary colon cancer sphere formation was inhibited [Kryczek et al. 2014]. SMYD3, the methyltransferase of H4K5 [Hamamoto et al. 2004] was found to be overexpressed in the majority of colorectal carcinomas [Van Aller et al. 2012; Xi et al. 2008]. Overexpression of SMYD3 was thought to be induced by KRAS mutation [Gaedcke et al. 2010]. RNAi-mediated SMYD3 knockdown inhibits CRC cell proliferation [Peserico et al. 2015]. WHSC1/MMSET/NSD2 is responsible for the methylation of H4K20 [Pei et al. 2011]. The WHSC1 protein is highly expressed in carcinomas of the gastrointestinal tract, including stomach, colon, anal canal, and the expression level was correlated with tumor aggressiveness [Hudlebusch et al. 2011]. WHSC1 could be a potential oncoprotein in CRC, but such a role remains to be established. CARM1, also known as PRMT4, methylates H3R17 and H3R26 [Di Lorenzo and Bedford, 2011]. CARM1 is overexpressed in human colon cancer cells and positively modulates β-catenin-mediated gene expression. Depletion of CARM1 by shRNA suppresses clonal survival and growth [Ou et al. 2011]. PRMT5 catalyzes symmetric dimethylation on histone 3 arginine 8 (H3R8me2s) and histone 4 arginine 3 (H4R3me2s), and induces transcriptional repression [Pal et al. 2004; Zhao et al. 2009]. It was found that PRMT5 was highly expressed in CRC tumor tissue and associated with poor patient survival. Knockdown of PRMT5 by siRNAs downregulated expression of oncogenes FGFR3 and eIF4E, led to inhibition of CRC cell proliferation and colony formation [Zhang et al. 2015a].
JARID2/JMJ, is found as a Polycomb-repressive complex-2-interacting component [Li et al. 2010]. JARID2 is involved in the TGF-β-induced epithelial–mesenchymal transition in HT29 colon cancer cells [Tange et al. 2014]. JARID2 could be a potential oncoprotein in CRC, but such a role remains to be established.
Histone-methylation enzymes as tumor suppressors in colorectal cancer
H3K4 methylation-associated enzymes
KMT2C/MLL3 catalyzes the methylation of H3K4 [Herz et al. 2012]. Frameshift mutations of KMT2C in both CRC cells and primary tumor were confirmed more commonly in cases with MSI [Watanabe et al. 2011]. Insertion mutation in the KMT2C was found in a pedigree with CRC and acute myeloid leukemia (AML). This insertion caused a premature truncation at codon 827 of KMT2C [Li et al. 2013b]. In line with these findings, an Single Nucleotide Polymorphism (SNP) in KMT2C had the strongest association with CRC risk and survival [Huhn et al. 2014]. These genetic alterations in KMT2C suggest that it is a potential tumor suppressor in CRC, but such a role remains to be established.
H3K9 methylation-associated enzymes
SETDB1 and PRDM2 are two H3K9 methyltransferases [Congdon et al. 2014; Schultz et al. 2002] involved in prevention of CRC development. SETDB1 mediates suppressing the expression of WNT target genes in human CRC cells [Kim et al. 2012a]. Consistent with this finding, SETDB1-mediated H3K9me3 repressed APAK and enhanced the hypoxia-induced p53-dependent apoptosis in CRC [Olcina et al. 2016]. SETDB1 could be a potential tumor suppressor in CRC, but such a role remains to be established. Many frameshift mutations of PRDM2 were revealed in hereditary and sporadic CRC; these mutations resulted in reduced or absent mRNA expression of PRDM2 [Chadwick et al. 2000]. In one study examining the MSI of Swedish patients, mutations of PRDM2 were detected in 31% of 29 MSI tumors [Emterling et al. 2004]. PRDM2 could be a potential tumor suppressor in CRC, but such a role remains to be established.
KDM3A, KDM3B/JMJD1B, and PHF2 are all H3K9 demethylases [Kim et al. 2012b; Wen et al. 2010; Yamane et al. 2006]. KDM3A is involved in the transcriptional reactivation of silenced 15-LOX-1 in CRC cells via demethylating H3K9me2 [Zuo et al. 2008]. Low expression of KDM3B was correlated with the lymph node status, Duke’s classification and TNM staging of CRC patients [Liu et al. 2013]. PHF2 was downregulated in human colon cancer tissue. PHF2 was also required for activation of the p53 pathway in the HCT116 xenograft model treated by oxaliplatin and doxorubicin [Lee et al. 2015].Taken together, KDM3A, KDM3B and PHF2 are all potential tumor suppressors in CRC, but such roles remain to be established.
Others
PRDM5/PFM2 is another tumor suppressor in CRC. Methylation of PRDM5 promoter was more frequently seen in BRAF mutant- than BRAF wild-type CRC [Bond et al. 2015]. Consistently, PRDM5 was found to be silenced in CRC and gastric cancer cell lines by DNA methylation; overexpression of PRDM5 suppressed cancer cell growth [Watanabe et al. 2007]. KDM6B/JMJD3 is responsible for dimethylation of H3K27 [Agger et al. 2007]. Decreased KDM6B was found to be an independent predictor for poor prognosis in 151 CRC patients. Knockdown of KDM6B in CRC cell lines resulted in increased proliferation, via apoptosis suppression and cell-cycle progression [Tokunaga et al. 2016]. These data suggested that KDM6B is a tumor suppressor in CRC.
Drugging histone-methylation enzymes for colorectal cancer
Given the fact that many histone-methylation enzymes play important roles in development of CRC, targeting histone-methylation enzymes by small-molecule modulators could be effective therapy for CRC. Currently, there are a number of small molecules targeting histone-methylation enzymes that have been used for CRC in preclinical studies (Figure 3 and Table 3).
Figure 3.

Histone methyltransferase and histone demethylase inhibitors in preclinical studies of colorectal cancer. The structure, name, target(s), potency, and the discoverers of inhibitors are shown.
HDM, histone demethylase; HMT, histone methyltransferase.
Table 3.
Inhibitors of histone-methylation enzymes in treating experimental colorectal cancer.
| Family | Enzyme | No. of potent ligands* | Inhibitors used in treating CRC | Structure of catalytic domain available | Reference |
|---|---|---|---|---|---|
| HMT | KMT2B | N/A | N/A | No | N/A |
| KMT2C | N/A | N/A | Yes | N/A | |
| KMT2D | N/A | N/A | No | N/A | |
| SETD1A | N/A | N/A | No | N/A | |
| SUV39H1 | 1 | Chaetocin | Yes | Yokoyama et al. [2013] | |
| EHMT2 | 36 | UNC0638; BIX01294 | Yes | Zhang et al. [2015b] | |
| PRDM2 | N/A | N/A | Yes | N/A | |
| PRDM16 | N/A | N/A | No | N/A | |
| SETDB1 | N/A | N/A | Yes | N/A | |
| EZH2 | 20 | GSK126;GSK343; DZNep |
Yes | Benoit et al. [2013a, 2013b]; Ferraro et al. [2014]; Maryan et al. [2015] | |
| DOT1L | 28 | EPZ004777 | Yes | Kryczek et al. [2014] | |
| SMYD3 | N/A | BCI-121 | Yes | Peserico et al. [2015] | |
| WHSC1 | N/A | N/A | Yes | N/A | |
| PRDM5 | N/A | N/A | No | N/A | |
| CARM1 | 17 | N/A | Yes | N/A | |
| PRMT5 | N/A | AMI-1 | No | Zhang et al. [2015a] | |
| HDM | KDM1A | 56 | Tranylcypromine | Yes | Ding et al. [2013] |
| KDM5B | N/A | N/A | Yes | N/A | |
| KDM3A | 5 | N/A | No | N/A | |
| KDM3B | N/A | N/A | Yes | N/A | |
| PHF2 | N/A | N/A | Yes | N/A | |
| KDM4B | N/A | N/A | Yes | N/A | |
| KDM4C | 10 | FLLL-32 | Yes | Lin et al. [2010] | |
| KDM6B | 1 | N/A | Yes | N/A | |
| JARID2 | N/A | N/A | No | N/A |
CRC, colorectal cancer; N/A, not applicable; HMT, histone methyltransferases; HDM, histone demethylase.
Data acquired from CHEMBL database (version 20).
EPZ00477 is a potent inhibitor of DOT1L with IC50 of 0.4 nmol [Daigle et al. 2011]. Treatment with EPZ004777 resulted in inhibited sphere formation in primary colon cancer and suppressed DLD-1 cell line in vitro at 10 µmol [Kryczek et al. 2014]. BCI-121, which was identified as SMYD3 inhibitor by virtual screening, suppressed the growth of CRC cells [Peserico et al. 2015]. Chaetocin is a fungal metabolite that potently inhibits SUV39H1 with IC50 of 800 nmol [Greiner et al. 2005, 2013]. Chaetocin inhibited the activity of SUV39H1 and the migration of CRC cells [Yokoyama et al. 2013]. BIX01294 and UNC0638 are two potent and selective EHMT2 inhibitors competing with substrates rather than cofactors [Kubicek et al. 2007; Vedadi et al. 2011]. BIX01294 and UNC0638 inhibited proliferation of CRC cell lines with IC50 ranging from 1 to 20 µmol [Zhang et al. 2015b]. EZH2 is the most promising PKMT target in experimental CRC, as validated by pharmacological inhibition. DZNep is an indirect EZH2 inhibitor [Tan et al. 2007], which increased apoptosis in CRC cell lines and colon cancer stem cells [Benoit et al. 2013a, 2013b]. EZH2 inhibitor GSK346 [Verma et al. 2012] reduced migration of CRC cells [Ferraro et al. 2014]. GSK126 is a highly specific inhibitor of EZH2 with subnanomolar potency [McCabe et al. 2012]. Treating Colo205 and HT-29 cell lines with GSK126 resulted in reduced level of H3K27me3 and increased CLDN23 mRNA and protein level [Maryan et al. 2015]. AMI-1 was initially reported as type I PRMT inhibitor [Castellano et al. 2010], which also demonstrated inhibition activity in PRMT5 [Zhang et al. 2015a]. AMI-1 inhibited proliferation of CRC cells and xenograft mouse models [Zhang et al. 2015a].
Tranylcypromine, previously used as an antidepressant drug, was discovered as a potent KDM1A inhibitor [Lee et al. 2006; Yang et al. 2007a, 2007b]. Treated with tranylcypromine at 2.5 mmol in SW620 cells, invasion and growth were significantly suppressed [Ding et al. 2013]. FLLL-32, one of the curcuminoids, inhibits KDM4C in vitro [Kim et al. 2014]. FLLL-32 inhibits STAT3 phosphorylation, resulting in the inhibition of cell proliferation of CRC cell lines [Lin et al. 2010].
We also noticed that among the 25 CRC-associated histone-methylation enzymes, only a few of them (nine) have potent chemical probes available (Table 3). Moreover, specific chemical probes, which can accurately modulate the enzymatic activity in vitro and in vivo, are also lacking. Both may hamper testing their roles in CRC development, or hinder utilizing their therapeutic value in CRC treatment.
Crystal structures of histone-modifying enzymes could provide opportunities to meet the requirements of developing selective and potent small-molecule modulators as novel epigenetic therapies for CRC. We searched all the available structures of CRC-associated histone-methylation enzymes in the PDB Data Bank. In total, 134 catalytic-domain-containing structures of 10 enzymes (including 5 HMTs and 5 HDMs) were found. These structures, either in apo form or in complex with substrates/cofactors/inhibitors/activators, provide fruitful insights into the structural basis of regulation of enzymatic activity.
Generally speaking, current structure-based drug-design efforts towards histone-modifying enzymes are primarily focused on the cofactor and substrate binding sites. PKMT and PRMT use the common cofactor SAM to catalyze the methylation of lysine and arginine. Except DOT1L, the catalytic domains of all PKMTs contain a conserved SET domain. The catalytic domain of PKMT is composed of several subdomains, including N-SET, I-SET, C-SET and post-SET. Along with the N-, C-SET domain, the I-SET and post-SET domains form the substrate and cofactor binding sites, where the substrate lysine and cofactor methyl meet at the catalytic channel. A potent and selective inhibitor of DOT1L, EPZ004777, occupies the SAM binding site [Figure 4(a), top left] [Basavapathruni et al. 2012]. A similar binding mode is adopted by sinefungin in SMYD3 [Figure 4(a), top right] [Sirinupong et al. 2011] or 4IK in CARM1 [Figure 4(a), bottom left]. The inhibitor can also occupy the substrate site of HMT. For example, CMPD-2 with IC50 of 27 nmol, binds CARM1 at the arginine cavity [Sack et al. 2011]. HDMs are classified into two subfamilies, the flavin-dependent LSD1 and LSD2, and the iron-dependent Jumonji C-domain-containing demethylases. The tranylcypromine derivative, MC2584, binds to LSD1 at the cofactor site [Figure 4(b)] [Binda et al. 2010].
Figure 4.

Crystal structures of histone methyltransferase and histone demethylase in complex with different inhibitors. (a) DOT1L binding to EPZ004777 at the cofactor site (top left); SMYD3 binding the Sinefungin at the cofactor site (top right); CARM1 binding to 4IK at the cofactor site (bottom left); CARM1 binding to CMPD-2 at the substrate site (bottom right). (b) LSD1 binding to MC2584 at the cofactor site.
SAM, S-adenosylmethionine cofactor; SAH, S-adenosylhomocysteine cofactor.
Development of selective and potent small-molecule modulators of histone-modifying enzymes should be emphasized in the near future. Firstly, the cofactor site is structurally conserved among family members. It is an ideal binding site for small-molecule inhibitors, like cofactor analogs, but the poor specificity is an increasing issue. To improve selectivity, bisubstrate inhibitor, which occupies both cofactor and substrate sites, might be a promising direction. Meanwhile, many crystal structures of these enzymes exhibit distinct conformers in crystal structures, like inactive or active states. It is possible to capture distinct intermediate states in transition pathways between inactive and active states by small molecules. The intermediate states are supposed to be specific for individual enzymes, which may raise hope in developing highly selective intermediate-bound inhibitors. Secondly, the allosteric site is also promising for specific inhibitors or activators. It requires thorough understanding of the regulatory domains in enzymes that are usually absent in the crystal structures. For histone-methylation enzymes as tumor suppressors in CRC, using an activator to target allosteric sites is an attractive way to confer tumors. Given the successful example in SIRT1 [Dai et al. 2015], it is possible to find small-molecule activators for these tumor suppressors. Thirdly, many histone-methylation enzymes are within multiprotein complexes in cells. Protein interfaces between proteins are also druggable sites for small molecules. New protein–protein interaction inhibitors for histone-modifying enzymes may be developed in the future, such as ICG-001, a good example of an inhibitor that disrupts the interaction between CBP and β-catenin [Emami et al. 2004].
Conclusion
Aberrant histone methylation, as well as associated enzymes have been widely linked with CRC. It is worth noting that some CRC-associated histone-methylation enzymes have not been validated as drug targets, including JARID2, KDM3A, KDM3B, KMT2C, KMT2D, PRDM2, PRDM16, SETDB1, and WHSC1. Modulating these proteins in CRC cells or animal models by overexpression, knockdown or pharmacological inhibition may shed light on their therapeutic values in CRC.
More attention should be paid on the mechanisms of histone-methylation enzymes in the development of CRC. We know that by regulating the histone-methylation profile, onco- or tumor-suppressor genes can be turned on or off. Nevertheless, current data suggest such regulation might be specific, and we should figure out exactly which genes are affected by deregulated histone-methylation enzymes. Moreover, histone-methylation enzymes can also modify nonhistone proteins and affect their functions in post-transcriptional level. Once these pathogenesis mechanisms can be elucidated, more precise treatment therapies can be expected.
The author noticed that current data of histone methylation in CRC is mainly preclinical. Intriguingly, EZH2 inhibitor, EPZ-6438 has entered into phase I/II trials for advanced solid tumor or B-cell lymphomas [ClinicalTrials.gov identifier: NCT01897571]. In this active field, we expect more histone-methylation therapies for CRC in clinical trials, identification of new histone-methylation enzymes as CRC drug targets, and discovery of new specific chemical probes of histone-methylation enzymes in coming years.
Acknowledgments
ZXB designed this study. TH and CYL researched literatures and analyzed data. TH, CYL and ZXB contributed in preparation draft of this manuscript. LDZ, LZ, GZ, APL, and JW made substantial contributions to discussion and content. All authors reviewed and approved the final manuscript. Tao Huang and Chengyuan Lin Contributed equally to this work.
Footnotes
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors have received funding from RGC, HKSAR (HKBU12104415).
Conflict of interest: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Contributor Information
Tao Huang, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China.
Chengyuan Lin, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China YMU-HKBU Joint Laboratory of Traditional Natural Medicine, Yunnan Minzu University, Kunming, PR China.
Linda L. D. Zhong, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China
Ling Zhao, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China.
Ge Zhang, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China.
Aiping Lu, Lab of Brain–Gut Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, PR China.
Jiang Wu, State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, PR China.
Zhaoxiang Bian, Room 307, Jockey Club School of Chinese Medicine Building, 7 Hong Kong Baptist University Road, Kowloon Town, Kowloon, Hong Kong.
References
- Agger K., Cloos P., Christensen J., Pasini D., Rose S., Rappsilber J., et al. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in Hox gene regulation and development. Nature 449: 731–734. [DOI] [PubMed] [Google Scholar]
- Allfrey V., Faulkner R., Mirsky A. (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A 51: 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sohaily S., Biankin A., Leong R., Kohonen-Corish M., Warusavitarne J. (2012) Molecular pathways in colorectal cancer. J Gastroenterol Hepatol 27: 1423–1431. [DOI] [PubMed] [Google Scholar]
- Ansari K., Kasiri S., Mishra B., Mandal S. (2012) Mixed lineage leukaemia-4 regulates cell-cycle progression and cell viability and its depletion suppresses growth of xenografted tumour in vivo. Br J Cancer 107: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A., Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res 21: 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardhan K., Liu K. (2013) Epigenetics and colorectal cancer pathogenesis. Cancers 5: 676–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavapathruni A., Jin L., Daigle S., Majer C., Therkelsen C., Wigle T., et al. (2012) Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des 80: 971–980. [DOI] [PubMed] [Google Scholar]
- Bedford M., Clarke S. (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard A., Goossens-Beumer I., Van Hoesel A., De Graaf W., Horati H., Putter H., et al. (2014) Histone trimethylation at H3K4, H3K9 and H4K20 correlates with patient survival and tumor recurrence in early-stage colon cancer. BMC Cancer 14: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard A., Van De Velde C., Lessard L., Putter H., Takeshima L., Kuppen P., et al. (2013) Epigenetic status of LINE-1 predicts clinical outcome in early-stage rectal cancer. Br J Cancer 109: 3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit Y., Laursen K., Witherspoon M., Lipkin S., Gudas L. (2013a) Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J Cell Physiol 228: 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit Y., Witherspoon M., Laursen K., Guezguez A., Beausejour M., Beaulieu J., et al. (2013b) Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res 319: 1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry W., Janknecht R. (2013) KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 73: 2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry W., Kim T., Janknecht R. (2014) Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int J Oncol 44: 1341–1348. [DOI] [PubMed] [Google Scholar]
- Binda C., Valente S., Romanenghi M., Pilotto S., Cirilli R., Karytinos A., et al. (2010) Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc 132: 6827–6833. [DOI] [PubMed] [Google Scholar]
- Bonasio R., Lecona E., Reinberg D. (2010) MBT domain proteins in development and disease. Semin Cell Dev Biol 21: 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond C., Bettington M., Pearson S., Mckeone D., Leggett B., Whitehall V. (2015) Methylation and expression of the tumour suppressor, PRDM5, in colorectal cancer and polyp subgroups. BMC Cancer 15: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghel G., Lin W., Whitehouse H., Brock I., Hammond D., Bury J., et al. (2013) Identification of candidate driver genes in common focal chromosomal aberrations of microsatellite stable colorectal cancer. PLoS ONE 8: e83859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano S., Milite C., Ragno R., Simeoni S., Mai A., Limongelli V., et al. (2010) Design, synthesis and biological evaluation of carboxy analogues of arginine methyltransferase inhibitor 1 (AMI-1). ChemMedChem 5: 398–414. [DOI] [PubMed] [Google Scholar]
- Chadwick R., Jiang G., Bennington G., Yuan B., Johnson C., Stevens M., et al. (2000) Candidate tumor suppressor RIZ is frequently involved in colorectal carcinogenesis. Proc Natl Acad Sci U S A 97: 2662–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B., Chen Y., Zhao Y., Bruick R. (2007) JMJD6 is a histone arginine demethylase. Science 318: 444–447. [DOI] [PubMed] [Google Scholar]
- Congdon L., Sims J., Tuzon C., Rice J. (2014) The PR-Set7 binding domain of Riz1 is required for the H4K20me1-H3K9me1 trans-tail ‘histone code’ and Riz1 tumor suppressor function. Nucleic Acids Res 42: 3580–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert G., Daujat S., Snowden A., Erdjument-Bromage H., Hagiwara T., Yamada M., et al. (2004) Histone deimination antagonizes arginine methylation. Cell 118: 545–553. [DOI] [PubMed] [Google Scholar]
- Dai H., Case A., Riera T., Considine T., Lee J., Hamuro Y., et al. (2015) Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat Commun 6: 7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S., Olhava E., Therkelsen C., Majer C., Sneeringer C., Song J., et al. (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissov S., Hofemeister H., Marks H., Kranz A., Ciotta G., Singh S., et al. (2014) Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141: 526–537. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo A., Bedford M. (2011) Histone arginine methylation. FEBS Lett 585: 2024–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J., Zhang Z., Xia Y., Liao G., Pan Y., Liu S., et al. (2013) LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br J Cancer 109: 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami K., Nguyen C., Ma H., Kim D., Jeong K., Eguchi M., et al. (2004) A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A 101: 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emterling A., Wallin A., Arbman G., Sun X. (2004) Clinicopathological significance of microsatellite instability and mutated RIZ in colorectal cancer. Ann Oncol 15: 242–246. [DOI] [PubMed] [Google Scholar]
- Esteller M. (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8: 286–298. [DOI] [PubMed] [Google Scholar]
- Fang R., Barbera A., Xu Y., Rutenberg M., Leonor T., Bi Q., et al. (2010) Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell 39: 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon E., Vogelstein B. (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759–767. [DOI] [PubMed] [Google Scholar]
- Ferraro A., Boni T., Pintzas A. (2014) EZH2 regulates cofilin activity and colon cancer cell migration by targeting ITGA2 gene. PLoS ONE 9: e115276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro A., Mourtzoukou D., Kosmidou V., Avlonitis S., Kontogeorgos G., Zografos G., et al. (2013) EZH2 is regulated by ERK/AKT and targets integrin alpha2 gene to control epithelial-mesenchymal transition and anoikis in colon cancer cells. Int J Biochem Cell Biol 45: 243–254. [DOI] [PubMed] [Google Scholar]
- Fluge O., Gravdal K., Carlsen E., Vonen B., Kjellevold K., Refsum S., et al. (2009) Expression of EZH2 and Ki-67 in colorectal cancer and associations with treatment response and prognosis. Br J Cancer 101: 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga M., Ballestar E., Villar-Garea A., Boix-Chornet M., Espada J., Schotta G., et al. (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400. [DOI] [PubMed] [Google Scholar]
- Gaedcke J., Grade M., Jung K., Camps J., Jo P., Emons G., et al. (2010) Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer 49: 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady W., Carethers J. (2008) Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135: 1079–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner D., Bonaldi T., Eskeland R., Roemer E., Imhof A. (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat Chem Biol 1: 143–145. [DOI] [PubMed] [Google Scholar]
- Greiner D., Bonaldi T., Eskeland R., Roemer E., Imhof A. (2013) Reply to ‘Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases’. Nat Chem Biol 9: 137. [DOI] [PubMed] [Google Scholar]
- Hamamoto R., Furukawa Y., Morita M., Iimura Y., Silva F., Li M., et al. (2004) SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol 6: 731–740. [DOI] [PubMed] [Google Scholar]
- He S., Zhou H., Zhou J., Zhou G., Han T., Wan D., et al. (2015) Inhibition of EZH2 expression is associated with the proliferation, apoptosis, and migration of SW620 colorectal cancer cells in vitro. Exp Biol Med 240: 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H., Mohan M., Garruss A., Liang K., Takahashi Y., Mickey K., et al. (2012) Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdermann I., Meyer N., Round A., Wild K., Sattler M., Sinning I. (2012) Chromodomains read the arginine code of post-translational targeting. Nat Struct Mol Biol 19: 260–263. [DOI] [PubMed] [Google Scholar]
- Hudlebusch H., Santoni-Rugiu E., Simon R., Ralfkiaer E., Rossing H., Johansen J., et al. (2011) The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res 17: 2919–2933. [DOI] [PubMed] [Google Scholar]
- Huhn S., Bevier M., Pardini B., Naccarati A., Vodickova L., Novotny J., et al. (2014) Colorectal cancer risk and patients’ survival: influence of polymorphisms in genes somatically mutated in colorectal tumors. Cancer Causes Control 25: 759–769. [DOI] [PubMed] [Google Scholar]
- Jie D., Zhongmin Z., Guoqing L., Sheng L., Yi Z., Jing W., et al. (2013) Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig Dis Sci 58: 1581–1589. [DOI] [PubMed] [Google Scholar]
- Jin L., Hanigan C., Wu Y., Wang W., Park B., Woster P., et al. (2013) Loss of LSD1 (lysine-specific demethylase 1) suppresses growth and alters gene expression of human colon cancer cells in a p53- and DNMT1(DNA methyltransferase 1)-independent manner. Biochem J 449: 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P., Baylin S. (2007) The epigenomics of cancer. Cell 128: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M., Lee B., Kim Y., Chang D., Kyu Park S., Chun H., et al. (2007) Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer. Int J Cancer 121: 2192–2197. [DOI] [PubMed] [Google Scholar]
- Khare S., Verma M. (2012) Epigenetics of colon cancer. Methods Mol Biol 863: 177–185. [DOI] [PubMed] [Google Scholar]
- Kim H., Koo B., Cho J., Kim Y., Seong J., Chang H., et al. (2012a) Notch1 counteracts WNT/beta-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest 122: 3248–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kim K., Eom G., Choe N., Kee H., Son H., et al. (2012b) KDM3B is the H3K9 demethylase involved in transcriptional activation of LMO2 in leukemia. Mol Cell Biol 32: 2917–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T., Fuchs J., Schwartz E., Abdelhamid D., Etter J., Berry W., et al. (2014) Pro-growth role of the JMJD2C histone demethylase in HCT-116 colon cancer cells and identification of curcuminoids as JMJD2 inhibitors. Am J Transl Res 6: 236–247. [PMC free article] [PubMed] [Google Scholar]
- Kinzler K., Vogelstein B. (1996) Lessons from hereditary colorectal cancer. Cell 87: 159–170. [DOI] [PubMed] [Google Scholar]
- Kodach L., Jacobs R., Heijmans J., Van Noesel C., Langers A., Verspaget H., et al. (2010) The role of EZH2 and DNA methylation in the silencing of the tumour suppressor RUNX3 in colorectal cancer. Carcinogenesis 31: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Kryczek I., Lin Y., Nagarsheth N., Peng D., Zhao L., Zhao E., et al. (2014) IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 40: 772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S., O’Sullivan R., August E., Hickey E., Zhang Q., Teodoro M., et al. (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 25: 473–481. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A., Jenuwein T., Tempst P., Reinberg D. (2004) Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell 14: 183–193. [DOI] [PubMed] [Google Scholar]
- Lee K., Park J., Sung H., Choi Y., Kim W., Lee H., et al. (2015) PHF2 histone demethylase acts as a tumor suppressor in association with p53 in cancer. Oncogene 34: 2897–2909. [DOI] [PubMed] [Google Scholar]
- Lee M., Wynder C., Schmidt D., Mccafferty D., Shiekhattar R. (2006) Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol 13: 563–567. [DOI] [PubMed] [Google Scholar]
- Li F., Mao G., Tong D., Huang J., Gu L., Yang W., et al. (2013a) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153: 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Margueron R., Ku M., Chambon P., Bernstein B., Reinberg D. (2010) Jarid2 and PRC2, partners in regulating gene expression. Genes Dev 24: 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Li Q., Xu S., Wei F., Ye Z., Cheng J., et al. (2013b) Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood 121: 1478–1479. [DOI] [PubMed] [Google Scholar]
- Lin L., Deangelis S., Foust E., Fuchs J., Li C., Li P., et al. (2010) A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol Cancer 9: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Ren L., Xiong H., Du W., Yu Y., Sun T., et al. (2013) Role of STAT3 and vitamin D receptor in EZH2-mediated invasion of human colorectal cancer. J Pathol 230: 277–290. [DOI] [PubMed] [Google Scholar]
- Liu Y., Zheng P., Liu Y., Ji T., Liu X., Yao S., et al. (2013) An epigenetic role for PRL-3 as a regulator of H3K9 methylation in colorectal cancer. Gut 62: 571–581. [DOI] [PubMed] [Google Scholar]
- Liu Y., Gao X., Jiang Y., Zhang G., Sun Z., Cui B., et al. (2015) Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J Cancer Res Clin Oncol 141: 661–669. [DOI] [PubMed] [Google Scholar]
- Lu C., Jain S., Hoelper D., Bechet D., Molden R., Ran L., et al. (2016) Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352: 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Wajapeyee N., Turker M., Glazer P. (2014) Silencing of the DNA mismatch repair gene MLH1 induced by hypoxic stress in a pathway dependent on the histone demethylase LSD1. Cell Rep 8: 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M. (2012) Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol 7: 443–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M., Ott H., Ganji G., Korenchuk S., Thompson C., Van Aller G., et al. (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492: 108–112. [DOI] [PubMed] [Google Scholar]
- Maryan N., Statkiewicz M., Mikula M., Goryca K., Paziewska A., Strzalkowska A., et al. (2015) Regulation of the expression of claudin 23 by the enhancer of zeste 2 polycomb group protein in colorectal cancer. Mol Med Rep 12: 728–736. [DOI] [PubMed] [Google Scholar]
- Mottamal M., Zheng S., Huang T., Wang G. (2015) Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20: 3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan T., Kallakury B., Sheehan C., Bartlett M., Ganesan N., Preet A., et al. (2010) Epigenetic regulator MLL2 shows altered expression in cancer cell lines and tumors from human breast and colon. Cancer Cell Int 10: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P., Bar-Sela G., Sun L., Bisht K., Cui H., Kohn E., et al. (2008) BAT3 and SET1A form a complex with CTCFL/BORIS to modulate H3K4 histone dimethylation and gene expression. Mol Cell Biol 28: 6720–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale K., Gendreizig S., White D., Bradbury C., Hollfelder F., Turner B. (2007) Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem 282: 4408–4416. [DOI] [PubMed] [Google Scholar]
- Ohta K., Haraguchi N., Kano Y., Kagawa Y., Konno M., Nishikawa S., et al. (2013) Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int J Oncol 42: 1212–1218. [DOI] [PubMed] [Google Scholar]
- Olcina M., Leszczynska K., Senra J., Isa N., Harada H., Hammond E. (2016) H3K9me3 facilitates hypoxia-induced p53-dependent apoptosis through repression of APAK. Oncogene 35: 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou C., Labonte M., Manegold P., So A., Ianculescu I., Gerke D., et al. (2011) A coactivator role of CARM1 in the dysregulation of beta-catenin activity in colorectal cancer cell growth and gene expression. Mol Cancer Res 9: 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S., Vishwanath S., Erdjument-Bromage H., Tempst P., Sif S. (2004) Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 24: 9630–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei H., Zhang L., Luo K., Qin Y., Chesi M., Fei F., et al. (2011) MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pek J., Anand A., Kai T. (2012) Tudor domain proteins in development. Development 139: 2255–2266. [DOI] [PubMed] [Google Scholar]
- Peserico A., Germani A., Sanese P., Barbosa A., Di Virgilio V., Fittipaldi R., et al. (2015) A SMYD3 small-molecule inhibitor impairing cancer cell growth. J Cell Physiol 230: 2447–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro I., Margueron R., Shukeir N., Eisold M., Fritzsch C., Richter F., et al. (2012) Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150: 948–960. [DOI] [PubMed] [Google Scholar]
- Pritchard C., Grady W. (2011) Colorectal cancer molecular biology moves into clinical practice. Gut 60: 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S., Min J. (2014) Structure and function of the nucleosome-binding PWWP domain. Trends Biochem Sci 39: 536–547. [DOI] [PubMed] [Google Scholar]
- Rea S., Eisenhaber F., O’carroll D., Strahl B., Sun Z., Schmid M., et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599. [DOI] [PubMed] [Google Scholar]
- Sack J., Thieffine S., Bandiera T., Fasolini M., Duke G., Jayaraman L., et al. (2011) Structural basis for CARM1 inhibition by indole and pyrazole inhibitors. Biochem J 436: 331–339. [DOI] [PubMed] [Google Scholar]
- Salz T., Li G., Kaye F., Zhou L., Qiu Y., Huang S. (2014) hSETD1A regulates WNT target genes and controls tumor growth of colorectal cancer cells. Cancer Res 74: 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D., Ayyanathan K., Negorev D., Maul G., Rauscher F., III (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16: 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secombe J., Eisenman R. (2007) The function and regulation of the JARID1 family of histone H3 lysine 4 demethylases: the Myc connection. Cell Cycle 6: 1324–1328. [DOI] [PubMed] [Google Scholar]
- Shah M., Denton E., Arrowsmith C., Lupien M., Schapira M. (2014) A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Lan F., Matson C., Mulligan P., Whetstine J., Cole P., et al. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953. [DOI] [PubMed] [Google Scholar]
- Sirinupong N., Brunzelle J., Doko E., Yang Z. (2011) Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3. J Mol Biol 406: 149–159. [DOI] [PubMed] [Google Scholar]
- Steger D., Lefterova M., Ying L., Stonestrom A., Schupp M., Zhuo D., et al. (2008) DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol 28: 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., et al. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takawa M., Masuda K., Kunizaki M., Daigo Y., Takagi K., Iwai Y., et al. (2011) Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci 102: 1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Yang X., Jiang X., Zhou J., Li Z., Lee P., et al. (2014) Integrative epigenome analysis identifies a Polycomb-targeted differentiation program as a tumor-suppressor event epigenetically inactivated in colorectal cancer. Cell Death Dis 5: e1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Yang X., Zhuang L., Jiang X., Chen W., Lee P., et al. (2007) Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 21: 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange S., Oktyabri D., Terashima M., Ishimura A., Suzuki T. (2014) JARID2 is involved in transforming growth factor-beta-induced epithelial-mesenchymal transition of lung and colon cancer cell lines. PLoS ONE 9: e115684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga R., Sakamoto Y., Nakagawa S., Miyake K., Izumi D., Kosumi K., et al. (2016) The prognostic significance of histone lysine demethylase JMJD3/KDM6B in colorectal cancer. Ann Surg Oncol 23: 678–685. [DOI] [PubMed] [Google Scholar]
- Torre L., Bray F., Siegel R., Ferlay J., Lortet-Tieulent J., Jemal A. (2015) Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108. [DOI] [PubMed] [Google Scholar]
- Tsukada Y., Fang J., Erdjument-Bromage H., Warren M., Borchers C., Tempst P., et al. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439: 811–816. [DOI] [PubMed] [Google Scholar]
- Vaiopoulos A., Athanasoula K., Papavassiliou A. (2014) Epigenetic modifications in colorectal cancer: molecular insights and therapeutic challenges. Biochim Biophys Acta 1842: 971–980. [DOI] [PubMed] [Google Scholar]
- Van Aller G., Reynoird N., Barbash O., Huddleston M., Liu S., Zmoos A., et al. (2012) Smyd3 regulates cancer cell phenotypes and catalyzes histone H4 lysine 5 methylation. Epigenetics 7: 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Engeland M., Derks S., Smits K., Meijer G., Herman J. (2011) Colorectal cancer epigenetics: complex simplicity. J Clin Oncol 29: 1382–1391. [DOI] [PubMed] [Google Scholar]
- Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., et al. (2011) A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol 7: 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S., Tian X., Lafrance L., Duquenne C., Suarez D., Newlander K., et al. (2012) Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett 3: 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Ye Y., Yuan J., Liu F., Zhang H., Wang S. (2010) EZH2 and STAT6 expression profiles are correlated with colorectal cancer stage and prognosis. World J Gastroenterol 16: 2421–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Castoro R., Kim H., North B., Oikawa R., Hiraishi T., et al. (2011) Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PLoS ONE 6: e23320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Toyota M., Kondo Y., Suzuki H., Imai T., Ohe-Toyota M., et al. (2007) PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin Cancer Res 13: 4786–4794. [DOI] [PubMed] [Google Scholar]
- Wen H., Li J., Song T., Lu M., Kan P., Lee M., et al. (2010) Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J Biol Chem 285: 9322–9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A., Johnstone R. (2014) New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J., Hawkins N., Ward R. (2007) Colorectal cancer: a model for epigenetic tumorigenesis. Gut 56: 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y., Formentini A., Nakajima G., Kornmann M., Ju J. (2008) Validation of biomarkers associated with 5-fluorouracil and thymidylate synthase in colorectal cancer. Oncol Rep 19: 257–262. [PubMed] [Google Scholar]
- Yamane K., Toumazou C., Tsukada Y., Erdjument-Bromage H., Tempst P., Wong J., et al. (2006) JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 125: 483–495. [DOI] [PubMed] [Google Scholar]
- Yang M., Culhane J., Szewczuk L., Gocke C., Brautigam C., Tomchick D., et al. (2007) Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol 14: 535–539. [DOI] [PubMed] [Google Scholar]
- Yang M., Culhane J., Szewczuk L., Jalili P., Ball H., Machius M., et al. (2007) Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46: 8058–8065. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y., Hieda M., Nishioka Y., Matsumoto A., Higashi S., Kimura H., et al. (2013) Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo . Cancer Sci 104: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Dong S., Zhu R., Hu C., Hou J., Li Y., et al. (2015a) Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget 6: 22799–22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., He P., Xi Y., Geng M., Chen Y., Ding J. (2015b) Down-regulation of G9a triggers DNA damage response and inhibits colorectal cancer cells proliferation. Oncotarget 6: 2917–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Rank G., Tan Y., Li H., Moritz R., Simpson R., et al. (2009) PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol 16: 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo X., Morris J., Shureiqi I. (2008) Chromatin modification requirements for 15-lipoxygenase-1 transcriptional reactivation in colon cancer cells. J Biol Chem 283: 31341–31347. [DOI] [PMC free article] [PubMed] [Google Scholar]