

Abstract
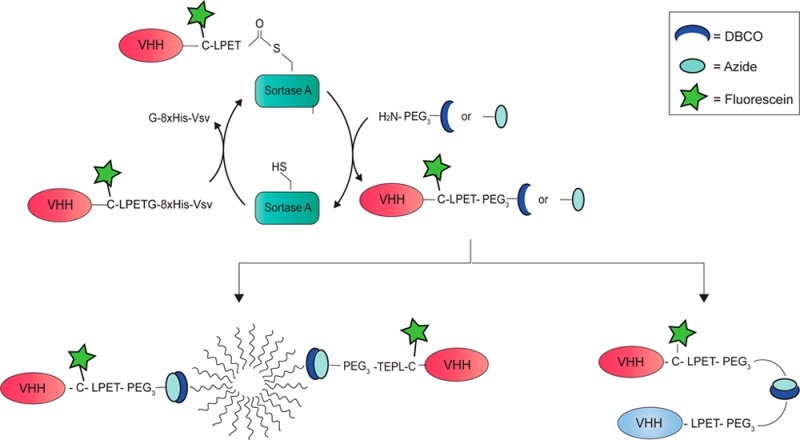
Conjugation of llama single domain antibody fragments (Variable Heavy chain domains of Heavy chain antibodies, VHHs) to diagnostic or therapeutic nanoparticles, peptides, proteins, or drugs offers many opportunities for optimized targeted cancer treatment. Currently, mostly nonspecific conjugation strategies or genetic fusions are used that may compromise VHH functionality. In this paper we present a versatile modular approach for bioorthogonal VHH modification and conjugation. First, sortase A mediated transPEGylation is used for introduction of a chemical click moiety. The resulting clickable VHHs are then used for conjugation to other groups employing the Cu+-independent strain-promoted alkyne–azide cycloadition (SPAAC) reaction. Using this approach, tail-to-tail bispecific VHHs and VHH-targeted nanoparticles are generated without affecting VHH functionality. Furthermore, this approach allows the bioconjugation of multiple moieties to VHHs for simple and convenient production of VHH-based theranostics.
Introduction
The challenge in cancer therapy is to specifically deliver therapeutic agents to tumor cells with minimal delivery to and effects on healthy tissues. Targeted delivery of drugs with antibody drug conjugates (ADCs) has received a lot of attention in the last decades. Also, nanoparticles have been used for drug delivery. Liposomes can carry hydrophilic drugs in the lumen1,2 while micelles are suited for carrying hydrophobic drugs.3 Liposomes and micelles, however, distribute relatively randomly in the body after intravenous injection,1 and introduction of tumor specificity in these nanoparticles could greatly increase local delivery and efficacy of cancer therapeutics.4−6
A number of therapeutic antibodies with relative tumor specificity are now applied clinically (e.g., trastuzumab and cetuximab against HER2 and EGFR7,8). Effects of treatments with these targeted drugs are often limited because of recurrences of drug-resistant tumors.9−12 The challenge should therefore be to develop a multispecific tumor-targeting nanoparticle platform that can deliver cytotoxic payload to all cancer cells in a tumor, resulting in specific and acute death of all cells in a tumor.
Conventional protein conjugation strategies mostly use relatively nonspecific methods, e.g., N-hydroxysuccinimide (NHS) chemistry utilizing ε-NH2 groups from lysines. Depending on the number and distribution of lysines in the active binding domain of the targeting agent, these conjugation protocols may compromise functionality. This specifically applies to single domain antibody fragments (VHHs, Nanobodies) due to their small size. VHHs are recombinant antigen-binding domains that are derived from cameloid heavy chain-only antibodies and receive increasing interest as therapeutic or diagnostic compounds.13,14 VHHs typically have a molecular weight of 15–20 kDa and may bind target antigens with pM to nM affinity, similar to conventional antibodies. The ease of genetic engineering and handling of VHHs, combined with a number of other advantages such as high water solubility, low production cost, small size, low immunogenicity in humans, and high thermo- and pH stability,15 makes this class of antibodies a highly interesting alternative to conventional antibodies.
A number of VHHs with high specificity and affinity against tumor targets (e.g., EGFR, HER2, MET) have been developed.16−18 Approaches to conjugate VHHs without compromising functionality include the site specific introduction of a carboxyterminal cysteine allowing maleimide chemistry19 and introduction of a carboxyterminal five-amino-acid sequence (LPXTG) allowing sortase A transpeptidation.20
Here we present a versatile modular approach for bioorthogonal VHH conjugations, using sortase A mediated transPEGylation to introduce a carboxyterminal click moiety, and subsequent Cu+-independent strain-promoted alkyne–azide cycloadition (SPAAC) for conjugation of a functional group that may be connected to another VHH and/or polymeric micelles. Furthermore, introduction of a cysteine before the LPXTG tag allows maleimide chemistry to introduce a second diagnostic or therapeutic compound. This method allows the production of highly uniform VHH-based conjugates that can consist of multiple modular moieties, examples being labeled bispecific VHHs, bivalent VHHs, and VHH-targeted nanoparticles. Because all chemistry occurs at the carboxyterminus, this method does not affect VHH functionality. Furthermore, concomitant with conjugation, carboxy-terminal tags that allow purification of the VHHs after bacterial expression are removed, a prerequisite for future clinical applications.
Results and Discussion
VHHs were successfully expressed and purified as VHH-C-LPETG-8xHis-Vsv or VHH-LPETG-8xHis-Vsv fusion proteins with yields of 5–10 mg/L E. coli culture (see Figure 1A for a representative Coomassie Brilliant Blue (CBB) stained SDS-PAGE gel of 7D12-C-LPETG-8xHis-Vsv, the anti-EGFR VHH that was used as a prototype in this study). Since the 8xHis-Vsv tags are substituted by compounds of interest during the sortase A reaction, loss of these tags was compensated for by introducing a cysteine residue directly upstream of the LPETG-8xHis-Vsv sequence, allowing maleimide-based labeling with alternative tags for detection. Liquid chromatography mass spectrometry (LC-MS) indicated the correct mass for expressed 7D12-C-LPETG-8xHis-Vsv and it showed that the free thiol group of this cysteine was oxidized, presumably by glutathione based on its molecular weight of 307 Da (Figure 1B). After a mild TCEP reduction, the thiol was available for conjugation (Figure 1C). The reaction of the 7D12-C-LPETG-8xHis-Vsv protein (from now on referred to as 7D12) with fluorescein-5-maleimide was efficient, resulting in a pure preparation of 7D12-C[Fluo]-LPETG-8xHis-Vsv (from now on referred to as 7D12[Fluo]) (Figure 2A+B, lane 1). LC-MS confirmed the conjugation of one fluorescein residue per VHH in 7D12[Fluo] (Figure 2C), demonstrating that the two native framework cysteine residues involved in an intramolecular disulfide bridge21 were not reactive toward fluorescein-5-maleimide under the conditions used. This was further confirmed in additional experiments that showed an absence of maleimide reaction with multiple VHHs lacking the C terminal cysteine (Figure S1).
Figure 1.

(A) CBB stained SDS-PAGE gel of samples obtained during 7D12-C-LPETG-8xHis-Vsv production. 1 = bacterial lysate, 2 = flow-through Ni-NTA purification, 3 = pre-eluate Ni-NTA purification, 4 = eluate post Ni-NTA purification and dialysis. (B) LC-MS characterization of 7D12-C-LPETG-8xHis-Vsv (with an expected mass of 17837 Da, observed 17962 and 18137 Da) before TCEP reduction. (C) LC-MS characterization of 7D12-C-LPETG-8xHis-Vsv after 20 min incubation with 20 mM TCEP. A pure product of 17831 Da was detected, suggesting that the thiol before TCEP reduction was oxidized by small molecules, among which was presumably glutathione. The left graphs show the ultraviolet absorbance chromatograms (at 215 nm) and the middle and right graphs show the total mass spectra and deconvoluted spectra of the UV-peaks bracketed by the arrowheads, respectively.
Figure 2.

(A) Fluorescent image and (B) CBB staining of an SDS-PAGE gel of samples obtained during the procedure of sortagging. 1 = 7D12[Fluo], 2 = 7D12[Fluo]/sortase A/H2N-PEG3-N3 reaction mixture after overnight reaction, 3 = purified 7D12[Fluo]-N3. Note the molecular shift between lanes 1 and 2, 3 that is a result of removal of the G-8xHis-Vsv tag and simultaneous addition of the small PEG3-N3 group. (C) LC-MS characterization of 7D12[Fluo] (expected 18265 Da, observed 18278 Da). (D) LC-MS characterization of 7D12[Fluo]-N3 (expected 15790 Da, observed 15806 Da). The left graphs show the ultraviolet absorbance chromatograms (at 215 nm) and the middle and right graphs show the total mass spectra and the deconvoluted spectra of the UV-peaks bracketed by the arrowheads, respectively.
The LPETG-8xHis-Vsv tag in 7D12[Fluo] allows sortase A mediated transpeptidation which releases the G-8xHis-Vsv tag in exchange for an H2N-GGG-containing peptide (the prototypical substrate of sortase A). This allows rapid and easy purification of the reaction product to homogeneity, because the G-8xHis-Vsv cleavage product and the 6xHis-tagged sortase A enzyme can be removed from the reaction mixture by Ni-bead depletion. Because a wide variety of chemically modified monodisperse PEG compounds is nowadays available, sortase A mediated conjugation of such compounds to the carboxyterminus is a highly attractive approach.
Using H2N-PEGx-X as a nucleophile for sortase A mediated PEGylation, a range of VHH-PEGx-X constructs can be generated in which X represents a drug or a reactive group. In this work we used the combination of H2N-PEG3-N3 and H2N-PEG3-DBCO in the SPAAC reaction.22 In an attempt to improve the reaction with H2N-PEGx-X, we tested recently described mutants of sortase A with optimized LPETG cleavage activity,23 and compared them to the variants with a N-terminal deletion of either 25 or 59 amino acids (Supporting Information Figure S8). These optimizations were performed using the anti-PlexinD1 VHH A12.24 Sortase A Δ59 was ultimately selected as most optimal, since despite increased LPETG cleavage rates, the low affinity of the mutant sortases for H2N-GGG or H2N-PEGx-X resulted in high levels of hydrolysis at the threonine. Furthermore, we found that the sortase A Δ25 variant, that contains an N-terminal 6xHis-tag, experienced unexplained proteolysis just downstream of the His-tag during the sortase A reaction (Supporting Information Figure S8D). Since this precluded complete removal of sortase A activity, we discarded this variant.
Further experimentation using different molar ratios of H2N-PEGx-X or H2N-GGG revealed that H2N-PEGx-X was incorporated less efficiently than H2N-GGG, requiring a 200-fold molar excess of H2N-PEGx-X over VHH-(C)LPETG-8xHis-Vsv as compared to a 25-fold excess of H2N-GGG (under conditions of 4 h reaction at 30 °C with sortase A Δ59). Lower concentrations of both nucleophiles in the reaction resulted in the formation of VHH-LPET-OH hydrolysis products, as observed with LC-MS (Supporting Information Figure S9A). These results were in line with those of Parthasarathy et al., who showed that conjugation of H2N-PEGx to EGFP in a ratio of 1:1 by sortase A Δ59 was highly inefficient.25 Yet, the use of H2N-PEGx-X nucleophiles instead of triglycine-containing substrates would have an important advantage: conjugation of H2N-PEGx-X yields VHH-(C)LPET-PEGx-X in a one-way reaction, unlike conjugation of H2N-GGG-X that reconstitutes a sortase A substrate site in the VHH-(C)LPET-GGG-X reaction product. Indeed we could show that VHH-LPET-PEGx-X is not a substrate for sortase A, in contrast to VHH-LPETGGG-peptide (see Figure S2).
Using the optimized conditions, 7D12[Fluo] was conjugated to H2N-PEG3-N3 with sortase A Δ59 to yield 7D12[Fluo]-N3 (Figure 2A+B). After removal of sortase A Δ59, G-8xHis-Vsv, and unreacted 7D12[Fluo] by Ni-NTA depletion, 7D12[Fluo]-N3 was obtained in a 66% yield with a >95% purity based on LC-MS (Figure 2D). Similar results were obtained when H2N-PEG3-DBCO was used in the reaction with sortase A Δ59 (Figure S3). Furthermore, the methodology has been successfully tested and validated for three more VHHs in our lab (Supporting Information Figure S10) demonstrating the versatility of this approach.
Bispecific or bivalent antibody conjugates are valuable compounds for targeted cancer therapy.26,27 Current approaches to make bispecific or bivalent VHHs often involve genetic fusion of VHH open reading frames, allowing production only in head-to-tail fusion format. It has already been shown that the affinity of the second VHH in such bispecific constructs may be dramatically affected by the presence of the first VHH.28 Sortase A technology allows the generation of tail-to-tail dimers, leaving antigen binding sites of the individual composing VHHs intact20 (Figures 3 and 4A). We tested the validity of this hypothesis by using sortase A Δ59 in combination with H2N-PEG3-X linkers to generate 7D12[Fluo]-N3 and 4E4-DBCO, 4E4 being a low-affinity Transferrin receptor (TfR)-binding VHH (unpublished results) that is used here as a control. After the click reaction (see Figure 4B+C for analysis of the reaction products), heterodimer N7D12C[Fluo]-C4E4N could be readily purified from residual monomers by size exclusion chromatography, resulting in >90% pure product of 36 kDa (Figure S4). In parallel, we generated conventional head-to-tail bispecific VHHs, with 7D12 and 4E4 separated by a [G4S]10 linker or a [G4S]20 linker and a C-terminal C-Flag-6xHis (compounds N7D12C-[G4S]n-N4E4C and N4E4C-[G4S]n-N7D12C; Figures 3 and 4A). E. coli expression of these bispecific VHHs was successful as shown with SDS-PAGE analysis (Figure 4D+E). Yields were 2–4 mg/L E. coli culture. The bispecifics were labeled on the C-terminal cysteine with fluorescein-5-maleimide as described for monomeric VHHs, and the correct mass was confirmed with LC-MS (Figure S5).
Figure 3.

Schematic representation of the monomeric VHH, head-to-tail bispecific VHH, and tail-to-tail bispecific VHH and their nomenclature in this report.
Figure 4.
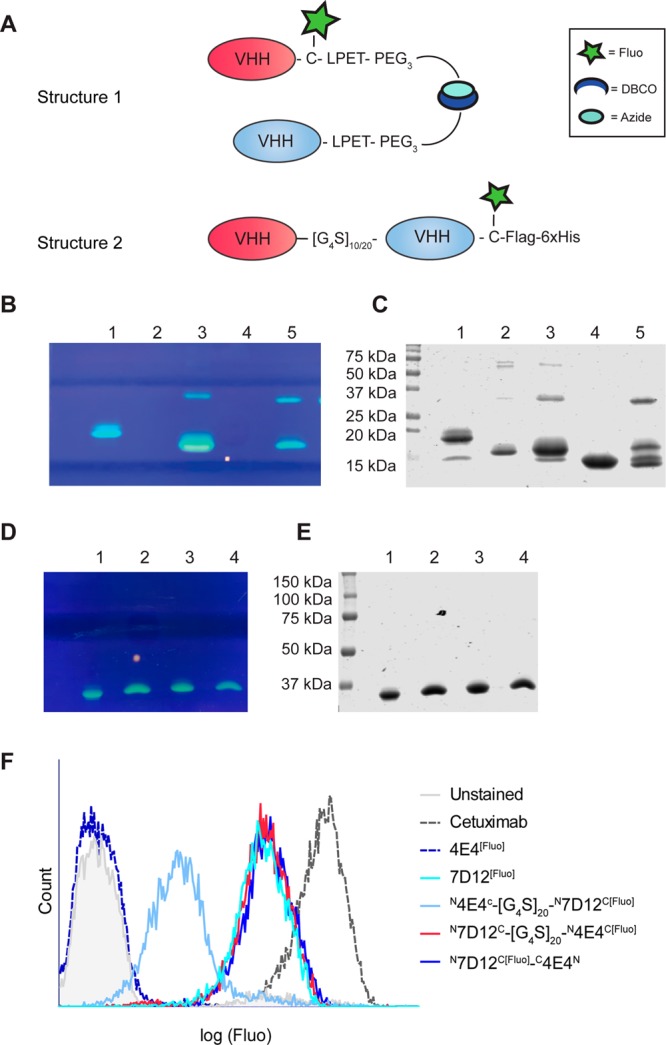
(A) Schematic overview of bispecific tail-to-tail (Structure 1) or head-to-tail (Structure 2) VHH constructs. In panels B and C, SDS-PAGE is shown (fluorescence signal and CBB staining, respectively) depicting the products of the sortase A reaction to produce bispecific N7D12C[Fluo]-C4E4N. 1 = 7D12[Fluo], 2 = 4E4, 3 = 7D12[Fluo]-DBCO, 4 = 4E4-N3, 5 = N7D12C[Fluo]-C4E4N before sephadex G75 purification. In panels D and E, the expression of bispecific head-to-tail VHHs is depicted with (D) fluorescence signal and (E) CBB staining of an SDS-PAGE gel. 1 = N7D12C-[G4S]10-N4E4C[Fluo], 2 = N4E4C-[G4S]10-N7D12C[Fluo], 3 = N7D12C-[G4S]20-N4E4C[Fluo], 4 = N4E4C-[G4S]20-N7D12C[Fluo]. (F) Flow cytometry histogram derived from A431 cells incubated with the various bispecific constructs and controls.
Importantly, in this context both the head-to-tail and tail-to-tail bispecific VHHs contain only one fluorescein molecule, allowing quantitative comparison of the EGFR-binding capacity of these formats with 7D12[Fluo]. Constructs were tested in flow cytometry analyses using the high EGFR expressing squamous carcinoma cell line A431 (Figure 4F). Compounds N7D12C-[G4S]n-N4E4C[Fluo] with both G4S linker lengths and N7D12C[Fluo]-C4E4N were equally effective as 7D12[Fluo] in binding to A431 cells. In contrast, binding to EGFR was reduced with a factor of 10 for compounds N4E4C-[G4S]n-N7D12C[Fluo] with both G4S linker lengths, confirming that, at least for VHH 7D12, epitope binding is hindered by the presence of a VHH at the amino-terminus. Thus, bispecific VHHs generated via tail-to-tail click fusion are more robust than head-to-tail bispecific VHHs.
PEGylation is an important modification of nanoparticles that is applied to increase half-life in the circulation by avoiding rapid clearance by spleen, liver, and kidney.29 PEGylation of VHHs, accomplished by clicking DBCO-PEG to VHH-N3, indeed results in better in vivo characteristics.30 Sortase A mediated conjugation of H2N-PEGx yields the interesting option to decorate PEGylated nanoparticles with functionally active VHHs. To test this hypothesis we applied click chemistry to conjugate 7D12[Fluo]-N3 to benzoyl-poly(ε-caprolactone)-methoxypoly(ethylene glycol) (ben-PCL7-mPEG2000) diblock-based micelles, equipped with a small percentage of DBCO groups. Ben-PCL7-mPEG2000 diblock-based micelles are biocompatible and biodegradable, and can be used as carriers of hydrophobic drugs that can be incorporated in the micellar core.31 The decoration of micelles with VHHs may increase affinity of the nanoparticles to targets by an avidity effect. Also, this approach allows the generation of multispecific drug-loaded targeting nanoparticles by simply decorating these with different tumor-targeting VHHs.
1H NMR spectra showed that synthesis of intermediate products (Figure S6) and end products ben-PCL7-mPEG2000 and ben-PCL7-mPEG2000-DBCO (Figure 5A+B) was successful. Micelles were prepared by film-hydration of a mixture of 90% ben-PCL7-mPEG2000 diblock polymers and 10% ben-PCL7-mPEG2000-DBCO, resulting in micelles that contain a calculated 20–30 DBCO click groups per particle. Dynamic light scattering (DLS) experiments of the resulting micelles revealed a mean particle size of 28 ± 2 nm and a polydispersity index (PDI) of 0.32 ± 0.02.
Figure 5.

Chemical structures and corresponding 1H NMR-spectra of (A) Ben-PCL7-mPEG2000 and (B) Ben-PCL7-PEG2000-DBCO measured in CDCl3..
DBCO-micelles were decorated with 7D12 via a click chemistry reaction with 7D12[Fluo]-N3 in a 2:1 DBCO:N3 ratio, to minimize amounts of residual unconjugated 7D12[Fluo]-N3 after the click reaction. To confirm that during handling and experimentation micelles remained intact, we included the hydrophobic photosensitizer meta-tetra(hydroxyphenyl)chlorin (mTHPC) during micelle formation, equipping the micelles with a fluorescent signal that is readily distinguishable from 7D12-associated fluorescein (Figure 6A). SDS-PAGE of the 7D12 conjugated micelles revealed a shift of ∼3 kDa as compared to the starting material 7D12[Fluo]-N3, indicating successful conjugation of 7D12[Fluo]-N3 to the DBCO-block-copolymer with a conjugation yield of >85% (Figure 6B,C). Again this conjugation method was verified for other available VHHs in our lab (Supporting Information, Figure S11A+B), showing its versatility.
Figure 6.

(A) Cartoon of VHH[Fluo]-decorated mTHPC loaded micelles. (B) Fluorescein visualization. (C) CBB staining of the SDS-PAGE analysis of SPAAC reaction between 7D12[Fluo]-N3 and DBCO-micelles. 1 = 7D12[Fluo], 2 = 7D12[Fluo]-N3, 3 = 10% DBCO-micelles with 5% mTHPC, and 4 = 10% DBCO-micelles with 5% mTHPC and 5% 7D12[Fluo]. (D) Median values of fluorescence intensity as measured in flow cytometry. Left bar graph = fluorescein total signal on A431 (EGFR+) and E98 (EGFR–) cells after incubation with 7D12[Fluo] as a positive control and 7D12[Fluo]-micelles. Right bar graph = mTHPC total signal on both A431 and E98 cells after incubation with targeted or nontargeted micelles. * indicates significance with p < 0.05. (E) Fluorescence microscopy of A431 cells immediately after incubation with 1 = 7D12[Fluo], 2 = 7D12[Fluo]-micelles, and 3 = nontargeted micelles. Cyan depicts mTHPC, incorporated in the micelles; green depicts fluorescein. The scale bar depicts 20 μM.
We next tested whether 7D12 on the decorated micelles had retained EGFR affinity by performing flow cytometry after incubation with EGFR-positive A431 squamous cell carcinoma or EGFR-negative E98 glioma cells,32 using 7D12[Fluo] as reference. 7D12[Fluo]-micelles showed efficient binding to A431 cells but negligible binding to E98 cells (Figure 6D). Importantly, binding of 7D12[Fluo]-micelles led to 70% higher cell-associated fluorescein-fluorescence than binding of monomeric 7D12[Fluo]. This may reflect the loading of individual micelles with multiple fluorescein-labeled 7D12 moieties or may be due to cooperative binding and an avidity effect. Increased binding of 7D12[Fluo]-micelles to A431 cells was confirmed with fluorescence microscopy, using the fluorescein signal as readout (Figure 6E, green signal in panel 2, compare to 7D12[Fluo] in panel 1). The increase in fluorescein signal was accompanied by an increased association of mTHPC fluorescence in 7D12[Fluo]-micelles as compared to nondecorated micelles (Figure 6D, right graph, and Figure 6E, cyan signal). There was no difference in binding of 7D12[Fluo]-micelles and nondecorated micelles to E98 cells, suggesting that 7D12[Fluo]-micelles bound as intact particles to A431 cells in an EGFR-dependent manner. It must however be noted that there was also some nonspecific association of mTHPC with both A431 cells and E98 cells. Whether this reflects nonspecific binding of micelles or of free mTHPC that is released from micelles is difficult to discriminate.
These results indicate that the multivalency induced by coupling multiple VHHs to a micelle, increased uptake of these particles and their payload in target positive cells. Although we restricted ourselves in this study to micelles, this conjugation approach is predicted to work with many other types of protein- or PEG-based nanoparticles (e.g., liposomes, polymersomes) once a controlled percentage of the building blocks is equipped with a chemical click group. Interestingly, this procedure will also allow the controlled synthesis of multispecific targeting nanoparticles, e.g., by preparing nanoparticles with a defined number of noncompatible clickable agents such as DBCO and tetrazine moieties, allowing simultaneous conjugation of VHH-PEG3-N3 and VHH-PEGx-TCO (trans-cyclooctene). Whereas the free thiol group in VHH-C-LPET-PEGx-X may be used for maleimide-based conjugation of hydrophilic drugs, micelles may be used to simultaneously carry hydrophobic drugs, making this to a versatile approach.
Conclusion
Conjugations of VHHs to diagnostic or therapeutic compounds should involve the VHH’s carboxy-terminus, distant from the antigen binding site, in order to retain full VHH functionality. Here, a bioorthogonal site-specific conjugation approach is presented based on sortase A and click chemistry, combined with cysteine–maleimide conjugation. This approach allows the preparation of molecularly defined targeted nanoparticles, preserving targeting potential and concomitantly removing unwanted tags from VHHs, a prerequisite for potential clinical applications. The potential to perform controlled dual labeling of proteins without the loss of protein function is an important next step to the preparation of optimized theranostics.
Experimental Procedures
VHH Production and Purification
VHH 4E4 is a low affinity VHH (Kd ∼ 500 nM) directed against transferrin receptor (TfR) and was used as an inert VHH in this study. VHH 7D12 against EGFR has been described in detail before.17,33 The VHH coding sequences were cloned in frame behind the pelB leader sequence in modified pHEN-IX vectors in which the sortase A recognition sequence LPETG, either or not preceded by a cysteine, was inserted just upstream of the 8xHis-Vsv tags, resulting in pHENIX-VHH-LPETG-8xHis-Vsv and pHENIX-VHH-C-LPETG-8xHis-Vsv. Plasmids were transformed in E. coli strain ER2566 for standard protein expression. Cells were grown in 2xTY medium containing 3.5% (w/v) glycerol and 50 μg/mL ampicillin at 37 °C. At an OD600 between 0.6 and 0.8, recombinant protein expression was induced with 1.0 mM isopropyl β-d-thiogalactoside (IPTG, Serva, Heidelberg, Germany) at 30 °C for 2.5 h. Cells were harvested by centrifugation at 2830 g for 20 min at 4 °C and the periplasmic protein fraction was isolated via osmotic lysis. Cells were resuspended in ice cold TES buffer (200 mM Tris pH 8.0, 0.5 mM EDTA, 20% w/v sucrose, protease inhibitors (Complete cocktail, Roche, Basel, Switzerland)) and incubated for 20 min on ice, followed by centrifugation (4424 g, 20 min, 4 °C). After collection of the supernatant the bacterial pellet was resuspended in TES buffer containing 15 mM MgSO4 and incubated on ice for 20 min. After centrifuging, both supernatants were pooled and incubated with Ni-NTA sepharose (IBA, Goettingen, Germany), pre-equilibrated with 50 mM phosphate pH 7.4, 500 mM NaCl, for 1 h at 4 °C. After washing of the beads with 50 mM phosphate pH 7.4, 500 mM NaCl, and 10 mM imidazole, 8xHis-tagged proteins were eluted with 500 mM imidazole in 50 mM phosphate pH 7.4 and 500 mM NaCl. The eluate was dialyzed against 50 mM TRIS pH 7.5 and 150 mM NaCl in a 3.5 kDa dialysis membrane (Spectrum laboratories, Los Angeles, CA, USA). VHHs were analyzed by SDS-PAGE under reducing conditions (Coomassie brilliant blue (CBB) staining), followed by analysis on the Odyssey CLx infrared imaging system (LI-COR, Lincoln, NE, USA)) and liquid chromatography and mass spectrometry (LC-MS, Shimadzu HPLC and Thermo Finnigan LCQ Fleet) on a C4 column. Protein concentrations were determined by absorbance at 280 nm using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Generation and Production of In Tandem VHH Dimers
In tandem bispecific N7D12C-N4E4C and the converse orientation N4E4C-N7D12C VHHs connected by [G4S]10 of [G4S]20 linkers and one C-terminal cysteine and Flag-8xHis-tags were synthesized as described before.32 In short, reading frames encoding 7D12 and 4E4 were PCR amplified with primer sets that allowed fusion of the two VHHs, separated by [G4S]10 or [G4S]20 linkers. The N-terminal VHH was amplified with M13rev and primer (Rev10GS agtaGGATCCGCCACCTCCACTGCCACCGCCACCtgaggagaccgtgacctgggtccc) annealing to the VTVSS sequence of the VHH thereby introducing the linker including a BamHI site. The C-terminal VHH was amplified with M13 and a primer (Forw10GS tcttGGATCCGGCGGGGGAGGTAGTGGGGGTGGGGGCTCAgaggtgcagctggtggagtctggg) annealing to EVQLV of the VHH introducing the rest of the linker and also including a BamHI site. The resulting fragments were respectively cut with SfiI/BamHI and BamHI/Eco91I and subsequently ligated into the vector (SffiI/Eco91I) in a three point ligation. Plasmids were validated by Sanger sequencing, and proteins expressed in E. coli strain TG1 and purified as described above. VHHs were analyzed by SDS-PAGE under reducing conditions.
Sortase A Production and Purification
Plasmid pGBMCS-SortA (Addgene, Cambridge, MA, USA, plasmid #21931), encoding sortase A with a deletion of amino acids 1–59 and an N-terminal Gb1 domain was transformed into E. coli ER2566 and protein expression induced using standard conditions as used for VHHs. After harvesting cells by centrifugation and resuspension in 50 mM TRIS pH 8.0, 150 mM NaCl, and Cocktail protease inhibitor (Roche), cells were lysed by sonication at 4 °C using a Bandalin Sonopuls HD2070 sonicator. The bacterial extracts were cleared by centrifugation and the 6xHis-tagged sortase A Δ59 was purified with Ni-NTA sepharose as described for VHHs. The purified sortase A Δ59 was dialyzed in a 3.5 kDa membrane against 50 mM TRIS pH 7.5 and 150 mM NaCl. The protein was analyzed by SDS-PAGE under reducing conditions (CBB staining) and LC-MS. Protein concentration was determined by absorbance at 280 nm using a Nanodrop spectrophotometer.
Fluorescein-5-Maleimide Conjugation
Monomeric VHH-C-LPETG-8xHis-Vsv and in tandem bispecific NVHHaC-[G4S]n-NVHHbC-C-Flag-6xHis (typically ∼50–250 μM) were incubated for 15 min at RT with 20 mM tris(2-carboxyethyl)phosphine (TCEP, Thermo Fisher Scientific, Waltham, MA, USA) to reduce the free thiol group. TCEP was removed by filtration over a 10 kDa MWCO centrifugal unit (Amicon, Millipore, Billerica, MS, USA) employing 5 washing cycles with 20 mM phosphate pH 7.0, 150 mM NaCl, and 5 mM EDTA. Fluorescein-5-maleimide (Thermo Fisher Scientific, Waltham, MA, USA) from a 10 mM stock in dimethylformamide was reacted with the reduced VHH construct in a 3:1 molar ratio, shielded from light and at RT for 2 h to yield VHH[Fluo] or NVHHAC-[G4S]10/20NVHHBC[Fluo]. Excess fluorescein-5-maleimide was removed by filtration in a 10 kDa MWCO centrifugal unit employing 4 washing cycles with 50 mM Tris pH 7.5 and 150 mM NaCl. Reaction products were analyzed by SDS-PAGE under reducing conditions and/or LC-MS. Fluorescein signal was visualized on a Chromato-vue TM-20 Transilluminator (UVP) after which the gel was stained with CBB. Protein concentration was determined by ultraviolet absorbance at 494 nm (ε494,7D12-FLUO = 70 000 M–1 cm–1) using a Nanodrop spectrophotometer.
Sortase A Mediated Conjugation of Click Moieties
To produce clickable fluorescent VHHs, 50 μM VHH[Fluo] was incubated overnight at RT in the dark with 50 μM sortase A Δ59 and 4.0 mM H2N-PEG3-N3 or H2N-PEG3-DBCO (Jena Biosciences, Jena, Germany) in 50 mM Tris pH 7.5, 150 mM NaCl supplemented with 10 mM CaCl2. This reaction induces the covalent linkage of PEG linkers via the amine group to the threonine in the LPETG tag, releasing G-8xHis-Vsv. Sortase A Δ59, the cleaved G-8xHis-Vsv tag from the reacted VHH, and residual intact VHH were removed by adsorption to Ni-NTA sepharose, pre-equilibrated with 50 mM phosphate pH 7.4 and 500 mM NaCl, for 1 h at 4 °C. Then, the excess of unreacted PEG linkers was removed by filtration in a 10 kDa MWCO centrifugal unit. The reaction product was washed three times with 50 mM phosphate pH 7.4 and 500 mM NaCl, and two times with 50 mM phosphate pH 7.4 and 500 mM NaCl containing 20% w/v glycerol. The protein constructs were analyzed by SDS-PAGE under reducing conditions and LC-MS, and protein concentration was determined by ultraviolet absorbance at 494 nm.
To check if LPET-PEG-X is a substrate for sortase A, 20 μM 7D12[Fluo], 20 μM 7D12[Fluo]-N3-DBCO-biotin, and 7D12-GGG-peptide were incubated with 50 μM sortase A Δ59 for 4 h at 30 °C. Reaction products were analyzed on a CBB stained SDS-PAGE gel and a Western blot stained with 1:10.000 Avidin-Alexa680 (Invitrogen).
Generation of VHH Dimers via C-to-C Conjugation
Tail to tail coupled bispecific VHHs were produced by incubating 4E4-N3 with 7D12[Fluo]-DBCO in a 1:1 molar ratio o/n at RT. Bispecific N7D12C[Fluo]-C4E4N was separated from single VHHs by separation on a G75 Sephadex (Pharmacia fine chemicals, Uppsala, Sweden) column. Fractions were analyzed on a SDS-PAGE gel under reducing conditions (CBB staining).
Synthesis of Diblock Copolymers
Ben-PCL7-PEG2000-methoxy
The amphiphilic diblock copolymer consisting of the hydrophobic poly(ε-caprolactone) (PCL) and the hydrophilic methoxy-polyethylene glycol (mPEG) were synthesized using a method described by Wennink et al. (submitted for publication). Briefly, Ben-PCL-OH macromers were prepared using benzyl alcohol as the initiator in the Tin(II) ethylhexanoate (Sn(Oct)2) catalyzed ring opening polymerization of ε-caprolactone (CL) (Figure 7A). The monomer to initiator ratio was chosen such that the average degree of polymerization was 7 CL units. The hydroxyl end groups of ben-PCL7-OH were subsequently reacted with p-nitrophenyl chloroformate to form p-nitrophenyl carbonate substituted polymers (PNC) (Figure 7B). The diblock copolymers were obtained by reacting ben-PCL7-PNC and mPEG2000-NH2 at 1:1 ratio in toluene at RT (Figure 7C). Average molecular weights of the polymers were determined by NMR.
Figure 7.

Synthesis routes for ben-PCL7-mPEG2000 and ben-PCL7-mPEG2000-DBCO. Synthesis of both molecules starts with (A) ring opening polymerization of ε-caprolactone with benzyl alcohol which affords ben-PCL7-OH and (B) activation of the hydroxyl end group with p-nitrophenylchloroformate. Then for synthesis of amphiphilic ben-PCL7-mPEG2000, (C) mPEG2000-NH2 is coupled to the activated ben-PCL7-PNC. For synthesis of clickable ben-PCL7-mPEG2000-DBCO, (D) NH2-PEG2000-NHBoc is coupled to the activated ben-PCL7-PNC, (E) the Boc protection group is removed with HCl gas, and (F) NHS-DBCO is conjugated to ben-PCL7-mPEG2000-NH2.
Ben-PCL7-PEG2000-DBCO
NH2-PEG2000-NH-Boc (1.00 g, 0.5 mmol) (Layson Bio, Arab, AL, USA) was added to a solution of ben-PCL7-PNC (0.707 g, 0.5 mmol) in 20 mL dry toluene. This mixture was stirred for 1 h at RT under nitrogen atmosphere (Figure 7D). The mixture was washed at least 6 times with diethyl ether to remove p-nitrophenol. The product was dried in a vacuum oven and obtained as a white powder (yield: 97%). Subsequently, 0.2 g of the powder was dissolved in 20 mL of DCM and the Boc group was removed by bubbling HCL gas through the solution for 15 min (Figure 7E). The product, ben-PCL7-PEG2000-NH2 (0.187 g, 0.063 mmol), was purified by precipitation in diethyl ether and subsequently dissolved in 20 mL of dry DCM. To produce clickable copolymers, dibenzocyclooctyne (DBCO)-NHS (0.025 g, 0.063 mmol) (Jena Biosciences, Jena, Germany) was added to this ben-PCL7-PEG2000-NH2 solution, and the reaction mixture was stirred for 24 h at RT under a nitrogen atmosphere (Figure 7F). The mixture was carefully washed with diethyl ether to remove unreacted DBCO-NHS. The product was dried in a vacuum oven and obtained as a white powder (yield: 61%).
1H (300 MHz) NMR spectra were recorded using a Gemini NMR spectrometer (Varian Associates Inc. NMR instruments, Palo Alto, CA). Polymers were dissolved in CDCl3 at a concentration of 0.015 g mL–1.
Dynamic Light Scattering
The size and the size distribution of empty particles were measured by dynamic light scattering (DLS) using a Malvern CGS-3 multiangle goniometer (Malvern ltd., Malvern), consisting of a HeNe laser source (λ = 632.8 nm, 22 mW output power), temperature controller (Julabo water bath) and a digital correlator ALV-5000/EPP. Time correlation functions were analyzed using the ALV-60X0 Software v 3.X provided by Malvern, to obtain the Z-average hydrodynamic diameter of the particles (Zave) and the particle size distribution (polydispersity index, PDI). The samples were analyzed at 25 °C.
7D12 Functionalized mTHPC Loaded Micelle Preparation
Micelles were formed by film-hydration. To prepare micelles with 10% functional groups on the surface, ben-PCL7-PEG2000 and ben-PCL7-PEG2000-DBCO were dissolved in dichloromethane (DCM, Sigma-Aldrich, Saint Louis, MS, USA) and mixed in a 9:1 weight ratio in a 5 mL glass vial (VWR, Radnor, PA, USA). To this mixture, the hydrophobic photosensitizer mTHPC (meta-tetra(hydroxyphenyl) chlorine, Biolitec AG, Jena, Germany) in tetrahydrofuran (THF, Sigma-Aldrich, Saint Louis, MS, USA) was added at a 5% w/w mTHPC/polymer ratio. DCM and THF were evaporated under vacuum. The obtained polymer film was hydrated to a 20 mg/mL solution in PBS for 2 h at RT, gently heated to 60 °C, and filtered through a 0.22 μm filter. To obtain micelles with 5% surface coverage with VHH 7D12[Fluo], the DBCO-micelles were reacted with 7D12[Fluo]-N3 in a 2:1 molar DBCO:N3 ratio. To ensure efficient click reaction kinetics, we kept the N3 and DBCO concentration >100 μM. Reactions were allowed to proceed overnight in the dark at 4 °C with continuous shaking at 450 rpm. The final product was analyzed by SDS-PAGE under reducing conditions (Fluorescein signal and CBB staining). Nontargeted mTHPC-ben-PCL7-PEG2000 micelles were prepared according to the same procedure, but without incubation with 7D12[Fluo]-N3.
Cell Culture
The EGFR-overexpressing squamous carcinoma cell line A431 and the EGFR-negative glioma cell line E9834 were cultured in DMEM (Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (FCS, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and 40 μg/mL gentamycin (Centrafarm, Etten-Leur, The Netherlands). Cells were incubated at 37 °C in 5% CO2 in a humidified atmosphere.
Flow Cytometry and Confocal Fluorescence Microscopy Analysis
Cells were dissociated from culture flasks with 10 mM EDTA, counted, and transferred to V-bottom-shaped 96-well microplates (BD Biosciences, Franklin Lakes, NJ, USA) at 5 × 105 cells/well. All subsequent steps were done on ice and all washing steps were executed by centrifugation of the plates at 1,500xg for 2 min. Cells were washed twice with PBS and aspecific binding sites were blocked by preincubating cells for 10 min with PBA (PBS, 0.5%BSA, 2% FCS). Subsequently cells were incubated with 1 μM fluorescein-labeled VHHs in PBA for 20 min, washed and resuspended in PBA, and analyzed using the CyAn ADP analyzer (Beckman Coulter, Fullerton, CA, USA).
To analyze uptake of VHH-micelles with flow cytometry, cells were grown to 80% confluency in 8-well chambered glass slides (NUNC, Thermo Fisher Scientific, Waltham, MA, USA) and incubated with 20 μM (=0.6 mg/mL) targeted or nontargeted mTHPC-micelles or equimolar VHH concentrations at 37 °C for 30 min. Cells were washed twice with warm DMEM and dissociated with trypsin at 37 °C. Then, cells were taken up in PBA and analyzed using the CyAn flow cytometer. mTHPC was quantified with parameter FL-8, fluorescein was quantified with parameter FL-1. To visualize binding and uptake with confocal microscopy A431 and E98 cells were grown in 8 wells Lab-Tek borosilicate coverglass chambers (Nunc, ThermoFisher Scientific, Waltham, MA, USA), and incubated with the micelles and controls as described for flow cytometry. After washing, cells were kept in phenol red free DMEM supplemented with 20 mM HEPES, and imaged on a TCS SP8 microscope (Leica Microsystems, Mannheim, Germany) equipped with a HC PL APO C5 40×/0.85 dry objective. During imaging, cells were maintained at 37 °C. The UV405 laser was used for excitation, and emission was collected between 500 and 600 nm for fluorescein and 630 and 730 nm for mTHPC.
Acknowledgments
We thank Prof. Floris van Delft for helpful discussions. The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under the CosmoPHOS-nano Project, grant agreement no 310337.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.6b00638.
Site specificity of maleimide labeling; reconstitution of the sortase A substrate site when conjugating with either H2N-PEG3-X or H2N-GGG; sephadex G75 purification of tail-to-tail dimers; LC-MS characterization of head-to-tail dimers and 1H NMR spectra of intermediate products of micellar building blocks; comparison of sortase A variants; comparison of H2N-PEG-N3 to H2N-GGG incorporation by sortase A; applicability of sortase A conjugations to other VHHs (PDF)
Author Contributions
# Sanne A. M. van Lith and Sander M. J. van Duijnhoven contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Barenholz Y. C. (2012) Doxil(R) - The first FDA-approved nano-drug: Lessons learned. J. Controlled Release 160 (2), 117–34. 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- Dawidczyk C. M.; Kim C.; Park J. H.; Russell L. M.; Lee K. H.; Pomper M. G.; Searson P. C. (2014) State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Controlled Release 187, 133–44. 10.1016/j.jconrel.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou M.; Zheng X.; Men K.; Zhang J.; Wang B.; Lv L.; Wang X.; Zhao Y.; Luo F.; Chen L.; et al. (2009) Self-assembled hydrophobic honokiol loaded MPEG-PCL diblock copolymer micelles. Pharm. Res. 26 (9), 2164–73. 10.1007/s11095-009-9929-8. [DOI] [PubMed] [Google Scholar]
- Molavi O.; Xiong X. B.; Douglas D.; Kneteman N.; Nagata S.; Pastan I.; Chu Q.; Lavasanifar A.; Lai R. (2013) Anti-CD30 antibody conjugated liposomal doxorubicin with significantly improved therapeutic efficacy against anaplastic large cell lymphoma. Biomaterials 34 (34), 8718–25. 10.1016/j.biomaterials.2013.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arachchige M. C.; Reshetnyak Y. K.; Andreev O. A. (2015) Advanced targeted nanomedicine. J. Biotechnol. 202, 88–97. 10.1016/j.jbiotec.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan M.; Liang X.; Yang D.; Pan X.; Li Z.; Wang H.; Shi B. (2016) Epidermal growth factor receptor-targeted peptide conjugated phospholipid micelles for doxorubicin delivery. J. Drug Targeting 24, 111. 10.3109/1061186X.2015.1058800. [DOI] [PubMed] [Google Scholar]
- Stemmler H. J.; Kahlert S.; Siekiera W.; Untch M.; Heinrich B.; Heinemann V. (2005) Prolonged survival of patients receiving trastuzumab beyond disease progression for HER2 overexpressing metastatic breast cancer (MBC). Oncol. Res. Treat. 28 (11), 582–6. 10.1159/000088296. [DOI] [PubMed] [Google Scholar]
- Grigorean V. T.; Ciuhu A. N.; Rahnea Nita G.; Strambu V.; Straja D. N.; Popescu M.; Sandu A. M.; Rahnea Nita R. A. (2014) Efficacy of cetuximab in metastatic colon cancer - case report. Chirurgia (Bucur) 109 (3), 383–9. [PubMed] [Google Scholar]
- de Paula Costa Monteiro I.; Madureira P.; de Vasconscelos A.; Pozza D. H.; de Mello R. A. (2015) Targeting HER family in HER2-positive metastatic breast cancer: potential biomarkers and novel targeted therapies. Pharmacogenomics 16 (3), 257–71. 10.2217/pgs.14.133. [DOI] [PubMed] [Google Scholar]
- Xie C.; Jin J.; Bao X.; Zhan W. H.; Han T. Y.; Gan M.; Zhang C.; Wang J. (2010) Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget 1 (1), 610–21. 10.18632/oncotarget.6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucheray M.; Capelletti M.; Pulido I.; Kuang Y.; Paweletz C. P.; Becker J. H.; Kikuchi E.; Xu C.; Patel T. B.; Al-Shahrour F.; et al. (2015) Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res. 75 (20), 4372–83. 10.1158/0008-5472.CAN-15-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Wang Y.; James M.; Jeong J. H.; You M. (2016) Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 55 (5), 991–1001. 10.1002/mc.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijanka M.; Dorresteijn B.; Oliveira S.; van Bergen en Henegouwen P. M. (2015) Nanobody-based cancer therapy of solid tumors. Nanomedicine (London, U. K.) 10 (1), 161–74. 10.2217/nnm.14.178. [DOI] [PubMed] [Google Scholar]
- Chakravarty R.; Goel S.; Cai W. (2014) Nanobody: the ″magic bullet″ for molecular imaging?. Theranostics 4 (4), 386–98. 10.7150/thno.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmsen M. M.; De Haard H. J. (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77 (1), 13–22. 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghimi S. M.; Rahbarizadeh F.; Ahmadvand D.; Parhamifar L. (2013) Heavy Chain Only Antibodies: A New Paradigm in Personalized HER2+ Breast Cancer Therapy. Bioimpacts 3 (1), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers R. C.; Laeremans T.; Huang L.; De Taeye S.; Verkleij A. J.; Revets H.; de Haard H. J.; van Bergen en Henegouwen P. M. (2007) Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol. Immunother. 56 (3), 303–317. 10.1007/s00262-006-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heukers R.; Altintas I.; Raghoenath S.; De Zan E.; Pepermans R.; Roovers R. C.; Haselberg R.; Hennink W. E.; Schiffelers R. M.; Kok R. J.; et al. (2014) Targeting hepatocyte growth factor receptor (Met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials 35 (1), 601–10. 10.1016/j.biomaterials.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Massa S.; Xavier C.; De Vos J.; Caveliers V.; Lahoutte T.; Muyldermans S.; Devoogdt N. (2014) Site-specific labeling of cysteine-tagged camelid single-domain antibody-fragments for use in molecular imaging. Bioconjugate Chem. 25 (5), 979–88. 10.1021/bc500111t. [DOI] [PubMed] [Google Scholar]
- Witte M. D.; Cragnolini J. J.; Dougan S. K.; Yoder N. C.; Popp M. W.; Ploegh H. L. (2012) Preparation of unnatural N-to-N and C-to-C protein fusions. Proc. Natl. Acad. Sci. U. S. A. 109 (30), 11993–8. 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbarizadeh F.; Ahmadvand D.; Sharifzadeh Z. (2011) Nanobody; an old concept and new vehicle for immunotargeting. Immunol. Invest. 40 (3), 299–338. 10.3109/08820139.2010.542228. [DOI] [PubMed] [Google Scholar]
- Debets M. F.; van Berkel S. S.; Schoffelen S.; Rutjes F. P.; van Hest J. C.; van Delft F. L. (2010) Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. (Cambridge, U. K.) 46 (1), 97–9. 10.1039/B917797C. [DOI] [PubMed] [Google Scholar]
- Chen I.; Dorr B. M.; Liu D. R. (2011) A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U. S. A. 108 (28), 11399–404. 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodink I.; Raats J.; van der Zwaag B.; Verrijp K.; Kusters B.; van Bokhoven H.; Linkels M.; de Waal R. M.; Leenders W. P. (2005) Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel target for diagnosis and therapy?. Cancer Res. 65 (18), 8317–23. [DOI] [PubMed] [Google Scholar]
- Parthasarathy R.; Subramanian S.; Boder E. T. (2007) Sortase A as a novel molecular ″stapler″ for sequence-specific protein conjugation. Bioconjugate Chem. 18 (2), 469–76. 10.1021/bc060339w. [DOI] [PubMed] [Google Scholar]
- Baeuerle P. A.; Reinhardt C. (2009) Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 69 (12), 4941–4. 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Kontermann R. E.; Brinkmann U. (2015) Bispecific antibodies. Drug Discovery Today 20 (7), 838–47. 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Els Conrath K.; Lauwereys M.; Wyns L.; Muyldermans S. (2001) Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 276 (10), 7346–50. 10.1074/jbc.M007734200. [DOI] [PubMed] [Google Scholar]
- Veronese F. M.; Mero A. (2008) The impact of PEGylation on biological therapies. BioDrugs 22 (5), 315–29. 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- Rashidian M.; Wang L.; Edens J. G.; Jacobsen J. T.; Hossain I.; Wang Q.; Victora G. D.; Vasdev N.; Ploegh H.; Liang S. H. (2016) Enzyme-Mediated Modification of Single-Domain Antibodies for Imaging Modalities with Different Characteristics. Angew. Chem., Int. Ed. 55 (2), 528–33. 10.1002/anie.201507596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman J. W.; Carstens M. G.; van Zeeland F.; Helwig C.; Flesch F. M.; Hennink W. E.; van Nostrum C. F. (2008) Photocytotoxicity of mTHPC (temoporfin) loaded polymeric micelles mediated by lipase catalyzed degradation. Pharm. Res. 25 (9), 2065–73. 10.1007/s11095-008-9590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navis A. C.; van Lith S. A.; van Duijnhoven S. M.; de Pooter M.; Yetkin-Arik B.; Wesseling P.; Hendriks W. J.; Venselaar H.; Timmer M.; van Cleef P.; et al. (2015) Identification of a novel MET mutation in high-grade glioma resulting in an auto-active intracellular protein. Acta Neuropathol. 130 (1), 131–44. 10.1007/s00401-015-1420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roovers R. C.; Vosjan M. J.; Laeremans T.; el Khoulati R.; de Bruin R. C.; Ferguson K. M.; Verkleij A. J.; van Dongen G. A.; van Bergen en Henegouwen P. M. (2011) A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int. J. Cancer 129 (8), 2013–24. 10.1002/ijc.26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes A.; Schuuring J.; Boots-Sprenger S.; Hendriks-Cornelissen S.; Dekkers M.; van der Kogel A. J.; Leenders W. P.; Wesseling P.; Jeuken J. W. (2008) Phenotypic and genotypic characterization of orthotopic human glioma models and its relevance for the study of anti-glioma therapy. Brain Pathol. 18 (3), 423–33. 10.1111/j.1750-3639.2008.00141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.