

Abstract
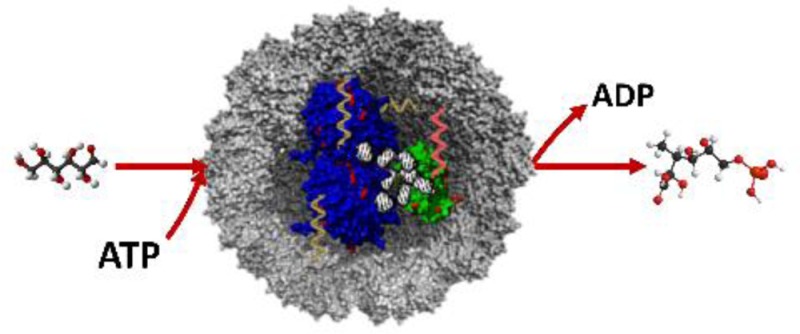
The packaging of proteins into discrete compartments is an essential feature for cellular efficiency. Inspired by Nature, we harness virus-like assemblies as artificial nanocompartments for enzyme-catalyzed cascade reactions. Using the negative charges of nucleic acid tags, we develop a versatile strategy to promote an efficient noncovalent co-encapsulation of enzymes within a single protein cage of cowpea chlorotic mottle virus (CCMV) at neutral pH. The encapsulation results in stable 21–22 nm sized CCMV-like particles, which is characteristic of an icosahedral T = 1 symmetry. Cryo-EM reconstruction was used to demonstrate the structure of T = 1 assemblies templated by biological soft materials as well as the extra-swelling capacity of these T = 1 capsids. Furthermore, the specific sequence of the DNA tag is capable of operating as a secondary biocatalyst as well as bridging two enzymes for co-encapsulation in a single capsid while maintaining their enzymatic activity. Using CCMV-like particles to mimic nanocompartments can provide valuable insight on the role of biological compartments in enhancing metabolic efficiency.
Introduction
In recent years, there is increasing evidence to suggest that prokaryotes adopt primitive organelle-like structures called bacterial microcompartments or nanocompartments, depending on their size. Such assemblies localize and compartmentalize multiple enzymes and substrates involved in specific metabolic pathways.1 The ability to mimic and understand enzymatic activity in confinement would provide ground-breaking insight into these assemblies and in organelles in general.2
One of the main challenges is to controllably package and coencapsulate different enzymes noncovalently within the same compartment as exemplified by Nature. There has been some success using coiled-coil helices,3 peptide tags,4 and protein–protein fusion constructs5 to direct enzymatic cargo encapsulation into protein cages. However, such approaches often lead to covalently connected protein cargo or inefficient loading (i.e., formation of empty cage assemblies). To circumvent these problems, we seek a versatile approach that would promote a noncovalent co-encapsulation of enzymes within a single protein cage in vitro.
The cowpea chlorotic mottle virus (CCMV) is an ideal candidate to mimic bacterial nanocompartments, owing to its size and biocompatibility. The CCMV capsid is 28 nm in diameter and is based on a T = 3 lattice (T, triangulation number), with 12 pentamers and 20 hexamers of identical monomers of capsid protein (CP) organized as 90 dimers.6 Similar to bacterial compartments,1c it has multiple pores in the capsid shell (around 2 nm),7 which allows molecules and substrates to diffuse in and out. The CP N-terminal region is enriched in positively charged residues, termed the arginine-rich motif (ARM), that face the capsid inner surface. After removal of native single-strand (ss) RNA cargo, the ARM can trigger reassembly of capsid protein dimers in the presence of an appropriate negatively charged template, resulting in the formation of monodisperse virus-like particles (VLP).3,8 Encapsulation of enzymes in VLPs has been shown to stabilize and protect the enzymes, and its ease of modification enables new applications.2d
In this contribution, single- and complementary-stranded DNA tags are chemically attached to the exterior of chosen enzymes, resulting in negatively charged complexes that induce the co-encapsulation inside CCMV capsids. Unlike covalent interactions, electrostatic interactions between the DNA tags and the interior of the capsid provide a tunable system, enabled, for instance, by changing the salt concentration or by varying the length of the DNA chains. Hence, this makes our system a model for natural bacterial compartments (e.g., the encapsulins) where the confined enzymes are not covalently bound but rather included in the protein cage by noncovalent, multivalent interactions.2e Noncovalent encapsulation mediated by nucleic acid tags has been reported with the use of genetic engineering9 and/or only focused on a single enzyme.10 Using this strategy, we were able to confine two separate cascade systems in vitro, for which the glucose oxidase (GOx), a 160 kDa dimeric enzyme, is chosen as the primary enzyme for both encapsulated cascades.11 GOx catalyzes the oxidation of glucose into gluconolactone (which undergoes spontaneous hydrolysis into gluconic acid)12 and produces hydrogen peroxide as the side product.11
In the first cascade system, hydrogen peroxide produced by GOx is consumed by the so-called DNAzyme, a peroxidase-mimic formed in situ by a specific sequence of ssDNA in the presence of hemin (Figure 1A).13 In the second cascade system, in the presence of ATP and NADP+, gluconic acid produced by GOx is consumed by a secondary enzyme, gluconokinase (GCK) that is coencapsulated inside the CCMV-like particles, followed by a nonencapsulated tertiary enzyme, 6-phosphogluconate dehydrogenase (6-PGDH), to form ribulose-5-phosphate and NADPH,14 the latter of which can be monitored spectroscopically (Figure 1B). Therefore, the nucleic acid tags in this work are useful both as a secondary biocatalyst (in cascade system I) and as negatively charged tags to trigger the encapsulation of the enzyme(s) (in cascade systems I and II).
Figure 1.

Schematic representation of the enzyme pathways (encapsulated processes shown in gray boxes). (A) Cascade reaction by GOx–DNAzyme. GOx oxidizes glucose to gluconic acid and produces H2O2, which DNAzyme uses for subsequent reaction with ABTS inside CCMV capsid. (B) Cascade reaction performed by co-encapsulated GOx and GCK. The conversion of glucose to d-gluconate-6-P occurs at the interior of the CCMV capsids, whereas the conversion of d-gluconate-6-P into ribulose-5-P occurs at the exterior of the CCMV capsid catalyzed by tertiary enzyme, 6-PGDH.
Results and Discussion
Encapsulation of Enzymes Promoted by DNA Anchor
We anticipated that functionalization and subsequent hybridization of GOx and GCK with (complementary) single-stranded DNA strands should promote their co-encapsulation into CCMV capsids (Figure 2). We suspect that the size of the enzyme cargo as well as the number and spatial distribution of negative charges anchored to the enzyme surface might play a role in determining the efficiency of the encapsulation. To modify and hybridize the relevant enzymes, the lysine residues of GOx and GCK were functionalized with a heterobifunctional linker using sulfo-NHS coupling followed by maleimide–thiol chemistry on the single stranded DNA (ssDNA) or its complementary sequence (csDNA), respectively (see Figures S1 and S2). All hybridized complexes were purified initially by spin-filtration to remove excess DNA. Furthermore, size-exclusion chromatography (SEC) was used for the GOx–GCK dual-enzyme complex to remove nonhybridized GCK and GOx prior to encapsulation (Figure S3).
Figure 2.
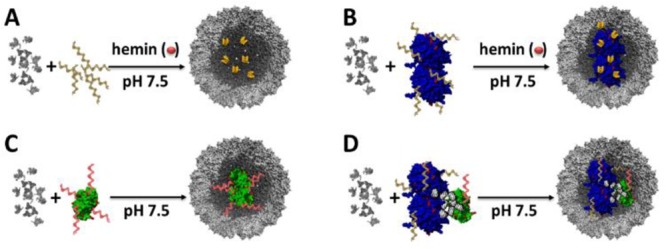
Encapsulation of different enzyme–DNA hybrids inside CCMV capsids (gray) at pH 7.5. Encapsulation of (A) ssDNA in yellow, (B) GOx, in blue, functionalized with ssDNA, (C) GCK, in green, functionalized with the complementary ssDNA in red, (D) GOx conjugated to GCK. The specific sequence of ssDNA is catalytically active in the presence of hemin.
Encapsulation of GOx–ssDNA, GCK–csDNA, or hybridized GOx–GCK in CCMV at pH 7.5 led to the formation of stable capsid-like assemblies, which were purified by SEC (Figure 3A–D). The elution volume (12 mL), together with the relative absorbance ratio (260 nm/280 nm) > 1, are characteristic features of intact CCMV capsids containing DNA-based cargo. Control experiments with no DNA tags (i.e., nonfunctionalized GOx and/or GCK) confirmed that enzymes lacking ssDNA or csDNA cannot be encapsulated (Figure S4). As anticipated, the DNA strands provide the required negative charges for reassembly into virus-like particles. This further ensures that no empty particles are obtained with this strategy and that formed particles always contain a negatively charged cargo.
Figure 3.

Purification and characterization of enzyme-filled CCMV capsids. Size-exclusion chromatograms for CCMV containing (A) ssDNA, (B) GOx–ssDNA, (C) GCK–csDNA, and (D) GOx–GCK, with monitoring at λ = 260 (red), 280 (blue), and 450 nm (black), for DNA, CCMV, and flavin (GOx), respectively. The schematic cartoons above each peak represent the corresponding particles. Negatively stained transmission electron microscopy of (E) ssDNA, (F) GOx–ssDNA, (G) GCK–csDNA, and (H) GOx–GCK encapsulated CCMV assemblies.
The assembled CCMV-like particles were characterized by negative staining transmission electron microscopy (TEM) (Figure 3E–H)15 and dynamic light scattering (DLS, Figure S5), showing spherical structures of around 20 nm in diameter. The size of around 20 nm indicates the formation of T = 1 icosahedral symmetry that is composed of 60 identical capsid subunits.16 Furthermore, co-encapsulation of GOx and GCK in a single particle was confirmed with SDS-PAGE and Western blot analyses (Figures S6 and S7). Additionally, their concentrations and relative ratios were estimated by gel densitometry, which suggested a GOx/GCK/capsid protein ratio of approximately 1:1.4:60. Since the capsid protein is composed of 60 identical subunits, we estimate that only a single GOx–ssDNA is confined inside CCMV-like particles for cascade system I and a hybrid of 1 GOx–DNA and 1 or 2 GCK–csDNA is confined for cascade system II.
Cryo-EM Reconstruction of Enzyme-Filled Particles
To confirm the assembly of CCMV-like particles with T = 1 icosahedral symmetry, we analyzed the GOx–ssDNA-loaded CCMV-like particles with cryo-electron microscopy (cryo-EM) to calculate their native three-dimensional reconstruction (3DR) (Figure 4A). The sample contained particles with spherical and elongated profiles as well as irregular assemblies (Figure 4A, inset). Two-dimensional classification followed by a three-dimensional classification using Relion software resulted in two sizes of icosahedral capsids with T = 1 architecture.16c Whereas class I capsids were 214 Å in diameter (Figure 4B), class II capsids were 226 Å (Figure 4C).
Figure 4.

Three-dimensional cryo-EM reconstructions of GOx–ssDNA-loaded CCMV capsids. (A) Cryo-electron micrograph of GOx–ssDNA-loaded CCMV capsids. Black arrows indicate elongated particles, and red arrowheads indicate irregular particles. Two-dimensional class averages derived from the final 15481 particle data set (inset). Bar, 50 nm. (B) Surface-shaded representation of the outer surface of the class I T = 1 capsid (diameter 21.4 nm) viewed along a 2-, 3-, and 5-fold axis of icosahedral symmetry (top to bottom). Models of the class I T = 1 capsid, with the front half of the cargo and protein shell removed (right). Protein shell is white, cargo is yellow. Arrow indicates a 2-fold axis of icosahedral symmetry. (C) Surface-shaded representations of the outer surface of the class II T = 1 capsid (diameter 22.6 nm) (as in B).
Capsids I and II made up 50% of the total particles in the sample (70% class I, 30% class II). Both capsids were built of 12 pentamers, with different compacting levels. The cryo-EM images analyzed for processing are in fact snapshots of the dynamic states of the sample; the ratio observed could be due to displacement of dynamic equilibrium toward class I T = 1 capsids (70% class I, 30% class II). The two particle sizes might be related to the reported dynamic swelling of the T = 3 CCMV native capsid, as the size difference of 5% involves a ∼7 Å outward radial expansion and widening of the pores, hinting at possible structural breathing. In both T = 1 capsids, the pentamer bases were strongly connected to the underlying GOx–ssDNA cargo, although the capsid surface pores were distinct. Whereas class I capsid pentamers barely left any space between their lateral contacts, those of class II capsids were clearly separated and left large pores, especially at the icosahedral 2-fold axes (Figure 4B, C, arrows).
Docking of CCMV capsid protein (CP) dimer into the cryo-EM density maps of GOx-ssDNA-loaded T = 1 VLP showed major structural differences of the two classes (Figure 5A,B). Connecting densities between pentamers and cargo were mediated by residues 42–50 of the CP N-terminal region (Figure 5C, dark blue), although the preceding region (residues 27–41) could also be involved (Figure 5C, pink). The CP C-terminal ends were responsible for CP dimer assembly in class I CCMV T = 1 capsids (Figure 5A, arrows; Figure 5D, red). The hinge angle formed between CP dimers in GOx–ssDNA-loaded T = 1 class I capsids was ∼60° (Figure 5D); it resembles that found at the quasi-2-fold axes of the swollen T = 3 CCMV capsid17 and in other CP dimers such as the phthalocyanine-loaded T = 1 VLP.18 CCMV CP dimers are the building blocks of native T = 3 virion capsids as well as of in vitro assembled structures such as tubes19 and icosahedral capsids with T = 1 (containing 30 CP dimers), T = 2 (60 dimers), and T = 3 (90 dimers) architecture.20 The outward expansion of the class II capsid pentamer entailed the disappearance of or a great reduction in dimeric contacts (Figure 5B, arrows) and indicated that these interactions contribute much less to class II capsid stability than to that of class I capsids. Assuming the same building block is involved, the class II capsids are based on pentamers bound weakly by the CP C-terminal ends (Figure 5E, red), which adhere strongly to the polyanionic cargo.
Figure 5.

Pseudoatomic model of GOx–ssDNA-loaded CCMV capsids. (A) T = 1 class I capsid viewed down a 3-fold axis from outside, with docked CCMV CP atomic coordinates. A pentamer is depicted in dark blue. Arrows indicate three dimeric contacts. (B) Class II T = 1 capsid viewed down a 3-fold axis from outside (as in B). (C) Pentamer contacts with the cargo mediated by residues 42–50 in the N-terminal region (dark blue, bottom view). The N-terminal region residues 27–41 might also contribute to cargo–pentamer contacts (pink). (D, E) CP dimers in class I (D) and class II T = 1 capsids (E). Side view (top), top view (bottom). The hinge dihedral angle is indicated. CP monomers in the class II dimer are 6.5 Å further apart than class I dimers (N-terminal 27–32 region is omitted).
After imposing icosahedral symmetry in class I and II capsids, we observed the packed cargo as a hollow sphere (9.6 × 105 and 1.2 × 106 Å3, respectively), with numerous connections to the T = 1 capsid inner surface. A significant difference in cargo packing between these two classes is therefore unlikely. Based on the atomic model of GOx (PDB 1GAL), several copies of GOx could be encapsulated in the capsid, although our biochemical analyses indicated the presence of a GOx dimer only. This discrepancy is probably due to the chemical modification of accessible Lys residues of GOx that are covalently bound to ssDNA. The cargo–capsid connections observed in the 3D cryo-EM maps probably represent the interaction of the ARM region with negatively charged DNA strands. GOx–ssDNA packaging resulted in a slightly disordered icosahedral capsid (also reflected in a limited map resolution), but this cargo enabled structural polymorphism with weak CP interactions in the dimer. Both T = 1 capsids coexist in dynamic equilibrium, probably enabled because the CP–ssDNA interactions are more flexible (or less well-defined) than the CP–ssRNA interactions. To our knowledge, this is the first demonstration of the formation of a T = 1 CCMV-like structure templated by a biological soft material that also displays an extreme capsid swelling.
Confined Catalysis by Enzymatic Pathways
Following structural characterization of enzyme–DNA complexes inside T = 1 CCMV-like particles, we proceeded to monitor the enzymatic activity of both cascade systems to examine whether the encapsulated complexes were still catalytically active. For cascade system I (Figure 1A and Figure 6A), we deliberately chose the ssDNA sequence coupled to the GOx to be that of a hemin-binding DNA quadruplex, the so-called DNAzyme. In the presence of hemin, the ssDNA spontaneously forms a scaffold that mimics the catalytic properties of horseradish peroxidase (HRP).13 The catalytic activity of GOx–DNAzyme was monitored via the production of ABTS+• at λ = 410 nm upon addition of glucose to the system.21 The activity plot obtained for encapsulated GOx–DNAzyme shows that both GOx and DNAzyme remained catalytically active after encapsulation (Figure 6B and Figure S8A). Furthermore, both systems displayed catalytic activity profiles that seem to follow Michaelis–Menten kinetics.22 An increase in both Km values (2.2-fold) and kcat values (1.7-fold) is observed when the system is encapsulated (summarized in Table S1).
Figure 6.

Enzymatic activity in CCMV confinement. (A) Schematic representation of cascade system I in the presence of a competing enzyme, catalase (encapsulated processes shown in gray boxes). (B) Kinetic measurements of cascade system I; the production of ABTS•+ was monitored at λ = 410 nm at different glucose concentrations. (C) Kinetic measurements of cascade system II; the production of NADPH was monitored at λ = 340 nm at different glucose concentrations. (D) Kinetic measurements for the production of ABTS+· in the presence of and after pH inactivation of the competing enzyme, catalase.
For cascade system II consisting of GOx, GCK, and 6-PGDH (Figure 1B), we monitored the formation of the end product, NADPH at λ = 340 nm, upon addition of glucose to the system.14 Both enzymes (GOx and GCK) are therefore required for the reaction and its visualization at 340 nm. The activity profile in Figure 6C and Figure S8B confirms that both enzymes were present in the system and still active upon hybridization and subsequent encapsulation. Comparable to system I, the catalytic profiles of both systems also follow Michaelis–Menten kinetics. While the Km values remain similar for both systems, the kcat values show a 2-fold increase for the encapsulated system (summarized in Table S1).
Based on the recurring trends, a slightly higher turnover number (kcat) upon pathway encapsulation is estimated, although the protein concentration determination by gel densitometry is expected to have a large deviation and consequently also the kcat. An eventual increase might be the result of a local enhancement in effective molarity due to confinement23 or of the channeling effect when multiple enzymes in a cascade pathway are brought to a close proximity inside a confined system.5a
The enzymatic activities observed for both cascade pathways indicated that the substrate glucose was able to diffuse into the capsid shell. We further investigated whether the intermediate of the cascade could also diffuse freely or was trapped inside the cagelike structure during the reaction. In order to confirm the state of the intermediate, we added a catalase enzyme to the bulk solution of encapsulated GOx–DNAzyme (Figure 6A). In a competitive pathway, H2O2 is broken down to water and oxygen by the enzyme catalase, and it can therefore act as an external competitor with the DNAzyme.24 In contrast, if the H2O2 intermediate is trapped inside CCMV (as proposed for the bacterial microcompartments),25 the kinetics of ABTS•+ production should remain unaltered. Instead, we observed almost complete suppression of ABTS•+ production in the presence of catalase (Figure 6D). Only upon lowering the buffer to pH 4 and hence inactivating catalase (t = 60 min) could the H2O2 conversion by DNAzyme be restored while maintaining the stability of the particles, as confirmed by SEC and TEM analyses in Figure S9. This strongly suggested that H2O2 can diffuse out of the CCMV capsid. Taken together, in agreement with a previous report,26 we also observed that the CCMV capsid shell is permeable to small molecules such as H2O2, which can diffuse out of the capsid shell and react with the competing enzyme. Nevertheless, it should be noted that the DNAzyme is likely to exhibit lower catalytic efficiency and lower affinity to H2O2 compared to catalase,27 which could also lead to the diffusion of H2O2 out of the CCMV capsid.
Conclusions
We have presented a highly effective strategy of using single-stranded DNA for the controlled noncovalent packing of enzyme cascades in a single protein capsid assembly. To demonstrate the versatility of this strategy, two different cascade systems based on glucose oxidase were assembled inside the protein shell of CCMV at pH 7.5 and were both shown to be catalytically active. This encapsulation strategy resulted in icosahedral structures of approximately 20 nm, which were further analyzed with 3D cryo-EM. The resulting 3D reconstruction provides the first-time demonstration of T = 1 structured assemblies of CCMV around a biological soft matter template. In addition, an extra-swelling phenomenon was indicated on the basis of the coexistence of two differently sized particles of similar structure and origin. The method presented for assembling virus-like particles can provide a structural and functional basis to analyze bacterial protein organelles and will further improve our understanding of their containment properties and biochemical function.
Experimental Section
Materials
Chemicals were purchased from Sigma-Aldrich unless stated otherwise. The ssDNA (5′-HS-(CH2)6-GGGTAGGGCGGGTTGGGTTTT-3′) and csDNA (5′-HS-(CH2)6-AAAACCCAACCCGCCCTACCC-3′) oligonucleotide sequences were synthesized by Eurofins MWG Operon. For the coupling of DNA to enzymes, the bifunctional cross-linker sulfo-EMCS (N-[ε-maleimidocaproyloxy] sulfosuccinimide ester) was purchased from Pierce. d-Gluconate/d-glucono-δ-lactone assay kit was purchased from Megazyme and used as provided. All other reagents were purchased from Sigma-Aldrich or Fluka unless stated otherwise and were used without further purification. A stock solution of hemin (5 mM) was prepared in DMSO and stored in the dark at 4 °C. Glucose was dissolved in 50 mM Tris–HCl, 100 mM KCl buffer at pH 7.5. All reactions were carried out at room temperature unless stated otherwise.
Encapsulation of ssDNA, GOx–ssDNA, csDNA–GCK, and GOx–GCK in CCMV Virus-like Particles
CCMV was dialyzed against assembly buffer (250 mM Tris–HCl, 500 mM NaCl, 50 mM MgCl2, 1 mM DTT, pH 7.5) to obtain CCMV dimer coat proteins (CCMV-CP) (500 μM). ssDNA, GOx–ssDNA, csDNA–GCK, and GOx–GCK were buffer exchanged against Milli-Q water using Amicon Ultra centrifugal filters (30 kDa or 10 kDa MWCO). For the encapsulation, ssDNA, GOx–ssDNA, csDNA–GCK, and GOx–GCK and CCMV–CP (in assembly buffer) were mixed in a 4:1 (v/v) ratio and incubated for 2 h at 4 °C before purification by size-exclusion chromatography (SEC) using a Superose 6 10/100 GL column, eluting with 50 mM Tris–HCl, 100 mM NaCl, 10 mM MgCl2, 0.5 mM DTT at pH 7.5. Protein fractions were collected and analyzed by SDS–PAGE, agarose gel, and Western blot analysis. For the complete procedure, see theSI.
Transmission Electron Microscopy Analysis
Samples (5 μL) were applied onto Formvar–carbon-coated grids. After 1 min, the excess liquid was drained. Uranyl acetate (5 μL, 1% w/v) was added and the excess liquid was drained after 20 s and dried for 30 min at room temperature. The samples were examined on a FEG-TEM (Phillips CM 30) operated at 300 kV acceleration voltages.
Cryo-electron Microscopy and Image Processing
GOx–ssDNA-loaded VLP (5 μL) were applied to one side of Quantifoil R 2/2 holey grids, blotted, and plunged into liquid ethane in a Leica EM CPC cryofixation unit. The grids were analyzed in a Tecnai G2 electron microscope equipped with a field emission gun operating at 200 kV, and images were recorded under low-dose conditions with a FEI Eagle CCD at a detector magnification of 69,444X (2.16 Å/pixel sampling rate). Image processing operations were performed using Xmipp28 and Relion,29 and graphic representations were produced with UCSF Chimera.30 The Xmipp automatic picking routine was used to select 15481 particles, and defocus was determined with CTFfind.31 Images were 2D-classified using the appropriate Relion routine and 7932 isometric particles were selected. The structure of phthalocyanine-loaded CCMV T = 1 capsid18 was filtered out to 30 Å, and the cargo density was masked. This map was used as an initial model for 3D classification of spherical particles, using Relion to select 5572 (class I) and 2318 (class II) particles; these data sets were used to obtain the final 3DRs using the Relion autorefinement routine. Resolution was assessed by gold standard FSC between two independently processed half-data sets. Applying a correlation limit of 0.5 (0.3), the resolution for class I and II 3D maps was 22.7 (22.2) and 25.6 (21.3) Å, respectively. The Chimera fitting tool was used to dock the atomic structure of a whole pentamer from the X-ray structure of CCMV17a (PDB entry 1CWP) into the cryoEM maps.
Kinetic Measurements of GOx–GCK: NADPH Assay
Substrate solutions containing various glucose concentrations (0–1 M, 180 μL) were prepared. An enzyme solution containing either (1) free GOx–GCK (60 μL) or (2) encapsulated GOx–GCK (60 μL) was used in these studies. 6-PGDH (55 U/mL, 2 μL) and 16.1 mM NADP+ containing 69.4 mM ATP were added to each reaction mixture at pH 7.5, according to the manufacturer’s instructions (Megazyme kit). The reaction was started upon addition of glucose (120 μL) to enzyme (82 μL), and formation of reduced NADPH was monitored at λ = 340 nm in 100 s time intervals over 2 h at 27 °C.
Kinetic Measurements of GOx–DNAzyme: ABTS Assay
Stock solutions containing both substrates glucose (ranging from 0–1 M) and 4 mM ABTS were freshly prepared at room temperature. Individual solutions containing either (1) free GOx–DNAzyme (30 μL) and hemin (30 μL, 5 μM) or (2) encapsulated GOx–DNAzyme (30 μL) and hemin (30 μL, 5 μM) were prepared and incubated at rt for 2 h. To each enzyme containing solution (60 μL) was added the substrate solution containing both glucose and ABTS (120 μL), and the reaction was monitored immediately at λ = 410 nm for the conversion of ABTS to ABTS•+ at 27 °C in 100 s time intervals over 2 h. Control experiments containing hemin, glucose, and ABTS were performed under the same reaction conditions.
Data Analysis
Experimental data were corrected for background absorbance (using the control experiment as a reference). The concentration of ABTS•+ or NADPH was determined using the Lambert–Beer law, assuming extinction coefficients of ABTS•+ (ε410 nm = 36000 M–1 cm–1) or NADPH (ε340 nm = 6300 M–1 cm–1) before plotting concentration (μM) vs time (min) curves, from which the velocity (v) was determined (μM/min). A dilution factor (DF) relative to the enzyme (GOx) and a proportionality factor of ABTS to substrate consumption (p = 1/2) were used to correct the velocity values as described in eq 1.
| 1 |
Acknowledgments
We acknowledge financial support from the ERC Consolidator Grant (Protcage) and the Indonesia Endowment Fund for Education (LPDP). This work was supported in part by grants from the Spanish Ministry of Economy and Competitivity (BFU2014-55475 to JRC) and the Comunidad Autónoma de Madrid (S2013/MIT-2807 to JRC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b10948.
Preparation and purification of enzyme–DNA hybrids, purification of GOx–GCK assemblies, control experiments, densitometry analysis, and Western blot analysis, kinetic profiles, and competing catalase pathway (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Agapakis C. M.; Boyle P. M.; Silver P. A. Nat. Chem. Biol. 2012, 8, 527. 10.1038/nchembio.975. [DOI] [PubMed] [Google Scholar]; b Bonacci W.; Teng P. K.; Afonso B.; Niederholtmeyer H.; Grob P.; Silver P. A.; Savage D. F. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 478. 10.1073/pnas.1108557109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tanaka S.; Sawaya M. R.; Yeates T. O. Science 2010, 327, 81. 10.1126/science.1179513. [DOI] [PubMed] [Google Scholar]; d Heinhorst S.; Williams E. B.; Cai F.; Murin C. D.; Shively J. M.; Cannon G. C. J. Bacteriol. 2006, 188, 8087. 10.1128/JB.00990-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Comellas-Aragones M.; Engelkamp H.; Claessen V. I.; Sommerdijk N. A.; Rowan A. E.; Christianen P. C.; Maan J. C.; Verduin B. J.; Cornelissen J. J. L. M.; Nolte R. J. M. Nat. Nanotechnol. 2007, 2, 635. 10.1038/nnano.2007.299. [DOI] [PubMed] [Google Scholar]; b Kang S.; Douglas T. Science 2010, 327, 42. 10.1126/science.1184318. [DOI] [PubMed] [Google Scholar]; c Uchida M.; Klem M. T.; Allen M.; Suci P.; Flenniken M.; Gillitzer E.; Varpness Z.; Liepold L. O.; Young M.; Douglas T. Adv. Mater. 2007, 19, 1025. 10.1002/adma.200601168. [DOI] [Google Scholar]; d O’Neil A.; Prevelige P. E. Jr.; Douglas T. Biomater. Sci. 2013, 1, 881. 10.1039/c3bm60063g. [DOI] [PubMed] [Google Scholar]; e Rurup W. F.; Snijder J.; Koay M. S. T.; Heck A. J. R.; Cornelissen J. J. L. M. J. Am. Chem. Soc. 2014, 136, 3828. 10.1021/ja410891c. [DOI] [PubMed] [Google Scholar]
- a Minten I. J.; Claessen V. I.; Blank K.; Rowan A. E.; Nolte R. J. M.; Cornelissen J. J. L. M. Chem. Sci. 2011, 2, 358. 10.1039/C0SC00407C. [DOI] [Google Scholar]; b Rurup W. F.; Verbij F.; Koay M. S. T.; Blum C.; Subramaniam V.; Cornelissen J. J. L. M. Biomacromolecules 2014, 15, 558. 10.1021/bm4015792. [DOI] [PubMed] [Google Scholar]
- Patterson D. P.; Prevelige P. E.; Douglas T. ACS Nano 2012, 6, 5000. 10.1021/nn300545z. [DOI] [PubMed] [Google Scholar]
- a Patterson D. P.; Schwarz B.; Waters R. S.; Gedeon T.; Douglas T. ACS Chem. Biol. 2014, 9, 359. 10.1021/cb4006529. [DOI] [PubMed] [Google Scholar]; b Schoonen L.; Pille J.; Borrmann A.; Nolte R. J. M.; van Hest J. C. M. Bioconjugate Chem. 2015, 26, 2429. 10.1021/acs.bioconjchem.5b00485. [DOI] [PubMed] [Google Scholar]
- Caspar D. L.; Klug A. Cold Spring Harbor Symp. Quant. Biol. 1962, 27, 1. 10.1101/SQB.1962.027.001.005. [DOI] [PubMed] [Google Scholar]
- Douglas T.; Young M. Science 2006, 312, 873. 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- a Brasch M.; de la Escosura A.; Ma Y. J.; Uetrecht C.; Heck A. J. R.; Torres T.; Cornelissen J. J. L. M. J. Am. Chem. Soc. 2011, 133, 6878. 10.1021/ja110752u. [DOI] [PubMed] [Google Scholar]; b Bode S. A.; Minten I. J.; Nolte R. J. M.; Cornelissen J. J. L. M. Nanoscale 2011, 3, 2376. 10.1039/c0nr01013h. [DOI] [PubMed] [Google Scholar]
- Fiedler J. D.; Brown S. D.; Lau J. L.; Finn M. G. Angew. Chem., Int. Ed. 2010, 49, 9648. 10.1002/anie.201005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow J. E.; Capehart S. L.; Francis M. B.; Tullman-Ercek D. ACS Nano 2012, 6, 8658. 10.1021/nn302183h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankar S. B.; Bule M. V.; Singhal R. S.; Ananthanarayan L. Biotechnol. Adv. 2009, 27, 489. 10.1016/j.biotechadv.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Salusjarvi T.; Kalkkinen N.; Miasnikov A. N. Appl. Environ. Microbiol. 2004, 70, 5503. 10.1128/AEM.70.9.5503-5510.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub E.; Freeman R.; Willner I. Angew. Chem., Int. Ed. 2011, 50, 11710. 10.1002/anie.201103853. [DOI] [PubMed] [Google Scholar]
- Cohen S. S. J. Biol. Chem. 1951, 189, 617. [PubMed] [Google Scholar]
- Johnson J. E.; Speir J. A. J. Mol. Biol. 1997, 269, 665. 10.1006/jmbi.1997.1068. [DOI] [PubMed] [Google Scholar]
- a Comellas-Aragones M.; de la Escosura A.; Dirks A. T.; van der Ham A.; Fuste-Cune A.; Cornelissen J. J. L. M.; Nolte R. J. M. Biomacromolecules 2009, 10, 3141. 10.1021/bm9007953. [DOI] [PubMed] [Google Scholar]; b Sikkema F. D.; Comellas-Aragones M.; Fokkink R. G.; Verduin B. J.; Cornelissen J. J. L. M.; Nolte R. J. M. Org. Biomol. Chem. 2007, 5, 54. 10.1039/B613890J. [DOI] [PubMed] [Google Scholar]; c Sun J.; DuFort C.; Daniel M.-C.; Murali A.; Chen C.; Gopinath K.; Stein B.; De M.; Rotello V. M.; Holzenburg A.; Kao C. C.; Dragnea B. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1354. 10.1073/pnas.0610542104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Speir J. A.; Munshi S.; Wang G.; Baker T. S.; Johnson J. E. Structure 1995, 3, 63. 10.1016/S0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu H.; Qu C.; Johnson J. E.; Case D. A. J. Struct. Biol. 2003, 142, 356. 10.1016/S1047-8477(03)00028-5. [DOI] [PubMed] [Google Scholar]
- Luque D.; de la Escosura A.; Snijder J.; Brasch M.; Burnley R. J.; Koay M. S. T.; Carrascosa J. L.; Wuite G. J. L.; Roos W. H.; Heck A. J. R.; Cornelissen J. J. L. M.; Torres T.; Caston J. R. Chem. Sci. 2014, 5, 575. 10.1039/C3SC52276H. [DOI] [Google Scholar]
- a Mukherjee S.; Pfeifer C. M.; Johnson J. M.; Liu J.; Zlotnick A. J. Am. Chem. Soc. 2006, 128, 2538. 10.1021/ja056656f. [DOI] [PubMed] [Google Scholar]; b de la Escosura A.; Janssen P. G.; Schenning A. P.; Nolte R. J. M.; Cornelissen J. J. L. M. Angew. Chem., Int. Ed. 2010, 49, 5335. 10.1002/anie.201001702. [DOI] [PubMed] [Google Scholar]; c Ng B. C.; Chan S. T.; Lin J.; Tolbert S. H. ACS Nano 2011, 5, 7730. 10.1021/nn202493w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lavelle L.; Gingery M.; Phillips M.; Gelbart W. M.; Knobler C. M.; Cadena-Nava R. D.; Vega-Acosta J. R.; Pinedo-Torres L. A.; Ruiz-Garcia J. J. Phys. Chem. B 2009, 113, 3813. 10.1021/jp8079765. [DOI] [PubMed] [Google Scholar]; b Cadena-Nava R. D.; Hu Y.; Garmann R. F.; Ng B.; Zelikin A. N.; Knobler C. M.; Gelbart W. M. J. Phys. Chem. B 2011, 115, 2386. 10.1021/jp1094118. [DOI] [PubMed] [Google Scholar]; c van Eldijk M. B.; Wang J. C.; Minten I. J.; Li C.; Zlotnick A.; Nolte R. J. M.; Cornelissen J. J. L. M.; van Hest J. C. M. J. Am. Chem. Soc. 2012, 134, 18506. 10.1021/ja308132z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyahovsky B.; Li D.; Katz E.; Willner I. Biosens. Bioelectron. 2007, 22, 2570. 10.1016/j.bios.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Johnson K. A.; Goody R. S. Biochemistry 2011, 50, 8264. 10.1021/bi201284u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Minton P. A. J. Biol. Chem. 2001, 276, 10577. 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]; b Chen Q.; Schonherr H.; Vancso G. J. Small 2009, 5, 1436. 10.1002/smll.200801455. [DOI] [PubMed] [Google Scholar]
- Kirkman H. N.; Gaetani G. F. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 4343. 10.1073/pnas.81.14.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penrod J. T.; Roth J. R. J. Bacteriol. 2006, 188, 2865. 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D. P.; McCoy K.; Fijen C.; Douglas T. J. Mater. Chem. B 2014, 2, 5948. 10.1039/C4TB00983E. [DOI] [PubMed] [Google Scholar]
- a Tao Z. M.; Raffel R. A.; Souid A. K.; Goodisman J. Biophys. J. 2009, 96, 2977. 10.1016/j.bpj.2008.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deisseroth A.; Dounce A. L. Physiol. Rev. 1970, 50, 319. [DOI] [PubMed] [Google Scholar]
- Marabini R.; Masegosa I. M.; San Martin M. C.; Marco S.; Fernandez J. J.; de la Fraga L. G.; Vaquerizo C.; Carazo J. M. J. Struct. Biol. 1996, 116, 237. 10.1006/jsbi.1996.0036. [DOI] [PubMed] [Google Scholar]
- Scheres S. H. J. Struct. Biol. 2012, 180, 519. 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Mindell J. A.; Grigorieff N. J. Struct. Biol. 2003, 142, 334. 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.