

Neuropathy of the enteric nervous system (ENS) is one of the major underlying causes of debilitating gastrointestinal (GI) motility disorders in diabetic patients. Recent studies suggest that diet–microbiome–host interactions – in particular, excess dietary calories, microbial metabolites, lipopolysaccharide (LPS) and disrupted mucosal barrier – play a fundamental role in the pathobiology of obesity and type II diabetes (Boulangé et al. 2016). Furthermore, the composition of the GI microbiome influences ENS physiology, neurochemistry and nerve cell health, as well as GI motility patterns, and vice versa (Kashyap et al. 2013). However, links between such interactions and the mechanisms underlying this neuropathy are not fully understood.
In this issue of The Journal of Physiology, Reichardt et al. (2017) address the question of whether ingesting a Western diet (WD) rich in saturated fatty acids and the associated alteration to the gut microbiome disrupts motility, and induces loss of nitrergic myenteric neurons (NMNs), the phenotype that is commonly damaged in diabetic neuropathy (Yarandi & Srinivasan, 2014). The rationale is that most studies have used a high fat diet (HFD; 60–72% kcal from fat), leading to little understanding of how a normal WD affects GI motility, the ENS and their role in the pathobiology of the metabolic syndrome and diabetes.
The authors used C57BL/6 mice fed WD (35% kcal from fat, enriched in palmitate) or a regular diet (RD, 16.9% kcal from fat, 4× less palmitate) for 3, 6, 9 and 12 weeks, and TLR4–/– and germ free mice fed WD and RD diets for 6 weeks. Gastrointestinal motility was measured, and damage to myenteric neurons and NMNs was studied in the ileum and proximal colon. Palmitate‐ and LPS‐induced damage to NMNs and the role of nitric oxide synthase (nNOS) in such injury were determined in vitro using immortalized myenteric neurons. Faecal metabolites, systemic and visceral fat and mucosal inflammation were analysed.
After ingesting WD for 6 weeks, mice were ‘overweight’, developed gut microbiota dysbiosis, altered faecal metabolites, increased intraluminal LPS and increased plasma free fatty acid (FFA) levels. Interestingly, unlike HFD, WD did not elicit hyperglycaemia, endotoxaemia and inflammation, suggesting the need to define key differences between the effect of HFD and WD on gut microbiome and metabolic profiles. Another important observation was that WD caused GI dysmotility before the overt loss of NMNs. This suggests that ENS functional changes occur before injuries can be seen microscopically. It highlights the need to study the effects of WD on the ENS and neuromuscular physiology.
HFD causes apoptotic loss of NMNs in the small and large intestine via an activated caspase‐3 signalling pathway. Reichardt et al.’s findings suggest that FFAs from WD trigger NMN cell death in colon before ileum. WD‐induced damage to NMNs is not associated with the activation of caspase‐3. Clearly, these data support the view that WD‐ and HFD‐induced ENS injuries have no easy answers, and instead need better understanding.
WD disruption of GI motility and the loss of NMNs depended on FFA‐ and LPS‐induced activation of TLR4, emphasizing the role of TLR4 in linking diet–microbiome–GI interactions to the development of ENS neuropathy and dysmotility. However, LPS and FFAs activate TLR4, and so can stimulate pro‐inflammatory pathways in a variety of cell types. One would expect elevated FFAs to activate TLR4 in adipose tissue and lead to inflammation and even insulin resistance (Kim & Sears, 2010). Therefore, the observation that increased FFA levels was not associated with inflammation or insulin resistance (indirectly shown by normoglycaemia) is interesting but needs further study. Noteworthy, LPS and palmitate acted synergistically to cause nerve cell death in vitro. Their actions depended on the generation of nitric oxide, because inhibition of nNOS with l‐NAME rescued NMNs from LPS/palmitate‐induced cell death. This is an interesting and provocative observation highlighting the importance of dietary and microbial product interaction in the pathobiology of enteric neuropathy and dysmotility. It raises many unanswered questions including the following. What are the factors triggering dysmotility in the whole GI tract prior to FFA‐induced loss of NMNs in colon? Does WD, and by extension FFAs, disrupt motility by damaging interstitial cells of Cajal or smooth muscle cells? Does elevated intraluminal LPS affect neurotransmission and motility by signalling to enteric neurons via intermediaries such as enterocytes or enteroendocrine cells?
Collectively, this paper improves our understanding of the pathophysiology of enteric nerve cell injuries and GI dysmotility associated with the onset of metabolic syndrome and diabetes (Fig. 1). It provides a new model to study the development of GI dysmotility and neuropathy associated with these conditions, and increases our need to understand GI neurodegenerative processes triggered by normal fat diets.
Figure 1. Excess saturated FFAs in WD causes dysbiosis of the gut microbiome, dysmotility and nerve cell injury in the myenteric plexus.
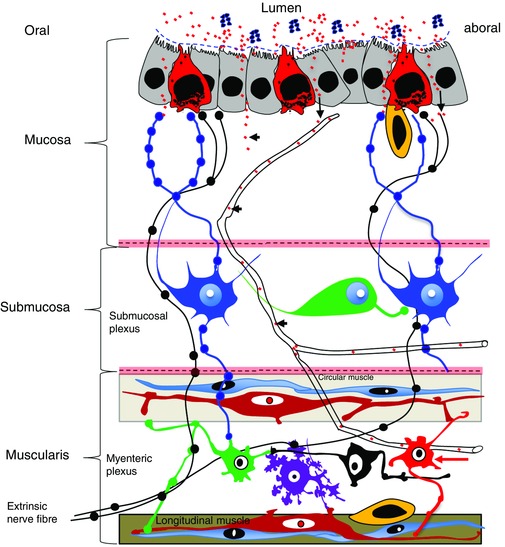
Elevated levels of FFAs (red dots) in the intestinal lumen and circulation (arrowheads) cause dysmotility prior to triggering microscopically visible damage to NMNs (red arrow). This suggests that FFAs alter the physiology of the ENS directly, or indirectly via interaction with enterocytes (grey), enteroendocrine cells (red), interstitial cells of Cajal (red cells in the muscularis), smooth muscle (light blue cells in the muscularis), gut macrophages (yellow) and enteric glia (not shown). WD‐induced dysbiosis causes an increase of intraluminal LPS (blue dots). Whether intraluminal LPS influences motility and ENS remains to be determined, because in vitro assays show that FFAs acts synergistically with LPS via TLR4 to damage NMNs. It is likely that dysmotility and damage to the ENS are crucial for disrupting mucosal barrier function, which is known to have a key role in causing endotoxaemia.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Linked articles This Perspective highlights an article by Reichardt et al. To read this paper, visit https://doi.org/10.1113/JP273269.
This is an Editor's Choice article from the 1 March 2017 issue.
References
- Boulangé CL, Neves AL, Chilloux J, Nicholson JK & Dumas M‐E (2016). Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million M, Tache Y, Pasricha PJ, Knight R, Farrugia G & Sonnenburg JL (2013). Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterol 144, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ & Sears DD (2010). TLR4 and insulin resistance. Gastroenterol Res Pract 2010, 212563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt F, Chassaing B, Nezami B, Li G, Tabatabavakili S, Mwangi S, Uppal K, Liang B, Vijay‐Kumar M, Jones D, Gewirtz A & Srinivasan S (2017). Western diet induces colonic nitrergic myenteric neuropathy and dysmotility in mice via saturated fatty acid‐ and LPS‐induced TLR4 signaling. J Physiol 595, 1831–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarandi SS & Srinivasan S (2014). Diabetic gastrointestinal motility disorders and the role of enteric nervous system: current status and future directions. Neurogastroenterol Motil 26, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]