

Abstract
Key points
The genetically encoded fluorescent calcium integrator calcium‐modulated photoactivatable ratiobetric integrator (CaMPARI) reports calcium influx induced by synaptic and neural activity. Its fluorescence is converted from green to red in the presence of violet light and calcium.
The rate of conversion – the sensitivity to activity – is tunable and depends on the intensity of violet light.
Synaptic activity and action potentials can independently initiate significant CaMPARI conversion.
The level of conversion by subthreshold synaptic inputs is correlated to the strength of input, enabling optical readout of relative synaptic strength.
When combined with optogenetic activation of defined presynaptic neurons, CaMPARI provides an all‐optical method to map synaptic connectivity.
Abstract
The calcium‐modulated photoactivatable ratiometric integrator (CaMPARI) is a genetically encoded calcium integrator that facilitates the study of neural circuits by permanently marking cells active during user‐specified temporal windows. Permanent marking enables measurement of signals from large swathes of tissue and easy correlation of activity with other structural or functional labels. One potential application of CaMPARI is labelling neurons postsynaptic to specific populations targeted for optogenetic stimulation, giving rise to all‐optical functional connectivity mapping. Here, we characterized the response of CaMPARI to several common types of neuronal calcium signals in mouse acute cortical brain slices. Our experiments show that CaMPARI is effectively converted by both action potentials and subthreshold synaptic inputs, and that conversion level is correlated to synaptic strength. Importantly, we found that conversion rate can be tuned: it is linearly related to light intensity. At low photoconversion light levels CaMPARI offers a wide dynamic range due to slower conversion rate; at high light levels conversion is more rapid and more sensitive to activity. Finally, we employed CaMPARI and optogenetics for functional circuit mapping in ex vivo acute brain slices, which preserve in vivo‐like connectivity of axon terminals. With a single light source, we stimulated channelrhodopsin‐2‐expressing long‐range posteromedial (POm) thalamic axon terminals in cortex and induced CaMPARI conversion in recipient cortical neurons. We found that POm stimulation triggers robust photoconversion of layer 5 cortical neurons and weaker conversion of layer 2/3 neurons. Thus, CaMPARI enables network‐wide, tunable, all‐optical functional circuit mapping that captures supra‐ and subthreshold depolarization.
Keywords: brain slices, CaMPARI, calcium indicator, connectivity, immediate early genes, mapping, optogenetics, thalamocortical
Key points
The genetically encoded fluorescent calcium integrator calcium‐modulated photoactivatable ratiobetric integrator (CaMPARI) reports calcium influx induced by synaptic and neural activity. Its fluorescence is converted from green to red in the presence of violet light and calcium.
The rate of conversion – the sensitivity to activity – is tunable and depends on the intensity of violet light.
Synaptic activity and action potentials can independently initiate significant CaMPARI conversion.
The level of conversion by subthreshold synaptic inputs is correlated to the strength of input, enabling optical readout of relative synaptic strength.
When combined with optogenetic activation of defined presynaptic neurons, CaMPARI provides an all‐optical method to map synaptic connectivity.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AP
action potential
- AP5
(2R)‐amino‐5‐phosphonopentanoic acid
- CaMPARI
calcium‐modulated photoactivatable ratiometric integrator
- ChR2
Channelrhodopsin‐2
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- GECI
genetically encoded calcium indicator
- MCPG
(RS)‐α‐methyl‐4‐carboxyphenylglycine
- S1
primary somatosensory cortex
- POm
posteromedial thalamic nucleus
Introduction
Genetically encoded calcium indicators (GECIs) are invaluable tools for measuring neuronal activity (Nakai et al. 2001; Tian et al. 2009; Lütcke et al. 2010; Akerboom et al. 2012; Chen et al. 2013). They can be used for real‐time measurement of activity in populations of neurons and/or within subcellular compartments (Chen et al. 2011; Petreanu et al. 2012; Andermann et al. 2013). However, GECIs provide a transient, local readout of activity that can require sophisticated and/or invasive imaging techniques for monitoring fluorescence in real time in predesignated neurons during behaviour. In addition, at the soma GECIs only faithfully report action potentials, not subthreshold synaptic input. Alternatively, immediate early gene (IEG) markers have been used to permanently tag active neurons (Morgan et al. 1987; Worley et al. 1993; Steiner and Gerfen, 1994; Staiger et al. 2000; Barth et al. 2004) but their induction requires minutes to hours and convolves numerous cellular processes, many unrelated to specific bouts of neural activity. These limitations of GECIs and IEG‐based tools can potentially be circumvented with the genetically encoded fluorescent calcium integrator CaMPARI (calcium‐modulated photoactivatable ratiobetric integrator), because it integrates calcium activity over reasonably short, user‐defined epochs.
CaMPARI is a fluorescent protein that photoconverts from green to red in the presence of user‐supplied violet light (∼400 nm), in a manner that dramatically increases with Ca2+ concentration. The resulting covalent fluorophore modification is permanent; thus the red signal remains for hours to weeks (governed by cellular protein turnover kinetics), providing a post hoc readout of active neurons.
CaMPARI has been used in vitro in cultured neurons and in vivo in mice, Drosophila and larval zebrafish to mark circuits activated during behaviour, and in Drosophila has been combined with CsChrimson stimulation of an upstream network to mark disynaptically separated active neurons (Fosque et al. 2015). As with any activity reporter, a critical aspect of interpreting experiments using CaMPARI is to carefully calibrate readout versus the underlying phenomenon under study; importantly, such calibrations should take place in the preparation of interest (when possible), as factors such as expression level, long‐term stability, and light delivery and collection can vary widely. Calcium entry into neurons is dynamic and triggered by many sources (Sabatini et al. 2002), including action potentials (APs) and synaptic activity.
In this study, we used whole‐cell patch‐clamp recordings to assess the activity‐dependent photoconversion of CaMPARI in acute mouse brain slices in response to activity, including synaptic potentials. Calibration in intact neural tissue constitutes an important step towards interpreting signals from behaving animals. Our experiments reveal that synaptic potentials can convert CaMPARI in a synaptic strength‐dependent manner. Thus, in any intact circuit at least a portion of the CaMPARI conversion can reflect subthreshold synaptic activity, i.e. where no action potentials are elicited. Moreover, and of particular importance to circuit mapping, it is possible to optically extract information about relative input strength, thus enabling large‐scale optical mapping of subthreshold inputs.
In addition to calibrating CaMPARI in an intact neural circuit, we explored the effect of photoconversion light intensities on photoconversion rate. We found that photoconversion is successful at low light intensities, which is appropriate for large tissue volume‐level photoconversion experiments and simultaneously reduces phototoxicity. Moreover, we show that conversion rate is linearly dependent on light intensity level. This property of CaMPARI can be strategically utilized for optimization of specific experimental objectives. In addition, with this relationship it is possible in principle to compare activity of neurons exposed to different illumination intensities, such as occurs most dramatically in vivo, where light delivery can be problematic.
Finally, we demonstrate that CaMPARI facilitates functional circuit mapping. Currently there are few effective, high‐throughput methods for accurately and rapidly estimating the synaptic drive from one brain region to another. Here, we show that CaMPARI, in conjunction with genetically targeted optogenetic effectors, is a powerful tool for the study of supra‐ as well as subthreshold connectivity in ex vivo acute brain slices. This technique can, in principle, be employed to measure functional synaptic connectivity between virtually any brain regions. By providing a paradigm for post hoc all‐optical anatomical and functional mapping this method can accelerate the study of functional connections throughout the brain.
Methods
All procedures were performed in accordance with protocols approved by the Charité Universitätsmedizin Berlin and the Berlin LAGeSo for the care and use of laboratory animals.
Slice preparation
Coronal slices, 300 μm thick, were prepared from wild‐type female C57/BL6J mice (supplied by the Charité Universitätsmedizin Berlin FEM; postnatal ages P30–P60). The artificial cerebrospinal fluid (ACSF) used for recordings and brain slicing contained (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2 and 25 d‐glucose, pH ∼7.4. Immediately following slice preparation, the slices were incubated at 32°C for 10 min in a solution containing (in mm): 93 N‐methyl‐d‐glucamine, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 Hepes, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, 0.5 CaCl2 and 25 d‐glucose, pH ∼7.4, followed by 20 min at 32°C in ACSF and then at room temperature, before recording in a submersion chamber at 32°C. All solutions were saturated with 95% O2/5% CO2 and had pH ∼7.4. In some experiments tetrodotoxin (TTX; 1 μm, Tocris Bioscience, Bristol, UK), (2R)‐amino‐5‐phosphonopentanoic acid (AP5; 50 μm, Sigma‐Aldrich, St Louis, MO, USA), 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX; 20 μm, Sigma‐Aldrich), and/or (RS)‐α‐methyl‐4‐carboxyphenylglycine (MCPG; 200 μm, Tocris) were bath applied to block sodium action potentials (TTX), ionotropic glutamate receptors (AP5 and CNQX block NMDA and AMPA/kainate receptors, respectively), and group I and II metabotropic glutamate receptors (MCPG). Calcium was replaced with magnesium in ‘zero calcium’ ACSF. Note that even in the ‘zero calcium’ solution, nanomolar calcium may be present.
Recording and stimulation
Whole‐cell current clamp recordings were made with high resistance (low dialysis) microelectrodes (> 16 MΩ). When using low resistance ∼6 MΩ pipettes there was a measurable loss of fluorescence > 10 min after break‐in. Pipettes were filled with an intracellular solution containing (in mm): 115 potassium gluconate, 20 KCl, 10 Hepes, 4 ATP‐Mg, 0.3 GTP‐Tris and 10 phosphocreatine. The pH was adjusted with KOH to 7.25–7.30 (285–295 mosmol l–1). A BVC‐700A amplifier (Dagan, Minneapolis, MN, USA) or a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) was used for recording and Igor Pro software (Wavemetrics, Lake Oswego, OR, USA) was used for acquisition and analysis.
For extracellular electrical stimuli we used theta glass bipolar electrodes. Postsynaptic potentials were elicited with 10–50 2‐ms pulses at 10 Hz per trial in standard ACSF (without blockers). Conversion light was applied from the start of the electrical stimuli to 1 s after the last stimulus.
Spikes generated by field stimulation were elicited with 100 1‐ms pulses at 50 Hz per trial. Conversion light was applied from the start of the stimulus train and continued for a total of 5 s. For these experiments, the bath contained CNQX, AP5 and MCPG (see above).
Virus injections
Animals were deeply anesthetized with ketamine/xylazine (7mg kg–1/1mg kg–1). The adeno‐associated viruses AAV2/1‐hSynapsin1‐hChR2 (H134R)‐EYFP‐WPRE and AAV2/1‐hSynapsin1‐CaMPARI (Forsque et al. 2015) were injected into P11–P13 mice. Seventy nanolitres of AAV2/1‐hSynapsin1‐hChR2 (H134R)‐EYFP‐WPRE was injected stereotactically into the posterior‐medial thalamic nucleus (POm). The stereotactic coordinates for POm were (mm from Bregma): lateral, 1.2; posterior, 1.2; depth, 2.7 below the pia. In all experiments except for two (where CaMPARI was injected into thalamus), 300–600 nl of AAV2/1‐hSynapsin1‐CaMPARI was injected into somatosensory cortex (S1), with coordinates (mm from Bregma): lateral, 3.0; posterior, 1.2; with multiple injections throughout the depth of cortex (0–0.9 mm from the pia, at ∼0.2 mm intervals). Viruses were allowed to express for > 18 days before slicing.
Optogenetics and photoconversion
For all experiments, violet light for optogenetic stimulation and CaMPARI photoconversion was delivered using an X‐cite 200 W mercury lamp (Excelitas Technologies, Mississauga, Ontario, Canada) and light guide through a 405/10 nm bandpass filter (Semrock, FF01‐405/10‐25). In one‐photon imaging experiments, photoconversion/stimulation light was delivered by a UPlanFL ×4/NA 0.13 objective. In two‐photon imaging experiments, photoconversion/stimulation light was delivered obliquely to the sample either by the light guide and filter only or with an additional collimator (Cairn Research, Faversham, UK) and lens (Thorlabs, Newton, NJ, USA; AC254‐030‐A‐ML, F = 30 mm). The light intensities used with acute brain slice samples ranged between ∼40 and 200 mW cm–2 and were measured by placing a photosensor in place of the slice (Thorlabs, S120VC sensor). The specific values for each experiment are indicated in the text.
Our standard protocol for optogenetic stimulation and CaMPARI conversion, including that used for circuit mapping, was (unless otherwise indicated) 10 pulses – 15 ms duration for each pulse – at 10 Hz, followed by a 5 s‐long pulse. The protocol was repeated 10 times at 15 s intervals. Variations on this protocol are detailed in the text.
CaMPARI photoconversion rate versus light intensity
To test CaMPARI photoconversion rate versus light intensity, CaMPARI was expressed in Escherichia coli T7 Express and purified by immobilized metal affinity chromatography, as before (Fosque et al. 2015). Purified CaMPARI in 500 μm CaCl2 buffered with 50 mm Tris, 150 mm NaCl at pH 8.0 was photoconverted using a 405 nm LED array (Loctite, Düsseldorf, Germany) over a range of light intensities. Red fluorescence was measured on a plate reader (Tecan, Männedorf, Zürich, Switzerland) at various time points after removal of free calcium by addition of EGTA to 10 mm. A single exponential rate was fitted to the appearance of red fluorescence (both appearance of red and disappearance of green fluorescence follow first‐order exponential kinetics; the red state saturates when the green state is depleted). Light intensities were measured on a power meter (Coherent, Santa Clara, CA, USA; no. 1098580) using a silicon photodiode (Coherent no. 1098313) and a neutral density filter (OD 1.0, Thorlabs NDUV10A). The rate constants for CaMPARI photoconversion were plotted as a function of light intensity and fitted to a simple linear regression. These experiments were performed in triplicate.
Imaging
A BX50WI microscope (Olympus Ltd, Tokyo, Japan) using a CoolSnap ES (Photometrics, Tucson, AZ, USA) CCD camera and oblique optics was used for one‐photon imaging. Red and green fluorescence were imaged with Texas Red (U‐MWIY2; 560/35 nm band‐pass excitation filter; 610 nm long‐pass emission filter) and FITC filter cubes (U‐MWIBA2; 475/30 nm band‐pass excitation filter, 530/40 nm band‐pass emission filter), respectively.
A Femto 2D two‐photon laser scanning system (Femtonics Ltd, Budapest, Hungary) was equipped with a femtosecond pulsed Chameleon Ti:Sapphire laser (Coherent, Santa Clara, CA, USA). Imaging was controlled by the Matlab‐based MES software package (Femtonics). For CaMPARI red and green fluorescence measurements the laser was tuned to λ = 820 nm. Fluorescence was detected in epifluorescence mode with a water immersion objective (LUMPLFL ×60/1.0 NA, Olympus, Hamburg, Germany). Trans‐fluorescence and transmitted infrared light were detected using an oil immersion condenser (Olympus; 1.4 NA). Fluorescence was divided by a dichroic mirror at ∼590–600 nm, and green and red signals were filtered using 525/50 and 650/50 bandpass filters, respectively. The red emission filter is red‐shifted in comparison to the filters used in (Fosque et al. 2015), and inefficiently collects the CaMPARI red emission (peak ∼585 nm), but it effectively eliminates any bleed‐through of the CaMPARI green fluorescence into the red channel.
Although we had planned to conduct analysis of CaMPARI‐expressing neurons in 4% paraformaldehyde (PFA)‐fixed brain slices under a confocal microscope, CaMPARI fluorescence, both red and green, was dramatically reduced as a result of the fixation. We therefore conducted our analysis in living brain slices primarily by two‐photon imaging. It is unclear why 4% PFA was less well tolerated in these preparations than in Fosque et al. (2015); nevertheless, there were key differences in tissue preparation. In this paper, brain slices used in experiments were subsequently fixed in PFA, whereas in Fosque et al. (2015), whole brains were fixed by perfusion of the animal with PFA. The history of the tissue (slicing, temperature variations, ionic environment, etc.) prior to fixation was dramatically different.
Analysis
The analysis of fluorescence was performed in ImageJ. For one‐photon experiments, red fluorescence intensity was determined from the pixel intensities of images taken with 200 ms of exposure. To help correct for out‐of‐focus CaMPARI red fluorescence that occurs in one‐photon imaging, we normalized the fluorescence level of each neuron chosen for analysis to an adjacent region: the red fluorescence level (R f) was determined by selecting a region of interest (ROI) within a cell's soma (R i), excluding the nucleus, and an adjacent region outside of the cell (R o; i.e. the out‐of‐focus background fluorescence level) and taking the difference of the average R i and average R o, such that: R f = R i – R o. Soma size was not considered in these measurements. In one‐photon imaging it was not possible to accurately measure green CaMPARI fluorescence in cells due to dense background green fluorescence from Channelrhodopsin‐2 (ChR2)–enhanced yellow fluorescent protein (EYFP)‐expressing axons and CaMPARI green in somata and processes of neighbouring neurons. Thus, in these one‐photon imaging experiments, CaMPARI was not used ratiometrically.
For two‐photon imaging, the analysis of the red fluorescence level was determined by the same method as used for one‐photon imaging. However, in these experiments the calculated R f value was divided by the green fluorescence (G i) level from the same region of interest in the neuron. This yielded a red/green ratio (R f/G i; F red/F green). Note that the F red/F green values displayed in the figures are normalized to F red/F green before conversion (except in Fig. 4 D). The key advantage of two‐photon imaging is that it does not suffer from out‐of‐focus light contaminating the region of interest, and thus accurate green fluorescence measurements are possible even in the presence of non‐specific background fluorescence, enabling the generation of an accurate red to green ratio.
Figure 4. CaMPARI for all‐optical functional connectivity mapping.
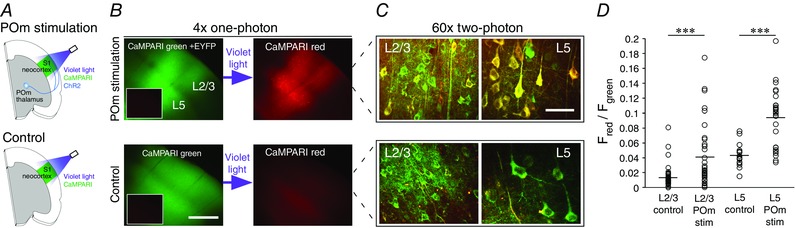
A, schematic representation of the experimental set‐up. In acute ex vivo brain slices, there were two conditions: CaMPARI expression in S1 with ChR2‐EYFP expression in POm thalamus (POm stimulation condition, top) or without ChR2‐EYFP expression in POm (control condition, bottom). Violet light (∼120 mW cm–2) applied to S1 cortex simultaneously activates ChR2 and drives conversion of connected, postsynaptic CaMPARI‐expressing neurons. B, CaMPARI green/ChR2‐EYFP under ×4 magnification one‐photon imaging (top left) or CaMPARI green only (bottom left). The inset in each panel shows red fluorescence background before stimulation, which is very low. After violet light illumination, red CaMPARI fluorescence (right panels) is evident in layers 2/3 and (most intensely) 5 only when POm input is stimulated. C, post‐stimulation/conversion ×60 magnification two‐photon images of CaMPARI red/green in layers 2/3 and 5, from the corresponding slices in B. D, red/green ratio measured from control and stimulated neurons; both L2/3 and L5 had significantly higher red/green ratios in stimulated neurons (P < 0.001; Student's t test). B, scale bar: 500 μm; C, scale bar: 30 μm. CL, conversion light.
For studying the effect of spikes on photoconversion by extracellular stimulation, cells that were most highly converted (in the top 50% of converted cells) and < 150 μm from the stimulating electrode were chosen for analysis. This selection criterion was used because, a priori, it cannot be known whether the extracellular stimulus generates spikes in a one‐to‐one relationship with the stimulus (there is no simultaneous postsynaptic recording). By considering only the top 50% of the converted neurons, the population of neurons that spiked with each stimulus pulse was strongly favoured and those that spiked intermittently or not at all were preferentially excluded. The effect of the stimulus in a recorded cell was used to tune the stimulus strength to ensure that some neurons would generate action potentials reliably.
The images of CaMPARI fluorescence were adjusted for clarity of display and for comparison within each experiment but do not represent absolute fluorescence values.
All values are expressed as the mean ± standard error of the mean. P‐values are from Student's two‐tailed t test, unless otherwise indicated.
Results
Action potentials and CaMPARI conversion
Neuronal activity – including action potential (AP) spiking and synaptic potentials – triggers calcium influx into neurons. To examine the response of CaMPARI to APs and synaptic potentials, we used whole‐cell patch‐clamp recording with high‐resistance patch pipettes to minimize dialysis of CaMPARI out of the recorded cells. A series of stimulation trials, each evoking 50–100 spikes, was applied to four CaMPARI‐expressing L2/3 pyramidal neurons in an acute slice preparation. Each trial consisted of a 2 s step depolarization with 5 s of conversion light separated by an interval of at least 10 s. The CaMPARI conversion level in the targeted and adjacent non‐stimulated neurons was measured periodically, after 2–10 trials (Fig. 1). Our results indicate that action potentials have a large effect on photoconversion of the targeted neuron. For example, the green fluorescent CaMPARI‐expressing neuron in Fig. 1 A, following ∼500 spikes (5 trials; Fig. 1 B shows 1 trial), converted into a strongly red fluorescent neuron (Fig. 1 C), even at the relatively low photoconversion light intensity of 40 mW cm–2.
Figure 1. Relationship between CaMPARI conversion and spiking.
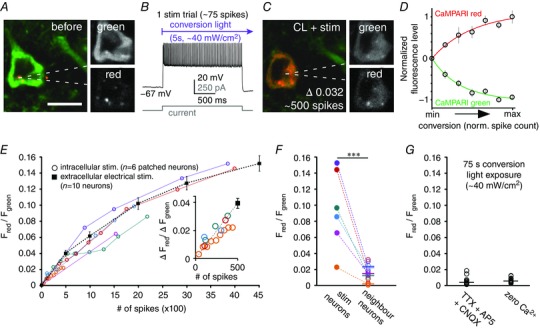
A, a patched neuron before conversion. The dotted lines indicate the location of the patch pipette. The two greyscale panels on the right show the raw fluorescence in the green (top) and red (bottom) channels. B, example recording of the APs elicited by a depolarizing current step during one stimulation trial. The violet arrow represents the presence of photoconversion light. C, the same neuron as in A after stimulation (∼500 spikes) and photoconversion light. The stimulus induced an increase in the red/green ratio (0.032). The two greyscale panels on the right show the raw fluorescence in the green (top) and red (bottom) channels. D, a comparison of the relative change in the red and green channels (n = 3 neurons). The red and green channels change equally and oppositely. Each data point from left to right represents an increasing number of spikes, which were used to drive calcium influx for conversion. The fitted lines are a one‐phase decay function (green) or a one‐phase association (red). E, relationship between increasing red/green ratio and increasing number of spikes. The graph displays data from whole‐cell recorded/stimulated neurons (blue, cyan, orange and red circles; n = 4 neurons) and neurons stimulated by a local field electrode in the presence of bath‐applied synaptic blockers (squares; n = 10 neurons; see Methods). The inset is an expansion of the first 500 spikes on the graph (for circles, R 2 = 0.77). F, the change in the red/green ratio in 4 stimulated neurons and their corresponding (indicated by identical colours) non‐stimulated neighbouring neurons. Same neurons as in E with corresponding colour code. Compared to the corresponding patched/stimulated neurons, neighbouring neurons on average converted ∼7‐fold less, only 15.1 ± 3.2% (P < 0.001; Student's t test). G, CaMPARI conversion when synaptic activity is blocked or extracellular calcium is removed is low. A total of 75 s of ∼40 mW cm–2 conversion light exposure was used. In the presence of TTX (1 μm), AP5 (50 μm) and CNQX (20 μm) the mean change in red/green ratio was 0.0036 ± 0.001 (n = 20 neurons; P = 0.06; Student's t test); in ‘zero calcium’ ACSF, the mean change in red/green ratio was 0.0047 ± 0.001 (n = 22 neurons; P = 0.003; Student's t test). Scale bar: 10 μm. CL, conversion light.
To examine the relationship between the red and green states of CaMPARI, we took fluorescence measurements in both channels at several points between periods of stimulation (n = 3 neurons). The results indicate a direct relationship between the decrease in green fluorescence and the increase in red in active neurons during CaMPARI photoconversion (Fig. 1 D). Thus, a two‐state model where green molecules convert directly to red, with no other photocycle states considered, seems sufficient to explain the observed data. These data also indicate that while red and green fluorescence may be used together to improve the signal to noise, each fluorescence channel can also, in principle, provide the same information independently. This becomes important when the imaging environment precludes accurate measurement of both the red and the green channels.
To quantify the relationship between photoconversion at low light levels (∼40 mW cm–2) and number of action potentials, we compared the red/green CaMPARI fluorescence ratio as a function of the number of evoked spikes by two experimental paradigms (Fig. 1 E). In one experiment, four whole‐cell patched neurons were directly recorded and intracellularly stimulated (Fig. 1 E circles). As additional support, we performed a second experiment using a field‐stimulating electrode to generate controlled spiking in a population (n = 10 neurons) of non‐patched (non‐dialyzed) neurons (Fig. 1 E squares; see Methods). Fewer than 100 spikes had a small effect on the red/green ratio (Fig. 1 E inset), and it was around the detection threshold on our two‐photon system. As the number of spikes increased, the red/green ratio also increased (Fig. 1 E). The relationship between spikes and red/green ratio did not appear to plateau, even after thousands of evoked spikes; this was probably related to the low intensity of the photoconversion light, which converted just a fraction of the available CaMPARI molecules. As a negative control, we compared the change in the red/green ratio of neighbouring (non‐stimulated) neurons to the red/green ratio of stimulated (patched) neurons (Fig. 1 F). Non‐stimulated neurons were ∼7‐fold less converted (15.1 ± 3.2% of the conversion observed in stimulated/patched neurons, n = 6, stim: 0.10 ± 0.022; neighbour: 0.015 ± 0.003; P < 0.001; Student's t test). Note that, in principle, neighbouring neurons could have been driven by the stimulated neuron through excitatory synaptic connections.
To test the level of conversion in the absence of activity or with extracellular calcium removed, we exposed slices to a total of 75 s of conversion light (∼40 mW cm–2, 1500 pulses, 50 ms pulses, at 5 Hz) in the presence of ACSF with TTX (1 μm), AP5 (50 μm) and CNQX (20 μm), or in ACSF without added calcium (Fig. 1 G). There was a negligible effect from conversion light exposure in both cases. The red/green ratio increased negligibly when synaptic activity and spikes were blocked (0.004 ± 0.001, n = 20 neurons) or when slices were bathed in zero calcium ACSF (0.005 ± 0.001; n = 22 neurons). Thus, there is little CaMPARI conversion in non‐stimulated neurons, and conversion requires extracellular calcium and spiking or synaptic activity.
Light intensity and CaMPARI conversion
Intense 400 nm light can be phototoxic to cells and tissue. Due to strong scattering and absorption of short wavelength light in brain tissue, experiments involving large‐scale and/or deep photoconversion may result in low levels of photoconverting light reaching cells. Thus, we opted to perform our experiments throughout this study at low photoconversion light intensities (∼40–200 mW cm–2). At low light intensities we consistently measured small changes in conversion from few spikes (Fig. 1 E), whereas Fosque et al. (2015) reported that a small number of APs was sufficient to trigger substantial CaMPARI photoconversion. The most obvious difference between the earlier experiments and the ones reported here is a large disparity in light intensity (40 mW cm–2 vs. > 1.5 W cm–2).
To determine how light intensity affects conversion rate, we examined whether increasing light intensity made it possible to integrate calcium signals over correspondingly shorter time scales. We first measured CaMPARI photoconversion at different light intensities in purified protein (Fig. 2 A and B). Conversion of purified protein that was exposed to a range of light intensities (up to ∼2.5 mW cm–2) showed a linear relationship between CaMPARI conversion rate and light intensity (Fig. 2 B). We also tested this relationship in neurons in acute brain slices. In a spiking neuron exposed to two different interleaved light intensities (∼50 and ∼200 mW cm–2), the conversion rate corresponded to the applied light intensity (Fig. 2 C, left panel). A 4‐fold increase in light intensity led to a 4‐fold increase in the initial rate of CaMPARI photoconversion (Fig. 2 C, right panel; n = 4 neurons). The responses of active neurons matched that of purified protein. Thus, we found that the initial rate of CaMPARI photoconversion was directly proportional to the light intensity in purified protein and active neurons.
Figure 2. The relationship between light intensity and photoconversion.
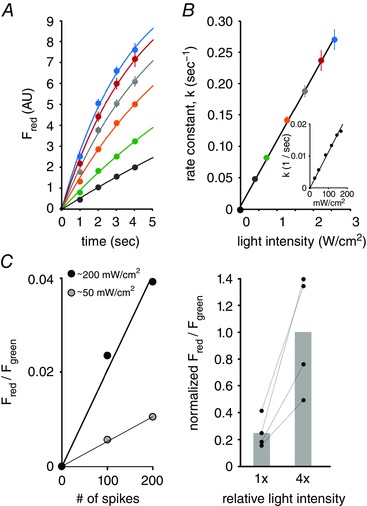
A, plots of red fluorescence formation from purified CaMPARI protein as a function of illumination time for various intensities of 405 nm light (light intensity from dark grey to blue (in mW cm–2): 392, 710, 1281, 1767, 2217, 2585; inset (left to right, in mW cm–2), 26, 47, 85, 116, 145, 167). The fitted lines are from a one‐phase association function, y = y 0 + (plateau – y 0)(1 – exp(–kx)), where y is red fluorescence at time x, y 0 is the initial red fluorescence, plateau is the maximum red fluorescence and k is the rate constant. B, the relationship between conversion rate (given by the rate constant, k) and light intensity is linear (R 2 = 0.99). Colour coded data taken from panel A. C, left, the change in red/green ratio relative to spike number and interleaved light intensity in a single neuron. Right, 4 neurons were illuminated by two different conversion light intensities (1× and 4×) and each given the same stimulus (spikes), resulting in a roughly 4‐fold faster conversion rate when the conversion light intensity was higher.
Synaptic input and CaMPARI conversion
Having calibrated CaMPARI conversion due to action potentials, we turned our attention to subthreshold excitatory synaptic potentials, another source of calcium entry. We used extracellular electrical stimulation adjusted to elicit strictly subthreshold postsynaptic potentials in CaMPARI‐expressing cortical neurons (Fig. 3 A). Postsynaptic potentials (Fig. 3 B) triggered CaMPARI conversion (at ∼40 mW cm–2 photoconversion light; Fig. 3 C) and followed a linear relationship with postsynaptic potential number (Fig. 3 D; n = 3 neurons). The change in the red/green ratio for one example neuron was ∼0.04 (∼2000 postsynaptic potentials), less conversion than results from a similar number of APs, but higher than from photoconversion in the absence of stimulation (Fig. 1 F and G). The extracellular stimulus also drove conversion in many neighbouring neurons and their processes (Fig. 3 C ).
Figure 3. CaMPARI conversion from postsynaptic potentials and rebound potentials.
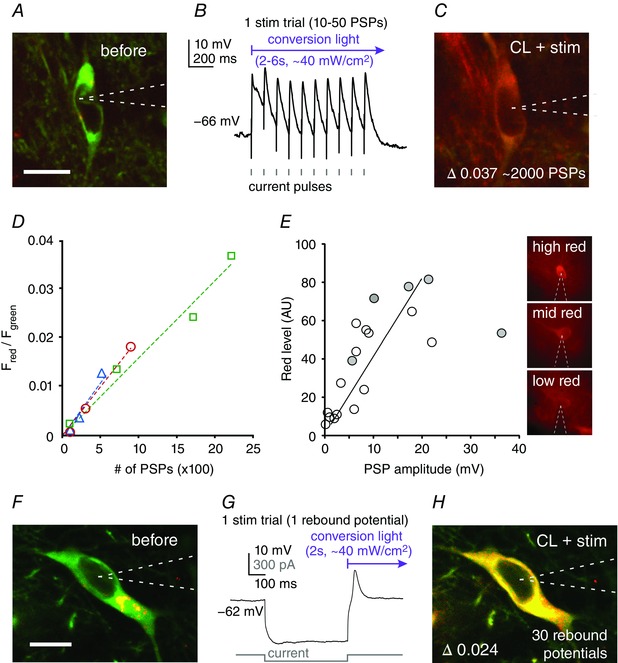
A–E, conversion by postsynaptic potentials. A, a patched CaMPARI‐expressing cortical neuron before conversion. B, local extracellular electrical stimulation elicits depolarizing postsynaptic potentials (PSPs). C, ∼2000 depolarizing PSPs produced a ∼0.04 increase in the red/green ratio. D, the red/green ratio increases linearly with increasing number of extracellularly evoked PSPs (n = 3 neurons, each with a separate colour; R 2 = 0.95). E, relationship between PSP amplitude and conversion (red fluorescence intensity), measured by post hoc whole‐cell recordings and one‐photon imaging (R 2 = 0.43). Monosynaptic PSPs were elicited by optogenetic activation of axon terminals in cortex. Red fluorescence intensity positively correlated with PSP amplitude (P = 0.002, R 2 = 0.443; linear regression). Shaded circles indicate neurons that reached spike threshold at least once due to the stimulus. The panels on the right are example neurons with high (top), mid (middle) and low (bottom) red fluorescence intensities. F–H, conversion by rebound potentials in thalamic neuron. F, a patched thalamic neuron before conversion. G, example recording of a hyperpolarizing current step triggering a T‐type calcium channel rebound potential (sodium spikes are blocked with 1 μm TTX). H) Thirty rebound potentials increased the red/green ratio (∆0.024). Scale bar: 10 μm. CL, conversion light.
We also used optogenetic stimulation of thalamocortical axons to elicit postsynaptic potentials and trigger CaMPARI conversion in primary whisker somatosensory cortex (S1). Optogenetic stimulation of thalamic axons and CaMPARI conversion were induced simultaneously by the same violet light illumination (∼120 mW cm–2, 10 Hz, 15 ms pulses, followed by 5 s of continuous light repeated 10 times; repeated high‐intensity (∼2 W cm–2), short‐duration pulses (100 ms) were also able to induce conversion from postsynaptic potentials (data not shown)). We targeted 19 neurons for whole‐cell recording that were previously photoconverted by the stimulus/conversion protocol, and measured their respective postsynaptic potential amplitudes. These measurements were used to establish the relationship between postsynaptic potential amplitude and photoconversion level (Fig. 3 E). For the epifluorescence measurements, we used only red CaMPARI fluorescence, as emission from the ChR2‐EYFP effector polluted the green channel (see above and Methods). The postsynaptic response to thalamocortical activation varied widely across our samples, providing a broad range of input strengths against which to relate the CaMPARI conversion values (Fig. 3 E ). APs were elicited by the optogenetic stimulation in 5/19 neurons (grey circles in Fig. 3 E), only one of which fired reliably with each stimulus. There was a positive relationship between postsynaptic potential amplitude and CaMPARI red conversion (P = 0.002, R 2 = 0.43; linear regression). These results indicate that the CaMPARI conversion level is well correlated with the postsynaptic potential amplitude, and thus provides a readout of input strength.
Calcium rebound potentials and CaMPARI conversion
Calcium‐dependent rebound potentials provide another source of calcium influx. Many thalamic neurons have two modes of activity, a tonic and a burst mode (Jahnsen and Llinas, 1984). The burst – a rebound from hyperpolarization – is generated by T‐type voltage‐dependent calcium channels (Coulter et al. 1989), which when released from inactivation trigger calcium entry into the neuron. To test the effect of rebound potentials on CaMPARI conversion, we recorded from a CaMPARI‐expressing thalamic neuron (Fig. 3 F–H) in the presence of TTX, thus isolating the rebound potential from sodium‐dependent APs. Here, 30 rebound potentials substantially increased the red/green ratio (0.024, n = 1; compare Fig. 3 F–H), and as few as 10 rebound potentials were sufficient to convert the cell (0.015). Thus, rebound potentials also effectively drive conversion of CaMPARI in the thalamus.
Functional circuit mapping with CaMPARI and ChR2 in brain slices
Having established the relationship between CaMPARI conversion and activity, we applied CaMPARI to map cortical circuit activity driven by optogenetically defined inputs in brain slices. This preparation is ideal for circuit mapping, since neurons in acute brain slices at rest are normally hyperpolarized and minimally active (as evidenced by negligible CaMPARI photoconversion in non‐stimulated neurons shown in Fig. 1 F), and despite severing some axons during slicing, axonal terminals remain viable and excitable by light when expressing ChR2 (Petreanu et al. 2007; Cruikshank et al. 2010). The slice can also be uniformly illuminated. The resulting postsynaptic activation pattern in an acute brain slice therefore faithfully reflects in vivo functional connectivity while providing the advantages of the in vitro preparation. Thus, this preparation provides a background of low spontaneous activity and uniform light intensity, against which to test the effects of specific, largely monosynaptic inputs reflecting the in vivo condition.
We examined the effect of activating POm thalamocortical axons on S1 neurons expressing CaMPARI. To achieve this, brain slices were obtained from mice with POm neurons expressing ChR2 and S1 neurons expressing CaMPARI, or, as a control, we used slices from mice only expressing CaMPARI in S1 – without ChR2 expression (Fig. 4 A; CaMPARI is driven by the Synapsin1 promoter, which primarily drives in L2/3 and L5 pyramidal cells). We applied light stimuli (∼120 mW cm–2, 405/10 nm; this light intensity was sufficient to induce strong conversion and maximize ChR2 activation for our protocol) for the simultaneous stimulation of ChR2 and conversion of CaMPARI, and used the same protocol for POm stimulated (ChR2‐expressing) and control slices. Stimulation/conversion and initial fluorescence imaging were performed under a one‐photon microscope through a ×4 objective.
Before the stimulation protocol, there was minimal red fluorescence in POm stimulation and control slices (Fig. 4 B, left panel insets). After stimulation, red fluorescence increased substantially only in the POm stimulation case (Fig. 4 B, right panels). POm axons target layers 1 and 5 in barrel cortex (Lu and Lin, 1993; Bureau et al. 2006; Kichula & Huntley, 2008). Photoconversion (red fluorescence) was most intense in L5 neurons (Fig. 4 B top right panel).
The slices were transferred to a two‐photon microscope for imaging and quantification of the red/green ratios of each neuron in the resulting z‐stack imaging fields (Fig. 4 C and D). We found that POm significantly drove CaMPARI conversion in both L2/3 (red/green ratio: 0.040 ± 0.007, n = 36 neurons; control: 0.013 ± 0.002, n = 46 neurons; P < 0.001) and L5 (red/green ratio: 0.094 ± 0.008, n = 27 neurons; control: 0.044 ± 0.004, n = 17 neurons; P < 0.001). The ratios acquired in these experiments represent the absolute red/green ratio following stimulation. Thus, comparison to control values (no ChR2) provides an appropriate picture of conversion (Fig. 4 D). In L5, 70% (19/27) of neurons were strongly marked by optogenetic activation of POm axons (with a red/green ratio greater than two standard deviations above the control mean) compared with 33% (12/36) of neurons in L2/3, suggesting that POm inputs drive L5 more strongly by inducing spiking and/or strong or numerous postsynaptic potentials.
Thus, CaMPARI provides a cortex‐wide snapshot of the functional synaptic connectivity triggered by specific optogenetically activated inputs in the acute brain slice preparation.
Practical considerations when using CaMPARI
Here we present a few considerations for working with CaMPARI. (1) Green‐to‐red photoconversion of individual CaMPARI molecules is irreversible. Each iteration of photoconversion increases the red/green ratio. A neuron will not convert further once the available reservoir of unconverted molecules is depleted. Decay of the red species will occur according to protein turnover by the cell. (2) CaMPARI has a non‐zero conversion rate in zero Ca2+, and a slightly higher ‘background’ conversion rate at resting Ca2+ levels. Furthermore, neurons that are damaged or unhealthy can have high intracellular calcium concentrations, and might therefore be converted without a stimulus. Moreover, since the green and red forms of CaMPARI are negative indicators (i.e. as calcium levels increase, fluorescence accordingly decreases), the fluorescence will appear dim when intracellular calcium is high (Fosque et al. 2015). (3) The resting level of calcium and composition of intracellular Ca2+ buffers can differ between classes of neurons (compare control values in Fig. 4 D). (4) The concentration (expression level) of CaMPARI molecules will have two effects. First, high expression provides a larger dynamic range (more molecules to convert), and second, high concentration implies greater calcium buffering capacity and thus potentially less fractional green‐to‐red conversion for the same calcium influx. Nevertheless, when averaging effects across a large cohort of neurons with variable expression levels, buffering should play a minimal role in the result. At reasonable expression levels such as those used in these experiments, there is no evidence of any adverse effects of CaMPARI on the expressing cells. (5) Weak conversion throughout the whole cell may appear equivalent to strong local conversion (e.g. within a dendritic branch) after red CaMPARI diffuses and mixes throughout the dendritic arbor with green CaMPARI (after ∼1–1.5 h for large neurons, such as L5 pyramidal neurons at 32°C). (6) The fluorescence of CaMPARI‐expressing neurons decreases as a whole‐cell pipette approaches and returns to baseline upon retraction of the pipette. This effect is presumably related to increased intracellular calcium (due to potassium extruded from the approaching pipette depolarizing nearby neurons) that triggers a decrease in green fluorescence. Both CaMPARI green and red fluorescence decrease as calcium concentration increases, but are reversible without conversion light illumination (Fosque et al. 2015).
Discussion
The results presented here demonstrate that photoconversion of CaMPARI reports action potentials, postsynaptic potentials, and calcium‐dependent rebound potentials in acute brain slices, indicating that CaMPARI conversion can be triggered by a wide range of activity. When activity is blocked or neurons are not stimulated, CaMPARI conversion in brain slices is minimal. Furthermore, we showed that the photoconversion rate increases linearly with light intensity; CaMPARI photoconversion is faster at high light intensities but is still effective at low levels. With these important characteristics of CaMPARI elucidated, we show that the combination of CaMPARI and channelrhodopsin constitutes a powerful approach for functional network mapping in brain slices. Our application of this method to test the influence of POm inputs on somatosensory cortical neurons revealed that optogenetic stimulation of POm axons drives significant calcium responses in both L5 and L2/3, consistent with results from in vivo electrophysiology and functional imaging (Gambino et al. 2014; Jouhanneau et al. 2014).
CaMPARI photoconversion and light intensity
We showed that CaMPARI conversion is effective across a broad range of light intensities and that the conversion rate correlates with the conversion light level. At low light intensities, CaMPARI is suited to capture the effects of many stimuli across a wide time span and dynamic range: hundreds or thousands of spikes or synaptic potentials can be acquired without saturation. In contrast, high light intensities cause fast conversion, triggering substantial conversion from only a few events, thus acquiring a temporally restricted, sensitive snapshot of activity. An important implication of these results is that light intensity level functions as a gain factor that can be tuned to suit the experiment. For example, a long‐duration event is best captured by low light intensity levels, whereas short‐duration events are best captured with high light intensity. In many experimental conditions, however, it may not be practical or possible to deliver high, isotropic light intensities throughout tissue, especially in vivo (although fibre optics, lenses or multiple light sources could improve light delivery (Murayama et al. 2007; Andermann et al. 2013)).
Understanding the relationship between conversion rate and light intensity also makes it possible to compare the CaMPARI signal among a population of neurons exposed to different light intensities. Thus, for example, in tissue exposed to anisotropic light intensity levels, such as in vivo, activity (i.e. conversion) levels can be compared among neurons from different regions as a linear function of exposed (either measured or simulated) light intensity.
Since adjusting the light intensity yielded commensurate changes in the initial conversion rate (Fig. 2), we conclude that the relationship between light intensity and gain (signal vs. spikes) is linear. By extension, there should also be a linear relationship between light intensity and dynamic range. Thus, CaMPARI is a tunable tool applicable to a wide range of experiments and objectives.
Effect of activity on CaMPARI conversion
Our work here and earlier work (Fosque et al. 2015) show that calcium and ∼400 nm light are necessary and sufficient to trigger substantial CaMPARI photoconversion: there is very little conversion when network activity is blocked or extracellular calcium is low (Fig. 1 F and G). Action potentials triggered substantial conversion of CaMPARI, and at low light levels, several thousand spikes could be measured without saturation of the red/green ratio (Fig. 1 E).
While sodium action potentials trigger large calcium influx, here we show that synaptic inputs and rebound potentials, which have very different time courses and calcium influx kinetics from APs, can also drive significant conversion of CaMPARI. The effective conversion of CaMPARI by subthreshold synaptic activity, which is correlated with amplitude, and somatic readout of the CaMPARI signal creates new opportunities for functional circuit mapping. It is now possible to deduce subthreshold synaptic input received by a neural population without electrical recordings. In contrast, GECIs require synaptic activation strong enough to result in action potentials for a somatic readout of neuronal activity. Therefore, imaging of local synaptically generated transient GECI responses for mapping of subthreshold inputs would require high‐resolution, high‐frequency whole‐neuron imaging of large volumes containing hundreds or thousands of neurons, which is technically challenging.
While the effect of action potentials on calcium entry and CaMPARI conversion is large in the soma and dendritic arbour of the neuron, synaptic inputs can generate large local calcium entry, by activating voltage‐dependent calcium channels and/or NMDA receptors, at distal locations on the dendritic arbour or in spines, while only generating small postsynaptic potentials at the soma. This is indeed likely to be the primary source of calcium influx from postsynaptic inputs. Since CaMPARI is not membrane‐bound, in principle, red CaMPARI could potentially diffuse from these distal locations to the soma (and vice versa), providing a readout of distal input.
Some additional considerations regarding conversion and synaptic input should also be noted: first, inhibition can truncate the excitatory synaptic potential at the soma, while having little effect on calcium entry (and thus conversion) induced by local excitatory input. Alternatively, inhibitory postsynaptic input can, in theory, trigger photoconversion in some neurons by activating calcium‐dependent rebound potentials (Fig. 3 F–H). Finally, synapses with a large NMDA receptor current or those that trigger voltage‐gated calcium channels, possibly leading to dendritic Ca2+ spikes (Larkum et al. 1999; Major et al. 2013), will in principle generate more conversion. Thus, careful consideration of the cells and network in question is advisable when interpreting the CaMPARI signal.
Nevertheless, it may be possible to hone the specific activity of interest (e.g. APs) by using a CaMPARI variant with the appropriate calcium binding affinity (Fosque et al. 2015). Low‐affinity variants, for example, would likely favour detection of high calcium concentration events.
CaMPARI for functional circuit mapping with ChR2‐defined inputs
In this study, we used CaMPARI in conjunction with ChR2 in acute brain slices to map functional inputs in the mammalian brain (Fig. 4). Combining calcium indicators with ChR2 activation is complicated by the fact that green GECI excitation overlaps with opsin activation (Akerboom et al. 2013; Wu et al. 2014). Although combining ChR2 with red‐shifted calcium indicators (Akerboom et al. 2013; Hoi et al. 2013; Wu et al. 2014; Shen et al. 2015) may be an alternative way to perform this experiment in the future, CaMPARI, functioning as a calcium integrator and permanent marker, still has advantages over transient calcium indicators. For example, measuring the CaMPARI signal after, rather than during, stimulation dramatically simplifies data acquisition, and potentially allows access to much greater tissue volumes. CaMPARI also has the ability to report subthreshold responses at the somatic level.
CaMPARI and ChR2 can be converted/stimulated by the same light source, providing a simple assay for rapid, all‐optical functional readouts of optogenetically specified inputs (Fig. 4). Acute brain slices offer a low background of spontaneous activity and the potential for uniform illumination across the sample, which would be complicated or impossible in vivo. By optogenetically activating presynaptic terminals directly, a relatively clear estimate of direct, (primarily) monosynaptic drive can be established from genetically defined inputs that reflect in vivo connectivity. This technique draws its potential from the advantages of acute brain slices, optogenetics and CaMPARI. Here, we performed the first functional circuit map in the mammalian cortex from an optogenetically specified input (from POm thalamus) using CaMPARI. With this technique, we revealed the functional (including weak, subthreshold) connections between POm and cortex, including an elusive connection with upper layer neurons suggested from in vivo recordings (Jouhanneau et al. 2014). We confirmed the connectivity between POm and L2/3 pyramidal neurons using direct recordings of postsynaptic L2/3 neurons (data not shown). Earlier work using scanning photostimulation for local glutamate un‐caging to stimulate POm neurons in brain slices did not reveal upper layer connectivity (Bureau et al. 2006). This study had a key technical limitation not true of the CaMPARI/ChR2 method: When uncaging in the thalamus, only a fraction of the thalamocortical projections are intact in the slice. Most of the thalamocortical axons traversing the slice were likely to have been severed before reaching L1, where they target L2/3 pyramidal neuron dendrites. Thus, we have demonstrated a novel thalamocortical connection in the brain using the CaMPARI/ChR2 method that was not detectable in a previous method in brain slices. The new approach presented here will make it possible to rapidly reveal previously undetectable connections in the brain. In our experiment, the network‐wide cortical expression of CaMPARI and ChR2‐expressing POm axons provided a nearly instant snapshot of in vivo‐like connections, which would otherwise require many repeated electrical recordings to achieve.
In principle, CaMPARI and ChR2 can be combined to study the connectivity between any neuronal populations in the brain. The technique provides a rapid readout of functional connectivity, screening hundreds or thousands of neurons at once, thus facilitating high‐throughput mapping and discovery of weak or previously unknown connections. The characteristics of postsynaptic neurons could also be easily studied relative to their input source. By targeting patch recordings to red or green postsynaptic neurons or applying various molecular techniques to these neurons, it may be possible to uncover neuronal characteristics definable by their input, thus identifying subnetworks or new subcategories of neurons.
Conclusion
CaMPARI is an effective tool for monitoring neuronal circuit activity in acute brain slices. CaMPARI's sensitivity to activity is linear with light intensity, thus enabling experiment‐specific fine‐tuning. Understanding this relationship also opens up the possibility of comparing activity across neurons exposed to different light intensities, which may prove important for in vivo applications. CaMPARI signals the strength of subthreshold synaptic input (as well as spikes and calcium rebound potentials), which can be measured at the soma. Finally, in combination with channelrhodopsin in ex vivo acute brain slices, CaMPARI provides a powerful circuit mapping method for mammalian brain circuits that can reveal connections and their strength.
Additional information
Competing interests
No competing interest declared.
Author contributions
Experiments were performed in the laboratory of Prof. Matthew Larkum at the Charité Universitätsmedizin Berlin, Berlin, Germany and the laboratory of Dr Eric Schreiter at the Janelia Research Campus, VA, USA. Conceptual design: T.A.Z., R.N.S.S., E.R.S., M.E.L., L.L.L. and F.S. Collection and assembly of data: T.A.Z., F.S. and E.R.S. Data analysis and interpretation: T.A.Z., F.S., E.R.S. and R.N.S.S. Manuscript writing: T.A.Z., R.N.S.S., M.E.L., E.R.S., L.L.L., F.S. and F.J. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The project was funded by Marie Curie and (Human Brain Project) HBP grants to R.N.S.S., Neurocure Center grant to T.A.Z., and Deutsche Forschungsgemeinschaft (DFG) and HBP grants to M.E.L. and DFG to F.J. Howard Hughes Medical Institute (HHMI) supported E.R.S., L.L.L. and F.S. This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No. 720270 (HBP SGA1).
Acknowledgements
We would like to thank the Berlin Vector Core (Thorsten Trimbuch) for assistance with some of the constructs and for useful discussions, and Anne‐Kathrin Theis for assistance with the two‐photon microscope and acquiring reagents. We also thank members of the Larkum lab, in particular Guy Doron, Naoya Takahashi, Julie Seibt and Christina Bocklish for helpful input and discussions.
Linked articles This article is highlighted by a Perspective by Chéreau & Holtmaat. To read this Perspective, visit http://dx.doi.org/10.1113/JP273733.
Contributor Information
Timothy A. Zolnik, Email: timothy.zolnik@charite.de.
Robert N. S. Sachdev, Email: robert.sachdev@charite.de.
References
- Akerboom J, Carreras Calderón N, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE, Macklin JJ, Patel R, Pulver SR, Wardill TJ, Fischer E, Schüler C, Chen TW, Sarkisyan KS, Marvin JS, Bargmann CI, Kim DS, Kügler S, Lagnado L, Hegemann P, Gottschalk A, Schreiter ER & Looger LL (2013). Genetically encoded calcium indicators for multi‐color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Carreras Calderón NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER & Looger LL (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32, 13819‐13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Gilfoy NB, Goldey GJ, Sachdev RN, Wölfel M, McCormick DA, Reid RC & Levene MJ (2013). Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron 80, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Gerkin RC & Dean KL (2004). Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J Neurosci 24, 6466–6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, von Saint Paul F & Svoboda K (2006). Interdigitated paralemniscal and lemniscal pathways in the mouse barrel cortex. PLoS Biol 12, e382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K & Kim DS (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Rochefort NL, Nelken I & Konnerth A (2011). Functional mapping of single spines in cortical neurons in vivo. Nature 475, 501–505. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR & Prince DA (1989). Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low‐threshold current. J Physiol 414, 587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV & Connors BW (2010). Pathway‐specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron 65, 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosque BF, Sun Y, Dana H, Yang CT, Ohyama T, Tadross MR, Patel R, Zlatic M, Kim DS, Ahrens MB, Jayaraman V, Looger LL & Schreiter ER (2015). Labeling of active neural circuits in vivo with designed calcium integrators. Science 347, 755–760. [DOI] [PubMed] [Google Scholar]
- Gambino F, Pages S, Khayas V, Baptista D, Tatt R, Carleton A & Holtmaat A (2014). Sensory evoked LTP driven by dendritic plateau potentials in vivo. Nature 515, 116–119. [DOI] [PubMed] [Google Scholar]
- Hoi H, Matsuda T, Nagai T & Campbell RE (2013). Highlightable Ca2+ indicators for live cell imaging. J Am Chem Soc 135, 46–49. [DOI] [PubMed] [Google Scholar]
- Jahnsen H & Llinás R (1984). Voltage‐dependent burst‐to‐tonic switching of thalamic cell activity: an in vitro study. Arch Ital Biol 122, 73–82. [PubMed] [Google Scholar]
- Jouhanneau JS, Ferrarese L, Estebanez L, Audette NJ, Brecht M, Barth AL & Poulet JF (2014). Cortical fosGFP expression reveals broad receptive field excitatory neurons targeted by POm. Neuron 84, 1065–1078. [DOI] [PubMed] [Google Scholar]
- Kichula EA & Huntley GW (2008). Developmental and comparative aspects of posterior medial thalamocortical innervation of the barrel cortex in mice and rats. J Comp Neurol 509, 239–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ & Sakmann B (1999). A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398, 338–341. [DOI] [PubMed] [Google Scholar]
- Lu SM & Lin RC (1993). Thalamic afferents of the rat barrel cortex: a light‐ and electron‐microscopic study using Phaseolus vulgaris leucoagglutinin as an anterograde tracer. Somatosens Mot Res 10, 1–16. [DOI] [PubMed] [Google Scholar]
- Lütcke H, Murayama M, Hahn T, Margolis DJ, Astori S, Zum Alten Borgloh SM, Göbel W, Yang Y, Tang W, Kügler S, Sprengel R, Nagai T, Miyawaki A, Larkum ME, Helmchen F & Hasan MT (2010). Optical recording of neuronal activity with a genetically‐encoded calcium indicator in anesthetized and freely moving mice. Front Neural Circuits 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major G, Larkum ME & Schiller J (2013). Active properties of neocortical pyramidal neuron dendrites. Annu Rev Neurosci 36, 1–24. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL & Curran T (1987). Mapping patterns of c‐fos expression in the central nervous system after seizure. Science 237, 192–197. [DOI] [PubMed] [Google Scholar]
- Murayama M, Pérez‐Garci E, Lüscher HR, Larkum ME (2007). Fiberoptic system for recording dendritic calcium signals in layer 5 neocortical pyramidal cells in freely moving rats. J Neurophysiol 98, 1791–1805. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M & Imoto K (2001). A high signal‐to‐noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol 19, 137–141. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Gutnisky DA, Huber D, Xu NL, O'Connor DH, Tian L, Looger L & Svoboda K (2012). Activity in motor‐sensory projections reveals distributed coding in somatosensation. Nature 489, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A & Svoboda (2007). Channelrhodopsin‐2‐assisted circuit mapping of long range cllaosal proejctions. Nat Neurosci 10, 663–668. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG & Svoboda K (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–52. [DOI] [PubMed] [Google Scholar]
- Shen Y, Lai T & Campbell RE (2015). Red fluorescent proteins (RFPs) and RFP‐based biosensors for neuronal imaging applications. Neurophotonics 2, 031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger JF, Bisler S, Schleicher A, Gass P, Stehle JH & Zilles K (2000). Exploration of a novel environment leads to the expression of inducible transcription factors in barrel‐related columns. Neuroscience 99, 7–16. [DOI] [PubMed] [Google Scholar]
- Steiner H & Gerfen CR (1994). Tactile sensory input regulates basal and apomorphine‐induced immediate‐early gene expression in rat barrel cortex. J Comp Neurol 344, 297–304. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K & Looger LL (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods 6, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Bhat RV, Baraban JM, Erickson CA, McNaughton BL & Barnes CA (1993). Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long‐term enhancement. J Neurosci 13, 4776–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Abdelfattah AS, Miracourt LS, Kutsarova E, Ruangkittisakul A, Zhou H, Ballanyi K, Wicks G, Drobizhev M, Rebane A, Ruthazer ES & Campbell RE (2014). A long Stokes shift red fluorescent Ca2+ indicator protein for two‐photon and ratiometric imaging. Nat Commun 5, 5262. [DOI] [PMC free article] [PubMed] [Google Scholar]