

Abstract
Key points
Smn +/− transgenic mouse is a model of the mildest form of spinal muscular atrophy.
Although there is a loss of spinal motoneurons in 11‐month‐old animals, muscular force is maintained.
This maintained muscular force is mediated by reinnervation of the denervated fibres by surviving motoneurons.
The spinal motoneurons in these animals do not show an increased susceptibility to death after nerve injury and they retain their regenerative capacity.
We conclude that the hypothesized immaturity of the neuromuscular system in this model cannot explain the loss of motoneurons by systematic die‐back.
Abstract
Spinal muscular atrophy (SMA) is a common autosomal recessive disorder in humans and is the leading genetic cause of infantile death. Patients lack the SMN1 gene with the severity of the disease depending on the number of copies of the highly homologous SMN2 gene. Although motoneuron death in the Smn +/− transgenic mouse model of the mildest form of SMA, SMA type III, has been reported, we have used retrograde tracing of sciatic and femoral motoneurons in the hindlimb with recording of muscle and motor unit isometric forces to count the number of motoneurons with intact neuromuscular connections. Thereby, we investigated whether incomplete maturation of the neuromuscular system induced by survival motoneuron protein (SMN) defects is responsible for die‐back of axons relative to survival of motoneurons. First, a reduction of ∼30% of backlabelled motoneurons began relatively late, at 11 months of age, with a significant loss of 19% at 7 months. Motor axon die‐back was affirmed by motor unit number estimation. Loss of functional motor units was fully compensated by axonal sprouting to retain normal contractile force in four hindlimb muscles (three fast‐twitch and one slow‐twitch) innervated by branches of the sciatic nerve. Second, our evaluation of whether axotomy of motoneurons in the adult Smn +/− transgenic mouse increases their susceptibility to cell death demonstrated that all the motoneurons survived and they sustained their capacity to regenerate their nerve fibres. It is concluded the systematic die‐back of motoneurons that innervate both fast‐ and slow‐twitch muscle fibres is not related to immaturity of the neuromuscular system in SMA.
Keywords: motoneuron counts, motor units, Smn+/− III transgenic mice, spinal muscular atrophy
Key points
Smn +/− transgenic mouse is a model of the mildest form of spinal muscular atrophy.
Although there is a loss of spinal motoneurons in 11‐month‐old animals, muscular force is maintained.
This maintained muscular force is mediated by reinnervation of the denervated fibres by surviving motoneurons.
The spinal motoneurons in these animals do not show an increased susceptibility to death after nerve injury and they retain their regenerative capacity.
We conclude that the hypothesized immaturity of the neuromuscular system in this model cannot explain the loss of motoneurons by systematic die‐back.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- CP
common peroneal
- EDL
extensor digitorum longus
- FG
fluorogold
- FR
fluororuby dextran tetramethylrhodamine
- ITS‐MUNE
incremental twitch subtraction motor unit number estimation
- MG
medial gastrocnemius
- MUNE
motor unit number estimation
- SMA
spinal muscular atrophy
- SMN protein
survival motoneuron protein
- SMN1
survival motoneuron gene 1
- SOL
soleus
- TA
tibialis anterior
- WT
wild type
Introduction
Spinal muscular atrophy (SMA) is the second most common autosomal recessive inherited disease in humans and is caused by mutations of the survival motoneuron gene (SMN1) (Lefebvre et al. 1995). SMN1 encodes for the ubiquitously expressed SMN protein. The protein is involved in the assembly of ribonucleoproteins that regulate pre‐mRNA splicing and translation for protein synthesis and may regulate the expression/trafficking of a number of mRNAs that are locally translated in axons (Monani et al. 1999; Zhang et al. 2008; Liu‐Yesucevitz et al. 2011; Fallini et al. 2012; Kye et al. 2014). It is duplicated alongside a highly homologous copy, SMN2, which may act as the phenotypic modifier of the disease but produces insufficient levels of SMN protein for normal motoneuron function and survival (Melki et al. 1994; Lefebvre et al. 1995; Burghes & Beattie, 2009). Based on age of onset and severity of the disease phenotype, SMA is classified into three types (Munsat & Davies, 1992). The most severe type I SMA (Wernig‐Hoffmann) is characterized by generalized weakness and hypotonia shortly after birth, with death usually occurring within 6 months. Type II is the intermediate form; affected children are able to sit but they cannot stand unaided and survive for more than 2 years. Type III is the relatively mild form where patients begin to develop muscle weakness after 18 months of age but survive into the adult years (Roberts et al. 1970; Pearn, 1978).
SMA is a neuromuscular disorder which affects all components of the motor unit, the motoneuron and the muscle fibres that it supplies, but the most prominent feature is the loss of spinal motoneurons resulting in progressive weakness and muscle atrophy (Crawford & Pardo, 1996). Pathology includes visible loss of nerves in ventral roots whereas the brain and spinal cord appear normal, and muscles are small and pale (Batten, 1911). Chromatolytic changes in motoneurons that are typically seen after cutting the axons (axotomy) in several species, including Aplysia, rats and cats (Lieberman, 1971; Gordon, 1983; Berdan et al. 1990), occur in conjunction with progressive degenerative changes in the peripheral nerves. The latter include loss of large myelinated fibres (Chou & Nonaka, 1978; Crawford & Pardo, 1996), and reduced calibre of spinal nerve roots (Simic et al. 2000). Active apoptotic death in motoneurons within fetal anterior horn sections was demonstrated by Soler‐Botija et al. (2002) who suggested that the cause of the selective neuromuscular degeneration in SMA may be over‐activation of naturally occurring apoptotic ‘pruning’ processes during development. Nonetheless, it is clear that in all three types of human SMA, motoneuron death occurs after neuromuscular connections are made (Soler‐Botija et al. 2002), indicating that the SMN deficiency negatively impacts these connections. Normally these connections are essential for the survival of immature motoneurons with extensive evidence of their demise by apoptosis in neonatal rats when the neuromuscular connections are disrupted in the first week of life but not thereafter (Lowrie et al. 1982; 1987; Greensmith & Vrbová, 1996; de Bilbao & Dubois‐Dauphin, 1996; Rossiter et al. 1996; Kemp et al. 2015). Possibly, the SMA motoneurons remain immature in the sense that they are susceptible to cell death after separation from their target muscles: once the ‘pruning’ of excess contacts at the neuromuscular junction occurs, the ‘immature’ SMA motoneurons may lose sufficient contact with their muscle fibres for each nerve fibre to retract completely from contact with muscle fibres, undergoing axonal die‐back (Greensmith & Vrbová, 1997, Hausmanowa‐Petrusewicz & Vrbová, 2005). In light of reported defects in motor axon growth (Rossoll et al. 2003; McWhorter et al. 2008), these motoneurons may fail to regenerate effectively and the susceptible motoneurons will die. Evidence that restoring SMN protein to SMA animal models is therapeutic and, importantly, that it is most effective during a relatively brief window in time early in the disease (Le et al. 2011; Lutz et al. 2011) was recently substantiated by findings that SMN is critical for the maturation of the neuromuscular junction during postnatal life (Kariya et al. 2014).
Presently, data are limited to findings of reduced numbers of surviving motoneurons in the lumbosacral cord of transgenic SMA mice at one or more time points in the lifespan of the mice (Jablonka et al. 2000; Monani et al. 2000, 2003; Balabanian et al. 2007). In order to determine if motor nerves progressively die back prior to death of the motoneurons in the ventral horn of the spinal cord, it is necessary to enumerate motoneurons that have functional connections and to establish the time course of the loss of functional motor units. It is also essential to document die‐back of motor axons by backlabelling motoneurons of the lumbosacral cord through intact axons located within the peripheral nerves that innervate hindlimb muscles. Thus the first objective of the present study was to use the backlabelling technique of applying a fluorescent dye to a hindlimb nerve to establish the time course of motor unit and motoneuron loss in the C57B6Smn +/− SMAIII transgenic mouse and to determine whether there is a temporal progression from disconnection of functional neuromuscular junctions to death of the motoneurons.
Normally, mature adult motoneurons, unlike immature neonatal motoneurons, survive the injuries that disrupt their neuromuscular contacts, namely axotomy (Xu et al. 2010). They undergo a change in gene expression from a chemically transmitting to a growth state that, in turn, supports the outgrowth and regeneration of their lost axons (Fu & Gordon, 1997). If indeed, SMN deficiency sustains a susceptibility of the motoneurons to cell death, death of at least some of the neurons might be expected when their neuromuscular contacts are disrupted. A second objective of this study was to determine whether the adult motoneurons in the C57B6Smn +/− SMAIII transgenic mouse model of SMA display an abnormal susceptibility to cell death following axotomy.
Data from transgenic mice in which the Smn gene was deleted (SMAII) indicate persistent muscle immaturity as evidenced by the loss of motoneurons, enduring muscle weakness, deficient axonal growth, and the continued expression of a neonatal muscle fibre phenotype (Frugier et al. 2000; Cifuentes‐Diaz et al. 2001; Fan & Simard, 2002; Arnold et al. 2004; Biondi et al. 2008). SMAII transgenic mice also display immature features in motor nerve terminals at the neuromuscular junction (Frugier et al. 2000; Cifuentes‐Diaz et al. 2001; Fan & Simard, 2002; Arnold et al. 2004; Biondi et al. 2008) that precede motoneuron death (Frugier et al. 2000; Monani et al. 2000; Soler‐Botija et al. 2002). A particularly notable feature is the impaired clustering of acetylcholine receptors revealing a persistent developmental interaction of peripheral motor nerves with their target muscle fibres (Grondard et al. 2005; Biondi et al. 2008; Kariya et al. 2008; Murray et al. 2008). Thus the third and final objective of the present study was to investigate axonal sprouting at motor nerve terminals to determine whether axonal sprouting occurred in the muscles of the C57B6Smn +/− SMAIII transgenic mice concurrent with loss of motoneurons.
Methods
Ethical approval
All surgical procedures and perioperative care measures were performed with strict accordance with the National Institutes of Health guidelines, The Canadian Council on Animal Care (CCAC) and were approved by the University of Alberta Health Sciences Animal Welfare and Policy Committee.
Animals
Breeding pairs of C57BL/6‐Smn +/− transgenic mice (Jablonka et al. 2000) and C57BL/6‐wild type (WT) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed in the Health Sciences Laboratory Animal Services (HSLAS) at the University of Alberta. Animals were housed in standard conditions, in soft bedding and artificial 12 h dark/light cycles, with food and water available ad libitum.
A total of 71 C57BL/6‐Smn +/− transgenic and 69 C57BL/6‐WT mice were used in the experiments. The mice were genotyped by PCR amplification of DNA extracted from ear biopsies according to protocols specified by Jackson Laboratories.
Anaesthetic protocols, monitoring and terminal procedures
All surgical procedures, application of retrograde dyes and in vivo recordings of muscle and motor unit isometric contractile force were made under deep anaesthesia and using a sterile surgical technique. The mice were anaesthetized with a ketamine/Atravet cocktail (100 mg ml−1 ketamine and 10 mg ml−1 acepromazine in sterile saline at a dose of 17.5 ml (kg body weight)−1 by intraperitoneal injection). If a supplementary dose was needed, one‐third of the dose was administered half an hour after the first dose. The depth of anaesthesia was determined by manually pinching the tail or the foot and the reflex response was monitored. The absence of a reflex had to be noted in order to proceed with the different procedures. After the surgery or the retrograde dye application, the wound was sutured and disinfected with iodine. Thereafter, the mice received a single dose of buprenorphine (0.05 mg (kg body weight)−1, by subcutaneous injection) and they were kept in a warm environment until they recovered fully from the anaesthesia. All the mice that were subjected to nerve surgery were periodically examined for signs of distress or autotomy at the denervated hindlimb. No signs of distress or autotomy were detected in any of the mice during the over survival periods of up to 4 months.
In the terminal experiments mice were either anaesthetized for hindlimb muscle contractile force recordings (see below) or they were deeply anaesthetized and perfused with 50 ml saline followed by 100 ml 4% paraformaldehyde at pH 7.4 through the aorta. Following perfusion, the lumbosacral spinal cord was removed to count backlabelled femoral or sciatic motoneurons. After the experiments in which force recordings were made under deep surgical anaesthesia, the mice were killed by cervical dislocation.
Confirmation of compliance
All the authors state that they understand the ethical principles under which The Journal of Physiology operates and their work complies with the animal ethics procedures outlined in Grundy (2015).
Backlabelling of motoneurons
Femoral and sciatic motoneuron pools
In 4‐month‐old C57BL/6‐Smn +/− (n = 6) and C57BL/6‐WT (n = 5) mice, the right sciatic nerve was exposed and crushed at the mid‐thigh. The left femoral nerve was exposed and crushed in the ventral part of the thigh. Five per cent fluororuby dextran tetramethylrhodamine (FR) and 4% fluorogold (FG; hydrostilbamidine bis(methane‐sulphonate); Sigma‐Aldrich no. 39286, St Louis. MO, USA) in distilled water (Molecular Probes, Eugene, OR, USA) were used to backlabel the motoneurons in the lumbosacral spinal cord, each of the two dyes identifying the motoneuron pool of the two nerves. About 100 μl of the dyes was injected at the crush site using a micropipette attached to a picospritzer (Intercel Picospritzer III, Parker Hennifin, Cleveland, OH, USA) as described previously (Brushart et al. 2002). The two dyes have been shown to yield similar counts of motoneurons (Al‐Majed et al. 2000; Boyd & Gordon, 2001) but they were alternated between contralateral nerves within animals to avoid variability in the motoneuron counts. The data obtained are shown in Fig. 3.
Figure 3. Number of femoral as compared to sciatic motoneurons in the lumbosacral (L2–L4) spinal cord in 4‐month‐old Smn +/− transgenic mice and wild type control mice.
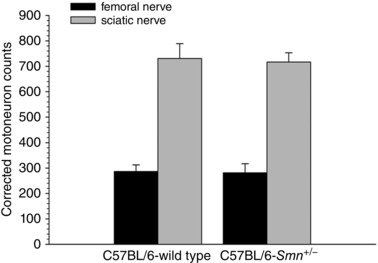
Motoneuron counts after backlabelling the femoral or sciatic nerves with either fluorogold or rubyred were completed on consecutive 40 μm thick longitudinal spinal cord sections between T11 and L2; motoneurons in all sections were counted. The data are corrected for thickness of the longitudinal sections. Each bar represents the mean ± SEM of data obtained from 5 (C57BL/6‐wild type) and 6 (C57BL/6‐Smn +/− transgenic) mice. Note that the number of motoneurons in the Smn +/− transgenic mouse is not different from the wild type control for either femoral (P > 0.91) or sciatic (P > 0.83) nerves at 4 months of age.
Four‐, 5‐, 7‐ and 12‐month‐old C57BL/6‐Smn +/− (n = 30) and C57BL/6‐WT (n = 30) mice were anaesthetized for exposure and crush of the sciatic nerve in order to backlabel and count the sciatic motoneurons that survive as a function of age. Right sciatic nerves were transected at mid‐thigh level just above the division of the sciatic into tibial and common peroneal (CP) nerves. The tip of the proximal sciatic nerve stump was dipped for 1 h in a solution of FG or FR that was contained in a Vaseline well (Al‐Majed et al. 2000; Boyd & Gordon, 2001). The exposure time was 1 h, after which the well was removed and the nerve stump washed with saline. The data that were obtained are plotted in Fig. 2.
Figure 2. Number of sciatic motoneurons in Smn +/− transgenic mice as compared to wild type mice at 4, 5, 7 and 12 months (m) of age.
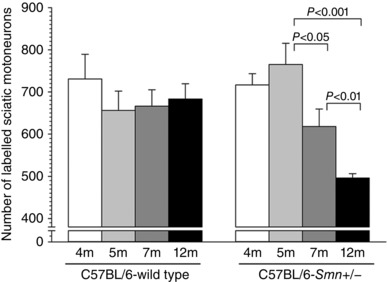
Each bar represents the mean ± SEM of data obtained from 6–8 mice. The numbers of motoneurons was counted after application of either fluorogold or rubyred to the sciatic nerve mid‐thigh. Note that the number of motoneurons in the Smn +/− transgenic mice began to decline significantly by 7 months.
Sciatic motoneurons after axotomy
To evaluate the survival of sciatic motoneurons after 2 and 4 months of prolonged axotomy, the sciatic nerve in the right hindlimb of Smn +/− SMA (n = 8 and 8, respectively) and WT (n = 7 and 8, respectively) 4‐month‐old mice was transected and axon regeneration was prevented by suturing the proximal nerve stump into the adjacent innervated muscles (Fu & Gordon, 1995). Two and 4 months later, the intact sciatic nerve of the left side was cut at the mid‐thigh level and the proximal nerve stump dipped for 1 h in a solution of FG or FR, contained in a Vaseline well. In the right operated hindlimb, either dye, depending on the dye applied on the left sciatic nerve, was applied to the right sciatic nerve proximal to the axotomy site. The dyes were alternated on the left and right sides in order to distinguish the neurons in the ventral horns of the spinal cord. The data obtained in these experiments are plotted in Fig. 8.
Figure 8. Mean (±SEM) numbers of motoneurons of C57BL/6‐Smn +/− transgenic and C57BL/6‐wild type control mice, back‐labelled from the sciatic nerve 2 months (A) and 4 months (B) after left sciatic nerve section and suture of the proximal nerve stump to an innervated muscle to prevent regeneration.
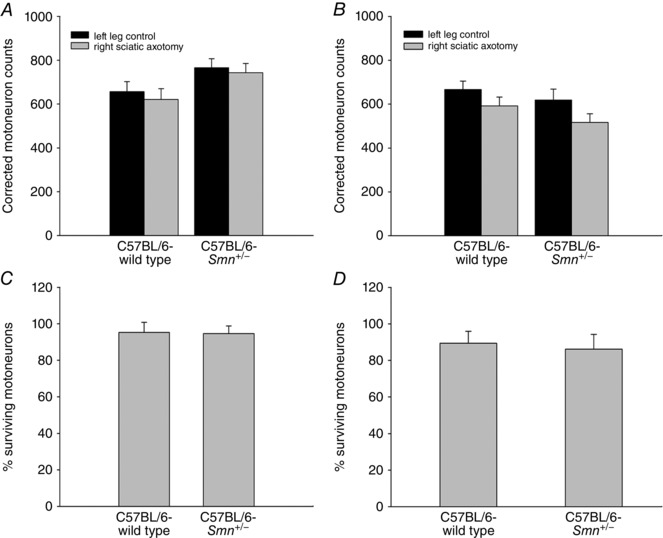
The corresponding proportions of surviving motoneurons after axotomy are shown in C and D. The left sciatic nerves served as intact internal controls. Two and 4 months after axotomy, the proximal stump of the sciatic nerve was cut and exposed to FR to back‐label the motoneurons that survived the injury. Mice were 5 and 7 months old at the end of experiments in A and B. Motoneuron counts were completed on consecutive 40 μm thick longitudinal spinal cord sections between T11 and L2; all sections were counted. The data are corrected for thickness of longitudinal sections. (n = 7 for C57BL/6‐wild type and n = 8 for C57BL/6‐Smn +/− transgenic mice).
Sciatic motoneurons after transection and surgical coaptation (resuture)
The capacity for axon regeneration was evaluated in the right hindlimbs of Smn +/− SMA (n = 8) and WT (n = 7) 4‐month‐old mice. The right sciatic nerve was exposed at the mid‐thigh, transected (40 mm proximal to the tip of the third digit) and surgically coapted by direct suture of the proximal and distal nerve stumps with two 9‐0 nylon sutures (Ethicon Inc., Somerville, NJ, USA). The wound was sutured closed and the mice allowed to recover from anaesthesia. Axonal regeneration was allowed for 3 weeks and afterwards the right CP nerve was cut prior to its entrance to the tibialis anterior (TA) muscle, distal to the CP nerve repair site. The tip of the proximal stump was dipped for 1 h in the 5% FR solution contained in a Vaseline well. The left CP nerve was also exposed for simultaneous backlabelling of the unoperated CP motoneurons. As described above, the dyes that were used, either FG or FR, were applied so that the neurons were backlabelled on left and right sides with separate dyes for identification. One week later, animals were killed and their spinal cords removed to visualize and count the motoneurons on both sides of the cord. The data are plotted in Fig. 9.
Figure 9. Numbers of backlabelled common peroneal (CP) motoneurons that regenerated their nerves after sciatic nerve transection and resuture in C57BL/6‐Smn +/− transgenic and C57BL/6‐wild type control mice.
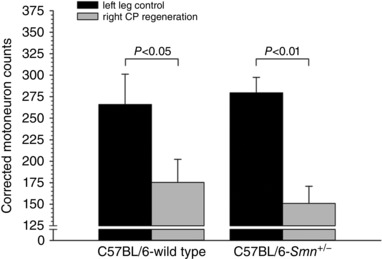
Left CP nerves served as intact internal controls. The right sciatic nerves of 4‐month‐old mice were cut and subsequently repaired (coapted) by direct suture of the proximal and distal stumps. Three weeks later, left control and the right axotomized CP motoneurons were backlabelled via the CP nerve at sites that were distal to the repair site of the right CP nerve. Motoneuron counts were completed on 20 μm thick longitudinal spinal cord sections between T11 and L2; every second section was counted through the entire spinal cord. Data are corrected for thickness of the longitudinal sections. Each bar represents the mean (±SEM) of data obtained from 8 C57BL/6‐Smn +/− transgenic and 7 C57BL/6‐wild type mice. The corrected motoneuron counts after right CP nerve regeneration were significantly different from counts from the left intact CP nerves but the counts were not different between the Smn +/− transgenic and the wild type mice.
Motoneuron counts
Seven days after the application of fluorescent dyes to backlabel motoneuronal cell bodies, the mice were deeply anaesthetized for perfusion via the aorta with 50 ml saline followed by 100 ml 4% paraformaldehyde at pH 7.4. After the perfusion, the spinal cord was dissected and the segment T11–L2 (containing the femoral, the sciatic and the common peroneal nerve motoneuron pools) was removed and post‐fixed with 30% sucrose in 4% paraformaldehyde solution overnight. The tissue was then frozen in liquid nitrogen after being embedded in Tissue‐Tek O.C.T. compound (Sakura Finetek, Torrance, CA, USA).
Frozen samples were sectioned in a cryostat (Jung CM3000, Leica, Wetzlar, Germany). Longitudinal spinal cord sections were cut at 40 μm thickness. The fluorescent bodies of the retrogradely labelled motoneurons of the different nerve pools were visualized and counted at ×40 under fluorescence at barrier filters of 580 nm for FR and an ultraviolet filter for FG. The number of motoneurons counted was corrected according to the thickness of the sections and the diameter of the bodies by the method of Abercrombie and Johnson (Abercrombie & Johnson, 1946).
Isometric contractile force recording
To evaluate the number of adult sciatic motoneurons that sustained their connections with skeletal muscles, the right hindlimb of 7‐month‐old (n = 6) and 11‐month‐old (n = 6) Smn +/− SMA transgenic mice and 7‐month‐old (n = 4) and 11‐month‐old (n = 4) WT mice were dissected under deep anaesthesia for in vivo isometric contractile force recording from each of four hindlimb muscles: the medial gastrocnemius (MG), soleus (SOL), extensor digitorum longus (EDL) and tibialis anterior (TA) muscles (Fig. 1 A). All other hindlimb muscles were denervated by cutting their nerve supplies.
Figure 1. In vivo isolation of the sciatic nerve and fast‐twitch medial gastrocnemius (MG), extensor digitorum longus (EDL) and tibialis anterior (TA) muscles and the slow‐twitch soleus (SOL) muscle for isometric contractile force recording.
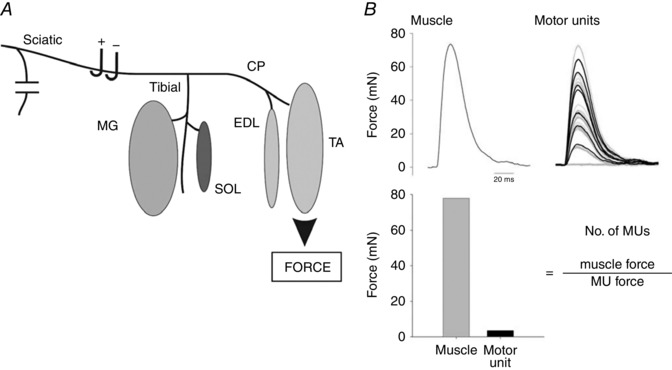
A, all nerves except the sciatic and its tibial and CP nerve branches to the MG and SOL muscles and to the EDL and SOL muscles were denervated. Each of the muscles was attached to a force transducer for the recording of maximal muscle twitch contractile force and, in response to all‐or‐none increments in stimulus intensity on the sciatic nerve, the progressive recruitment of single motor units (B). The number of motor units (MUs) was obtained from the ratio of muscle and MU forces as described in the text.
Each of the muscles was attached in random order to a force transducer for recording isometric contractile forces. Evoked isometric forces were amplified and visualized on an oscilloscope and digitized using Axoscope (Version 8.0, Axon Instruments, Union City, CA, USA). Muscle length was adjusted for maximal isometric twitch force in response to stimulation of the sciatic nerve. Whole muscle twitch and tetanic forces were recorded in response to single and repetitive suprathreshold (twice threshold voltage at 100 ms duration) stimulation of the sciatic nerve at 0.5 and 100 Hz, respectively. The tetanic contractions lasted for 210 ms and the maximum tetanic force was measured. An example of muscle and motor unit force recordings from a 60 day control mouse TA muscle is shown in Fig. 1 B. To estimate the number of motor units in the hindlimb muscles of the mice, we used a modified version of the mechanical motor unit number estimation (MUNE) that was first described by McComas et al. (1971) for EMG measurements of single motor units. The validity of this new method, the incremental twitch subtraction motor unit number estimation (ITS‐MUNE) method of counting motor units, was described previously from our laboratory (Major et al. 2007). In this study, we used the ITS‐MUNE method to overcome potential problems of alternation, namely that motor units can be recruited in numerous combinations by stimulating a motor nerve (Galea et al. 1991), which, in turn, may result in overestimation of the numbers of motor units (Stein & Yang, 1990). An interactive software program was used that randomly selects from a set of stimulus‐evoked, incremental muscle twitch force waveforms (Major et al. 2007). The basic principle that underlies all MUNE techniques is the division of the total muscle response by the estimate of the average motor unit response. The difference between the different MUNE techniques is the method used to estimate the average single motor unit response. With the ITS‐MUNE method, 8–20 isometric twitch responses were selected randomly from ∼100 incremental force waveforms. The waveforms were recorded from each of the four muscles in response to a manually controlled incremental rise in the stimulus voltage of 100 μs duration to the sciatic nerve from 0 to 10 V at a frequency of 0.5 Hz until the recorded twitch force was ∼66% of the whole muscle twitch force. This resulted in incremental increases in muscle twitch force that have been shown to reflect a random distribution of axon thresholds (Major et al. 2007). The custom computer program – a graphical user interface based in MATLAB called ITS‐MUNE – filtered and aligned incremental twitch force traces, randomly selected twitch responses, rejected non‐physiological motor unit twitches with regions of negative force, subtracted two rank‐ordered force traces to generate a candidate motor unit twitch response, and finally averaged motor unit twitch samples to estimate mean twitch force and to calculate ITS‐MUNE (Major et al. 2007) (Fig. 1 B). The data are plotted in Figs 4 and 6.
Figure 4. Mean (±SEM) isometric contractile forces developed by fast‐twitch tibialis anterior (TA), medial gastrocnemius (MG) and extensor digitorum longus (EDL) muscles, and slow‐twitch soleus (SOL) muscles.
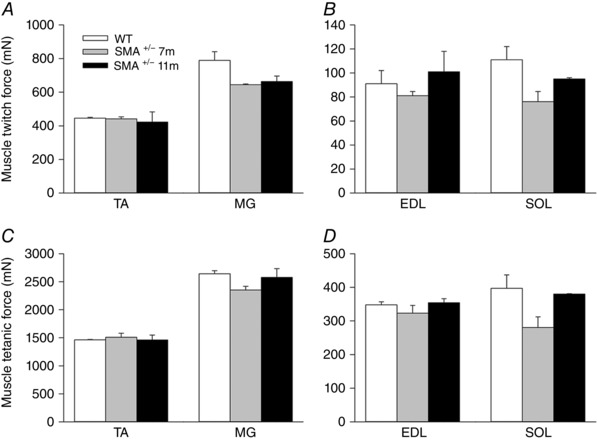
Maximal twitch forces in response to 2× threshold electrical stimulation of the sciatic nerve at 0.5 Hz. A and B, and, at 100 Hz, maximal tetanic forces (C and D) forces in C57BL/6‐Smn +/− transgenic and C57BL/6‐wild type mice. The twitch forces of Smn +/− transgenic mouse muscles were 89.6 ± 17.8% (P > 0.16) of those of the wild type controls; muscle tetanic forces of the Smn +/− transgenic mice were 93.6 ± 3.3% (P > 0.20) of wild type controls.
Figure 6. Mean ± SEM of the estimated numbers of intact motor units (MUNE) in hindlimb muscles of C57BL/6‐Smn +/− transgenic mice expressed as a percentage of corresponding muscles in C57BL/6‐wild type control mice.
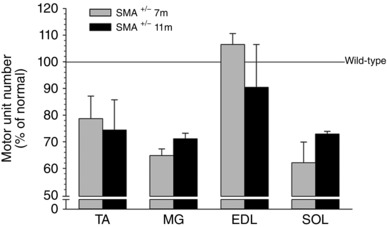
The mean number of motor units was 30% lower (P < 0.01) in TA, MG and SOL of the Smn +/− transgenic mice as compared with wild type control muscles.
After completion of the muscle force and motor unit force recordings, the muscles were excised, blotted dry, and weighed. The data are plotted in Fig. 5.
Figure 5. Mean ± SEM of the wet weights of fast‐twitch TA, MG and EDL muscles and slow‐twitch SOL muscles of C57BL/6‐Smn +/− transgenic and C57BL/6‐wild type mice.
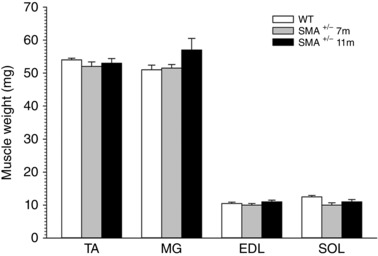
Muscle weights of Smn +/− were 97 ± 2.7% (P > 0.44) of control wild type.
Combined silver/cholinesterase histochemical staining
Under deep anaesthesia, 11‐month‐old Smn +/− SMA transgenic mice (n = 6) and WT mice (n = 4) were perfused with 50 ml saline followed by 100 ml 4% formalin at pH 7.4 through the aorta. The TA muscles from both hindlimbs were dissected, cryoprotected by subsequent overnight incubation in gum sucrose solution and then frozen in isopentane at −74°C. Cryostat longitudinal sections at 100 μm were cut and stained using combined silver/cholinesterase (Ag/AChE) histochemical staining to visualize the motor axons, sprouts and the muscle endplates. For AChE staining, 100 μm cryostat longitudinal sections were collected in distilled water and incubated for 25–30 min at room temperature in a mixture of 0.01 m Tris‐HCl buffer, pH 7.2, bromoindoxyl acetate, 1.65% potassium ferricyanide, and 2.11% calcium chloride. For silver staining, sections from AChE staining were first incubated in 20% silver nitrate for 15 min, washed in distilled water, incubated in 3% sodium sulfite for 10 min and washed again, and finally, developed in a mixture of silver nitrate and physical developer. The number of axon sprouts (having visible axonal attachment to muscle endplates) and free endplates (having no visible axonal attachment) were subsequently counted from the longitudinal muscle sections. A sample of 500 endplates from the middle portion of each TA muscle was examined under light microscopy at a total magnification of ×160 or ×400 and the axonal sprouts categorized as intranodal, terminal and ultraterminal sprouts with intranodal sprouts originating from the last node of Ranvier (Fig. 7 A), terminal sprouts originating from the axons after the last node of Ranvier (Fig. 7 B), and the ultraterminal sprouts originating from the endplate area and contacting another endplate (Fig. 7 C). The quantity of endplates that were reinnervated by axonal sprouts and those that were not contacted by an axon (free endplates) was determined and expressed as a percentage of all the endplates counted. The motor axons were stained brown and the motor endplates were revealed by greenish‐blue staining of the AChE enzyme at the synaptic lamina. Unlike acetylcholine receptors, AChE remains indefinitely after denervation.
Figure 7. Photomicrographs of longitudinal sections of the TA muscle from an 11‐month‐old C57BL/6‐Smn +/− transgenic mouse stained with the combined silver/cholinesterase stain to visualize axons and motor endplates (EPs), respectively.
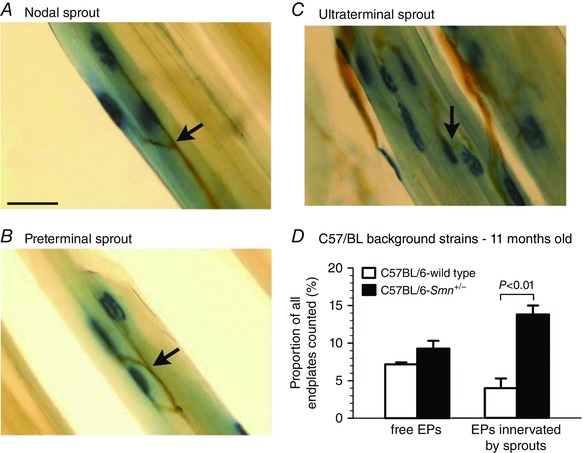
Examples of nodal (A), preterminal (B) and ultraterminal (C) sprouts observed in muscles are shown. D, mean (±SEM) proportions of endplates that were free of nerve endings (free EPs) or were innervated by axon sprouts (EPs innervated by sprouts) are plotted for C57BL/6‐Smn +/− transgenic and C57BL/6‐wild type control mice. Scale bar is 50 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Statistics
Data are reported as mean ± the standard error of the mean (SEM). Group means were compared using an independent samples Student's t test. Differences were considered significant at P < 0.05. P values are reported.
Results
Survival of motoneurons in Smn +/− transgenic mice
The numbers of labelled sciatic motoneurons in the ventral horn of the lumbosacral (L4–L5) spinal cord of the Smn +/− transgenic mouse model of SMAIII did not show any decline until 7 months when a significant reduction of 19% was detected between 5 and 7 months (P < 0.05), continuing to a 35% reduction (P < 0.001) at 12 months of age (Fig. 2). When direct comparisons are made at each age between the C57BL/6‐wild type transgenic mice and the corresponding Smn +/− type III transgenic mice, it becomes clear that the number of lumbosacral motoneurons with intact axons in the sciatic nerve declined significantly only in the 12‐month‐old mice. The numbers of motoneurons in the wild type mice did not change with age.
Counts of the number of femoral motoneurons in 4‐month‐old Smn +/− and wild type control transgenic mice were in agreement with the data for the sciatic motoneurons with there being no detectable difference between the smaller number of femoral motoneurons innervating more proximal muscles in the Smn +/− transgenic and wild type mice as was observed for the sciatic motoneurons at the same age (Fig. 3). That the numbers of surviving sciatic motoneurons at 4 months of age was the same whether the motoneurons were backlabelled with retrograde dyes applied to the nerve at a crush site by a picosprizer (Fig. 3) or by dipping the cut proximal stump into a pool of the dye, verifies the validity of the two methods.
Reduced numbers of intact motor units in Smn +/− transgenic mice
To address whether the number of lumbosacral motoneurons that were counted by backlabelling via the sciatic nerve have intact neuromuscular connections or whether the motor axons die‐back, we recorded maximal contractions of representative hindlimb muscles innervated by the sciatic nerve and counted the number of intact motor units using isometric force recording as described in the Methods. These recordings were made at 7 and 11 months of age, the ages when we had noted significant declines in motoneuron numbers (Fig. 2). As shown in Fig. 4, the fast‐twitch tibialis anterior (TA), medial gastrocnemius (MG) and extensor digitorum longus (EDL) and the slow‐twitch soleus (SOL) hindlimb muscles in the Smn +/− transgenic mice developed the same maximal twitch and tetanic isometric forces at 7 and 11 months of age as the corresponding wild type control mice. The weights of the muscles for both the larger TA and MG muscles as well as the smaller EDL and SOL muscles were also the same, consistent with the sustained muscle forces (cf. Figs 4 and 5). Yet the average number of intact TA, MG and SOL motor units at both 7 and 11 months of age in the Smn +/− transgenic mice had declined by 30% (P < 0.01) compared with the corresponding muscles of the wild type control transgenic mice (Fig. 6). The findings that the maximal nerve stimulation resulted in the development of normal contractile forces indicated that the remaining intact motor units had enlarged by axon sprouting to reinnervate the muscle fibres denervated by the loss of axon connections.
Sprouting of axons in hindlimb skeletal muscles of Smn +/− transgenic mice
In order to evaluate sprouting directly, we visualized axons and the muscle endplates in the TA muscle of Smn +/− and wild type control transgenic mice. Nodal and terminal sprouting has been reported in the homozygous mouse model of SMA/III at 4 months of age when muscle fibres become denervated in association with a reduction of ∼30% in the numbers of viable motoneurons (Monani et al. 2003). In light of our findings of reductions of ∼19% (P < 0.01) in numbers of motoneurons with nerve fibres traversing the sciatic nerve between 7 and 12 months of age (Fig. 2), we visualized the nerves and their innervation of end‐plates in longitudinal muscle sections with Ag/AChE histochemistry in 11‐month‐old mice. As described previously (Tam et al. 2001), ∼10% of endplates are free of silver stained axons as a result of the cryostat sectioning in both the Smn +/− and wild type control transgenic mice (Fig. 7 D). In line with the findings of significant reductions in motoneuron numbers (Fig. 2), we observed sprouting in the muscles of the Smn +/− transgenic mice (Fig. 7). As shown in the typical examples of intramuscular nerves stained with silver chloride and their innervation of muscle endplates stained for cholinesterase in Fig. 7 A–C, all types of sprouts – nodal, preterminal and ultraterminal – typical of partially denervated muscles (Tam et al. 2001) were seen in the muscles. The mean number of axon sprouts increased by 4‐fold (P < 0.01), being sufficient to reinnervate all the denervated fibres in the partially denervated muscles (Fig. 7 D) and for full reinnervation of the partially denervated muscles and recovery of their muscle contractile forces and muscle weights (Figs 5 and 6).
Motoneuron survival after axotomy in Smn +/− transgenic mice
If mature motoneurons that make functional connections in SMA nevertheless fail to make the complete transition from a growth to a transmitting phenotype, we would expect that these motoneurons would be highly susceptible to cell death after axotomy, as has been demonstrated for immature motoneurons at postnatal ages of 1–5 days in rats (Lowrie et al. 1987). We cut the sciatic nerve in the right hindlimb of Smn +/− SMA transgenic and wild‐type control mice at 3 months of age (prior to any significant motoneuron death, see Fig. 3) and evaluated survival of axotomized sciatic motoneurons 2 and 4 months post‐axotomy. We prevented nerve fibre regeneration in these experiments by suturing the proximal nerve stump into innervated muscle. We did not observe any difference between the Smn +/− and wild type control mice in the survival of their motoneurons that were backlabelled from the sciatic nerve 2 months (P > 0.60) and 4 months (P > 0.16) post‐axotomy (Fig. 8). The number of motoneurons in the lumbar spinal cord was the same whether the motor nerves were intact or subjected to prolonged axotomy. The slow age‐dependent decline in motoneuron numbers (see Fig. 2) did not affect the survival after axotomy (P > 0.83), the percentage of motoneurons that survived the axotomy being ∼90% in both the Smn +/− transgenic and the wild type control mice after both 2 and 4 months (Fig. 8).
Motoneuron capacity for nerve regeneration in Smn +/− transgenic mice
We went on to address the possibility that a sustained ‘growth’ mode of motoneurons, in Smn +/− mice, that were encouraged to regenerate their axons, may be sufficient to promote death of motoneurons. This possibility was again negated by our data. We found that the number of CP motoneurons that regenerated their axons was not reduced as compared to wild type controls 3 weeks after axotomy and resuture of the proximal and distal sciatic nerve stumps (P > 0.47) (Fig. 9). Hence the axotomized motoneurons that regenerated their axons survived as well as those axotomized motoneurons that were prevented from regenerating their axons. These findings indicate that the sustained ‘growth’ mode did not detrimentally affect the survival of the motoneurons in the SMAIII C57BL/6‐Smn +/− transgenic mice.
Discussion
The major findings of the present study were threefold. (1) There were surprisingly small reductions in the number of motoneurons with peripheral axons that survived during 12 months in the Smn +/− SMAIII transgenic mouse. (2) The sprouting capacity of the remaining motor nerve fibres accounted for the maintained weights of both fast‐ and slow‐twitch muscles and the capacity of the muscles to generate normal muscle contractile forces for up to 12 months in the normal lifespan of 28 months in the mice. (3) Motoneurons survived after axotomy in the adult mouse, demonstrating a normal capacity for nerve regeneration. These data indicate that, in the mild form of SMAIII disease, minimal motoneuron loss occurs in a die‐back fashion. Moreover, the survival of all motoneurons after axotomy negates the hypothesis that incomplete maturation of the neuromuscular system induced by SMN defects is responsible for axon die‐back and death of motoneurons.
Significant reductions in the numbers of surviving motoneurons were reported for the same mouse using a Kluver Barrera stain to identify the surviving motoneurons in cross‐sections of the ventral horn of the lumbosacral spinal cord (L1–L6) (Balabanian et al. 2007). Importantly though, our counts of motoneurons are those motoneurons with nerve fibres that project as far as the sciatic nerve in the thigh. These counts of motoneurons with nerve fibres being substantially higher than the counts of Nissl‐ and Kluver Barrera‐stained motoneurons suggest that the published reduced motoneuron counts probably overestimated the death of motoneurons in these type III SMA mouse models. Importantly, the die‐back of motor nerve fibres shown by the small but significant decline in numbers of dye‐labelled motoneurons at 7 and 12 months of age in the motoneurons of Smn +/− SMAIII transgenic mice indicates that motoneuron death (Fig. 2), if it occurs at all in this strain of mice, is likely to be later than 7 months of age. Reductions in surviving motoneurons were reported as early as 5 weeks of age (Balabanian et al. 2007), but in the same mouse strain losses of ∼40 and 54% were reported at 6 and 12 months, respectively (Jablonka et al. 2000). Whilst it could be argued that there is more severe loss of motoneurons that supply the more proximal musculature, our backlabelling of femoral motoneurons via the femoral nerve in the groin at 4 months of age did not reveal an early loss of these motoneurons (Fig. 3). Similarly, only a 23% reduction in facial motoneurons was described in these mice at 12 months (Jablonka et al. 2000).
Thus, reduction in SMN protein content in these mice does not impact severely on the survival of motoneurons. Hence the time course of motoneuron loss is clearly slow in this mouse, and considerably slower than in the Smn −/− type III transgenic mouse; the reduction in spinal motoneurons of 35% by 12 months in the Smn +/− type III mice (Fig. 2) compares with the equivalent reduction of ∼29% reported already by 3.5 months of age in the Smn −/− type III mice (Monani et al. 2003). Indeed the rate of motoneuron loss correlates with the onset of disease symptoms, with symptoms being minor in the Smn +/− type III transgenic mouse in contrast to more severe symptoms of muscle weakness as early as 1 month of age in the Smn −/− type III transgenic mouse.
This mouse model of SMAIII with a normal lifespan (Jablonka et al. 2000) is consistent with the normal lifespan of patients with SMA type III (Lunn & Wang, 2008). However, the full reinnervation of hindlimb musculature in the mouse accounts for the findings of relatively normal movement in these mice in contrast to the patients, many of whom are wheelchair bound in and/or after teenage years (Lunn & Wang, 2008). The progressive weakness in the patients may be associated with continuing loss of functional motor units until the numbers decline below 15–20% of normal when the upper limit of fivefold increase in motor unit size by sprouting fails to reinnervate all the denervated muscle fibres, and muscle contractile force, in turn, declines (Gordon et al. 1993, 2004).
It is interesting that we did not observe any contractile force deficits in the partially denervated muscles at 7 and 11 months of age in the Smn +/− SMAIII transgenic mice relative to their wild type control muscles (Fig. 4). In addition, there was no observable muscle atrophy with muscle weights being the same in the Smn +/− SMAIII and the wild type control mice (Fig. 5). Impaired acetylcholine receptor clustering at the endplate region of the muscles in SMAII transgenic mice suggested that neuromuscular transmission might be compromised (Biondi et al. 2008). This was not the case in the Smn +/− transgenic mouse model of SMAIII where stimulation of the sciatic nerve effectively elicited twitch and tetanic contractions of the same force as the muscles from the corresponding wild type control mice. This was so for both fast‐ and slow‐twitch muscles (Fig. 4).
The ∼30% reduction in numbers of motoneurons with intact neuromuscular contacts in representative hindlimb fast‐ and slow‐twitch muscles (Fig. 6) was the same as for the surviving sciatic motoneurons that were enumerated after their backlabelling (Fig. 2), the equivalency attesting to the validity of the ITS‐MUNE method of counting motor units in rodents (Major et al. 2007). Despite the motor axon die‐back, the partially denervated muscles retained their normal contractile force capability in the Smn +/− III transgenic mouse (Fig. 4). Enlargement of motor units with increased motor unit contractile forces is the likely explanation with our histological evidence for substantial axon sprouting to reinnervate denervated muscle fibres (Fig. 7). This explanation is consistent with previous findings of significant sprouting in the triceps surae muscles of 4‐month‐old type III Smn −/− transgenic mice (Monani et al. 2003). In our study, the axonal sprouting was effective in reinnervating all denervated motor endplates because the muscle contractile forces were not reduced in line with reduced numbers of intact motor units, as in partially denervated rat and cat muscles (Rafuse et al. 1992; Tam et al. 2001, Tam & Gordon, 2003). Hence, the sprouting effectively compensated for the loss of the innervation by a proportion of motoneurons in the Smn +/− III transgenic mice. It is likely that the sprouting resulted in some but not extensive fibre‐type clumping in the partially denervated muscles because clumping becomes obvious only after extensive partial denervation (Gordon & Tyreman, 2010).
The capacity for sprouting in SMA differs fundamentally from findings in transgenic mouse models of amyotrophic lateral sclerosis (ALS). In the latter ALS model, the large motoneurons with intact neuromuscular connections fail to sprout and reinnervate the type IIB muscle fibres that are progressively denervated by selective die‐back of their motor axons (Frey et al. 2000; Hegedus et al. 2007, 2008), in association with the expression of the inhibitory semaphorin 3A that is present at the endplates of these muscle fibres (Frey et al. 2000; Pun et al. 2006; De Winter et al. 2006). The greater resilience of the small motoneurons and their sustained capacity to sprout and reinnervate denervated muscle fibres resulted in delayed susceptibility of these motoneurons to die‐back (Hegedus et al. 2008). In association with this preferential die‐back of axons from the type IIB fast glycolytic muscle fibres, a progressive conversion of the remaining reinnervated muscle fibres occurs probably associated with compensatory hyperactivity of the motor units (Hegedus et al. 2008). In the Smn +/− III transgenic mouse, where axon die‐back occurs very slowly indeed and is not substantial, even at 12 months of age, there was no evidence of preferential die‐back of axons from fast‐twitch as opposed to slow‐twitch muscles, the extent of motor unit loss being indistinguishable in the fast‐ and slow‐twitch muscles (Fig. 6).
Whilst the basis for the motor axon die‐back in both SMA and ALS is not understood, the gene defect in the two motoneuron diseases is fundamentally different and the onset of the disease is early in life in SMA and later in life in ALS (Al Chalabi & Leigh, 2000; Nicole et al. 2002; Iannaccone et al. 2004; Monani, 2005; Russman, 2007). That motoneuron survival is prolonged in the mouse model of ALS by axotomy is intriguing with the capacity of axonal transport to sustain small rather than large axons being a possible explanation (Kong & Xu, 1999). In contrast, in SMA where axonal transport may also be compromised, we determined whether axotomy might increase the loss of motoneurons in the spinal cord. We hypothesized that, if indeed the motoneuron is in an immature state and thereby more susceptible to cell death, akin to the susceptibility of neonatal immature motoneurons in the rat being highly susceptible to cell death (Greensmith et al. 1996), we might observe increased loss of motoneurons after axotomy as has been observed with axotomy in neonatal rats at 1–3 days (Lowrie et al. 1987; de Bilbao & Dubois‐Dauphin, 1996; Rossiter et al. 1996). The axon transection to axotomize sciatic motoneurons did not promote any motoneuron loss and hence our findings did not support the hypothesis (Fig. 8). We must conclude from these findings that there is a very gradual loss of motoneurons that project their axons to the sciatic nerve, that the die‐back of axons precedes motoneuron loss and that the immaturity of the neuromuscular system is not, in of itself, an explanation for the progressive die‐back in SMA. Moreover, the evidence that the axotomized common peroneal motoneurons regenerated their axons normally when regeneration was encouraged by sciatic nerve transection and surgical repair (Fig. 9), demonstrates the normal capacity of these SMA motoneurons to regenerate as well as to sprout under conditions of nerve injury and partial denervation, respectively.
It is important to note that in both SMA and ALS, there is evidence to support the possibility that increased neuromuscular activity may enhance the survival of intact motor units and/or the transgenic mice. In SMA where a deficiency in expression of the NR2A subunit of the NMDA receptor was reported in conjunction with defective clustering of acetylcholine receptors at the endplate regions of skeletal muscle in SMA, exercise of the mice upregulated the gene for the subunit in conjunction with enhanced survival of the mice (Biondi et al. 2008). Similarly in the G93A transgenic mouse model of ALS, our findings demonstrated that all remaining intact motor units in the affected hindlimb muscles survived in conjunction with the compensatory hyperactivity of the remaining motor units (Gordon et al. 2010).
Additional information
Competing interests
None declared.
Author contributions
Experiments were performed in the laboratories of T.G. and C.T.P. E.U. and N.T. participated in the acquisition, analysis and interpretation of data and drafted and critically revised the work. L.R.H. and V.E.C. participated in the acquisition, analysis and interpretation of data and critically revised the work. T.G. and C.T.P. designed the work, participated in the analysis and interpretation of data, and drafted and critically revised the work. All authors approved the final version of the manuscript and ensure the accuracy and integrity of any part of the work presented, and all qualify for authorship. All those who qualify for authorship are listed.
Funding
We appreciate the financial support by the Spinal Muscular Atrophy Foundation of America of an operating grant to T.G. and C.T.P. (C59935), by the Natural Sciences and Engineering Council of Canada (NSERC) of an operating grant to C.T.P. (RGPIN217643), and a NSERC postgraduate scholarship to V.E.C. C.T.P. is a Senior Scholar and T.G. was a Senior Investigator of the Alberta Heritage Foundation for Medical Research.
References
- Abercrombie M & Johnson ML (1946). Quantitative histology of Wallerian degeneration: I. Nuclear population in rabbit sciatic nerve. J Anat 80, 37–50. [PubMed] [Google Scholar]
- Al Chalabi A & Leigh PN (2000). Recent advances in amyotrophic lateral sclerosis. Curr Opin Neurol 13, 397–405. [DOI] [PubMed] [Google Scholar]
- Al‐Majed AA, Neumann CM, Brushart TM & Gordon T (2000). Brief electrical stimulation promotes the speed and accuracy of motor axonal regeneration. J Neurosci 20, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold AS, Gueye M, Guettier‐Sigrist S, Courdier‐Fruh I, Coupin G, Poindron P & Gies JP (2004). Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab Invest 84, 1271–1278. [DOI] [PubMed] [Google Scholar]
- Balabanian S, Gendron NH & MacKenzie AE (2007). Histologic and transcriptional assessment of a mild SMA model. Neurol Res 29, 413–424. [DOI] [PubMed] [Google Scholar]
- Batten FE (1911). Progressive spinal muscular atrophy of infants and young children. Brain 33, 433–463. [PMC free article] [PubMed] [Google Scholar]
- Berdan RC, Hauser G & Bulloch AG (1990). Ultrastructure of an identified molluscan neuron in organ culture and cell culture following axotomy. J Comp Neurol 296, 437–446. [DOI] [PubMed] [Google Scholar]
- Biondi O, Grondard C, Lecolle S, Deforges S, Pariset C, Lopes P, Cifuentes‐Diaz C, Li H, della GB, Chanoine C & Charbonnier F (2008). Exercise‐induced activation of NMDA receptor promotes motor unit development and survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 28, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JG & Gordon T (2001). The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J Neurobiol 49, 314–325. [DOI] [PubMed] [Google Scholar]
- Brushart TM, Hoffman PN, Royall RM & Gordon T (2002). Electrical stimulation synchronizes motoneuron regeneration without increasing its speed or conditioning the neuron. J Neurosci 22, 6631–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH & Beattie CE (2009). Spinal muscular atrophy: why do low levels of surival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SM & Nonaka I (1978). Werdnig‐Hoffmann disease: proposal of a pathogenetic mechanism. Acta Neuropathol 41, 45–54. [DOI] [PubMed] [Google Scholar]
- Cifuentes‐Diaz C, Frugier T, Tiziano FD, Lacene E, Roblot N, Joshi V, Moreau MH & Melki J (2001). Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol 152, 1107–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TO & Pardo CA (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3, 97–110. [DOI] [PubMed] [Google Scholar]
- de Bilbao F & Dubois‐Dauphin M (1996). Time course of axotomy‐induced apoptotic cell death in facial motoneurons of neonatal wild type and bcl‐2 transgenic mice. Neuroscience 71, 1111–1119. [DOI] [PubMed] [Google Scholar]
- De Winter F, Vo T, Stam FJ, Wisman LA, Bar PR, Niclou SP, van Muiswinkel FL & Verhaagen J (2006). The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci 32, 102–117. [DOI] [PubMed] [Google Scholar]
- Fallini C, Bassell GL & Rossoll W (2012). Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res 1462, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L & Simard LR (2002). Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum Mol Genet 11, 1605–1614. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W & Caroni P (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20, 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frugier T, Tiziano FD, Cifuentes‐Diaz C, Miniou P, Roblot N, Dierich A, Le Meur M & Melki J (2000). Nuclear targeting defect of SMN lacking the C‐terminus in a mouse model of spinal muscular atrophy. Hum Mol Genet 9, 849–858. [DOI] [PubMed] [Google Scholar]
- Fu SY & Gordon T (1995). Contributing factors to poor functional recovery after delayed nerve repair: prolonged axotomy. J Neurosci 15, 3876–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SY & Gordon T (1997). The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol 14, 67–116. [DOI] [PubMed] [Google Scholar]
- Galea V, de Bruin H, Cavasin R & McComas AJ (1991). The numbers and relative sizes of motor units estimated by computer. Muscle Nerve 14, 1123–1130. [DOI] [PubMed] [Google Scholar]
- Gordon T (1983). Dependence of peripheral nerves on their target organs In Somatic and Autonomic Nerve–Muscle Interactions, ed. Burnstock G, O'Brien R. & Vrbová G, chap.10, pp 289–316. Elsevier, Amsterdam. [Google Scholar]
- Gordon T, Hegedus J & Tam SL (2004). Adaptive and maladaptic motor axonal sprouting in aging and motoneuron disease. Neurol Res 26, 174–185. [DOI] [PubMed] [Google Scholar]
- Gordon T & Tyreman N (2010). Sprouting capacity of lumbar motoneurons in the normal and hemisected spinal cord of the rat. J Physiol 588, 2745–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Tyreman N, Li S, Putman CT & Hegedus J (2010). Functional over‐load saves motor units in the SOD1‐G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 37, 412–422. [DOI] [PubMed] [Google Scholar]
- Gordon T, Yang JF, Ayer K, Stein RB & Tyreman N (1993). Recovery potential of muscle after partial denervation: a comparison between rats and humans. Br Res Bull 30, 477–482. [DOI] [PubMed] [Google Scholar]
- Greensmith L, Dick J, Emanuel AO & Vrbová G (1996). Induction of transmitter release at the neuromuscular junction prevents motoneuron death after axotomy in neonatal rats. Neuroscience 71, 213–220. [DOI] [PubMed] [Google Scholar]
- Greensmith L & Vrbová G (1996). Motoneurone survival: a functional approach. Trends Neurosci 19, 450–455. [DOI] [PubMed] [Google Scholar]
- Greensmith L & Vrbová G (1997). Disturbances of neuromuscular interaction may contribute to muscle weakness in spinal muscular atrophy. Neuromuscul Disord 7, 369–372. [DOI] [PubMed] [Google Scholar]
- Grondard C, Biondi O, Armand AS, Lécolle S, Della Gaspera B, Pariset C, Li H, Gallien CL, Vidal PP, Chanoine C & Charbonnier F (2005). Regular exercise prolongs survival in a type 2 spinal muscular atrophy model mouse. J Neurosci 25, 7615–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmanowa‐Petrusewicz I & Vrbová G (2005). Spinal muscular atrophy: a delayed development hypothesis. Neuroreport 16, 657–661. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT & Gordon T (2007). Time course of preferential motor unit loss in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28, 154–164. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N & Gordon T (2008). Preferential motor unit loss in the SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586, 3337–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannaccone ST, Smith SA & Simard LR (2004). Spinal muscular atrophy. Curr Neurol Neurosci Rep 4, 74–80. [DOI] [PubMed] [Google Scholar]
- Jablonka S, Schrank B, Kralewski M, Rossoll W & Sendtner M (2000). Reduced survival motor neuron (Smn) gene dose in mice leads to motor neuron degeneration: an animal model for spinal muscular atrophy type III. Hum Mol Genet 9, 341–346. [DOI] [PubMed] [Google Scholar]
- Kariya S, Obis T, Carone C, Akay T, Sera F, Iwata S, Homma S & Monani UR (2014). Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 124, 785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno‐Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT & Monani UR (2008). Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17, 2552–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp SW, Chiang CD, Liu EH, Wood MD, Willand MP, Gordon T & Borschel GH (2015). Characterization of neuronal death and functional deficits following nerve injury during the early postnatal developmental period in rats. Dev Neurosci 37, 66–77. [DOI] [PubMed] [Google Scholar]
- Kong J & Xu Z (1999). Peripheral axotomy slows motoneuron degeneration in a transgenic mouse line expressing mutant SOD1 G93A. J Comp Neurol 412, 373–380. [DOI] [PubMed] [Google Scholar]
- Kye MJ, Niederst ED, Wertz MH, Gonçalves IdoCG, Akten B, Dover KZ, Peters M, Riessland M, Neveu P, Wirth B, Kosik KS, Sardi SP, Monani UR, Passini MA & Sahin M (2014). SMN regulates axonal local translation via miR‐183‐mTOR pathway. Hum Mol Genet 23, 6318–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, McGovern VL, Alwine IE, Wang X, Massoni‐Laporte A, Rich MM & Burghes AH (2011). Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet 20, 3578–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P & Zeviani M (1995). Identification and characterization of a spinal muscular atrophy‐determining gene. Cell 80, 155–165. [DOI] [PubMed] [Google Scholar]
- Lieberman AR (1971). The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol 14, 49–124. [DOI] [PubMed] [Google Scholar]
- Liu‐Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST & Wolozin B (2011). Local RNA translation at the synapse and in disease. J Neurosci 31, 16086–16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie MB, Krishnan S & Vrbová G (1982). Recovery of slow and fast muscles following nerve injury during early post‐natal development in the rat. J Physiol 331, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie MB, Krishnan S & Vrbová G (1987). Permanent changes in muscle and motoneurones induced by nerve injury during a critical period of development of the rat. Brain Res 428, 91–101. [DOI] [PubMed] [Google Scholar]
- Lunn MR & Wang CH (2008). Spinal muscular atrophy. Lancet 371, 2120–2133. [DOI] [PubMed] [Google Scholar]
- Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK, Pellizzoni L, Rojas, J , Valenzuela DM, Murphy AJ, Winberg ML & Monani UR (2011). Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest 121, 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComas AJ, Fawcett PR, Campbell MJ & Sica RE (1971). Electrophysiological estimation of the number of motor units within a human muscle. J Neurol Neurosurg Psychiatry 34, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter ML, Boon KL, Horan ES, Burghes AH & Beattie CE (2008). The SMN binding protein Gemin2 is not involved in motor axon outgrowth. Dev Neurobiol 68, 182–194. [DOI] [PubMed] [Google Scholar]
- Major LA, Hegedus J, Weber DJ, Gordon T & Jones KE (2007). Method for counting motor units in mice and validation using a mathematical model. J Neurophysiol 97, 1846–1856. [DOI] [PubMed] [Google Scholar]
- Melki J, Lefebvre S, Burglen L, Burlet P, Clermont O, Millasseau P, Reboullet S, Benichou B, Zeviani M & Le Paslier D (1994). De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Science 264, 1474–1477. [DOI] [PubMed] [Google Scholar]
- Monani UR (2005). Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron‐specific disease. Neuron 48, 885–896. [DOI] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH & McPherson JD (1999). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2 . Hum Mol Genet 8, 1177–1183. [DOI] [PubMed] [Google Scholar]
- Monani UR, Pastore MT, Gavrilina TO, Jablonka S, Le TT, Andreassi C, DiCocco JM, Lorson C, Androphy EJ, Sendtner M, Podell M & Burghes AH (2003). A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol 160, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE & Burghes AH (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn −/− mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9, 333–339. [DOI] [PubMed] [Google Scholar]
- Munsat TL & Davies KE (1992). International SMA consortium meeting. (26‐28 June 1992, Bonn, Germany). Neuromuscul Disord 2, 423–428. [DOI] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K & Gillingwater TH (2008). Selective vulnerability of motor neurons and dissociation of pre‐ and post‐synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet 17, 949–962. [DOI] [PubMed] [Google Scholar]
- Nicole S, Diaz CC, Frugier T & Melki J (2002). Spinal muscular atrophy: recent advances and future prospects. Muscle Nerve 26, 4–13. [DOI] [PubMed] [Google Scholar]
- Pearn J (1978). Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 15, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L & Caroni P (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9, 408–419. [DOI] [PubMed] [Google Scholar]
- Rafuse VF, Gordon T & Orozco R (1992). Proportional enlargement of motor units after partial denervation of cat triceps surae muscles. J Neurophysiol 68, 1261–1275. [DOI] [PubMed] [Google Scholar]
- Roberts DF, Chavez J & Court SD (1970). The genetic component in child mortality. Arch Dis Child 45, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter JP, Riopelle RJ & Bisby MA (1996). Axotomy‐induced apoptotic cell death of neonatal rat facial motoneurons: time course analysis and relation to NADPH‐diaphorase activity. Exp Neurol 138, 33–44. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kroning AK, Karle K, Monani UR & Sendtner M (2003). Smn, the spinal muscular atrophy‐determining gene product, modulates axon growth and localization of β‐actin mRNA in growth cones of motoneurons. J Cell Biol 163, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russman BS (2007). Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol 22, 946–951. [DOI] [PubMed] [Google Scholar]
- Simic G, Seso‐Simic D, Lucassen PJ, Islam A, Krsnik Z, Cviko A, Jelasic D, Barisic N, Winblad B, Kostovic I & Kruslin B (2000). Ultrastructural analysis and TUNEL demonstrate motor neuron apoptosis in Werdnig‐Hoffmann disease. J Neuropathol Exp Neurol 59, 398–407. [DOI] [PubMed] [Google Scholar]
- Soler‐Botija C, Ferrer I, Gich I, Baiget M & Tizzano EF (2002). Neuronal death is enhanced and begins during foetal development in type I spinal muscular atrophy spinal cord. Brain 125, 1624–1634. [DOI] [PubMed] [Google Scholar]
- Stein RB & Yang JF (1990). Methods for estimating the number of motor units in human muscles. Ann Neurol 28, 487–495. [DOI] [PubMed] [Google Scholar]
- Tam SL, Archibald V, Tyreman N, Jassar B & Gordon T (2001). Increased neuromuscular activity reduces sprouting in partially denervated muscles. J Neurosci 21, 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SL & Gordon T (2003). Mechanisms controlling axonal sprouting at neuromuscular junction. J Neurocytol 32, 961–974. [DOI] [PubMed] [Google Scholar]
- Xu Q‐G, Forden J, Walsh SK, Gordon T & Midha R (2010). Motoneuron survival in chronic and sequential peripheral nerve injuries in the rat. J Neurosurg 112, 890–899. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M & Dreyfuss G (2008). SMN deficiency causes tissue‐specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]