

Abstract
Key points
Our objective was to quantify endothelial function (via brachial artery flow‐mediated dilatation) at sea level (344 m) and high altitude (3800 m) at rest and following both maximal exercise and 30 min of moderate‐intensity cycling exercise with and without administration of an α1‐adrenergic blockade.
Brachial endothelial function did not differ between sea level and high altitude at rest, nor following maximal exercise.
At sea level, endothelial function decreased following 30 min of moderate‐intensity exercise, and this decrease was abolished with α1‐adrenergic blockade. At high altitude, endothelial function did not decrease immediately after 30 min of moderate‐intensity exercise, and administration of α1‐adrenergic blockade resulted in an increase in flow‐mediated dilatation.
Our data indicate that post‐exercise endothelial function is modified at high altitude (i.e. prolonged hypoxaemia). The current study helps to elucidate the physiological mechanisms associated with high‐altitude acclimatization, and provides insight into the relationship between sympathetic nervous activity and vascular endothelial function.
Abstract
We examined the hypotheses that (1) at rest, endothelial function would be impaired at high altitude compared to sea level, (2) endothelial function would be reduced to a greater extent at sea level compared to high altitude after maximal exercise, and (3) reductions in endothelial function following moderate‐intensity exercise at both sea level and high altitude are mediated via an α1‐adrenergic pathway. In a double‐blinded, counterbalanced, randomized and placebo‐controlled design, nine healthy participants performed a maximal‐exercise test, and two 30 min sessions of semi‐recumbent cycling exercise at 50% peak output following either placebo or α1‐adrenergic blockade (prazosin; 0.05 mg kg −1). These experiments were completed at both sea‐level (344 m) and high altitude (3800 m). Blood pressure (finger photoplethysmography), heart rate (electrocardiogram), oxygen saturation (pulse oximetry), and brachial artery blood flow and shear rate (ultrasound) were recorded before, during and following exercise. Endothelial function assessed by brachial artery flow‐mediated dilatation (FMD) was measured before, immediately following and 60 min after exercise. Our findings were: (1) at rest, FMD remained unchanged between sea level and high altitude (placebo P = 0.287; prazosin: P = 0.110); (2) FMD remained unchanged after maximal exercise at sea level and high altitude (P = 0.244); and (3) the 2.9 ± 0.8% (P = 0.043) reduction in FMD immediately after moderate‐intensity exercise at sea level was abolished via α1‐adrenergic blockade. Conversely, at high altitude, FMD was unaltered following moderate‐intensity exercise, and administration of α1‐adrenergic blockade elevated FMD (P = 0.032). Our results suggest endothelial function is differentially affected by exercise when exposed to hypobaric hypoxia. These findings have implications for understanding the chronic impacts of hypoxaemia on exercise, and the interactions between the α1‐adrenergic pathway and endothelial function.
Keywords: endothelial function, exercise, flow‐mediated dilatation, high‐altitude, sympathetic nervous activity
Key points
Our objective was to quantify endothelial function (via brachial artery flow‐mediated dilatation) at sea level (344 m) and high altitude (3800 m) at rest and following both maximal exercise and 30 min of moderate‐intensity cycling exercise with and without administration of an α1‐adrenergic blockade.
Brachial endothelial function did not differ between sea level and high altitude at rest, nor following maximal exercise.
At sea level, endothelial function decreased following 30 min of moderate‐intensity exercise, and this decrease was abolished with α1‐adrenergic blockade. At high altitude, endothelial function did not decrease immediately after 30 min of moderate‐intensity exercise, and administration of α1‐adrenergic blockade resulted in an increase in flow‐mediated dilatation.
Our data indicate that post‐exercise endothelial function is modified at high altitude (i.e. prolonged hypoxaemia). The current study helps to elucidate the physiological mechanisms associated with high‐altitude acclimatization, and provides insight into the relationship between sympathetic nervous activity and vascular endothelial function.
Abbreviations
- 2RM‐ANOVA
two‐way repeated measures ANOVA
- CO
cardiac output
- FEV
forced expiratory volume
- FMD
flow‐mediated dilatation
- FVC
forced vital capacity
- HR
heart rate
- MAP
mean arterial pressure
- SNS
sympathetic nervous system
peripheral oxyhaemoglobin saturation
- SRAUC
shear rate area under the curve
- SV
stroke volume
- TPR
total peripheral resistance
Introduction
Flow‐mediated dilatation (FMD) is a commonly used, non‐invasive measurement of conduit artery diameter in response to an imposed change in shear rate; it is widely accepted to be an index of endothelial function (Thijssen et al. 2011), and is largely mediated by nitric oxide (Green et al. 2014). It has been demonstrated that immediately after cycling exercise, endothelial function measured by brachial (i.e. non‐exercising limb) FMD is transiently reduced (Jones et al. 2010; Birk et al. 2013; Dawson et al. 2013; Atkinson et al. 2015), and the reduction in FMD is inversely related to exercise intensity (Birk et al. 2013). Although the precise mechanisms responsible for this acute effect of exercise on FMD remain unclear, several possibilities exist, including: (1) elevated oxidative stress (Silvestro et al. 2002; Goel et al. 2007 a); (2) altered haemodynamics (i.e. shear rate, shear pattern and blood pressure) (Lamping and Dole, 1987; Millgard and Lind, 1998; Dawson et al. 2008; Johnson et al. 2012 a; Birk et al. 2013); (3) changes in baseline artery diameter following exercise (Padilla et al. 2007; Atkinson et al. 2013); and (4) elevations in sympathetic nervous system (SNS) activity (Hijmering et al. 2002; Dyson et al. 2006; Atkinson et al. 2015). The role of the SNS was recently examined in a study which observed that the exercise‐mediated reduction in FMD following exercise was abolished after administration of an α1‐adrenergic receptor blockade (Atkinson et al. 2015). Interestingly, the relationship between increased SNS activity and post‐exercise FMD has not previously been explored under conditions where resting SNS activity is chronically elevated, such as in ageing, pathology (e.g. obstructive sleep apnoea, heart failure) or hypoxia (i.e. normobaric and hypobaric) (Saito et al. 1988; Duplain et al. 1999; Xie et al. 2001; Hansen and Sander, 2003).
Exposure to hypoxia is associated with arterial stiffening, but the effects of hypoxia on endothelial function remain unclear (Rhodes et al. 2011; Boos et al. 2012; Lewis et al. 2014 a). For example, different studies have reported that exposure to simulated high altitude (i.e. acute normobaric hypoxia) results in no change in FMD [∼4000 m, = 0.13 (Iglesias et al. 2015)], and decreased FMD [∼5000 m, = 0.11 (Lewis et al. 2014 a)]. Upon ascent to high altitude between 3700 and 5050 m (i.e. hypobaric hypoxia), a decrease in FMD has been observed in most (Lewis et al. 2014 a; Bakker et al. 2015), but not all studies (Bruno et al. 2015; 2016). The disparity within this literature might be due to different methodological approaches between investigations. For example, the two studies that reported reductions in FMD involved ascent to high altitude over 7–10 days of trekking for several hours each day (Lewis et al. 2014 a; Bakker et al. 2015). In contrast, the study that reported no change in FMD involved participants ascending rapidly to high altitude via cable car (Bruno et al. 2015). The methodological difference between these studies raises the possibility of a moderating impact of trekking exercise at altitude, and total acclimatization time [e.g. 3–10 days of exposure (FMD reduced) vs. 1 day of exposure (no change in FMD)] on vascular responses at high altitude.
By employing a double‐blinded, counterbalanced, randomized and placebo‐controlled design, the primary purposes of the current study were to investigate: (1) the effects of non‐trekking (i.e. passive) ascent to high altitude on endothelial function (via brachial FMD) and shear patterns, and (2) the effects of post‐exercise‐related increases in SNS activity on FMD and shear patterns at both sea level and high altitude (3800 m). We hypothesized that: (1) at rest, FMD would be impaired at high altitude compared to sea level, (2) FMD would be reduced to a greater extent at sea level compared to high altitude after maximal exercise and (3) reductions in endothelial function following moderate‐intensity exercise at both sea level and high altitude are mediated via an α1‐adrenergic pathway.
Methods
Ethical approval
All experimental procedures and protocols were approved by the clinical research ethics board at the University of British Columbia and conformed to the Declaration of Helsinki. All participants provided written informed consent prior to participation in this study. This study was part of a larger research expedition conducted in October 2015. As such, participants took part in a number of studies conducted at the University of British Columbia (Kelowna, BC; 344 m) and during 2 weeks at the Barcroft high‐altitude research station (White Mountain, California, USA; 3800 m). However, the a priori, primary research questions addressed in the current paper are novel and are exclusively dealt within this study alone – there is no overlap between this investigation and others completed on the research expedition.
Participants
Recruited participants (n = 11; three female) were normotensive (systolic blood pressure <140 and diastolic pressure <90 mmHg) at rest, and completed a medical history questionnaire. Two of the recruited participants were excluded from all mean data analysis; one participant due to illness (i.e. syncope, light‐headedness, nausea) caused by our drug intervention (prazosin), and one participant due to illness at high altitude. The participants (n = 9; two female) included in the data analysis were non‐smokers, had no previous history of cardiovascular, cerebrovascular or respiratory diseases, and were not taking any medications during testing besides oral contraceptives (n = 1). All participants arrived at the Barcroft high‐altitude research facility (elevation = 3800 m) on the same day, and approximately at the same time (i.e. within 1 h). Ten of these participants drove to the Barcroft high‐altitude research facility after staying overnight in Palm Springs, CA, USA (elevation = 146 m), while one participant stayed overnight in Bishop, CA, USA (elevation = 1265 m). Maximal exercise testing for this study occurred on day 3, while moderate‐intensity exercise experimentation occurred between days 4 and 7 at high altitude.
Experimental design
This study was conducted in two parts: sea‐level and high‐altitude investigations. Prior to each experiment, all participants abstained from exercise, alcohol and caffeine for at least 12 h. Additionally, participants were asked to consume a light meal at least 2 h prior to experimentation, and to keep their diet consistent between experimentation days. To determine whether our participants had normal healthy lung function, at sea level we conducted a forced vital capacity (FVC) test to measure lung function, a vital capacity and inspiratory capacity manoeuvre to measure lung volumes, and a single breath carbon monoxide test to quantify diffusing capacity on each individual. All testing procedures were conducted in accordance with the American Thoracic Society and European Respiratory Society's joint guidelines (Macintyre et al. 2005; Miller et al. 2005). For each of these tests, participants sat within a body plethysmography box (V6200, Vmax Sensormedics, Yorba Linda, CA, USA) with a rigid upright posture and their feet flat on the ground, whilst breathing through a spirometer and bacteriological filter while wearing a nose‐clip. All pulmonary function measurements were compared against population‐based predictions.
Exercise testing was then conducted on three separate lab visits at both sea level and high altitude while participants lay in the semi‐recumbent position. The time of day of the testing sessions were kept the same for each participant, with a minimum of 24 h between testing sessions.
Maximal exercise protocols
Prior to the prolonged moderate‐intensity exercise testing at sea level and high altitude, participants were required to conduct a maximal exercise protocol in order to obtain peak power output. In the semi‐recumbent position, participants rested during a quiet baseline period on the cycle ergometer (Lode Ergometer, Lode, Groningen, Netherlands) for 15 min. Immediately after the baseline period a brachial artery FMD was performed (Thijssen et al. 2011). Exercise began with a 2 min warm‐up period (40 W for females, 60 W for males), followed by staged exercise with workload increased by 20 W every minute. This maximal exercise protocol was terminated when participants either (1) reached volitional exhaustion, or (2) cycling cadence could no longer be maintained. Immediately after the maximal exercise protocol was completed, another brachial artery FMD was performed.
Moderate‐intensity exercise protocol
Following at least 24 h after the maximal exercise test, participants performed two moderate‐intensity exercise protocols on two independent days separated by at least 24 h. Upon arrival on each of these testing sessions, participants ingested a capsule containing either oral prazosin (α1‐adrenergic receptor blocker; 0.05 mg kg−1), or placebo (i.e. sugar pill). The order of condition (placebo and prazosin) was counterbalanced and randomized at both sea level and high altitude. The dosage of prazosin used has been demonstrated to provide ∼80% α1‐adrenergic blockade, and has been used in other studies by our research group (Lewis et al. 2014 b; Atkinson et al. 2015). Seventy‐five minutes after ingestion, participants were instrumented on the semi‐recumbent cycle ergometer (see details below in section Experimental measurements). Participants were asked to sit in the semi‐recumbent position quietly for 15 min, and afterwards a baseline FMD on the left brachial artery was conducted (90 min after ingestion). Immediately after the baseline FMD, participants completed 30 min of cycle exercise at 50% of their peak exercise workload. At sea level, this workload was 137.0 ± 6.9 W, while at high altitude the average workload was 114.3 ± 5.9 W. Every 5 min during exercise a 1 min measurement of brachial artery shear rate and blood flow was recorded (total of six recordings during the 30 min of exercise). After completion of the 30 min of moderate‐intensity exercise, a brachial FMD was conducted immediately, and then following 60 min of post‐exercise rest.
Experimental measurements
Cardiovascular measurements
All continuously recorded cardiovascular measurements were acquired at 200 Hz using an analog‐to‐digital converter (Powerlab/16SP ML 880; ADInstruments, Colorado Springs, CO, USA) interfaced with a personal computer. Commercially available software was used to analyse cardiovascular variables (LabChart V7.1, ADInstruments). Electrocardiogram electrodes were placed in lead II configuration (Bioamp, ML132, ADInstruments) to measure heart rate. Beat‐by‐beat arterial pressure, cardiac output, stroke volume and total peripheral resistance were measured by finger photoplethysmography (Finometer Pro, Finapres medical systems, Amsterdam, Netherlands). Prior to baseline data collection, the Finometer was calibrated using the return‐to‐flow function, and blood pressure from the Finometer was confirmed with automated brachial blood pressure readings (HEM‐775CAN, Omron Healthcare, Bannockburn, IL, USA). Mean, systolic and diastolic arterial pressure were quantified from the raw Finometer recordings.
Brachial artery imaging
With the participants left arm extended perpendicular (i.e. 80 deg) from their body while seated on the semi‐recumbent cycle ergometer, an inflation/deflation cuff was placed on the participants left forearm, and their arm was fixed into position on a table. Brachial artery image acquisition was obtained using a 10 MHz multifrequency linear array probe attached to a high‐resolution ultrasound machine (15L4, Terason t3200, Burlington, MA, USA). All brachial artery images were taken by the same experienced ultrasonographer (J.T.), who has a between‐day coefficient of variation in FMD of 8.3 ± 2.1% (n = 10, unpublished data). Following optimal image acquisition, and 1 min of baseline recordings, the forearm was occluded by inflating the cuff to 220–250 mmHg for 5 min. Recordings of diameter and velocity continued 30 s prior to cuff deflation and continuously for 3 min thereafter (Thijssen et al. 2011).
Data analysis
Brachial artery diameter and blood flow analysis
Ultrasound recordings were continuously screen captured and saved for offline analysis. Blood flow analysis of the brachial artery was performed using automated edge detection and wall tracking software, which allows for the integration of synchronous diameter and velocity measurements to continuously determine flow, shear, diameter and velocity at 30 Hz, independent of investigator bias (Woodman et al. 2001). Antegrade, retrograde and mean shear rates were calculated as four times the mean blood velocity, divided by vessel diameter. The FMD was calculated as the per cent increase in vessel diameter from resting baseline diameter, where baseline and peak diameters were automatically detected from the continuous data described above.
Statistics
All statistical analyses were performed using SigmaStat V11 (Systat, Chicago, IL, USA), and are reported as mean ± SEM. Statistical significance was assumed at P < 0.05. When significant F‐ratios were detected, post hoc comparisons were made using Tukey's post hoc test for pairwise comparisons. Our study is an extension of a previous study from our laboratory (Atkinson et al. 2015), which found a ∼2% decrease in FMD after 30 min of exercise. To detect a 2% decrease in post‐exercise FMD with a standard deviation of 2%, we calculated that a sample size of 10 participants was required to achieve a statistical power >0.80 with a statistical difference of P < 0.05. Based on this power calculation, we recruited 11 participants for the sea‐level and high‐altitude arms of the study.
Maximal exercise
For the cardiovascular data obtained at sea level and high altitude, baseline measurements were averaged over 1 min immediately prior to exercise, peak maximal exercise data were averaged over the last 30 s of the maximal exercise protocol, and post‐maximal exercise data were averaged over 1 min, immediately prior to FMD cuff release after maximal exercise was terminated (i.e. approximately 4 min after exercise). Differences between peak output and maximal exercise time at sea level and high altitude were determined using a paired Student's t‐test. For the cardiovascular data, a two‐way repeated measures ANOVA (2RM‐ANOVA) was used to detect differences across time (baseline, maximal exercise, post‐maximal exercise) and conditions (sea level and high altitude). For the brachial artery data measured during FMD, a 2RM‐ANOVA was used to detect differences across time (baseline and post‐maximal exercise) and conditions (sea level and high altitude).
Moderate‐intensity exercise
For the cardiovascular data obtained at sea level and high altitude, baseline measurements were averaged over 1 min immediately prior to exercise. During moderate‐intensity exercise, cardiovascular data were averaged over 30 s at every 5 min time‐point (i.e. time = 5, 10, 15, 20, 25 and 30 min). Post‐exercise cardiovascular data were averaged during the 1 min prior to FMD cuff release, immediately after, and 60 min after, moderate‐intensity exercise. For the cardiovascular data, a 2RM‐ANOVA was used to detect differences across time [baseline, exercise (six time‐points) and post‐exercise (two time‐points)] and conditions (placebo and prazosin), at sea level and high altitude, separately. For the brachial artery FMD data, a 2RM‐ANOVA was used to detect differences across time (baseline, post‐exercise, 60 min post‐exercise) and conditions (placebo and prazosin), at sea level and high altitude, separately.
Adjusted flow‐mediated dilatation
The effects of time and condition were analysed within and between sea level and high altitude for FMD. To determine if our FMD results were different due to changes in baseline arterial diameter and/or shear rate area under the curve (SRAUC), we included these variables as covariates in a logarithmic‐linked generalized linear model, where FMD was the dependent variable. This approach has been used to account for any changes in FMD that may be related to differences in baseline diameter or shear rate between conditions (i.e. time and condition) (Atkinson et al. 2013).
Results
Participants and resting FMD data
The nine participants included in the sea‐level and high‐altitude protocol data analysis had a mean ± SEM age of 26.9 ± 1.8 years, height of 176.3 ± 1.5 cm and weight of 71.1 ± 2.5 kg. Participants had normal pulmonary health with an FVC of 5.5 ± 0.2 l (109.0 ± 3.2% of predicted), forced expiratory volume in 1 s (FEV1) of 4.3 ± 0.2 litres (100.6 ± 2.8% of predicted), FEV1/FVC of 78.7 ± 1.0 (no individuals reported an FEV1/FVC <75), total lung capacity of 6.7 ± 0.3 litres (101.9 ± 0.3% of predicted), and had a diffusing capacity of the lung for carbon monoxide of 32.1 ± 5.7 ml min−1 mmHg −1 (94.1 ± 4.7% of predicted). Recruited participants did not demonstrate any signs of small or large airway obstruction characterized by an irregular expiratory flow tracing during the FVC manoeuvre. Additionally, participants were normotensive (systolic blood pressure = 130.6 ± 2.5 mmHg, diastolic blood pressure 68.7 ± 1.9 mmHg).
No differences were observed in FMD between sea level and high altitude during the placebo (P = 0.287) and prazosin (P = 0.110) trials at baseline (Fig. 1).
Figure 1. FMD data collected during baseline on placebo and prazosin at sea level and high altitude.
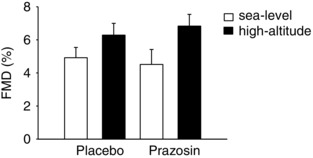
White bars represent sea‐level data ± SEM, and black bars represent high‐altitude data ± SEM in nine participants. FMD, flow‐mediated dilatation. These findings illustrate that there were no differences in resting FMD between sea level and high altitude after passive ascent to 3800 m.
Maximal exercise data
Figure 2 illustrates the cardiorespiratory data collected during the maximal exercise protocol at sea level and high altitude. At sea level, peak output and maximal exercise protocol time (min) were greater than at high altitude (275.4 ± 12.4 W vs. 228.8 ± 11.2 W, P < 0.001; 13.0 ± 0.5 min vs. 10.6 ± 0.5 min, P < 0.001). Cardiac output and stroke volume were higher at sea level compared to high altitude (P = 0.047 and P = 0.032, respectively). In contrast, heart rate at high altitude was elevated compared to sea level (P = 0.038). No differences were observed between sea level and high altitude for mean arterial pressure or total peripheral resistance at baseline, peak exercise and post‐exercise (P = 0.130 and P = 0.055, respectively). As expected, a main effect was observed for , as it was elevated at sea level compared to high altitude (P < 0.001).
Figure 2. Cardiovascular data during baseline, maximal exercise and post‐maximal exercise at sea level and high altitude.
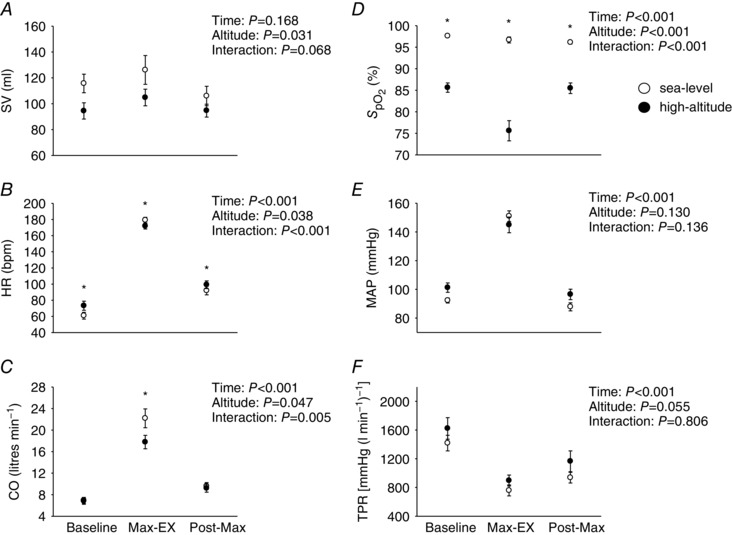
Open circles represent sea‐level data ± SEM, and closed circles high‐altitude data ± SEM in nine participants. * P < 0.05, for interaction effects. Statistics for main effects and interactions are displayed on the top right of each panel. BL, baseline; Max‐Ex, maximal exercise; Post‐Max, post‐maximal exercise; , per cent oxygen saturation of haemoglobin; SV, stroke volume, HR, heart rate; CO, cardiac output; MAP, mean arterial pressure; TPR, total peripheral resistance. Collectively, these findings reveal that SV, HR, CO and were all elevated at sea level compared to high altitude during a maximal exercise test.
Table 1 illustrates brachial diameter, shear and FMD data during baseline and post‐maximal exercise at sea level and high altitude. Brachial artery diameter was greater at sea level compared to high altitude across all time‐points (P = 0.030). At both sea level and high altitude, brachial artery diameter was reduced post‐maximal exercise compared to baseline by 4.8 ± 2.5 and 8.7 ± 1.2%, respectively (P = 0.004). Mean shear rate increased post‐maximal exercise compared to baseline at sea level by 211.2 ± 26.5 s−1, and at high altitude by 101.7 ± 20.9 s−1 (P < 0.001). Although a main effect was not present for mean shear rate between sea level and high altitude (P = 0.244), an interaction effect was observed post‐maximal exercise as mean shear was higher at sea level by 36.1 ± 19.0% compared to high altitude (P = 0.003). Antegrade shear rate increased post‐maximal exercise compared to baseline at sea level by 236.2 ± 20.2 s−1, and at high altitude by 163.8 ± 20.3 s−1 (both P < 0.001); however, similar to mean shear rate, altitude had no effect (P = 0.833). Retrograde shear rate increased (i.e. became more negative) between baseline and post‐maximal exercise at sea level by −25.3 ± 7.8 s−1 and at high altitude by 67.4 ± 11.7 s−1 (P < 0.001). Retrograde shear was also greater (i.e. more negative) at high altitude compared to sea level (P = 0.031).
Table 1.
Brachial artery diameter, shear and flow‐mediated dilatation data during baseline and post‐maximal exercise at sea level and high altitude
| Sea level | High altitude | |||
|---|---|---|---|---|
| Baseline | Post‐max | Baseline | Post‐max | |
| Diameter (mm) | 4.4 ± 0.2 | 4.2 ± 0.2* | 4.3 ± 0.2 | 3.9 ± 0.2* |
| Time: P = 0.004, Altitude: P = 0.030, Interaction: P = 0.328 | ||||
| Mean shear (s−1) | 86.4 ± 13.5 | 297.6 ± 26.2* , † | 111.8 ± 20.1 | 213.5 ± 27.7* |
| Time: P < 0.001, Altitude: P = 0.244, Interaction: P = 0.003 | ||||
| Antegrade shear (s−1) | 101.7 ± 13.7 | 337.9 ± 20.5* , † | 144.5 ± 17.3 | 308.3 ± 24.3* , † |
| Time: P < 0.001, Altitude: P = 0.833, Interaction: P = 0.012 | ||||
| Retrograde shear (s−1) | ‐15.2 ± 6.2 | −40.5 ± 8.6* | −22.1 ± 7.1 | −89.6 ± 14.9* |
| Time: P = 0.001, Altitude: P = 0.031, Interaction: P = 0.009 | ||||
| Change in diameter (mm) | 0.23 ± 0.05 | 0.25 ± 0.05 | 0.26 ± 0.04 | 0.32 ± 0.05 |
| Time: P = 0.453, Altitude: P = 0.380, Interaction: P = 0.581 | ||||
| FMD (%) | 5.0 ± 1.1 | 6.2 ± 1.2 | 6.3 ± 1.3 | 8.5 ± 1.6 |
| Time: P = 0.282, Altitude: P = 0.244, Interaction: P = 0.532 | ||||
| FMD SRAUC (103 s−1) | 23.3 ± 1.9 | 30.3 ± 2.8* | 23.2 ± 2.7 | 22.6 ± 1.9* |
| Time: P = 0.025, Altitude: P = 0.312, Interaction: P = 0.185 | ||||
FMD, flow‐mediated dilatation; SRAUC, shear rate area under the curve.
* P < 0.05, pre‐max data vs. post‐max data.
† P < 0.05, interaction effect baseline vs. post‐max.
No differences were found in absolute (mm) or relative changes (FMD) in brachial artery diameter between baseline and post‐maximal exercise at sea level or high altitude (P = 0.453 and P = 0.282, respectively), or between sea level and high altitude (P = 0.380 and P = 0.244, respectively). Flow‐mediated dilatation SRAUC was greater post‐maximal exercise compared to baseline (P = 0.025), but there was no difference between sea level and high altitude (P = 0.312). When taking into account baseline diameter and SRAUC as covariates, the FMD results remained the same as there was no effect of time (P = 0.614) or altitude (P = 0.291), nor was there an interaction between these effects (P = 0.717) (refer to Fig. 3 for individual FMD data at baseline and post‐maximal exercise).
Figure 3. Individual FMD data collected at baseline and post‐maximal exercise at sea level and high altitude.
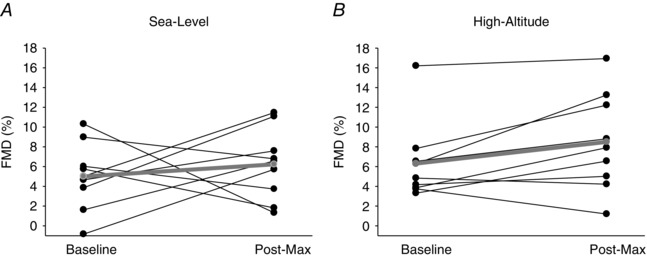
Mean data (n = 9) are represented by the grey line plot. BL, baseline; Post‐Max, post‐maximal exercise; FMD, flow‐mediated dilatation. These data reveal that we found no change in FMD at post‐maximal exercise at sea level and high altitude.
Moderate intensity exercise at sea level
Figure 4 A–F illustrates the cardiovascular data collected during moderate‐intensity exercise on placebo and prazosin at sea level. No differences were detected for cardiac output between placebo and prazosin during baseline, exercise and post‐exercise (P = 0.444). In contrast, prazosin increased heart rate (P < 0.001), and decreased stroke volume (P = 0.026), across all time‐points compared to placebo. prazosin had no effect on mean arterial pressure (P = 0.701) or on total peripheral resistance (P = 0.492) during baseline, exercise and post‐exercise time‐points. Additionally, there was no difference in between placebo and prazosin trials (P = 0.237).
Figure 4. Cardiovascular data during baseline, moderate‐intensity exercise and post‐exercise time‐points on placebo and prazosin at sea level and high altitude.
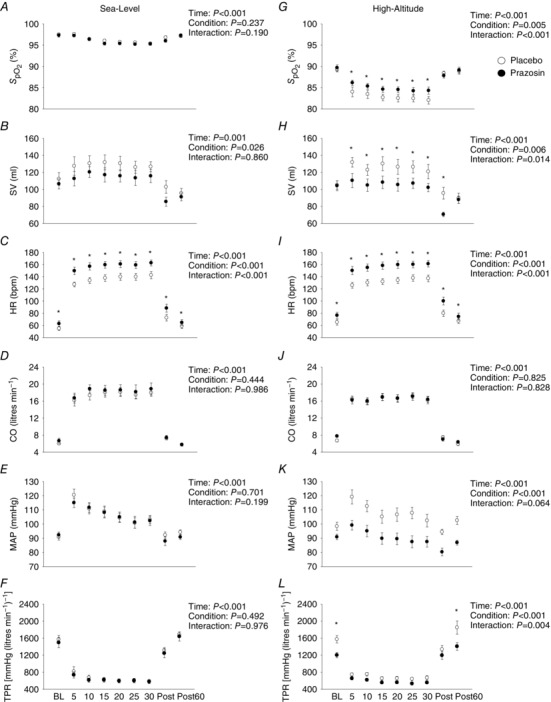
Open circles represent placebo data ± SEM, and closed circles represent prazosin data ± SEM in nine participants. * P < 0.05, for interaction effects. Statistics for main effects and interactions are displayed on the top right of each panel. BL, baseline; Post, immediately after exercise; Post 60, 60 min after exercise; , per cent oxygen saturation of haemoglobin; SV, stroke volume, HR, heart rate; CO, cardiac output; MAP, mean arterial pressure; TPR, total peripheral resistance. These findings demonstrate that, at sea level, SV was elevated, while HR was reduced on placebo compared to prazosin during 30 min of moderate‐intensity exercise. At high altitude, and HR were lower, while MAP and HR were higher on placebo compared to prazosin during 30 min of moderate‐intensity exercise.
Figure 5 A–C highlights the brachial antegrade, retrograde and mean shear rate data collected during baseline, exercise and post‐exercise time‐points on placebo and prazosin at sea level. Mean and antegrade shear rate were not different between conditions (P = 0.567 and P = 0.156, respectively). Retrograde shear rate was lower (i.e. more negative) during the prazosin trial compared to placebo (P = 0.037).
Figure 5. Brachial artery shear rate data during baseline, moderate‐intensity exercise and post‐exercise time‐points on placebo and prazosin at sea level and high altitude.
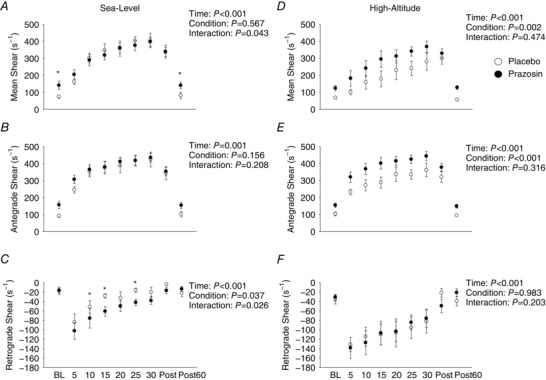
Open circles represent placebo data ± SEM, and closed circles represent prazosin data ± SEM in nine participants. * P < 0.05, for interaction effects. Statistics for main effects and interactions are displayed on the top right of each panel. BL, baseline; Post, immediately after exercise; Post 60, 60 min after exercise. These data reveal that retrograde shear rate was greater after prazosin administration at sea level, while at high altitude, mean and antegrade shear rate were greater after prazosin administration compared to placebo during 30 min of moderate‐intensity exercise.
Figure 6 A–D illustrates brachial artery velocity, diameter, blood flow and conductance data collected during baseline, exercise and post‐exercise points on placebo and prazosin at sea level. No differences were found in mean blood flow (P = 0.285), forearm vascular conductance (P = 0.294), artery diameter (P = 0.623) or blood velocity (P = 0.400) between the placebo and prazosin trials.
Figure 6. Brachial artery velocity, diameter, blood flow and conductance data collected during baseline, moderate‐intensity exercise and post‐exercise time‐points on placebo and prazosin at sea level and high altitude.
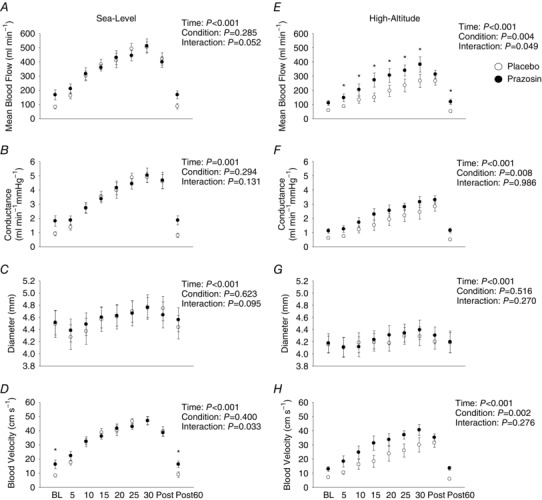
Open circles represent placebo data ± SEM, and closed circles represent prazosin data ± SEM in nine participants. * P < 0.05, for interaction effects. Statistics for main effects and interactions are displayed on the top right of each panel. BL, baseline; Post, immediately after exercise; Post 60, 60 min after exercise. These findings illustrate that there were no differences found in brachial artery velocity, diameter, blood flow and conductance at sea level between placebo and prazosin trials; however, at high altitude, we found brachial artery velocity, blood flow and conductance were reduced while participants were on placebo compared to prazosin during the 30 min moderate‐intensity exercise test.
Table 2 and Figure 7 A and B displays FMD data collected at the baseline, post‐exercise and 60 min post‐exercise time‐points on placebo and prazosin at sea level. No differences in FMD were detected between placebo and prazosin (P = 0.916), but there was a time effect during the placebo trial, where FMD was reduced immediately after exercise compared to baseline (P = 0.043), and at the 60 min post‐exercise point (P < 0.001). Additionally, an interaction effect was present between placebo and prazosin immediately post‐exercise, where FMD was greater during the prazosin trial by 2.9 ± 1.3% compared to the placebo trial (P = 0.039). No differences were found in FMD between placebo and prazosin trials during baseline (P = 0.762), or at 60 min post‐exercise (P = 0.107). When taking into account baseline diameter and SRAUC as covariates, the FMD results remained the same as there was still a main effect for time (P = 0.016), no effect between conditions (i.e. placebo vs. prazosin) (P = 0.450) and an interaction effect (P = 0.026).
Table 2.
Brachial artery diameter and flow‐mediated dilatation data during baseline and after moderate‐intensity exercise at sea level and high altitude
| Sea level | High altitude | ||||||
|---|---|---|---|---|---|---|---|
| Pre | Post | Post‐60 | Pre | Post | Post‐60 | ||
| Diameter (mm) | Placebo | 4.5 ± 0.2 | 4.8 ± 0.2* | 4.4 ± 0.2* | 4.2 ± 0.1 | 4.2 ± 0.1 | 4.2 ± 0.2 |
| Prazosin | 4.5 ± 0.2 | 4.6 ± 0.2 | 4.6 ± 0.2 | 4.2 ± 0.2 | 4.3 ± 0.1 | 4.2 ± 0.2 | |
| Time: P = 0.015, Condition: P = 0.783, | Time: P = 0.496, Condition: P = 0.398, | ||||||
| Interaction: P = 0.008 | Interaction: P = 0.501 | ||||||
| Change in | Placebo | 0.21 ± 0.03 | 0.12 ± 0.02 | 0.30 ± 0.05† | 0.26 ± 0.03 | 0.22 ± 0.04 | 0.25 ± 0.03 |
| diameter (mm) | Prazosin | 0.20 ± 0.04 | 0.22 ± 0.05 | 0.22 ± 0.03† | 0.29 ± 0.03 | 0.31 ± 0.05 | 0.35 ± 0.05 |
| Time: P = 0.022, Condition: P = 0.954, | Time: P = 0.502, Condition: P = 0.093, | ||||||
| Interaction: P = 0.074 | Interaction: P = 0.501 | ||||||
| FMD (%) | Placebo | 4.9 ± 0.7 | 2.1 ± 0.8* | 7.0 ± 1.2† | 6.3 ± 0.8 | 5.3 ± 1.0 | 5.8 ± 0.6 |
| Prazosin | 4.5 ± 1.0 | 4.9 ± 1.3‡ | 4.0 ± 0.9 | 6.8 ± 0.8 | 7.3 ± 1.2 | 8.5 ± 1.2 | |
| Time: P = 0.013, Condition: P = 0.916, | Time: P = 0.474, Condition: P = 0.099, | ||||||
| Interaction: P = 0.033 | Interaction: P = 0.455 | ||||||
| FMD SRAUC | Placebo | 19.8 ± 2.4 | 37.9 ± 4.5* , † | 18.3 ± 2.7 | 26.9 ± 2.8 | 40.2 ± 5.3* , † | 25.5 ± 1.7 |
| (103 s−1) | Prazosin | 27.4 ± 2.1 | 39.8 ± 4.7* , † | 23.7 ± 2.5 | 30.5 ± 2.5 | 50.2 ± 3.0* , † | 37.6 ± 3.2 |
| Time: P < 0.001, Condition: P = 0.060, | Time: P < 0.001, Condition: P = 0.005, | ||||||
| Interaction: P = 0.448 | Interaction: P = 0.287 | ||||||
FMD, flow‐mediated dilatation; SRAUC, shear rate area under the curve.
* P < 0.05, vs. pre‐exercise data.
† P < 0.05, Post‐60 vs. Post.
‡ P < 0.05, interaction within Post FMD. Condition: P = 0.099, when accounted for baseline diameter and shear rate, P = 0.032.
Figure 7. Individual FMD data collected during baseline and post‐exercise time‐points on placebo and prazosin at sea level and high altitude.
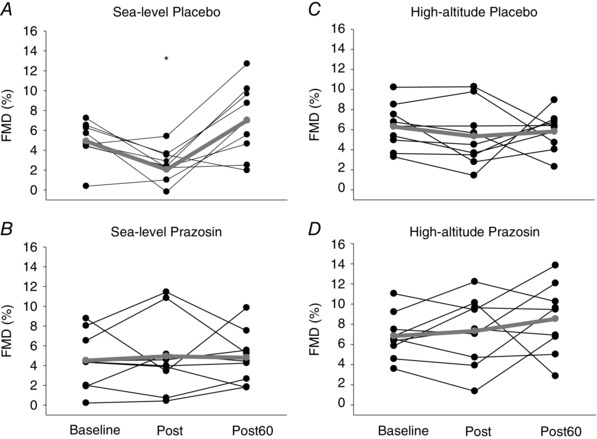
Mean data (n = 9) are represented by the grey line plot. * P < 0.05, represents time effect, where FMD Post‐EX was lower compared to baseline, and Post‐60 (for more details, see Results section). Post, immediately after exercise; Post 60, 60 min after exercise; FMD, flow‐mediated dilatation. Collectively, these findings reveal that at sea level, FMD was reduced immediately after 30 min of moderate‐intensity exercise while on placebo, and the reduction in FMD was abolished after administration of prazosin. At high altitude, there was no observed reduction in FMD immediately after 30 min of moderate‐intensity exercise after administration of placebo or prazosin. Additionally, after taking into account changes in brachial baseline diameter and shear rate between placebo and prazosin trials, FMD was elevated after administration of prazosin compared to placebo at high altitude.
Moderate‐intensity exercise at high altitude
Figure 4 G–L outlines the cardiovascular data collected during baseline, moderate‐intensity exercise and post‐exercise on placebo and prazosin at high altitude. No differences were detected for cardiac output between placebo and prazosin during baseline, exercise and post‐exercise (P = 0.825). In contrast, heart rate was elevated (P < 0.001), and stroke volume was decreased (P = 0.006), while on prazosin compared to placebo. Prazosin resulted in a lower mean arterial pressure (P < 0.001) and total peripheral resistance (P < 0.001) compared to the placebo trial. Interestingly, was elevated by 2.5 ± 0.7% (P = 0.005) during the prazosin trial compared to placebo during the moderate‐intensity exercise.
Figure 5 D–F illustrates brachial shear rate data collected during baseline, moderate‐intensity exercise and following exercise on placebo and prazosin at high altitude. Mean and antegrade shear rates were elevated during the prazosin trial compared to the placebo trial across all time‐points (P = 0.002 and P < 0.001, respectively). However, retrograde shear rate was not different between placebo and prazosin (P = 0.983).
Figure 6 E–H highlights the brachial artery velocity, diameter, blood flow and conductance data collected during baseline, exercise and post‐exercise time‐points on placebo and prazosin at high altitude. During the prazosin trial, mean blood flow, conductance and blood velocity were elevated compared to the placebo trial (P = 0.004, P = 0.008 and P = 0.002, respectively). No differences were found to brachial artery diameter between placebo and prazosin trials (P = 0.516).
Table 2 and Figure 7 C and D illustrates FMD data collected at baseline, post‐exercise and 60 min post‐exercise points on placebo and prazosin at high altitude. No differences in FMD were detected between time‐points (i.e. baseline, post‐exercise and 60 min post‐exercise points) (P = 0.474), between placebo and prazosin (P = 0.099). When taking into account baseline diameter and SRAUC as covariates, no differences were detected between time‐points (P = 0.681), nor were any interactions present (P = 0.474), which was consistent with our original results. However, we found that there was a main effect for condition (i.e. prazosin vs. placebo), where FMD was higher with prazosin compared to placebo (P = 0.032).
Discussion
Using a double‐blinded, counterbalanced, randomized and placebo‐controlled design, this is the first study to examine the role of the SNS on post‐exercise peripheral vascular endothelial function at both sea level (344 m) and high altitude (3800 m). Our main findings were (1) at rest, brachial artery FMD remained unchanged between sea level and high altitude on both placebo and prazosin conditions, (2) FMD remained unchanged after maximal exercise at both sea level and high altitude, and (3) FMD decreased immediately after moderate‐intensity exercise at sea level, but not at high altitude. Prazosin abolished the observed post‐exercise FMD decrease at sea level, and resulted in an overall increase in FMD at high altitude compared to placebo when SRAUC and baseline diameter were considered as covariates. These data demonstrate that hypobaric hypoxia counteracts the effect of moderate‐intensity exercise on FMD.
Endothelial function between sea level and high altitude at rest
Acclimatization to high altitude results in several physiological changes, but the effect of high altitude on endothelial function remains unclear. At sea level, recent work has established that increases in SNS activity (Hijmering et al. 2002; Dyson et al. 2006; Atkinson et al. 2015), and altered haemodynamics (e.g. increased retrograde shear) (Lamping and Dole, 1987; Millgard and Lind, 1998; Dawson et al. 2008; Johnson et al. 2012 a; Birk et al. 2013) can negatively affect vascular endothelial function as assessed by FMD. Since both of these physiological changes occur during high‐altitude exposure, it is logical to hypothesize that endothelial function would be reduced at high altitude, yet we found that there was no change. Comparisons of FMD between sea level and high altitude have been made previously, but studies have reported contradictory results such as reduced FMD (Lewis et al. 2014 a; Bakker et al. 2015), or no change in FMD upon acclimatization to high altitude (Bruno et al. 2016; 2015).
The major difference between these studies is the mode of transport to altitude – exercise versus cable car ascent. We sought to further investigate the effects of non‐trekking arrival to high altitude on endothelial function, and consistent with the results of Bruno et al. (2015), we found no change in endothelial function between sea level and high altitude at rest. This finding opposes our hypothesis that endothelial function would be impaired at high altitude due to elevated resting SNS activity. Rather, our data indicate that exercise at high altitude (i.e. trekking) may directly affect endothelial function, and we speculate the mechanism(s) for this may be due to increased vascular inflammation (Bruno et al. 2016), oxidative stress (Quindry et al. 2016) and reductions in nitric oxide bioavailability (Lewis et al. 2014 a).
Endothelial function before and after maximal exercise at sea level and high altitude
The degree of observed post‐exercise reduction in FMD has been reported to be exercise intensity‐dependent (for a review see Birk et al. 2013); however, few studies have measured FMD after maximal exercise in healthy individuals in an inactive limb (i.e. free from exercise) (Thijssen et al. 2006; Hwang et al. 2012; McClean et al. 2015). Moreover, no studies to date have reported FMD after maximal exercise at high altitude. Based on previous reports, FMD following maximal exercise remains unchanged in the majority of studies (Thijssen et al. 2006; Hwang et al. 2012; McClean et al. 2015) but this is not a universal finding (Hwang et al. 2012). Studies with a similar experimental design compared to ours (Thijssen et al. 2006; Hwang et al. 2012; McClean et al. 2015) have demonstrated and further confirm that at sea level, and for the first time at high altitude – FMD remained unchanged post‐maximal exercise. Collectively, these findings oppose the idea that post‐exercise related decreases in FMD are inversely related to exercise intensity (Birk et al. 2013). A likely explanation for this discrepancy is that the exercise duration of the maximal exercise test was too short in duration (i.e. exercise volume) to induce a reduction in FMD (Johnson et al. 2012 b; Dawson et al. 2013). Interestingly, at high altitude, FMD was elevated in a positive direction at post‐maximal exercise (6.3 to 8.5%); however, this observation was not statistically significant.
Endothelial function before and after moderate‐intensity exercise at sea level and high altitude
Sea level
In a different group of subjects, Atkinson et al. (2015) performed a similar investigation to the sea‐level component of this study and demonstrated that FMD was transiently impaired immediately after 30 min of moderate‐intensity exercise; these changes improved back to pre‐exercise values 60 min after moderate‐intensity exercise. They attributed this reduction in FMD to exercise related increases in SNS activity. Congruent with Atkinson et al. (2015) and other studies with similar modern methodology to assess FMD (Goel et al. 2007 b; Dawson et al. 2008; Jones et al. 2010; Johnson et al. 2012 a; Birk et al. 2013), we found similar results. We acknowledge, however, that such transient reductions in FMD following moderate‐intensity exercise (at least at sea level) should not necessarily be interpreted as endothelial dysfunction, but rather that FMD is influenced by a myriad of factors that may well reflect physiological adaptation in other vascular beds, including resistance and conduit arteries.
High altitude
Although there has been a recent investigation of post‐exercise endothelial function after moderate‐intensity exercise in acute normobaric hypoxia (Katayama et al. 2016), our experiment is the first to investigate endothelial function after moderate‐intensity exercise at high altitude (i.e. 3800 m). In the study by Katayama et al. (2016), endothelial function was not different after 30 min of moderate‐intensity (i.e. 60% ) exercise between normoxia and hypoxia ( = 0.12–0.13) trials. However, longer exposure to hypoxia (i.e. 4–7 days at high altitude) may well yield differential results due to cardiovascular adaptation (Rhodes et al. 2011; Boos et al. 2012; Lewis et al. 2014 a), and perhaps differential tonic SNS activity (Hansen and Sander, 2003). There is evidence suggesting increased SNS activity may be responsible for vascular dysfunction directly (Hijmering et al. 2002), or indirectly by increasing retrograde shear rate (Thijssen et al. 2014). Due to high‐altitude‐related increases in SNS activity, and based on previous reports that endothelial function (via brachial FMD) is reduced upon arrival at high altitude in some (Lewis et al. 2014 a; Bakker et al. 2015), but not all studies (Bruno et al. 2016), we anticipated a reduction in FMD immediately after moderate‐intensity exercise at high altitude. Our original hypothesis was not supported as no reduction in FMD was observed after moderate‐intensity exercise, indicating that SNS‐related blood vessel regulation is different between sea level and high altitude. This latter notion suggests that post‐exercise associated elevations in SNS activity may have a differential transduction to the peripheral vasculature compared to sea level.
Effects of α1‐adrenergic blockade on endothelial function at sea level and high altitude
α1‐Adrenergic blockade results in vasodilatation of the peripheral vasculature by reducing SNS transduction directly to smooth muscle cells. At sea level, prazosin resulted in an increased heart rate and decreased stroke volume during exercise. However, the differential heart rate response between placebo and prazosin trials did not result in a different blood flow or mean and antegrade shear rate response to exercise. Similar to Atkinson et al. (2015), we reported that prazosin administration abolished the reduction in endothelial function immediately after 30 min of moderate‐intensity exercise.
At rest, SNS blockade has no effect on at sea level (Liu et al. 2007) or at high altitude (Ainslie et al. 2012). During exercise, remained the same at sea level between conditions; however, at high altitude, an unexpected observation was that participants on prazosin had a higher compared to the placebo trial. This was probably due to increased ventilation during exercise on the prazosin trial, perhaps due to baroreflex–chemoreflex interaction, supported by the increased heart rate, and reduced stroke volume and mean arterial pressure responses to exercise between placebo and prazosin trials. The ventilatory response to changes in arterial blood pressure is related to converging baroreceptor and chemoreceptor afferents at the nucleus tractus solitarius (Richter and Seller, 1975; Eckberg and Orshan, 1977). A reduction in arterial blood pressure potentiates the chemoreflex and results in an increase in ventilation, while an increase in arterial blood pressure dampens ventilation (Heistad et al. 1974).
In contrast to our sea level data, prazosin resulted in an increased mean and antegrade shear rate at high altitude. These changes were probably facilitated by blockade of SNS vasoconstriction, which was likely to be enhanced at altitude. Increases in antegrade shear rate have been demonstrated to have a positive effect on endothelial function (Thijssen et al. 2014). This enhanced antegrade shear response at altitude versus sea level may explain the lack of post‐exercise FMD impairment, and could be in part responsible for the increase in endothelial function observed during the prazosin trial at altitude. The increase in FMD observed at high altitude during the prazosin trial compared to the placebo trial suggests that there is indeed some SNS‐related vascular constraint. These data collected at high altitude indicate that SNS activity is, in part, responsible for the FMD response.
Methodological considerations
The intervention to reduce SNS activity was an α1‐specific adrenergic receptor blockade, and the dose of administration has been used to establish ∼80% blockade in SNS activity, but this was not confirmed on an individual basis. An important methodological consideration for the current study is that prazosin, a specific α1‐adrenergic receptor blockade, was used; therefore, it is possible that during prazosin administration there is unopposed upregulation of α2‐adrenergic and β‐adrenergic receptor activity (Weisbrod et al. 2001). These net effects probably result in competing vasoconstriction and vasodilatation effects, respectively. Future studies investigating the role of the SNS on endothelial function should consider using both non‐selective α‐ and β‐adrenergic receptor blockades, along with a combined α‐ and β‐adrenergic receptor blockade. Second, we cannot exclude the possibility that the vascular response to changes in vasodilatatory substances is also altered (Calbet et al. 2014), and could hence influence our findings. Additionally, we achieved adequate power (>0.80) in nearly all of our measurements at sea level and high altitude. Although our small sample size was smaller than our calculated sample size (n = 9 vs. n = 10), our sea level data replicate previous findings by Atkinson et al. (2015), and we achieved a statistical effect. Moreover, additional novelty and statistical power of our study is within the nature of the double‐blinded, counterbalanced, randomized and placebo‐controlled design.
A final consideration is that we did not directly measure SNS activity directly via microneurography. Obviously, obtaining muscle SNS measurements in the leg (via peroneal nerve) would not be possible during exercise; however, past studies have reported such data in humans in the arm (via radial nerve) (Rea and Wallin, 1989). This approach would not have been feasible given our small sample size and the amount of measurements needed in each participant (i.e. twice at both sea level and high altitude). Nevertheless, both at sea level and at high altitude, we have for the first time used a powerful double‐blinded, counterbalanced, randomized and placebo‐controlled study to examine the role of SNS blockade both at rest and during exercise.
Conclusion
Our findings illustrate that at sea level exercise‐related increases in SNS activity reduce FMD immediately following moderate‐intensity exercise, but not at high altitude. These findings indicate that differential governing mechanisms for endothelial function between sea level and high altitude. Together, our findings have implications for better understanding the chronic impacts of hypoxaemia and exercise, and the interactions on sympathetic activity and vascular function.
Additional information
Competing interests
The authors have no conflict of interest.
Author contributions
M.M.T., J.C.T. and P.N.A. were responsible for conception and design of the current study. All authors contributed to the collection, assembly, analysis and interpretation of the data, along with drafting the article or revising it critically for important intellectual content. All authors approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the Natural Sciences and Engineering Research Council of Canada (PNA), and the Canadian Foundation for Innovation and a Canada Research Chair (PNA).
Acknowledgements
This study was carried out within the framework of the Barcroft high‐altitude research expedition; we thank the research stations staff for their friendly accommodation. The authors are grateful to the members of the UBC International Research expedition and to the Barcroft research facility for invaluable help with organization and implementation of this research study.
References
- Ainslie PN, Lucas SJ, Fan JL, Thomas KN, Cotter JD, Tzeng YC & Burgess KR (2012). Influence of sympathoexcitation at high altitude on cerebrovascular function and ventilatory control in humans. J Appl Physiol (1985) 113, 1058–1067. [DOI] [PubMed] [Google Scholar]
- Atkinson CL, Lewis NC, Carter HH, Thijssen DH, Ainslie PN & Green DJ (2015). Impact of sympathetic nervous system activity on post‐exercise flow‐mediated dilatation in humans. J Physiol 593, 5145–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson G, Batterham AM, Thijssen DH & Green DJ (2013). A new approach to improve the specificity of flow‐mediated dilation for indicating endothelial function in cardiovascular research. J Hypertens 31, 287–291. [DOI] [PubMed] [Google Scholar]
- Bakker E, Engan H, Patrician A, Schagatay E, Karlsen T, Wisloff U & Gaustad SE (2015). Acute dietary nitrate supplementation improves arterial endothelial function at high altitude: a double‐blinded randomized controlled cross over study. Nitric Oxide 50, 58–64. [DOI] [PubMed] [Google Scholar]
- Birk GK, Dawson EA, Batterham AM, Atkinson G, Cable T, Thijssen DH & Green DJ (2013). Effects of exercise intensity on flow mediated dilation in healthy humans. Int J Sports Med 34, 409–414. [DOI] [PubMed] [Google Scholar]
- Boos CJ, Hodkinson P, Mellor A, Green NP & Woods DR (2012). The effects of acute hypobaric hypoxia on arterial stiffness and endothelial function and its relationship to changes in pulmonary artery pressure and left ventricular diastolic function. High Alt Med Biol 13, 105–111. [DOI] [PubMed] [Google Scholar]
- Bruno RM, Ghiadoni L & Pratali L (2016). Vascular adaptation to extreme conditions: the role of hypoxia. Artery Res 14, 15–21. [Google Scholar]
- Bruno RM, Giardini G, Malacrida S, Catuzzo B, Armenia S, Ghiadoni L, Brustia R, Laveder P, Salvi P, Cauchy E & Pratali L (2015). Role of altered vascular reactivity in the pathophysiology of acute mountain sickness. Artery Res 12, 29. [Google Scholar]
- Calbet JA, Boushel R, Robach P, Hellsten Y, Saltin B & Lundby C (2014). Chronic hypoxia increases arterial blood pressure and reduces adenosine and ATP induced vasodilatation in skeletal muscle in healthy humans. Acta Physiol (Oxf) 211, 574–584. [DOI] [PubMed] [Google Scholar]
- Dawson EA, Green DJ, Cable NT & Thijssen DH (2013). Effects of acute exercise on flow‐mediated dilatation in healthy humans. J Appl Physiol (1985) 115, 1589–1598. [DOI] [PubMed] [Google Scholar]
- Dawson EA, Whyte GP, Black MA, Jones H, Hopkins N, Oxborough D, Gaze D, Shave RE, Wilson M, George KP & Green DJ (2008). Changes in vascular and cardiac function after prolonged strenuous exercise in humans. J Appl Physiol (1985) 105, 1562–1568. [DOI] [PubMed] [Google Scholar]
- Duplain H, Vollenweider L, Delabays A, Nicod P, Bartsch P & Scherrer U (1999). Augmented sympathetic activation during short‐term hypoxia and high altitude exposure in subjects susceptible to high altitude pulmonary edema. Circulation 99, 1713–1718. [DOI] [PubMed] [Google Scholar]
- Dyson KS, Shoemaker JK & Hughson RL (2006). Effect of acute sympathetic nervous system activation on flow‐mediated dilation of brachial artery. Am J Physiol Heart Circ Physiol 290, H1446–453. [DOI] [PubMed] [Google Scholar]
- Eckberg DL & Orshan CR (1977). Respiratory and baroreceptor reflex interactions in man. J Clin Invest 59, 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R, Majeed F, Vogel R, Corretti MC, Weir M, Mangano C, White C, Plotnick GD & Miller M (2007). Exercise‐induced hypertension, endothelial dysfunction, and coronary artery disease in a marathon runner. Am J Cardiol 99, 743–744. [DOI] [PubMed] [Google Scholar]
- Green DJ, Dawson EA, Groenewoud HM, Jones H & Thijssen DH (2014). Is flow‐mediated dilation nitric oxide mediated?: a meta‐analysis. Hypertension 63, 376–382. [DOI] [PubMed] [Google Scholar]
- Hansen J & Sander M (2003). Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol 546, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL & Schmid PG (1974). Interaction of baroreceptor and chemoreceptor reflexes. Modulation of the chemoreceptor reflex by changes in baroreceptor activity. J Clin Invest 53, 1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijmering ML, Stroes ES, Olijhoek J, Hutten BA, Blankestijn PJ & Rabelink TJ (2002). Sympathetic activation markedly reduces endothelium‐dependent, flow‐mediated vasodilation. J Am Coll Cardiol 39, 683–688. [DOI] [PubMed] [Google Scholar]
- Hwang IC, Kim KH, Choi WS, Kim HJ, Im MS, Kim YJ, Kim SH, Kim MA, Sohn DW & Zo JH (2012). Impact of acute exercise on brachial artery flow‐mediated dilatation in young healthy people. Cardiovasc Ultrasound 10, 39‐7120‐10‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias D, Gomez Rosso L, Vainstein N, Merono T, Lezon C & Brites F (2015). Vascular reactivity and biomarkers of endothelial function in healthy subjects exposed to acute hypobaric hypoxia. Clin Biochem 48, 1059–1063. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Mather KJ, Newcomer SC, Mickleborough TD & Wallace JP (2012. a). Brachial artery flow‐mediated dilation following exercise with augmented oscillatory and retrograde shear rate. Cardiovasc Ultrasound 10, 34‐7120‐10‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BD, Padilla J & Wallace JP (2012. b). The exercise dose affects oxidative stress and brachial artery flow‐mediated dilation in trained men. Eur J Appl Physiol 112, 33–42. [DOI] [PubMed] [Google Scholar]
- Jones H, Green DJ, George K & Atkinson G (2010). Intermittent exercise abolishes the diurnal variation in endothelial‐dependent flow‐mediated dilation in humans. Am J Physiol Regul Integr Comp Physiol 298, R427–432. [DOI] [PubMed] [Google Scholar]
- Katayama K, Yamashita S, Iwamoto E & Ishida K (2016). Flow‐mediated dilation in the inactive limb following acute hypoxic exercise. Clin Physiol Funct Imaging 36, 60–69. [DOI] [PubMed] [Google Scholar]
- Lamping KG & Dole WP (1987). Acute hypertension selectively potentiates constrictor responses of large coronary arteries to serotonin by altering endothelial function in vivo. Circ Res 61, 904–913. [DOI] [PubMed] [Google Scholar]
- Lewis NC, Bailey DM, Dumanoir GR, Messinger L, Lucas SJ, Cotter JD, Donnelly J, McEneny J, Young IS, Stembridge M, Burgess KR, Basnet AS & Ainslie PN (2014. a). Conduit artery structure and function in lowlanders and native highlanders: relationships with oxidative stress and role of sympathoexcitation. J Physiol 592, 1009–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NC, Bain AR, MacLeod DB, Wildfong KW, Smith KJ, Willie CK, Sanders ML, Numan T, Morrison SA, Foster GE, Stewart JM & Ainslie PN (2014. b). Impact of hypocapnia and cerebral perfusion on orthostatic tolerance. J Physiol 592, 5203–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Smith TG, Balanos GM, Brooks J, Crosby A, Herigstad M, Dorrington KL & Robbins PA (2007). Lack of involvement of the autonomic nervous system in early ventilatory and pulmonary vascular acclimatization to hypoxia in humans. J Physiol 579, 215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, Gustafsson P, Hankinson J, Jensen R, McKay R, Miller MR, Navajas D, Pedersen OF, Pellegrino R & Wanger J (2005). Standardisation of the single‐breath determination of carbon monoxide uptake in the lung. Eur Respir J 26, 720–735. [DOI] [PubMed] [Google Scholar]
- McClean C, Harris RA, Brown M, Brown JC & Davison GW (2015). Effects of exercise intensity on postexercise endothelial function and oxidative stress. Oxid Med Cell Longev 2015, 723679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J & ATS/ERS Task Force (2005). Standardisation of spirometry. Eur Respir J 26, 319–338. [DOI] [PubMed] [Google Scholar]
- Millgard J & Lind L (1998). Acute hypertension impairs endothelium‐dependent vasodilation. Clin Sci (Lond) 94, 601–607. [DOI] [PubMed] [Google Scholar]
- Padilla J, Harris RA & Wallace JP (2007). Can the measurement of brachial artery flow‐mediated dilation be applied to the acute exercise model?. Cardiovasc Ultrasound 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quindry J, Dumke C, Slivka D & Ruby B (2016). Impact of extreme exercise at high altitude on oxidative stress in humans. J Physiol 594, 5093–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea RF & Wallin BG (1989). Sympathetic nerve activity in arm and leg muscles during lower body negative pressure in humans. J Appl Physiol (1985) 66, 2778–2781. [DOI] [PubMed] [Google Scholar]
- Rhodes HL, Chesterman K, Chan CW, Collins P, Kewley E, Pattinson KT, Myers S, Imray CH, Wright AD & Birmingham Medical Research Expeditionary Society (2011). Systemic blood pressure, arterial stiffness and pulse waveform analysis at altitude. J R Army Med Corps 157, 110–113. [DOI] [PubMed] [Google Scholar]
- Richter DW & Seller H (1975). Baroreceptor effects on medullary respiratory neurones of the cat. Brain Res 86, 168–171. [DOI] [PubMed] [Google Scholar]
- Saito M, Mano T, Iwase S, Koga K, Abe H & Yamazaki Y (1988). Responses in muscle sympathetic activity to acute hypoxia in humans. J Appl Physiol (1985) 65, 1548–1552. [DOI] [PubMed] [Google Scholar]
- Silvestro A, Scopacasa F, Oliva G, de Cristofaro T, Iuliano L & Brevetti G (2002). Vitamin C prevents endothelial dysfunction induced by acute exercise in patients with intermittent claudication. Atherosclerosis 165, 277–283. [DOI] [PubMed] [Google Scholar]
- Thijssen DH, Atkinson CL, Ono K, Sprung VS, Spence AL, Pugh CJ & Green DJ (2014). Sympathetic nervous system activation, arterial shear rate, and flow‐mediated dilation. J Appl Physiol (1985) 116, 1300–1307. [DOI] [PubMed] [Google Scholar]
- Thijssen DH, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME & Green DJ (2011). Assessment of flow‐mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol 300, H2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijssen DH, de Groot P, Kooijman M, Smits P & Hopman MT (2006). Sympathetic nervous system contributes to the age‐related impairment of flow‐mediated dilation of the superficial femoral artery. Am J Physiol Heart Circ Physiol 291, H3122–3129. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR (2001). Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol 537, 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman RJ, Playford DA, W GF, Cheetham C, Reed C, Taylor RR, Puddey IB, Beilin LJ, Burke V, Mori TA & Green D (2001). Improved analysis of brachial artery ultrasound using a novel edge‐detection software system. J Appl Physiol (1985) 91, 929–937. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS & Morgan BJ (2001). Exposure to hypoxia produces long‐lasting sympathetic activation in humans. J Appl Physiol (1985) 91, 1555–1562. [DOI] [PubMed] [Google Scholar]