

Abstract
Key points
The functional importance of residues in loop G of the GABAA receptor has not been investigated. D43 and T47 in the α1 subunit are of particular significance as their structural modification inhibits activation by GABA.
While the T47C substitution had no significant effect, non‐conservative substitution of either residue (D43C or T47R) reduced the apparent potency of GABA.
Propofol potentiated maximal GABA‐evoked currents mediated by α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. Non‐stationary variance analysis revealed a reduction in maximal GABA‐evoked P open, suggesting impaired agonist efficacy.
Further analysis of α1(T47R)β2γ2 receptors revealed that the efficacy of the partial agonist THIP (4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridine‐3‐ol) relative to GABA was impaired.
GABA‐, THIP‐ and propofol‐evoked currents mediated by α1(T47R)β2γ2 receptors deactivated faster than those mediated by α1β2γ2 receptors, indicating that the mutation impairs agonist‐evoked gating.
Spontaneous gating caused by the β2(L285R) mutation was also reduced in α1(T47R)β2(L285R)γ2 compared to α1β2(L285R)γ2 receptors, confirming that α1(T47R) impairs gating independently of agonist activation.
Abstract
The modification of cysteine residues (substituted for D43 and T47) by 2‐aminoethyl methanethiosulfonate in the GABAA α1 subunit loop G is known to impair activation of α1β2γ2 receptors by GABA and propofol. While the T47C substitution had no significant effect, non‐conservative substitution of either residue (D43C or T47R) reduced the apparent potency of GABA. Propofol (1 μm), which potentiates sub‐maximal but not maximal GABA‐evoked currents mediated by α1β2γ2 receptors, also potentiated maximal currents mediated by α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. Furthermore, the peak open probabilities of α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors were reduced. The kinetics of macroscopic currents mediated by α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors were characterised by slower desensitisation and faster deactivation. Similar changes in macroscopic current kinetics, together with a slower activation rate, were observed with the loop D α1(F64C) substitution, known to impair both efficacy and agonist binding, and when the partial agonist THIP (4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridine‐3‐ol) was used to activate WT or α1(T47R)β2γ2 receptors. Propofol‐evoked currents mediated by α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors also exhibited faster deactivation than their WT counterparts, revealing that these substitutions impair gating through a mechanism independent of orthosteric binding. Spontaneous gating caused by the introduction of the β2(L285R) mutation was also reduced in α1(T47R)β2(L285R)γ2 compared to α1β2(L285R)γ2 receptors, confirming that α1(T47R) impairs gating independently of activation by any agonist. These findings implicate movement of the GABAA receptor α1 subunit's β1 strand during agonist‐dependent and spontaneous gating. Immobilisation of the β1 strand may provide a mechanism for the inhibition of gating by inverse agonists such as bicuculline.
Keywords: cys‐loop receptors, pentameric ligand‐gated ion channels, site‐directed mutagenesis, spontaneous gating
Key points
The functional importance of residues in loop G of the GABAA receptor has not been investigated. D43 and T47 in the α1 subunit are of particular significance as their structural modification inhibits activation by GABA.
While the T47C substitution had no significant effect, non‐conservative substitution of either residue (D43C or T47R) reduced the apparent potency of GABA.
Propofol potentiated maximal GABA‐evoked currents mediated by α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. Non‐stationary variance analysis revealed a reduction in maximal GABA‐evoked P open, suggesting impaired agonist efficacy.
Further analysis of α1(T47R)β2γ2 receptors revealed that the efficacy of the partial agonist THIP (4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridine‐3‐ol) relative to GABA was impaired.
GABA‐, THIP‐ and propofol‐evoked currents mediated by α1(T47R)β2γ2 receptors deactivated faster than those mediated by α1β2γ2 receptors, indicating that the mutation impairs agonist‐evoked gating.
Spontaneous gating caused by the β2(L285R) mutation was also reduced in α1(T47R)β2(L285R)γ2 compared to α1β2(L285R)γ2 receptors, confirming that α1(T47R) impairs gating independently of agonist activation.
Abbreviations
- GluCl
glutamate‐activated Cl− channel
- HEK‐293
human embryonic kidney 293 cell
- MD
molecular dynamic
- MTSEA
2‐aminoethyl methanethiosulfonate
- pLGIC
pentameric ligand‐gated ion channel
- THIP
4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridine‐3‐ol
- TM
transmembrane
- τw
weighted tau
Introduction
GABA type A (GABAA) receptors are cysteine‐loop receptors of the pentameric ligand‐gated ion channel (pLGIC) family. They mediate fast inhibitory neurotransmission. Nineteen different genes encode GABAA receptor subunits, providing considerable heterogeneity in GABAA receptor composition. The most abundant GABAA receptors in the brain comprise α1, β2 and γ2 subunits (Whiting et al. 1995).
In common with other pLGICs, GABAA receptors have an N‐terminal extracellular domain, which contains the orthosteric agonist binding site and four transmembrane (TM) domains (TM1–4) containing additional binding sites for positive modulators and allosteric agonists, such as the general anaesthetic propofol. The Cl−‐selective channel pore, containing the gate, is encompassed by five TM2 domains arranged pseudo‐symmetrically (Miller & Aricescu, 2014). The intracellular domains of pLGICs are generally large, being mostly composed of the TM3–4 loop (Baptista‐Hon et al. 2013). TM domains are most highly conserved across pLGICs, while the intracellular loops are highly heterogeneous.
The orthosteric binding site of the GABAA receptor is located in the N‐terminal domain between adjacent α‐ and β‐ subunits (Smith & Olsen, 1995; Cromer et al. 2002). In this region, seven non‐contiguous loops (A–G) line the orthosteric site, where they participate in agonist binding (Boileau et al. 1999; Holden & Czajkowski, 2002; Wagner et al. 2004; Goldschen‐Ohm et al. 2011; Tran et al. 2011) and/or gating (Boileau et al. 2002; Newell & Czajkowski, 2003; Venkatachalan & Czajkowski, 2008; Szczot et al. 2014; Baptista‐Hon et al. 2016). These loops are contained within an anti‐parallel β sandwich structure. In the canonical α1β2γ2 GABAA receptor, loops A, B and C are contributed by the primary interface, from the β2 subunit, while loops D, E, F and G are contributed by the complementary interface, from the α1 subunit.
Arg37 in loop G was recently identified for its role in glutamate binding to the Caenorhabditis elegans glutamate‐activated Cl− (GluCl) pLGIC (Hibbs & Gouaux, 2011). However, residues in this region are not implicated in binding agonists to other pLGICs, such as the GABAA β3 homopentamer (Miller & Aricescu, 2014), the glycine receptor (Du et al. 2015) or the α4β2 nicotinic acetylcholine receptor (Morales‐Perez et al. 2016). Despite a lack of involvement in binding to GABAA receptors, there is considerable homology among loop G amino acids between α subunits, consistent with a conserved role in receptor function (Fig. 1 A).
Figure 1. Loop G and loop D of C. elegans GluCl and GABAA receptor α subunits.
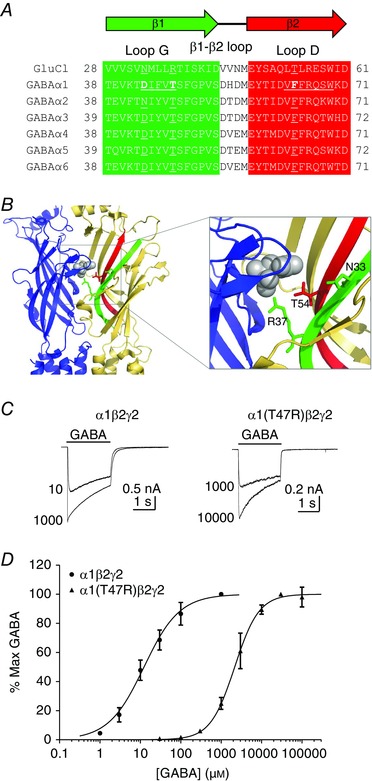
A, amino acid sequence alignment of the mouse GABAA α1–α6 subunits and the GluCl α subunit. The β1 and β2 strands are highlighted in green and red, respectively. Residues in loop G and D of the GABAA α1 subunit are underlined. Also underlined are the homologous residues on the GABAA α2–α6 and GluCl which are relevant to this study. B, the C. elegans GluCl (Hibbs & Gouaux, 2011) model, showing the interface between two subunits (blue, primary interface; yellow, complementary interface) with the β1 and β2 strands highlighted (green and red, respectively). Bound glutamate is shown in grey. Inset shows the highlighted area in more detail. Asn33 and Arg37 are shown on the β1 strand as stick rendering in green. Thr54 is shown on the β2 strand as stick rendering in red. C, representative examples of whole‐cell currents evoked by a maximal and an approximate EC50 concentration of GABA (indicated) mediated by α1β2γ2 or α1(T47R)β2γ2 GABAA receptors. The bar indicates GABA application (2 s). D, concentration–response relationships for α1β2γ2 (circles) or α1(T47R)β2γ2 (triangles) receptors. Current amplitudes were expressed as a percentage of the maximum current amplitude recorded from each cell. The sigmoidal curve represents the logistic function fitted to the data points. [Color figure can be viewed at wileyonlinelibrary.com]
The potential importance of loop G during gating is highlighted by comparisons of apo‐ and agonist‐bound structures of the glycine receptor (Du et al. 2015) and GluCl (Althoff et al. 2014). The β1 strand containing loop G, the β2 strand containing loop D and the interconnecting β1–β2 loop move towards the TM2–3 loop in the presence of bound agonist. This movement appears to precede the structural rearrangement of the TM domains that leads to channel opening (Calimet et al. 2013).
We recently demonstrated the involvement of specific GABAA receptor α1 subunit loop G residues in function using cysteine‐scanning mutagenesis (Baptista‐Hon et al. 2016). D43C was the only one of five loop G substituents to reduce the apparent potency of GABA. Furthermore, its modification by positively charged 2‐aminoethyl methanethiosulfonate (MTSEA) caused additional functional impairment. By contrast, while T47C did not significantly affect GABA's potency, its modification by MTSEA inhibited GABA‐evoked currents. Cysteines substituted at three other positions in loop G of the α1 subunit (residues 44, 45 and 46) either were inaccessible to MTSEA or their modification was without functional consequence (Baptista‐Hon et al. 2016).
In this study we investigated whether amino acid substitutions at positions 43 and 47 that impair function do so by impairing gating. To replicate MTSEA‐modified α1(T47C), which impairs function (Baptista‐Hon et al. 2016), we replaced Thr47 by Arg. We compared the efficacy of GABA as an activator of α1(D43C)β2γ2, α1(T47R)β2γ2 and α1β2γ2 receptors using analysis of channel open probability (P open) and potentiation by the positive allosteric modulator propofol. We investigated the kinetics of THIP‐ (4,5,6,7‐tetrahydroisoxazolo[5,4‐c]pyridine‐3‐ol) and propofol‐evoked currents mediated by α1(T47R)β2γ2 receptors. We also examined the influence of the α1(T47R) substitution on receptors containing the β2(L285R) substitution, which causes enhanced spontaneous gating. Using these approaches, we established that non‐conservative substitutions at two key positions in loop G reduce gating efficacy.
Methods
Cell culture and transfection
Human embryonic kidney 293 (HEK‐293) cells were grown and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 μg ml−1 penicillin and 100 units ml−1 streptomycin at 37 °C and 5% CO2. Cells were seeded at low density in either poly‐l‐lysine‐coated, or uncoated 35 mm dishes for outside‐out patch recordings or whole‐cell recordings, respectively. Transfections were performed by calcium phosphate precipitation, using 1 μg total cDNA per dish, as described previously (Baptista‐Hon et al. 2016). cDNAs encoding wild type (WT) and mutant mouse GABAA subunits were in the pRK5 mammalian expression vector. For heteromeric expression of GABAA α1β2γ2 subunits, a 1:1:1 transfection ratio was used. cDNA encoding enhanced green fluorescence protein (in pEGFP vector, 0.1 μg) was included to identify successfully transfected cells using fluorescence microscopy. Transfected cells were functionally examined using voltage‐clamp electrophysiology after 48–72 h. All tissue culture reagents were obtained from Invitrogen (Paisley, UK).
Mutagenesis of GABAA α1 and β2 subunits
Single point mutations were performed by overlap extension polymerase chain reaction (PCR) (Heckman & Pease, 2007). For α1 subunits, PCR products were digested using SmaI restriction endonuclease and EcoRI (5′) and BamHI (3′) restriction endonucleases were used for β2 subunits. All GABAA receptor subunits were ligated into the pRK5 vector. All mutagenesis reactions and ligations were verified using agarose gel electrophoresis and sequenced prior to functional characterisation (Genetics Core Services, University of Dundee). All PCR and molecular cloning reagents were obtained from Fermentas (Thermo‐Fisher, Loughborough, UK).
Electrophysiology
The whole‐cell or excised outside‐out patch configurations of the patch‐clamp technique were used to record GABA‐evoked currents from HEK‐293 cells expressing WT, or mutant, GABAA receptors. Recording electrodes were fabricated from borosilicate glass capillaries, and when filled with intracellular solution had resistances of 1.3–2.3 MΩ for whole‐cell recordings, and 3.0–5.0 MΩ for outside‐out patch recordings. The electrode solution contained (in mm): 140 CsCl, 2 MgCl2, 1.1 EGTA, 3 Mg‐ATP and 10 HEPES (pH 7.4 with CsOH). The extracellular solution contained (in mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 HEPES and 10 glucose (pH 7.4 with NaOH). Cells were voltage clamped at an electrode potential of −60 mV. For whole‐cell recordings, currents were evoked by rapid application of agonists using the three‐pipe Perfusion Fast Step system (Warner Instruments, Hamden, CT, USA), as described previously (Baptista‐Hon et al. 2016).
For outside‐out patch recordings, maximally efficacious and saturating concentrations of agonists were applied also using the Perfusion Fast Step system, with heat pulled and bevelled (Narashige, London, UK) theta and three‐barrelled pipes (Othman et al. 2012). This allows rapid solution exchange consistently of < 500 μs around an open pipette tip (Hinkle & Macdonald, 2003). Solution exchange times were measured using liquid junction currents arising from moving the open pipette tip into an extracellular solution diluted by 10%. The 10–90% rise time of the liquid junction current was used as a measure of solution exchange rate. Liquid junction currents were routinely measured at the end of every outside‐out patch recording, to ensure the fidelity of fast solution exchange. For experiments examining the macroscopic current kinetics of propofol‐evoked currents, the concentration of propofol used was chosen on the basis that the evoked current does not contain a surge following propofol removal, which is mediated by propofol blockade (Adodra & Hales, 1995).
All electrophysiological data were recorded using an Axopatch 200B amplifier. Data were low pass filtered at 2 kHz for whole cell currents and 10 kHz for outside‐out patch currents. Analog data were digitised at 20 kHz for whole cell currents and 100 kHz for outside‐out patch currents using a Digidata 1320A interface and acquired using pCLAMP8 software (all from Molecular Devices, Sunnyvale, CA, USA).
Data analysis
The peak amplitudes of agonist‐evoked currents were measured using Clampfit10 software (Molecular Devices), using averaged current traces from at least five agonist‐evoked currents. The potentiating effects of propofol on peak GABA‐evoked currents were analysed as percentage potentiation, using the formula:
where I pot and I GABA represent the potentiated and control peak current amplitudes, respectively.
Non‐stationary variance analysis was performed as described by Szczot et al. (2014). A minimum of 10 consecutive responses to a 5 ms application of maximally efficacious and saturating concentration of GABA were recorded from outside‐out patches, using the rapid application method described above. The currents were analysed using a custom‐written script in MatLab (The Mathworks, Inc., Natick, MA, USA). The rising phase of each current was excluded from the analysis. The starting point for analysis was therefore set at the peak of the current. Mean current (I) and variance (σ2) at each time point were divided into 30 equally spaced bins and plotted as I vs σ2, which were fitted with the following function:
where i is the single channel amplitude, N is the number of channels and C is the background variance. From these parameters, the maximal open probability (P open) was calculated by:
Single channel conductance was calculated by dividing the single channel amplitude (i) by the membrane holding potential (−60 mV).
Activation rates were measured using the 10–90% rise time of agonist‐evoked inward current. Macroscopic desensitisation and deactivation kinetics were measured by fitting multi‐exponential functions to the decaying phase of the inward current, during or following agonist application, respectively. The multi‐exponential function is defined by:
where τN are time constants and AN represent the proportion of the particular τ. The best‐fit number of exponential terms (one to three terms) were determined using an F‐test with the confidence set at the 95% level. Rates of desensitisation and deactivation are provided as weighted τ (τw) values, calculated using:
Individual GABA concentration vs current amplitude relationships were fitted with a logistics equation:
from which GABA EC50 and Hill slope (n H) values were determined.
Statistics
Data are presented as mean ± SEM. Differences in means of three or more groups were compared using one‐way ANOVA, with a post hoc Dunnet's or Tukey's comparison. Pairwise comparisons were performed using Student's t‐test. In all cases P < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA).
Results
Residues in loop G and loop D influence the apparent potency of GABA
Loop G of the GABAA receptor α subunit contributes residues to the complementary interface near the orthosteric binding site in heteropentameric GABAA receptors. The alignment reveals that the α1 subunit Thr47 is entirely conserved among all GABAA α subunits at the position equivalent to that of Arg37 in the C. elegans GluCl α subunit (Fig. 1 A). Asp43, an α1 subunit residue we previously demonstrated to be important for GABAA receptor function (Baptista‐Hon et al. 2016), is also conserved in all but the α2 subunit, which contains an Asn at the equivalent position. Loop G runs adjacent and anti‐parallel to loop D on the β2 strand. The location of a critical loop D residue, Phe64, known to participate in GABA binding and efficacy is also shown in the alignment (Boileau et al. 1999; Szczot et al. 2014). Residues in these positions are highlighted on the GluCl structure in relation to bound glutamate (Fig. 1 B) (Hibbs & Gouaux, 2011). GluCl Arg37 in loop G forms part of the glutamate binding site and is involved in receptor activation (Hibbs & Gouaux, 2011). We investigated the influence of the T47R substitution on GABAA α1β2γ2 receptor function. Our previous study demonstrated that a non‐conservative substitution to Asp43 (D43C) in loop G of the GABAA α1 subunit significantly reduced the apparent potency of GABA (Baptista‐Hon et al. 2016).
WT α1 or α1(T47R) subunits were transiently expressed with β2 and γ2 subunits in HEK‐293 cells. Representative examples of GABA‐evoked currents recorded under voltage‐clamp at −60 mV from cells expressing α1β2γ2 or α1(T47R)β2γ2 receptors are shown in Fig. 1 C. GABA‐evoked current amplitudes were expressed as a percentage of the maximum and plotted as a concentration–response relationship (Fig. 1 D). A logistic function fitted to the data points reveals that the α1(T47R) substitution caused a rightward shift in the GABA concentration–response relationship (Fig. 1 D). The EC50 for GABA activation of WT α1β2γ2 receptors was 17 ± 5 μm (n = 7). The EC50 for GABA activation of α1(T47R)β2γ2 receptors was 1500 ± 320 μm (n = 7). The non‐conservative D43C substitution in loop G of the GABAA α1 subunit produced a similar reduction in the apparent potency of GABA (Baptista‐Hon et al. 2016).
Phe64, which is located on the adjacent and anti‐parallel loop D, is involved in GABA binding and gating (Boileau et al. 1999; Szczot et al. 2014). The α1(F64C) substitution causes a significant reduction in the apparent potency of GABA (Boileau et al. 1999; Szczot et al. 2014; Baptista‐Hon et al. 2016). Substitution of Phe64 with the equivalent residue found at this position in GluCl (F64T) caused a similar impairment in the apparent potency of GABA. The EC50 of α1(F64T)β2γ2 receptors was 16 ± 1 mm (n = 3). A one‐way ANOVA revealed a significant difference between the GABA EC50 values (P < 0.0001). A post hoc Tukey's comparison revealed statistically significant differences in GABA EC50 between WT and α1(T47R)β2γ2 receptors (P < 0.05), between WT and α1(F64T)β2γ2 receptors (P < 0.0001) and also between α1(T47R)β2γ2 and α1(F64T)β2γ2 receptors (P < 0.0001). There were no statistically significant differences in the Hill slope values of the GABA concentration–response relationships (P = 0.24; one‐way ANOVA). This observation suggests that the number of GABA binding sites remains the same and argues against altered stoichiometry caused by the α1 subunit substitutions. We also evaluated the peak current density of α1β2γ2, α1(T47R)β2γ2 and α1(F64T)β2γ2 receptors, by normalising the current amplitude evoked by maximally efficacious concentrations of GABA, to cell capacitances. The mean (± SEM) current densities are 1510 ± 264 pA pF−1 (n = 18), 684 ± 119 pA pF−1 (n = 24) and 401 ± 155 pA pF−1 (n = 5), for α1β2γ2, α1(T47R)β2γ2 and α1(F64T)β2γ2 receptors, respectively. There was a statistically significant difference in peak current density (P = 0.003; one‐way ANOVA). A post hoc Tukey's comparison revealed significant reductions in mean current densities for α1(T47R)β2γ2 (P < 0.001) and α1(F64T)β2γ2 receptors (P < 0.05), when compared with α1β2γ2 receptors. We have previously reported a reduction in current density for α1(D43C)β2γ2 and α1(F64C)β2γ2 receptors, compared to WT receptors (Baptista‐Hon et al. 2016).
The identities of residues in loop G at positions 43 and 47 influence agonist efficacy
Apparent potencies, determined from concentration–response relationships, are composites of agonist binding affinity and gating efficacy (Colquhoun, 1998). Therefore, reductions in EC50 values for α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors compared to WT could be caused by reduced GABA affinity and/or efficacy. We investigated the possibility that D43C and T47R substitutions might impair the efficacy of GABA using propofol, a general anaesthetic that acts as a positive allosteric modulator of GABAA receptors (Hales & Lambert, 1991). Co‐application of propofol (1 μm) with an EC50 concentration of GABA (10 μm) to HEK‐293 cells expressing α1β2γ2 receptors caused current amplitudes to increase by 62 ± 18% (n = 4), relative to currents evoked by GABA (10 μm) alone. The application of propofol (1 μm) alone had no effect (data not shown). We subsequently tested the ability of propofol (1 μm) to potentiate currents evoked by maximally efficacious concentrations of GABA mediated by α1β2γ2 (1 mm), α1(D43C)β2γ2 (100 mm), α1(T47R)β2γ2 (30 mm) and α1(F64C)β2γ2 receptors (300 mm). GABA concentrations were chosen based on the concentration–response relationship shown in Fig. 1 D for α1β2γ2 and α1(T47R)β2γ2 receptors, and our previously published concentration–response relationships for α1(D43C)β2γ2 and α1(F64C)β2γ2 receptors (Baptista‐Hon et al. 2016). Figure 2 A shows representative examples of maximal GABA‐evoked currents in the presence or absence of propofol. Co‐application of propofol with GABA to cells expressing α1β2γ2 receptors did not cause any potentiation, consistent with GABA having full efficacy. By contrast, recordings from cells expressing α1(D43C)β2γ2 or α1(T47R)β2γ2 receptors revealed that the co‐application of GABA with propofol increased current amplitudes relative to GABA alone (Fig. 2 A). This suggests that GABA lacks full efficacy at α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. The loop D α1(F64C) substitution is known to reduce the efficacy of GABA (Szczot et al. 2014). Consistent with this observation, we found that co‐application of GABA (300 mm) with propofol (1 μm) also increased the current amplitude relative to GABA (300 mm) alone at α1(F64C)β2γ2 receptors (Fig. 2 A). We quantified the effect of propofol on maximal GABA‐evoked currents (Fig. 2 B). The graph shows data expressed as mean percentage potentiation by propofol. Currents mediated by α1β2γ2 receptors were unaffected by propofol (−0.72 ± 1.6%; n = 5). By contrast, propofol (1 μm) potentiated maximal GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors by 19 ± 5% (n = 9), 23 ± 5% (n = 9) and 47 ± 8% (n = 4), respectively (Fig. 2 B). The differences in mean percentage potentiation were statistically significant (P < 0.0001; one‐way ANOVA). A post hoc Tukey's comparison revealed a statistically significant difference in propofol potentiation of maximal GABA‐evoked currents between α1β2γ2 and α1(D43C)β2γ2 (P < 0.05), α1(T47R)β2γ2 (P < 0.001) and α1(F64C)β2γ2 receptors (P < 0.0001). There was also a significant difference in propofol potentiation between α1(F64C)β2γ2 and either α1(D43C)β2γ2 (P < 0.001) or α1(T47R)β2γ2 receptors (P < 0.05). These data indicate that D43C and T47R substitutions impair the efficacy of GABA, albeit to a lesser extent than the F64C substitution.
Figure 2. The identity of Loop G and loop D residues influences agonist efficacy.
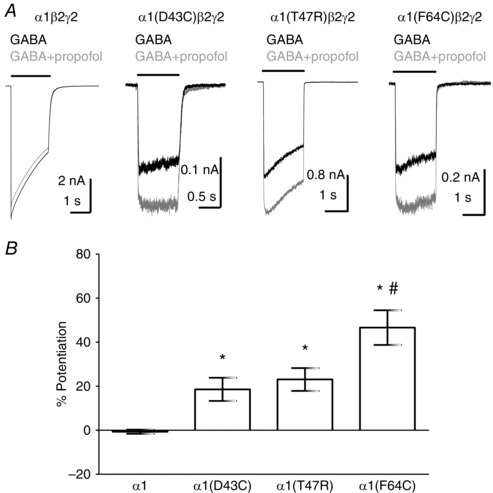
A, representative examples of whole‐cell currents mediated by α1β2γ2, α1(D43C)β2γ2, α1(T47R)β2γ2 or α1(F64C)β2γ2 receptors evoked by a maximal concentration of GABA alone (black traces) or in the presence of 1 μm propofol (grey traces). The concentration of GABA used was 1 mm for α1β2γ2, 300 mm for α1(D43C)β2γ2, 30 mm for α1(T47R)β2γ2 and 300 mm for α1(F64C)β2γ2 receptors. Propofol potentiated GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. B, bar graph showing mean percentage potentiation by propofol. Propofol significantly potentiated GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors (* P < 0.05, P < 0.001 and P < 0.0001, respectively; one‐way ANOVA post hoc Tukey's comparison). There was also a statistically significant difference between α1(F64C)β2γ2 receptors and α1(D43C)β2γ2 or α1(T47R)β2γ2 receptors (# P < 0.05 and P < 0.001, respectively; one‐way ANOVA post hoc Tukey's comparison).
We examined whether the reduced efficacy of GABA as an agonist at α1(D43C)β2γ2 or α1(T47R)β2γ2 was associated with reduced maximal open probabilities (P open) of GABA‐activated channels, as would be expected for mutations that affect gating. We calculated P open using non‐stationary variance analysis of maximal GABA‐evoked currents. This approach has been used previously to demonstrate that the P open of GABAA receptors containing the α1(F64C) substitution was reduced (Szczot et al. 2014). Currents evoked by brief (5 ms) rapid applications of maximally efficacious concentrations of GABA were recorded from outside‐out patches containing α1β2γ2, α1(D43C)β2γ2 or α1(T47R)β2γ2 receptors. A minimum of 10 consecutive GABA‐evoked currents were used to calculate mean current (I) and variance (σ2) for each time point. Representative examples of variance and mean current are shown for α1β2γ2 and α1(T47R)β2γ2 receptors (Fig. 3 A). The rising phases of the currents were not included in the analysis (grey portion in Fig. 3 A). A parabolic function was fitted to the plot of mean current versus variance to determine the single channel amplitude and the number of channels (Fig. 3 B). From these values, the single channel conductance (γ) and maximal P open were calculated (see Methods). Neither the α1(D43C) nor the α1(T47R) substitutions significantly altered γ (Fig. 3 C; P = 0.96; one‐way ANOVA). The mean maximum P open values for α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors were 0.78 ± 0.05 (n = 8), 0.50 ± 0.09 (n = 5) and 0.56 ± 0.05 (n = 7), respectively (Fig. 3 D). One‐way ANOVA revealed a statistically significant difference in mean maximal P open (P = 0.0083). A post hoc Tukey's comparison determined a statistically significant difference in mean maximal P open between α1β2γ2 and α1(D43C)β2γ2 receptors (P < 0.05), and between α1β2γ2 and α1(T47R)β2γ2 receptors (P < 0.05). These data agree well with the results of propofol potentiation experiments and indicate that the α1(D43C) and α1(T47R) substitutions reduce the efficacy of GABA by reducing the maximal P open of GABAA receptors to a similar extent.
Figure 3. Maximal P open is reduced in α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors.

A, representative examples of variance and mean current calculated from 10 consecutive GABA applications to α1β2γ2 or α1(T47R)β2γ2 receptors. The application of GABA is indicated by the liquid junction current above each trace. Only the variance and mean current values following the peak of the current (black) were used in the analysis. B, mean current versus variance plot for α1β2γ2 or α1(T47R)β2γ2 receptors. The dotted line represents the parabolic function fitted to the data points. C, bar graph of mean single channel conductances. The single channel conductances for α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors were 26 ± 3.2 pS (n = 8), 27 ± 2.9 pS (n = 7) and 26 ± 2.3 pS (n = 5), respectively. There was no significant difference between the means (P = 0.96; one‐way ANOVA). D, bar graph of mean maximal P open for α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. There was a statistically significant difference in P open between α1β2γ2 and α1(D43C)β2γ2 receptors and between α1β2γ2 and α1(T47R)β2γ2 receptors (* P < 0.05; one‐way ANOVA; post hoc Tukey's comparison).
Substitutions in loop G and loop D have common effects on GABA‐evoked gating kinetics
We used rapid GABA application to outside‐out patches excised from HEK‐293 cells to investigate the kinetics of macroscopic currents mediated by recombinant α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. Figure 4 A shows representative examples of currents evoked by maximal concentrations of GABA, recorded at −60 mV from patches containing α1β2γ2, α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. Macroscopic currents mediated by α1(F64T)β2γ2 receptors resemble those of α1(F64C)β2γ2 receptors and are not shown. Traces in Fig. 4 B are amplitude‐normalised rising phases mediated by α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. Current activation rates were quantified as 10–90% rise‐times. Mean activation times are plotted in the bar graph (Fig. 4 B). Comparison of the mean rise‐times reveals a statistically significant difference between these receptor subtypes (P < 0.0001; one‐way ANOVA; Fig. 4 B; Table 1). Currents mediated by both α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors were activated more slowly than α1β2γ2 (P < 0.0001) and also when compared with α1(D43C)β2γ2 (P < 0.0001) and α1(T47R)β2γ2 receptors (P < 0.0001, post hoc Tukey's comparison). The activation rates between α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors did not differ significantly. However, there was a trend towards a slower activation rate for both loop G substitutions.
Figure 4. The kinetics of GABA‐evoked currents mediated by α1β2γ2, α1(D43C)β2γ2, α1(T47R)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors.
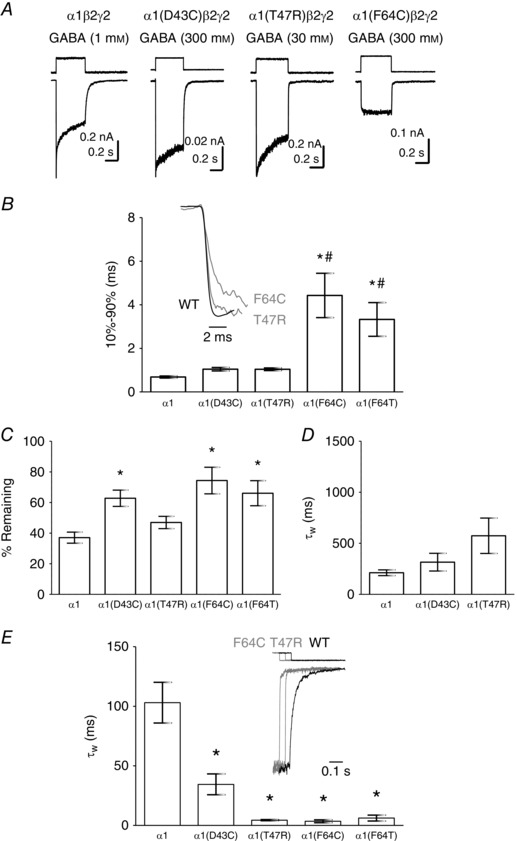
A, representative examples of GABA‐evoked currents mediated by α1β2γ2, α1(D43C)β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors recorded from outside‐out patches. The associated square pulses indicate the junction currents corresponding to agonist application. B, (inset) representative examples of amplitude‐normalised activation phases of GABA‐evoked currents mediated by α1β2γ2 (black), α1(T47R)β2γ2 (grey) or α1(F64C)β2γ2 (grey) receptors. Bar graph shows mean 10–90% rise time of current activation. Both α1(F64C) and α1(F64T) substitutions significantly slowed the GABA activation rate when compared with α1β2γ2 receptors (* P < 0.0001; one‐way ANOVA post hoc Tukey's comparison) and with α1(D43C)β2γ2 or α1(T47R)β2γ2 receptors (# P < 0.0001; one‐way ANOVA post hoc Tukey's comparison). C, bar graph shows the mean percentage current remaining. GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors desensitise significantly less, when compared with α1β2γ2 receptors (* P < 0.05; P < 0.001 and P < 0.001, respectively; one‐way ANOVA post hoc Tukey's comparison). D, mean desensitisation τw of α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. There was no statistically significant difference in mean τw (P = 0.08; one‐way ANOVA). The individual components of the multi‐exponential fit are summarised in Table 1. E, (inset) representative examples of amplitude‐normalised current deactivation following agonist removal. The superimposed traces are time‐shifted for clarity. The step above each trace indicates the liquid junction current corresponding to agonist removal. The bar graph shows mean deactivation τw. GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47R)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors deactivated significantly faster when compared to those mediated by α1β2γ2 receptors (* P < 0.0001; one‐way ANOVA post hoc Tukey's comparison). The individual components of the multi‐exponential fits are summarised in Table 2.
Table 1.
Summary of desensitisation components
| Agonist | τf | %f | τm | %m | τs | %s | τw | |
|---|---|---|---|---|---|---|---|---|
| α1β2γ2 | GABA | 3.2 ± 0.34 | 27 ± 3.0 | 36 ± 6.3 | 30 ± 6.0 | 410 ± 49 | 43 ± 4.5 | 190 ± 25 |
| THIP | 4.6 | 7.5 ± 5.8# | 33 ± 5.9 | 31 ± 5.0 | 350 ± 34 | 61 ± 6.5# | 240 ± 35 | |
| α1(D43C)β2γ2 | GABA | 6.7 ± 1.9* | 21 ± 6.1 | 42 ± 7.0 | 11 ± 4.3 | 460 ± 110 | 68 ± 7.0* | 320 ± 87 |
| α1(T47R)β2γ2 | GABA | 4.6 ± 0.94 | 13 ± 3.7* | 50 ± 7.7 | 18 ± 3.7 | 790 ± 220 | 69 ± 4.6* | 570 ± 170 |
# P < 0.05 (unpaired t‐test) compared with the equivalent WT GABA‐evoked current value.
* P < 0.05 (one‐way ANOVA post hoc Tukey's comparison) compared with GABA‐evoked currents mediated by α1β2γ2 receptors.
GABA‐evoked currents decay in the continued presence of the agonist through the process of desensitisation. We quantified desensitisation by measuring current remaining at the end of GABA application and expressing it as a percentage of peak current amplitude. The averaged values are plotted in Fig. 4 C. Comparison of these values revealed a significant difference between the receptor subtypes (P = 0.0003; one‐way ANOVA; Fig. 4 C). GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors desensitised significantly less than those mediated by α1β2γ2 receptors (P < 0.001, post hoc Tukey's comparison). The extent of desensitisation did not significantly differ between α1β2γ2 and α1(T47R)β2γ2 receptors.
It was possible to fit exponential functions to evaluate the apparent desensitisation rates for α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. An F‐test (see Methods) revealed that a three‐component exponential function consistently yielded the best fit to the desensitising current. The components of the multi‐exponential function were used to calculate τw (see Methods) and the mean values of τw are plotted in Fig. 4 D. There was no significant difference in mean τw values (P = 0.08; one‐way ANOVA), although visual inspection of the exemplar traces in Fig. 4 A shows apparent differences in the time course of desensitisation between α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. Indeed, analysis of the individual components of desensitisation reveals significant differences (Table 1). The α1(D43C) and α1(T47R) substitutions influence the fastest component of apparent desensitisation by reducing the rate and the percentage, respectively. There was also an increase in the contribution of the slowest time constant to apparent desensitisation for α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. The intermediate component was not affected by the substitutions. These analyses indicate that the loop G α1(D43C) and α1(T47R) substitutions cause similar changes in desensitisation kinetics.
Traces in Fig. 4 E show amplitude‐normalised deactivation time courses of α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. The deactivation traces for α1(D43C)β2γ2, α1(T47R)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors are indistinguishable from each other. Therefore, for clarity, examples of α1(D43C)β2γ2 and α1(F64T)β2γ2 receptors are not shown. An F‐test revealed that a three‐component exponential function best describes the deactivation time course in α1β2γ2, α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors. For α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors, the slowest component was lost and the deactivation time courses were best described with a two‐exponential function. Comparison of the mean τw values for deactivation of GABA‐evoked currents mediated by the different receptor subtypes revealed a statistically significant difference (P < 0.0001; one‐way ANOVA; Fig. 4 E; Table 2). There were significant differences between the τw values for α1β2γ2 and α1(D43C)β2γ2, α1(T47R)β2γ2, α1(F64C)β2γ2 and α1(F64T)β2γ2 receptors (all P < 0.0001, post hoc Tukey's comparison). The individual components of the multi‐exponential fits are summarised in Table 2. Macroscopic currents mediated by GABAA receptors containing the loop D α1(F64C) and α1(F64T) substitutions were more severely affected, in terms of activation rates and extent of apparent desensitisation, than were those containing the α1(D43C) and α1(T47R) substitutions. By contrast, deactivation rates were similarly affected by all substitutions in loops G and D.
Table 2.
Summary of deactivation components
| Agonist | τf | %f | τm | %m | τs | %s | τw | |
|---|---|---|---|---|---|---|---|---|
| α1β2γ2 | GABA | 11 ± 2.0 | 8.1 ± 3.7 | 47 ± 5.6 | 61 ± 5.2 | 270 ± 54 | 30 ± 5.0 | 100 ± 17 |
| THIP | 3.2 ± 0.89# | 40 ± 8.4# | 12 ± 1.7# | 61 ± 7.1 | 68 ± 13# | 6.1 ± 1.5# | 12 ± 1.4# | |
| α1(D43C)β2γ2 | GABA | 7.9 ± 1.8 | 64 ± 3.1* | 45 ± 12 | 28 ± 2.6* | 280 ± 110 | 7.5 ± 2.6* | 34 ± 8.7* |
| α1(T47R)β2γ2 | GABA | 0.91 ± 0.094* | 73 ± 5.4* | 8.4 ± 2.2 | 25 ± 5.3* | 140 ± 27 | 2.3 ± 0.32* | 4.4 ± 0.47* |
| THIP | 0.93 ± 0.076 | 93 ± 1.6# | 20 ± 4.5# | 7.0 ± 1.6# | NA | NA | 2.2 ± 0.46# | |
| α1(F64C)β2γ2 | GABA | 1.9 ± 0.62* | 91 ± 5.4* | 42 ± 15 | 8.5 ± 5.4* | NA | NA | 3.5 ± 1.3* |
| α1(F64T)β2γ2 | GABA | 0.83 ± 0.071* | 93 ± 1.5* | 72 ± 24 | 6.6 ± 1.5* | NA | NA | 6.2 ± 2.5* |
# P < 0.05 (unpaired t‐test) compared with the equivalent GABA‐evoked current value.
* P < 0.05 (one‐way ANOVA post hoc Tukey's comparison) compared with GABA‐evoked currents mediated by α1β2γ2 receptors.
NA, not applicable due to a loss of this component.
Kinetics of currents activated by the partial agonist THIP
In the preceding investigation of GABA efficacy and gating kinetics, the loop G α1(D43C) and α1(T47R) substitutions had similar effects. We therefore restricted our subsequent analysis to the α1(T47R) substitution using the loop D α1(F64C) substitution as a comparator.
Propofol potentiation and maximal P open data suggest that the efficacy of GABA is impaired by non‐conservative substitutions in loop G. These changes in efficacy were associated with dramatically increased deactivation rates and changes in desensitisation kinetics. We investigated currents evoked by THIP, a partial agonist at WT GABAA α1β2γ2 receptors, to determine whether these are kinetic hallmarks of partial agonism.
We verified that THIP is a partial agonist when applied rapidly (see Methods) to excised outside‐out patches containing α1β2γ2 receptors. Maximally effective concentrations of GABA and THIP were chosen on the basis of concentration–response relationships (Fig. 1 D) and the observation that higher agonist concentrations did not further increase current amplitude when applied rapidly to outside‐out patches (data not shown). Figure 5 A shows representative examples of maximally effective GABA‐ and THIP‐evoked currents recorded from outside‐out patches containing α1β2γ2 and α1(T47R)β2γ2 receptors. THIP‐evoked currents mediated by α1β2γ2 receptors were smaller than those activated by GABA (Fig. 5 B). The exemplar data reveal a further reduction in the relative THIP‐evoked current amplitude mediated by α1(T47R)β2γ2 receptors, consistent with our data above that this non‐conservative substitution reduces agonist efficacy. THIP did not evoke measurable currents at α1(F64C)β2γ2 receptors (data not shown). The relative efficacy of THIP at α1β2γ2 receptors was on average 77 ± 5% of that of GABA (n = 5; Fig. 5 B). The relative efficacy of THIP as an agonist of α1(T47R)β2γ2 receptors was reduced to 17 ± 3% of that of GABA (n = 6). The reduction in THIP efficacy at α1(T47R)β2γ2 receptors was statistically significant (P = 0.0001; t‐test; Fig. 5 B).
Figure 5. The kinetics of GABA‐ and THIP‐evoked currents mediated by α1β2γ2 or α1(T47R)β2γ2 receptors.
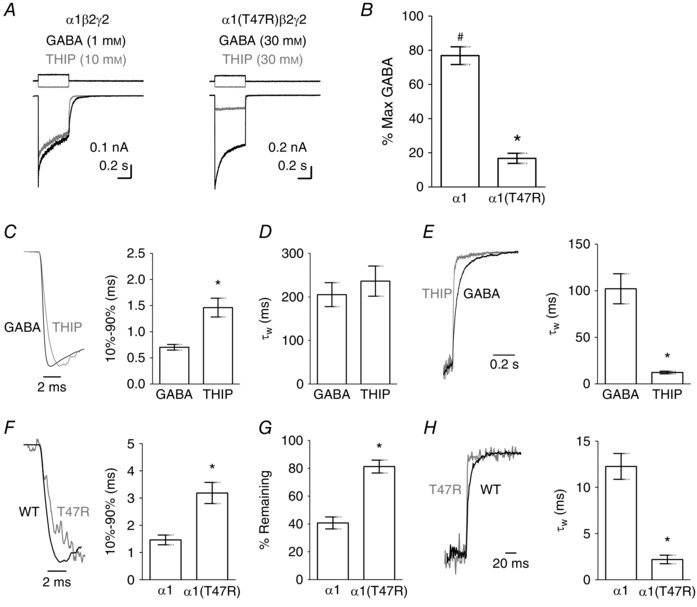
A, representative examples of maximal GABA‐ (black) and THIP‐evoked (grey) currents recorded from excised outside‐out patches containing α1β2γ2 or α1(T47R)β2γ2 receptors. The upward (black) and downward (grey) steps above each correspond to the liquid junction current for GABA and THIP application, respectively. B, the mean maximum THIP‐evoked current amplitude (as a percentage of maximal GABA current amplitude) was significantly less than the maximum GABA‐evoked current amplitude at α1β2γ2 receptors (# P = 0.002; paired t‐test). The efficacy of THIP was significantly reduced at α1(T47R)β2γ2 receptors (* P = 0.0001; t‐test). C, representative examples of the activation phases of GABA‐ (black) and THIP‐evoked (grey) currents recorded from excised outside‐out patches containing α1β2γ2 receptors. The associated graph illustrates the mean 10–90% rise times. THIP‐evoked currents activated significantly slower than GABA‐evoked currents (* P = 0.0001; t‐test). D, bar graph shows mean desensitisation τw. E, representative examples show amplitude‐normalised deactivation phases of GABA‐ (black) and THIP‐evoked (grey) currents. Bar graph shows mean deactivation τw. THIP‐evoked currents deactivated significantly faster than GABA‐evoked currents (* P = 0.0001; t‐test). F, representative activation phases of THIP‐evoked currents from outside‐out patches containing α1β2γ2 (black) and α1(T47R)β2γ2 (grey) receptors. Bar graph shows mean 10–90% rise times. The α1(T47R) substitution significantly slowed the activation rates of THIP‐evoked currents, as compared to WT receptors (* P = 0.002; t‐test). G, bar graph shows mean percentage current remaining. THIP‐evoked currents mediated by α1(T47R)β2γ2 receptors show less desensitisation as compared to α1β2γ2 receptors (* P = 0.0001; t‐test). H, representative examples of amplitude‐normalised deactivation phases of THIP‐evoked currents mediated by α1β2γ2 (black) and α1(T47R)β2γ2 (grey) receptors. Bar graph shows mean deactivation τw. The α1(T47R) substitution significantly increased deactivation rate (* P = 0.0001; t‐test).
Inspection of α1β2γ2 receptor‐mediated currents reveals that those activated by THIP and GABA exhibit differing kinetics (Fig. 5 A). The normalised current traces in Fig. 5 C illustrate the rising phases of GABA‐ and THIP‐evoked currents mediated by WT receptors. We compared their activation rates by measuring the time for currents to increase from 10 to 90% of their peaks (Fig. 5 C). THIP‐evoked currents activated significantly more slowly than did GABA‐evoked currents (P < 0.0001; t‐test).
The apparent desensitisation time course for THIP‐evoked currents mediated by α1β2γ2 receptors was best described using a three‐component exponential function. THIP‐evoked currents showed subtle differences in desensitisation kinetics compared with GABA‐evoked currents. There was no statistically significant difference in the τw between GABA‐evoked and THIP‐evoked currents (Fig. 5 D; P = 0.29; t‐test). The individual time constants and their relative proportions are summarised in Table 1. There was a significant reduction in the proportion of the fastest desensitisation component, and a concurrent significant increase in the slowest component. The effect of the partial agonist on desensitisation time course at α1β2γ2 receptors was similar to those caused by the loop G α1(D43C) and α1(T47R) substitutions, which impaired efficacy.
Traces in Fig. 5 E (normalised to the amplitude at the end of a 500 ms GABA application) are examples of GABA‐ and THIP‐evoked current deactivation mediated by WT receptors. The deactivation time course for THIP‐evoked currents were best described using a triple exponential function, similar to GABA‐evoked currents. Comparison of mean τw values reveal that THIP‐evoked currents deactivate significantly faster than GABA‐evoked currents (P = 0.0008, t‐test; Fig. 5 E). The individual components of the deactivation kinetics are summarised in Table 2. The increase in the τw of deactivation can be attributed to a significant reduction in all three time constants and a shift in the proportion from the slowest to the fastest component. These data agree well with our findings above that partial agonism is associated with an increase in deactivation rate, but also, to a lesser extent, with slower activation and desensitisation kinetics. These findings support a previous report of kinetics associated with partial efficacy using single‐channel recording (Mortensen et al. 2004) and imply that slow activation, slow desensitisation and fast deactivation are associated with weaker agonists.
In the presence of the α1(T47R) substitution, the efficacy of THIP is further reduced relative to GABA (Fig. 5 A). We therefore compared the macroscopic kinetics of THIP‐evoked currents mediated by α1β2γ2 and α1(T47R)β2γ2 receptors. The current traces in Fig. 5 F show amplitude‐normalised rising phases of THIP‐evoked currents mediated by α1β2γ2 and α1(T47R)β2γ2 receptors. The bar graph shows mean 10–90% rise times. THIP‐evoked currents mediated by α1(T47R)β2γ2 receptors were activated significantly more slowly than those mediated by α1β2γ2 receptors (P = 0.002; t‐test; Fig. 5 F; Table 1).
Currents mediated by α1(T47R)β2γ2 receptors desensitised too slowly to allow fitting with an exponential function (see Fig. 5 A). Therefore, we examined desensitisation by measuring the current remaining at the end of the THIP application as a percentage of that at its peak. This approach revealed a significant reduction in the extent of THIP‐evoked current desensitisation mediated by α1(T47R)β2γ2 receptors compared to those mediated by α1β2γ2 receptors (P = 0.0001; t‐test; Fig. 5 G; Table 1).
The current traces in Fig. 5 H show amplitude‐normalised deactivation phases of THIP‐evoked currents at α1β2γ2 and α1(T47R)β2γ2 receptors. The deactivation of THIP‐evoked currents mediated by α1(T47R)β2γ2 receptors was consistently best described using two exponential terms, compared to three terms necessary to adequately describe deactivation mediated by α1β2γ2 receptors. Analysis of the mean τw values reveals a significant reduction of the τw values for the activation of THIP‐evoked currents mediated by α1(T47R)β2γ2 receptors compared to WT receptors (P = 0.0001; t‐test; Fig. 3 G; Table 2). There was a significant reduction in the fastest time constant, as well as a significant increase in its proportion. As mentioned above, the slowest deactivation time constant present in recordings of THIP‐evoked currents mediated by WT receptors was not seen in recording of currents mediated by α1(T47R)β2γ2 receptors. These data agree well with the other macroscopic current kinetic data showing progressively increasing effects on activation and deactivation rates with reductions in agonist efficacy.
Deactivation kinetics of propofol‐evoked currents
It is possible that the changes in current kinetics quantified above (slow activation, slow desensitisation and fast deactivation), caused by mutations in the vicinity of the orthosteric binding site, are composites of altered rates of agonist association and dissociation, agonist efficacy and desensitisation. This might be expected particularly for the α1(F64C) mutation, which affects agonist binding. To examine the effect of amino acid substitutions in loop G and loop D on receptor gating, independent of orthosteric agonist binding, we examined the kinetics of currents activated by the allosteric agonist propofol mediated by α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. The concentration of propofol used, which evoked robust currents without evidence of block, was either 30 or 100 μm, and did not differ between the receptor subtypes, suggesting that the potency for propofol was not dramatically affected by the amino acid substitutions. The activation time course of propofol‐ (30 or 100 μm) evoked currents mediated by α1β2γ2 receptors was independent of concentration (data not shown).
Figure 6 A shows representative examples of propofol‐evoked currents recorded from outside‐out patches containing α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. The current traces in Fig. 6 B are amplitude‐normalised rising phases of propofol‐evoked currents mediated by WT and mutant receptors. We measured activation rate as the 10–90% rise time. The means are plotted in the bar graph (Fig. 6 B). The α1(T47R) and α1(F64C) substitutions did not significantly affect the 10–90% rise time of propofol‐evoked currents (P = 0.51; one‐way ANOVA).
Figure 6. The kinetics of propofol‐evoked currents mediated by α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors.
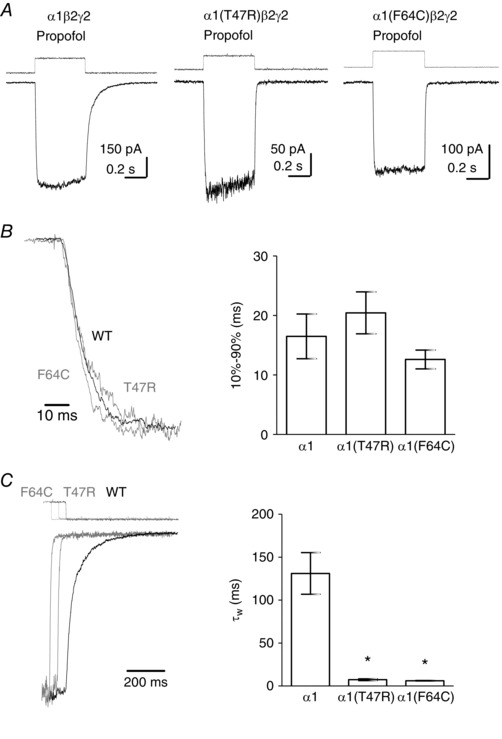
A, representative examples of propofol‐evoked currents mediated by α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors recorded from excised outside‐out patches. The square pulse above each trace indicates the time‐course of solution exchange. B, representative examples of amplitude‐normalised activation phases of propofol‐evoked currents mediated by α1β2γ2 (black), α1(T47R)β2γ2 (grey) and α1(F64C)β2γ2 (grey) receptors. The bar graph shows the mean 10–90% rise time of current activation. There were no statistically significant differences in the activation rates of propofol‐evoked currents. C, representative examples of amplitude‐normalised decaying phases of the currents depicted in A, following propofol removal. The superimposed traces are time‐shifted for clarity. The step above each trace indicates the liquid junction current corresponding to agonist removal. The bar graph shows mean deactivation τw. Propofol‐evoked currents mediated by α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors deactivated significantly faster when compared to those mediated by α1β2γ2 receptors (* P < 0.0001 for both; one‐way ANOVA post hoc Tukey's comparison). Mean kinetic parameters are summarised in Table 1.
Propofol‐ (30 or 100 μm) evoked currents did not exhibit measurable apparent desensitisation and therefore this parameter was not analysed (Fig. 6 A). The traces in Fig. 6 C are amplitude‐normalised deactivation phases of propofol‐evoked currents mediated by α1β2γ2, α1(T47R)β2γ2 and α1(F64C)β2γ2 receptors. A double exponential best describes the deactivation time course and the mean τw is plotted in the bar graph (Fig. 6 C). Comparison of the τw values for deactivation revealed significant differences (one‐way ANOVA) between α1β2γ2, α1(T47R)β2γ2 (P < 0.0001) and α1(F64C)β2γ2 (P < 0.0001, post hoc Tukey's comparison). The τw of deactivation did not differ between different mutant receptors. These data demonstrate that the deactivation rate for the allosteric agonist propofol is affected by the T47R and F64C substitutions, suggesting that loop D Phe64 and loop G Thr47 are involved in gating of α1β2γ2 GABAA receptors independent of occupation of the orthosteric binding site.
α1 subunit T47R reduces spontaneous gating
Our observation that propofol‐evoked currents were influenced by the α1(T47R) substitution suggests that loop G Thr47 influences gating regardless of the occupancy of the orthosteric binding site. To examine the influence of the α1(T47R) and α1(F64C) substitutions on GABAA receptor gating independent of any agonist activation, we studied their effects on GABAA receptors that display enhanced spontaneous gating. Leu285 is a conserved residue in all β‐subunits located in TM3. The GABAA β1(L285R) substitution is associated with alcohol preference and enhanced spontaneous gating (Anstee et al. 2013).
We first determined whether GABAA α1β2(L285R)γ2 receptors also displayed enhanced spontaneous gating by applying picrotoxin (100 μm) to block spontaneous currents recorded in the absence of agonist (I Spont). Maximal GABA‐evoked currents (I GABA) were then recorded. We quantified the extent of spontaneous gating as the percentage of I Spont to I GABA (I Spont/I GABA). The β2(L285R) substitution conferred enhanced I Spont (Fig. 7). We investigated the effect of the α1(T47R) and α1(F64C) substitutions on these currents. However, GABA failed to activate α1(F64C)β2(L285R)γ2 receptors, while picrotoxin inhibited standing currents (n = 3; data not shown). Therefore, our subsequent experiments were restricted to the α1(T47R) substitution. Figure 7 A shows representative examples of I Spont (grey traces) and I GABA (black traces) mediated by α1β2γ2, α1β2(L285R)γ2 and α1(T47R)β2(L285R)γ2 receptors. The presence of the α1(T47R) substitution reduced the amplitude of I Spont relative to I GABA (Fig. 7 A). The mean extent of spontaneous gating is plotted in Fig. 7 B. WT α1β2γ2 receptors had negligible I Spont/I GABA (0.0008 ± 0.0002%; n = 6). By contrast, α1β2(L285R)γ2 receptors had enhanced I Spont/I GABA (65 ± 12%; n = 4) and that of α1(T47R)β2(L285R)γ2 receptors was only 12 ± 5% (n = 4). A one‐way ANOVA revealed a statistically significant difference in mean I Spont/I GABA (P < 0.0001). A post hoc Tukey's comparison revealed statistically significant differences between the mean I Spont/I GABA of α1β2γ2 and α1β2(L285R)γ2 receptors (P < 0.0001), and between α1β2(L285R)γ2 and α1(T47R)β2(L285R)γ2 receptors (P < 0.0001). Our data therefore demonstrate that the β2(L285R) substitution confers greatly enhanced spontaneous activity, but the α1(T47R) substitution attenuated this effect. This demonstrates that the α1(T47R) substitution reduces the gating of GABAA α1β2γ2 receptors independent of any agonist occupancy.
Figure 7. Spontaneous gating mediated by α1β2γ2, α1β2(L285R)γ2 and α1(T47R)β2(L285R)γ2 receptors.
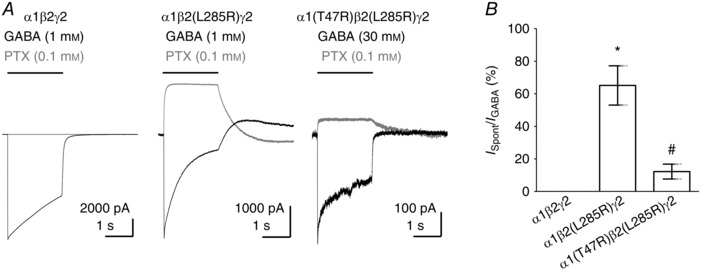
A, representative examples of GABA‐evoked currents (black traces) and inhibition of spontaneous currents by picrotoxin (grey traces) mediated by α1β2γ2, α1β2(L285R)γ2 and α1(T47R)β2(L285R)γ2 receptors. B, bar graph shows mean percentage spontaneous current (expressed as I Spont/I GABA). One‐way ANOVA revealed a statistically significant difference in mean spontaneous current (P < 0.0001). Mean spontaneous current amplitudes differed between α1β2γ2 and α1β2(L285R)γ2 receptors (* P < 0.0001) and between α1β2(L285R)γ2 and α1(T47R)β2(L285R)γ2 receptors (# P < 0.0001; post hoc Tukey's comparison).
Discussion
Data from this study and our previous study respectively demonstrate that two substitutions, T47R and D43C, at strategic locations in loop G reduce the apparent potency of GABA (Baptista‐Hon et al. 2016). Changes in apparent potency can be caused by reduced binding affinity and/or gating efficacy (Colquhoun, 1998). Both mechanisms contribute to the reduced apparent potency of GABA caused by the α1(F64C) loop D substitution (Szczot et al. 2014; Baptista‐Hon et al. 2016). Loop D located in the β2 strand is critical for agonist binding in the GABAA receptor and, consistent with this, modification of Cys64 by MTSEA is hindered by GABA, but not by the allosteric agonists, propofol and pentobarbital (Holden & Czajkowski, 2002; Baptista‐Hon et al. 2016). By contrast, we previously demonstrated that both GABA and propofol reduce the rate of MTSEA modification of Cys43 and Cys47 in α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors, respectively (Baptista‐Hon et al. 2016). This is consistent with movement associated with gating being the cause of reduced accessibility, rather than agonist binding. Movement of loop G during gating may be responsible for reduced accessibility to MTSEA. Furthermore, restriction of movement by non‐conservative substitutions of amino acids at positions 43 and 47 may impair gating, leading to the observed dextral shift in the GABA concentration–response relationship for α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors relative to WT.
We directly explored whether the α1(D43C) and α1(T47R) substitutions reduce GABA efficacy using propofol at a concentration below that required for activation, but sufficient for positive allosteric modulation of sub‐maximal GABA‐evoked currents mediated by WT receptors (Hales & Lambert, 1991). Propofol potentiated maximal GABA‐evoked currents mediated by both α1(D43C)β2γ2 and α1(T47R)β2γ2, but not WT receptors. This suggests that propofol restores the efficacy of GABA lost through the D43C and T47R substitutions in loop G. Using non‐stationary variance analysis, we also demonstrated that the maximal P open is reduced for α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors, without a change in single channel conductance, indicating that loop G does indeed play a role in efficacy. Using the same approach, it was demonstrated that the loop D α1(F64C) substitution also reduces maximal P open, albeit to a greater extent (Szczot et al. 2014). Our data showing that the magnitude of propofol potentiation of a maximally efficacious concentration of GABA was greater in α1(F64C)β2γ2 receptors, than in α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors, agree well with the relative impairments of P open associated with these substitutions. Furthermore, the efficacy of THIP was also reduced by loop G and loop D substitutions, providing additional evidence that orthosteric agonist efficacy is indeed impaired.
Mounting structural evidence suggests that the anti‐parallel β1 and β2 strands, which contain loop G and loop D, respectively, move during gating. Comparisons of apo‐ and ligand‐bound structures of the glycine receptor (Du et al. 2015) and the C. elegans GluCl (Althoff et al. 2014) reveal movements in the β1 and β2 strands. Molecular dynamic (MD) simulations of C. elegans GluCl deactivation show that the loop connecting β1 and β2 strands moves towards the TM2–3 loop (Calimet et al. 2013). This movement appears to precede conformational changes within the TM domains which lead to gating. Furthermore, mutations that prevent the electrostatic interaction between the β1–β2 loop and the TM2–3 loop in the GABAA α1 subunit reduce GABA efficacy (Kash et al. 2003). We have also previously demonstrated that substitution of the conserved α1 subunit TM2–3 Lys278 by methionine reduces the efficacy of GABA (Hales et al. 2006). These functional, structural and simulation data, together with our current findings, suggest that the movement of loop G residues is involved in a series of structural rearrangements required for normal channel gating.
GABA‐evoked currents mediated by α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors exhibit a dramatically increased deactivation rate compared to WT receptors. There were also changes to apparent desensitisation kinetics and a tendency towards slowed activation rate. α1(F64C) and α1(F64T) substitutions in loop D caused more substantial changes in macroscopic current kinetics, with activation, apparent desensitisation and deactivation rates all significantly affected. This is consistent with the dual binding and gating role played by Phe64 in loop D (Boileau et al. 1999; Szczot et al. 2014). These changes in kinetics, particularly the observed increase in deactivation rates, are consistent with those we have previously observed for the α1(K278M) substitution, at the TM2–3 loop location, far removed from the agonist binding site, which also impairs agonist efficacy (Hales et al. 2006; Othman et al. 2012). In this case faster deactivation reflected a reduced mean open time. Reduced mean open time may also account for the increase in deactivation rates for α1(D43C)β2γ2 and α1(T47R)β2γ2 receptors as both substitutions caused reduced P open compared to WT receptors.
Ligand binding experiments indicate that GABA cannot dissociate from homomeric ρ1 receptors in the open channel conformation (Chang & Weiss, 1999). Similarly, GABA becomes trapped in its binding sites on α1β3γ2 receptors and does not unbind until the channel closes (Bianchi & Macdonald, 2001). Therefore, the deactivation time course is largely dictated by the mean open time of the receptor. Our data demonstrate that THIP‐evoked currents deactivate faster than GABA‐evoked currents, consistent with single channel data demonstrating a shorter mean open time associated with activation by the partial agonist (Mortensen et al. 2004).
Our data indicate that the efficacy of THIP is severely impaired in the presence of the α1(T47R) substitution. It is worth noting that the macroscopic current kinetics of THIP‐evoked currents in α1(T47R)β2γ2 receptors begin to resemble those with the loop D α1(F64C) and α1(F64T) substitutions. This may suggest that as agonist efficacy becomes progressively impaired, there may be incremental changes to activation, apparent desensitisation and deactivation rates.
We have previously observed that reduced efficacy associated with the α1(K278M) substitution was not restricted to GABA but also generalised to activation by propofol (Hales et al. 2006). Consistent with a similar scenario, propofol‐evoked currents mediated by α1(T47R)β2γ2 receptors also deactivate faster than their WT counterparts, suggesting that the mean open times for propofol‐activated channels were also reduced by the α1(T47R) substitution. These data confirm that the α1(T47R) substitution impairs gating through a mechanism independent of orthosteric binding and suggest a common conformational rearrangement associated with activation by orthosteric and allosteric agonists.
We also examined the effect of α1(T47R) substitution on spontaneous gating to further explore the generality of its ability to impair gating. Our previous work demonstrates that WT GABAA receptors exhibit a low level of spontaneous gating that can be blocked by the non‐competitive inhibitor picrotoxin and the inverse agonist bicuculline (McCartney et al. 2007). The α1(K278M) substitution, which inhibits GABA and propofol efficacy, also reduces spontaneous gating (Othman et al. 2012).
To examine the effects of the loop G and loop D substitutions on gating that is independent of any agonist, we used the L285R substitution in the β2 subunit to enhance spontaneous gating. A previous study of β1(L285R) demonstrated that the substitution caused a large increase in GABA‐independent gating (Anstee et al. 2013). Consistent with this, α1β2(L285R)γ2 receptors also exhibited marked spontaneous currents that were inhibited by picrotoxin. Spontaneous gating was reduced in α1(T47R)β2(L285R)γ2 compared to α1β2(L285R)γ2 receptors confirming that, like α1(K278M), α1(T47R) impairs gating independently of agonist activation.
In summary, this study suggests that movement of loop G in the β1 strand of the GABAA receptor α1 subunit is involved in a conformational rearrangement associated with channel activation. The non‐conservative replacements of Asp43 and Thr47 reduce efficacy presumably by impeding movement of the entire β1–β2 antiparallel loop structure. The importance of this region of pLGICs in activation by orthosteric agonists might have been predicted by structural data and MD simulations (Calimet et al. 2013; Althoff et al. 2014; Du et al. 2015).
Kinetic models of glycine and nicotinic acetylcholine receptor gating incorporate pre‐open shut states, termed flip and prime, respectively (Burzomato et al. 2004; Mukhtasimova et al. 2009). The rates of transition into these states correlate with agonist efficacy (Lape et al. 2008). It is therefore not a surprise that the α1(F64C) substitution in loop D, which reduces the efficacy of GABA, also reduces the transition rate constants into the flip state (Szczot et al. 2014). It is possible that the D43C and T47R substitutions also hinder these pre‐open transitions. Perhaps more surprisingly, however, the T47R substitution also impairs gating that is independent of orthosteric (or indeed allosteric) agonist activation.
A requirement for the movement of loop G residues during agonist‐independent gating may provide a mechanism for the negative efficacy that is characteristic of inverse agonism. It is possible that bicuculline inhibits GABAA receptor gating, caused by either allosteric or spontaneous activation, by immobilising the anti‐parallel β1–β2 strand structure (McCartney et al. 2007). Additional structural studies will be required to test this hypothesis.
Additional Information
Competing interests
The authors declare no competing financial interests.
Author contributions
D.T.B‐H. and T.G.H. designed the study. D.T.B‐H. and S.G. acquired the data. D.T.B‐H., S.G. and T.G.H. analysed and interpreted the data. D.T.B‐H. and T.G.H. drafted the work and revised it critically for intellectual content. All authors approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy and integrity of the work are appropriately investigated and resolved. All authors qualify for authorship and only those who qualify for authorship are listed.
Funding
This work was supported by funding from Tenovus Scotland.
References
- Adodra S & Hales TG (1995). Potentiation, activation and blockade of GABAA receptors of clonal murine hypothalamic GT1‐7 neurones by propofol. Br J Pharmacol 115, 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoff T, Hibbs RE, Banerjee S & Gouaux E (2014). X‐ray structures of GluCl in apo states reveal a gating mechanism of Cys‐loop receptors. Nature 512, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstee QM, Knapp S, Maguire EP, Hosie AM, Thomas P, Mortensen M, Bhome R, Martinez A, Walker SE, Dixon CI, Ruparelia K, Montagnese S, Kuo YT, Herlihy A, Bell JD, Robinson I, Guerrini I, McQuillin A, Fisher EM, Ungless MA, Gurling HM, Morgan MY, Brown SD, Stephens DN, Belelli D, Lambert JJ, Smart TG & Thomas HC (2013). Mutations in the Gabrb1 gene promote alcohol consumption through increased tonic inhibition. Nat Commun 4, 2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista‐Hon DT, Deeb TZ, Lambert JJ, Peters JA & Hales TG (2013). The minimum M3‐M4 loop length of neurotransmitter‐activated pentameric receptors is critical for the structural integrity of cytoplasmic portals. J Biol Chem 288, 21558–21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista‐Hon DT, Krah A, Zachariae U & Hales TG (2016). A role for loop G in the β1 strand in GABA receptor activation. J Physiol 594, 5555–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT & Macdonald RL (2001). Agonist trapping by GABAA receptor channels. J Neurosci 21, 9083–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Evers AR, Davis AF & Czajkowski C (1999). Mapping the agonist binding site of the GABAA receptor: evidence for a beta‐strand. J Neurosci 19, 4847–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Newell JG & Czajkowski C (2002). GABAA receptor beta 2 Tyr97 and Leu99 line the GABA‐binding site. Insights into mechanisms of agonist and antagonist actions. J Biol Chem 277, 2931–2937. [DOI] [PubMed] [Google Scholar]
- Burzomato V, Beato M, Groot‐Kormelink PJ, Colquhoun D & Sivilotti LG (2004). Single‐channel behavior of heteromeric α1β glycine receptors: an attempt to detect a conformational change before the channel opens. J Neurosci 24, 10924–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calimet N, Simoes M, Changeux JP, Karplus M, Taly A & Cecchini M (2013). A gating mechanism of pentameric ligand‐gated ion channels. Proc Natl Acad Sci USA 110, E3987–E3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y & Weiss DS (1999). Channel opening locks agonist onto the GABAC receptor. Nature neuroscience 2, 219–225. [DOI] [PubMed] [Google Scholar]
- Colquhoun D (1998). Binding, gating, affinity and efficacy: the interpretation of structure–activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125, 924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ & Parker MW (2002). Anxiety over GABAA receptor structure relieved by AChBP. Trends Biochem Sci 27, 280–287. [DOI] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y & Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschen‐Ohm MP, Wagner DA & Jones MV (2011). Three arginines in the GABAA receptor binding pocket have distinct roles in the formation and stability of agonist‐ versus antagonist‐bound complexes. Mol Pharmacol 80, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Deeb TZ, Tang H, Bollan KA, King DP, Johnson SJ & Connolly CN (2006). An asymmetric contribution to γ‐aminobutyric type A receptor function of a conserved lysine within TM2‐3 of α1, β2, and γ2 subunits. J Biol Chem 281, 17034–17043. [DOI] [PubMed] [Google Scholar]
- Hales TG & Lambert JJ (1991). The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol 104, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman KL & Pease LR (2007). Gene splicing and mutagenesis by PCR‐driven overlap extension. Nat Protoc 2, 924–932. [DOI] [PubMed] [Google Scholar]
- Hibbs RE & Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle DJ & Macdonald RL (2003). β Subunit phosphorylation selectively increases fast desensitization and prolongs deactivation of α1β1γ2L and α1β3γ2L GABAA receptor currents. J Neurosci 23, 11698–11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JH & Czajkowski C (2002). Different residues in the GABAA receptor α1T60‐α1K70 region mediate GABA and SR‐95531 actions. J Biol Chem 277, 18785–18792. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR & Harrison NL (2003). Coupling of agonist binding to channel gating in the GABAA receptor. Nature 421, 272–275. [DOI] [PubMed] [Google Scholar]
- Lape R, Colquhoun D & Sivilotti LG (2008). On the nature of partial agonism in the nicotinic receptor superfamily. Nature 454, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney MR, Deeb TZ, Henderson TN & Hales TG (2007). Tonically active GABAA receptors in hippocampal pyramidal neurons exhibit constitutive GABA‐independent gating. Mol Pharmacol 71, 539–548. [DOI] [PubMed] [Google Scholar]
- Miller PS & Aricescu AR (2014). Crystal structure of a human GABA receptor. Nature 512, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Perez CL, Noviello CM & Hibbs RE (2016). X‐ray structure of the human α4β2 nicotinic receptor. Nature 538, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Kristiansen U, Ebert B, Frolund B, Krogsgaard‐Larsen P & Smart TG (2004). Activation of single heteromeric GABAA receptor ion channels by full and partial agonists. J Physiol 557, 389–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL & Sine SM (2009). Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 459, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell JG & Czajkowski C (2003). The GABAA receptor β1 subunit Pro174‐Asp191 segment is involved in GABA binding and channel gating. J Biol Chem 278, 13166–13172. [DOI] [PubMed] [Google Scholar]
- Othman NA, Gallacher M, Deeb TZ, Baptista‐Hon DT, Perry DC & Hales TG (2012). Influences on blockade by t‐butylbicyclo‐phosphoro‐thionate of GABAA receptor spontaneous gating, agonist activation and desensitization. J Physiol 590, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB & Olsen RW (1995). Functional domains of GABAA receptors. Trends Pharmacol Sci 16, 162–168. [DOI] [PubMed] [Google Scholar]
- Szczot M, Kisiel M, Czyzewska MM & Mozrzymas JW (2014). α1F64 Residue at GABAA receptor binding site is involved in gating by influencing the receptor flipping transitions. J Neurosci 34, 3193–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PN, Laha KT & Wagner DA (2011). A tight coupling between β2Y97 and β2F200 of the GABAA receptor mediates GABA binding. J Neurochem 119, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalan SP & Czajkowski C (2008). A conserved salt bridge critical for GABAA receptor function and loop C dynamics. Proc Natl Acad Sci USA 105, 13604–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C & Jones MV (2004). An arginine involved in GABA binding and unbinding but not gating of the GABAA receptor. J Neurosci 24, 2733–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ, McKernan RM & Wafford KA (1995). Structure and pharmacology of vertebrate GABAA receptor subtypes. Int Rev Neurobiol 38, 95–138. [DOI] [PubMed] [Google Scholar]