

Abstract
Staphylococcus aureus infections present a serious challenge to healthcare practitioners due to the emergence of resistance to numerous conventional antibiotics. Due to their unique mode of action, peptide nucleic acids are novel alternatives to traditional antibiotics to tackle the issue of bacterial multidrug resistance. In this study, we designed a peptide nucleic acid covalently conjugated to the HIV-TAT cell penetrating peptide (GRKKKRRQRRRYK) in order to target the RNA polymerase α subunit gene (rpoA) required for bacterial genes transcription. We explored the antimicrobial activity of the anti-rpoA construct (peptide nucleic acid-TAT) against methicillin-resistant S. aureus, vancomycin-intermediate S. aureus, vancomycin-resistant S. aureus, linezolid-resistant S. aureus, and methicillin-resistant S. epidermidis in pure culture, infected mammalian cell culture, and in an in vivo Caenorhabditis elegans infection model. The anti-rpoA construct led to a concentration-dependent inhibition of bacterial growth (at micromolar concentrations) in vitro and in both infected cell culture and in vivo in C. elegans. Moreover, rpoA gene silencing resulted in suppression of its message as well as reduced expression of two important methicillin-resistant S. aureus USA300 toxins (α-hemolysin and Panton-Valentine leukocidin). This study confirms that rpoA gene is a potential target for development of novel antisense therapeutics to treat infections caused by methicillin-resistant S. aureus.
Keywords: Peptide nucleic acid, antisense agent, MRSA, alpha-hemolysin, Panton-Valentine leukocidin, antimicrobials
Introduction
One of the most pressing current medical issues facing healthcare practitioners and researchers is the rapid emergence of bacterial pathogens exhibiting resistance to traditional antibiotics. Of these pathogens, methicillin-resistant Staphylococcus aureus (MRSA) has received considerable attention due to the diverse array of diseases it causes (ranging from superficial skin infections to invasive syndromes such as pneumonia and sepsis); MRSA was linked to nearly half of all fatalities in the USA attributed to infections caused by antibiotic-resistant bacteria in 2013.1 The morbidity and mortality associated with MRSA infections is due in large part to the bacteria's ability to secrete a large number of virulence factors including toxins that induce pores in the plasma membrane of host cells leading to leakage of intracellular contents and lysis.2,3 Though a large arsenal of traditional antibiotics was once capable of treating MRSA infections, resistance to these antibiotics (including agents of last resort such as vancomycin) has emerged indicating alternative therapeutic agents need to be explored in order to circumvent this challenge.4,5 One such alternative that has received more attention in recent years is the use of peptide nucleic acids (PNAs) to target and suppress essential genes in bacterial pathogens.
PNAs are nucleic acid analogues that are capable of forming strong and stable complexes with RNA and DNA thus permitting targeted inhibition of specific genes.6 One target of significant interest is bacterial RNA polymerase given it is highly conserved among bacterial species and is a key enzyme in gene transcription, directly interfering with its function is postulated to negatively impact bacterial viability.7 Recently, RNA polymerase has emerged as a promising target for development of antisense agents, including PNAs, against both Gram-negative and Gram-positive pathogens.8,9,10,11,12,13 However, one of the deficiencies of these specific PNAs is their limited uptake by host cells due to their high molecular weight and nonionic structure.8 To overcome this limitation, researchers have examined the impact of conjugating PNAs to different cell penetrating peptides (CPPs), in order to significantly enhance PNA delivery to host tissues.14
In this study, we designed a peptide nucleic acid covalently conjugated to the TAT CPP in order to target a specific region of the rpoA gene in drug-resistant staphylococci. The HIV-1 TAT-based peptide has been previously shown to potentiate the antisense effect of anti-gyrA PNA against Streptococcus pyogenes.15 We examined our anti-rpoA PNA-TAT construct's ability to disrupt growth of staphylococci (including MRSA) in pure culture, cell culture, and in vivo in a Caenorhabditis elegans model. Results garnered from this study confirm that the rpoA gene in MRSA is a promising antisense target and C. elegans AU37 is a suitable in vivo model to utilize for screening PNAs.
Results
PNA target site selection
The Basic Local Alignment Search Tool was utilized to perform sequence alignment of the rpoA 5′ terminal region across different Staphylococcus species (Supplementary Table S1). Based on the bioinformatics analysis, we set out to design a peptide nucleic acid covalently conjugated to HIV-1 TAT to target a specific conserved region with 100% identity to the rpoA gene's mRNA across different Staphylococcus species. The conserved region selected included the translation start codon and the 5′ terminal region, as this area is accessible for ribosome assembly and consistent success has been experienced targeting this particular region.16,17 To confirm the efficiency of the designed PNA, the sequence was analyzed using the OligoWalk program.
Antimicrobial activity of PNA in vitro
Minimum inhibitory concentration. The minimum inhibitory concentrations (MICs) of PNA-TAT, free PNA, and TAT CPP were examined against a panel of Staphylococcus species (Table 1) in pure culture. The MIC of PNA-TAT, required to inhibit growth of different drug-resistant Staphylococcus clinical isolates, were found to be in the range of 8 to 32 µmol/l (Table 2). The PNA demonstrated good activity against multiple clinical isolates of MRSA, particularly MRSA USA300-0114, a community-associated strain responsible for outbreaks of staphylococcal skin and soft-tissue infections in the USA.18 Similarly, bactericidal activity was observed in other important clinical MRSA isolates (USA100, USA500, USA700, and USA800) that exhibit resistance to various antibiotic classes including macrolides, aminoglycosides, lincosamides, and fluoroquinolones. In addition PNA demonstrated consistent activity against multidrug-resistant clinical isolates including vancomycin-intermediate S. aureus, vancomycin-resistant S. aureus, linezolid-resistant S. aureus, and methicillin-resistant S. epidermidis.
Table 1. Staphylococcal isolates used in this study.
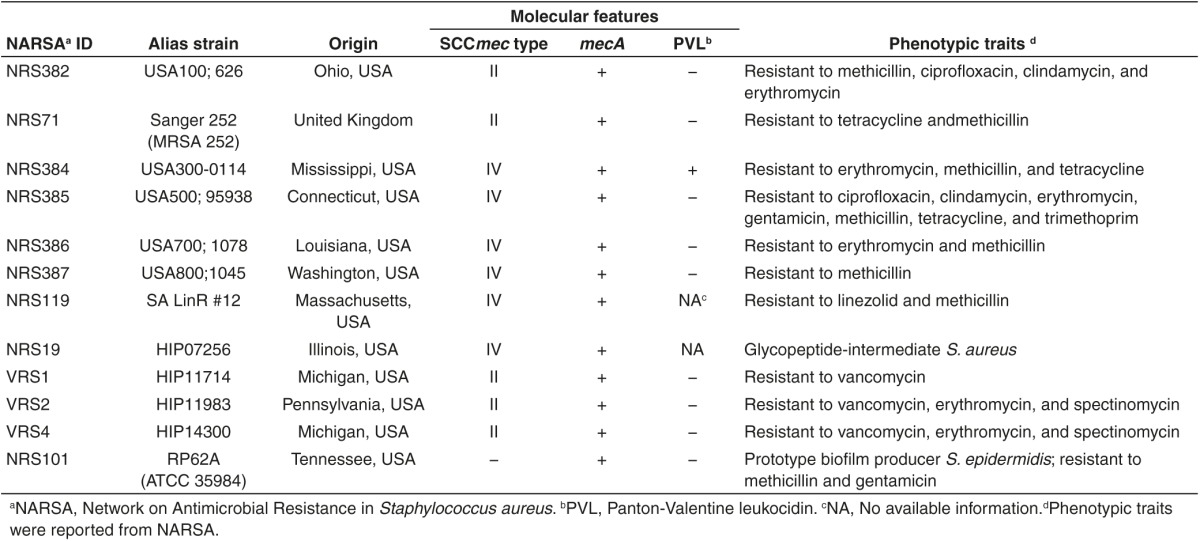
Table 2. Minimum inhibitory concentration (MIC) of conjugated PNA and TAT CPP against staphylococcal species.
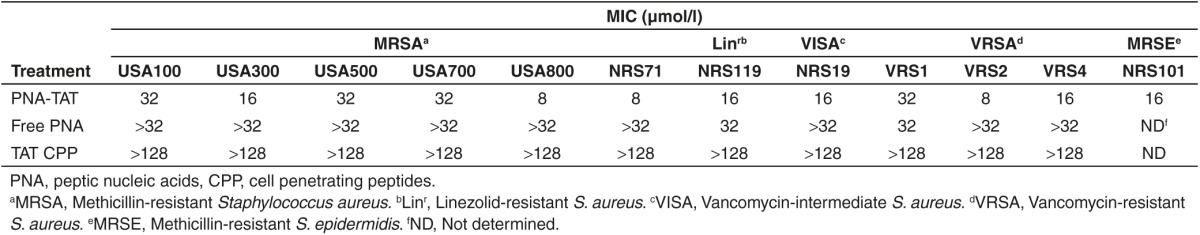
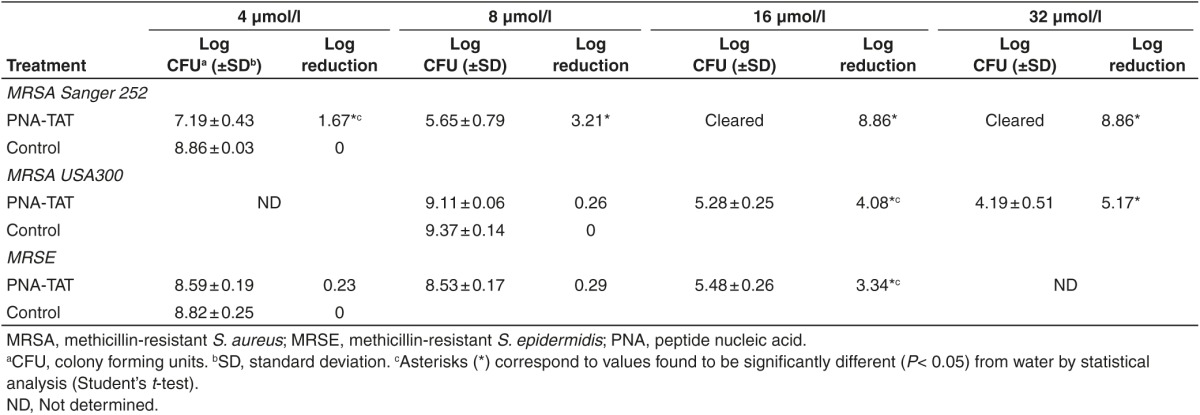
Bacterial reduction. Concentration-dependent bacterial reduction was determined against MRSA Sanger 252, MRSA USA300 and methicillin-resistant S. epidermidis. Against MRSA Sanger 252 cultures, the PNA-TAT cleared bacteria (produced an 8.86 log10 reduction) at the MIC (16 μmol/l); at 4 μmol/l and 8 μmol/l concentrations, the PNA-TAT produced a 1.67 and 3.21 log10 reduction, respectively (Table 3). With regards to MRSA USA300, significant bacterial reduction was achieved at the MIC value of 16 μmol/l (observed a 4.08 log10 reduction) and at 32 μmol/l (observed a 5.17 log10 reduction), respectively (Table 3). Additionally, at the MIC (16 µmol/l), significant bacterial reduction (3.34 log10 reduction) was observed for the PNA-TAT against methicillin-resistant S. epidermidis (Table 3).
Table 3. Effect of PNA-TAT on pure culture of MRSA Sanger 252, MRSA USA300, and MRSE.
PNA-TAT displays rapid bactericidal activity against MRSA
After confirming the antimicrobial activity of the designed PNA-TAT construct against different staphylococcal species, we investigated how rapidly the construct kills MRSA in pure culture. MRSA Sanger 252 was exposed to 16 μmol/l of the PNA, vancomycin, linezolid, and a control (water) over a 24 hours period. Samples were collected every 2 hours and the number of viable colony forming units (CFU) was determined. As presented in Figure 1, the PNA-TAT construct exhibits a bactericidal effect and completely eradicates a high starting inoculum of MRSA (~2.75 × 105 CFU/ml) within 6 hours. Vancomycin requires 24 hours to achieve the same effect. Linezolid, in contrast to PNA-TAT and vancomycin, exhibits bacteriostatic activity.
Figure 1.

Time-kill kinetics of PNA-TAT against MRSA Sanger 252. Anti-rpoA PNA–TAT at 16 μmol/l (tested in triplicate) was incubated for 24 hours at 37°C with shaking and samples were collected for every 2 hours, serially diluted, and plated onto tryptic soy agar. Viable colony forming units per ml were determined after incubation at 37°C for 16 hours. Vancomycin and linezolid (both at 16 μmol/l) were used as positive controls. The results are presented as mean ± SD from two independent experiments. PNA, peptide nucleic acid; MRSA, methicillin-resistant S. aureus.
PNA-TAT suppresses rpoA gene expression and expression of MRSA toxin genes
Real-time quantitative reverse-transcriptase polymerase chain reaction (PCR) was used to examine the impact of PNA on rpoA gene expression in MRSA USA300 and the subsequent suppression of gene expression for two important toxins, α-hemolysin (hla) and Panton-Valentine leukocidin (PVL). As depicted in Figure 2a PNA-TAT produced a 53.16% (at 8 μmol/l), 78.1% (at 16 μmol/l) and 86% (at 32 μmol/l) reduction in rpoA gene expression in MRSA USA300 indicating a concentration-dependent inhibition of gene expression. Interestingly, suppression of rpoA gene expression led to significant down regulation of the toxin genes hla and PVL in MRSA USA300 (Figure 2b,c). The suppression of toxins gene expression was found to be concentration-dependent (at 32 µmol/l, there was negligible expression of both hla and PVL).
Figure 2.

Dose-dependent down regulation of MRSA USA300 rpoA, hla, and pvl genes after incubation with anti-rpoA PNA-TAT. Bacterial cultures were maintained in tryptic soy broth with 8, 16, and 32 µmol/l PNA for 16 hours at 37°C with shaking. Total RNA was extracted from the treated and untreated cultures. The levels of transcripts were determined using RT-PCR. (a) The level of rpoA expression. (b) The level of hla (α-hemolysin) expression. (c) The level of pvl (Panton-Valentine leukocidin) expression. 16s rRNA was used as an internal control. *P ≤ 0.05 using two-tailed Student's t-test was considered as significant. PNA, peptide nucleic acid; MRSA, methicillin-resistant S. aureus; RT-PCR, reverse transcriptase polymerase chain reaction.
Anti-rpoA PNA-TAT constructs significantly reduces intracellular MRSA in infected cell culture
As PNA-TAT exhibited potent anti-MRSA activity against extracellular bacteria, we were interested to explore the ability of PNA-TAT to eliminate MRSA harboring inside macrophages. To examine the ability of anti-rpoA PNA-TAT to eradicate MRSA harboring inside host cells, murine macrophage J774 cells were initially infected with MRSA Sanger 252. Cells were subsequently exposed to either the PNA-TAT construct or vancomycin and the reduction in MRSA cells present inside the infected macrophages was determined. As presented in Figure 3, PNA-TAT outperformed vancomycin, eradicating 89% (at 8 μmol/l) and 93.1% (at 16 μmol/l) of intracellular MRSA, respectively. No reduction in MRSA CFU was observed with vancomycin at the same test concentrations. These findings suggest that PNA-TAT is a potential valuable treatment option for challenging MRSA infections (such as pneumonia) where MRSA resides inside host cells.
Figure 3.

Examining PNA-TAT's ability to eradicate MRSA Sanger 252 present inside infected murine macrophages (J774). J774 cells were incubated with ~2 × 106 CFU/ml of bacteria for 45 minutes. After removal of the extracellular bacteria, the infected cells were treated with anti-rpoA PNA – TAT (at 8 and 16 μmol/l) for 4 hours with 5% CO2. Vancomycin and sterile water were used as positive and negative controls. The results are presented as mean ± SD from two independent experiments. Data without error bars indicate that the SD is too small to be seen. *P ≤ 0.05 using two-tailed Student's t-test are deemed significant. PNA, peptide nucleic acid; MRSA, methicillin-resistant S. aureus; CFU, colony forming units.
Anti-rpoA PNA-TAT construct did not in vitro cytotoxicity analysis of rpoA-TAT against J774 cells
Using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium) (MTS) assay with murine macrophage J774 cells, it was confirmed that Anti-rpoA PNA-TAT construct was not toxic at a concentration of 16 μmol/l (Supplementary Figure S1).
Efficacy of PNA-TAT in vivo in a C. elegans animal model
In order to validate the in vitro results confirming PNA-TAT's anti-MRSA activity, the temperature-sensitive sterile mutant strain C. elegans AU37 (sek-1(km4); glp-4(bn2) I) was utilized as a whole animal model to assess the ability of the antisense construct to work in a living system. This strain is sterile at room temperature and capable of laying eggs only at 15°C. Additionally, this strain is more susceptible to infection due to a mutation in the sek-1 gene of the p38 mitogen-activated protein kinase pathway.19,20 Worms infected with MRSA Sanger 252 were treated with PNA-TAT at 1 × MIC, 2 × MIC, or 3 × MIC, vancomycin, or water for 18 hours. The reduction in MRSA CFU in infected worms was subsequently determined. In comparison to sterile water, PNA-TAT produced a concentration-dependent significant reduction in MRSA with no microscopically observed toxicity to the worms. A 30, 77.4, and 93.4% log10 MRSA CFU reduction was observed at 1 × MIC, 2 × MIC and 3 × MIC of PNA-TAT, respectively (Figure 4). A 65.1 and 74.2% reduction in MRSA CFU was observed in vancomycin-treated worms at concentrations = 2 × MIC and 3 × MIC, respectively.
Figure 4.

Efficiency of PNA-TAT in treatment of MRSA Sanger 252 infected C. elegans. L4-stage worms were grown on tryptic soy agar plates seeded with a lawn of bacteria for 2 hours. Worms were treated with anti-rpoA PNA-TAT at 1 × MIC, 2 × MIC or 3 × MIC for 18 hours using vancomycin and sterile water as controls. Worms were lysed, colony forming units (CFUs) were counted, and the percent bacterial reduction per worm in treated groups was calculated (relative to the untreated control groups). 10 worms (tested in triplicate) were used for each treatment. The results are presented as mean ± SD from two independent experiments. Data without error bars indicate that the SD is too small to be seen. *P ≤ 0.05 using two-tailed Student's t-test are deemed significant. PNA, peptide nucleic acid; MRSA, methicillin-resistant S. aureus.
PNA-TAT does not bind to the major component of human serum
Several antibiotics have limited use systemically due to their proficiency in binding to components of human serum, in particular the protein albumin. In order to examine if the antimicrobial activity of the PNA-TAT construct would be impaired in the presence of serum, MRSA Sanger 252 was incubated with 4 × MIC of PNA-TAT, daptomycin, or vancomycin in the presence and absence of 4% human serum albumin (HSA). As presented in Figure 5, daptomycin, an antibiotic known to bind strongly to HSA, is unable to inhibit MRSA growth in the presence of 4% HSA. In contrast, vancomycin's antimicrobial activity is unaffected by the presence of HSA. The PNA-TAT construct mimics the behavior of vancomycin (a slight decrease in bacterial inhibition from 99.99 (in HSA-free media, Figure 2a) to 94.34% (in HSA-supplemented media, Figure 2b) is observed)). The result indicates the construct's antimicrobial activity is not impacted by the presence of HSA.
Figure 5.

Impact of human serum albumin (HSA) on the efficacy of PNA-TAT inhibition of MRSA Sanger 252. Anti-rpoA PNA–TAT at 4 × MIC was incubated with ~5 × 105 CFU/ml in Mueller-Hinton broth II, in the presence and absence of 4% HSA. Daptomycin and vancomycin 4 × MIC served as positive and negative controls, respectively. The experiment was carried out in triplicate for 12 hours at 37 ºC. (a) MRSA Sanger 252 inhibition in the absence of HSA. (b) MRSA Sanger 252 inhibition in the presence of 4% HSA. The results are presented as mean ± SD. *P ≤ 0.05 using two-tailed Student's t-test are deemed significant. PNA, peptide nucleic acid; MRSA, methicillin-resistant S. aureus; MIC, minimum inhibitory concentration; CFU, colony forming units.
Discussion
MRSA infections continue to impose a significant problem both in the healthcare and community settings. MRSA has been associated with an array of different superficial and invasive diseases including skin infections, pneumonia, osteomyelitis, and sepsis.21,22,23,24 These infections impact a diverse patient demographic ranging from individuals with weakened immune systems (namely neonates and geriatric patients) to healthcare workers to healthy athletes.25,26,27 The challenge of treating patients infected with MRSA has been compounded in recent years with the rise in the pathogen's resistance to many conventional antibiotics, including agents of last resort such as vancomycin and linezolid.28,29 This points to a critical need to find alternative antibacterial agents that can be used to address the burden of MRSA infections.
One alternative to conventional antibiotics that has been recently explored by our research group and others is developing antisense agents (such as PNAs) to silence expression of essential genes in bacterial pathogens.8,9,10,11,12 This approach has been successfully employed in previous studies to target the RNA polymerase α subunit (rpoA) and RNA polymerase sigma 70 (rpoD) in both Salmonella enterica serovar Typhimurium and Listeria monocytogenes indicating the viability of these genes as novel targets for antimicrobial agents. In addition, MRSA RNA polymerase sigma 70 encoded by rpoD gene has been shown as a promising target for PNA inhibition.10,12 However, thus far, no published reports have examined the capability of direct inhibition of rpoA in MRSA using PNAs. One significant limitation of PNAs is their inability to cross cell membranes to attack their target inside the bacterial cytoplasm. This issue can be mitigated by conjugating the PNA to a suitable cell penetrating peptide. The aim of this study therefore was to examine the impact of conjugating the HIV-1 TAT cell penetrating peptide to a PNA designed to silence expression of the essential gene rpoA in MRSA.
We demonstrated that the presence of the TAT CPP did enhance the antimicrobial activity of the anti-rpoA PNA (the construct successfully inhibited MRSA growth at a concentration of 16 µmol/l). Additionally, using reverse transcriptase polymerase chain reaction, we confirmed that the PNA-TAT construct suppressed the expression of both rpoA and genes (hla and PVL) encoding for two key toxins, hla and PVL. These two toxins are responsible for promoting pathogenesis of MRSA infections and impairing the host immune response from clearing an infection. For example, PVL promotes lysis of leukocytes and is associated with more invasive forms of MRSA skin infections and community-acquired pneumonia.30 We surmise that the ability of the PNA-TAT to suppress the expression of hla and PVL will alleviate the morbidity associated with infections (such as necrotizing pneumonia) caused by PVL- and hla positive MRSA strains, such as MRSA USA300 and MRSA USA400. This would correlate with a previous report that demonstrated suppression of Hla and PVL toxins, by the antibiotic linezolid, improved the treatment outcome of rabbits infected with MRSA USA300.31
Confirmation of the anti-rpoA PNA-TAT construct's antimicrobial activity against MRSA in pure culture led us next to investigate if the presence of the CPP would permit the construct to clear an intracellular MRSA infection. Bacterial pathogens that harbor inside host cells pose a difficult challenge for both the host immune response and many traditional antibiotics to clear. For example, β-lactam antibiotics and aminoglycosides are not able to effectively penetrate host cells to clear intracellular pathogens.32,33 While antibiotics from the macrolide and fluoroquinolone classes are capable of penetrating host cells, they are “poorly retained” and thus limited in their ability to clear an infection.32,33 The inability of many traditional antibiotics to clear intracellular MRSA is postulated to contribute to the high rate of recurring infection present in the healthcare setting.34,35,36 Accordingly, treatment with conventional drugs of choice such as vancomycin and aminoglycosides is often associated with high clinical failures that exceeded 40% in intracellular MRSA infections due to poor intracellular penetration of drugs.37,38 Our investigation revealed the anti-rpoA PNA-TAT construct, at the same concentration as its MIC (16 µmol/l), was capable of eradicating more than 90% of intracellular MRSA present inside infected macrophages. This proved far superior to the antibiotic vancomycin (16 µmol/l), which was unable to kill MRSA harboring inside macrophages. The result indicates that the PNA-TAT construct possesses a selective advantage over antibiotics such as vancomycin in its ability to clear intracellular MRSA, thus potentially limiting the possibility of a recurring infection.
In vivo studies are critical to perform in order to validate in vitro results obtained for promising therapeutic agents. However, given the high cost of synthesizing and purifying a small quantity of PNA, it is very expensive to test PNAs in traditional animal models such as rodents. This requires the use of alternative animal models in order to examine the effect of PNAs on pathogens in vivo. C. elegans has been used as a whole animal model for more than four decades to examine critical questions in the field of biology; however, more recently, this model has gained traction as an alternative method to screen antimicrobial agents for suitable in vivo activity.39,40 Thus we postulate that C. elegans is a good animal model to use as a proof-of-concept to confirm the antimicrobial activity of promising PNAs in vivo prior to examining these agents in more complex animal models such as rodents. Utilizing the C. elegans AU37 strain, which is more sensitive to the effects of pathogens, we confirmed that the anti-rpoA PNA-TAT construct does possess potent antimicrobial activity in vivo. At 2 × MIC, the antisense construct reduced the presence of MRSA in infected worms by >75% (compared to 64% for vancomycin). Interestingly, at 3 × MIC, the anti-rpoA PNA-TAT construct eradicated more than 93% of MRSA in infected worms in contrast to vancomycin (which produced a 74% reduction). These results further support the notion that rpoA is a good target for developing novel anti-MRSA agents and the anti-rpoA PNA-TAT construct is a promising antisense agent that warrants further investigation.
Several antibiotics used to treat staphylococcal infections, such as daptomycin, rifampicin, and clindamycin, bind strongly to components in human serum, thus limiting the amount of free/unbound drug available to treat an infection.41,42,43 One limitation of using C. elegans as an animal model is the worm lacks plasma/blood (similar to vertebrate mammals) thus restricting a researcher's ability to examine the potential impact of PNA (or antimicrobial) binding to components of human serum.41,42 This limitation can be addressed by examining the ability of PNAs to inhibit bacterial growth in the presence of major components of human serum (such as the protein albumin). Utilizing this method, we demonstrated there was no significant impairment in the PNA-TAT construct's ability to inhibit MRSA Sanger 252 growth in the presence of a physiological concentration of HSA. Thus, the PNA-TAT construct does not appear to bind to HSA in a manner similar to daptomycin.
RNA polymerase plays a critical role in DNA transcription. The enzyme is composed of multiple subunits, including α subunit (encoded by rpoA). The α subunit helps to stabilize the transcription complex, helps with promoter recognition, functions to assemble the core enzyme, and regulates the initiation of gene transcription.44 Interfering with rpoA would be deleterious to the function of RNA polymerase as it would severely impair the functionality of the transcription complex. One of the negative consequences of this effect for bacteria would be decreasing the production of key virulence factors that promote MRSA pathogenesis (such as toxin production as demonstrated in this study). Furthermore, irreversible inhibition of RNA polymerase function (through inhibition of rpoA) in the target organism would ultimately lead to cell death.45 In addition, suppression of rpoA leads to destabilization of the RNA polymerase complex. Destabilization of this complex will directly interfere with transcription of genes to mRNA and subsequently translation to the effector protein. One of the key systems involved in expression of virulence factors (including toxin production) in S. aureus is the accessory gene regulation (agr) quorum sensing system. High expression of agr is suspected to be a main contributor to the virulence of the MRSA USA300 strain.46
Interestingly, agr encodes two divergent promoters, one of which directs the synthesis of the small regulatory molecule RNAIII.47 RNAIII is the effector for most genes of the agr regulon and RNAIII has been implicated in the increased production of many secreted S. aureus toxins.46,47,48 RNAIII regulates the expression of many mRNAs both at the transcription and translation level.49 For example, it has been shown that the 5′ domain of RNAIII is responsible for translation of hla mRNA to the effector protein.50 Thus, decreased transcription of hla and rnaIII (due to downregulation of rpoA) limits RNAIII's ability to promote hla mRNA translation to the toxin. Decreased expression of agr leads to decreased expression of virulence gene expression, namely toxin genes like hla, saeB (enterotoxin B), and tst (toxic shock syndrome toxin-1).51 This observation coincides with compounds isolated from Photobacterium halotolerans that inhibit the quorum sensing system in MRSA. Decreased expression of both rnaIII and hla were observed due to interference with the S. aureus agr quorum sensing system.46 We suspect that the decrease in expression of toxin genes observed from our reverse transcriptase PCR experiment is due directly to the suppression of rpoA gene expression. We postulate that suppression of rpoA expression leads to downregulation of key genes involved in virulence factor production including rnaIII and the agr quorum sensing system.
The multifaceted role of the RNA polymerase α subunit makes it a very good target for designing antisense agents for antibiotic-resistant pathogens. Few reports have discussed the promise of using PNAs as a novel therapeutic to target essential genes in MRSA. In addition to targeting rpoA in the RNA polymerase complex, the rpoD gene (encoding the RNA polymerase σ70 subunit) has been shown to be another viable target for designing antisense agents. Bai et al., constructed PNAs (against a conserved region for rpoD mRNA) covalently conjugated to the (KFF)3K -cell penetrating peptide. The most promising antisense construct (anti-rpoD PPNA2332) was able to inhibit growth of four MRSA strains at a concentration of 12.5 µmol/l.45 This is similar to the MIC obtained in our study with the anti-rpoA-TAT construct (MIC of 16 µmol/l). Substitution of the (KFF)3K CPP with an alternative peptide ((RXR)4XB) resulted in an improvement in anti-MRSA activity (growth inhibition observed at 6.25 µmol/l); this demonstrates the impact altering the CPP can have on the antibacterial effect of the conjugated PNA. However, one drawback of PNAs conjugated to the (RXR)4XB peptide is that, these agents can permeabilize the bacterial membrane resulting in nonspecific inhibition of growth (as was observed in Bai et al.'s study).45 The anti-rpoD PPNA2332 exhibited a concentration-dependent bactericidal activity against MRSA and inhibited rpoD expression in a similar concentration-dependent manner (with complete gene suppression observed at 40 µmol/l).
In addition to Bai et al.'s study, investigators have examined the promise of PNAs targeting other essential genes in S. aureus including fmhB (involved in cell wall synthesis) and gyrA (involved in DNA synthesis).52 As in Bai et al.'s study, the antisense constructs (which were also conjugated to the (KFF)3K CPP) designed against fmhB and gyrA exhibited a concentration-dependent bactericidal effect (with complete inhibition of bacterial growth observed at 10 µmol/l). A recent study conducted by Liang et al. further demonstrated the potential of using PNAs to target essential genes in MRSA.53 In this study, the authors designed PNAs (conjugated to the (RXR)4XB CPP) to target a critical gene (ftsZ) involved in bacterial cell division. Two antisense constructs, designated as PPNA1 and PPNA2, were able to inhibit growth of MRSA CY11 at 30 µmol/l and 40 µmol/l, respectively. Though both constructs were bactericidal against MRSA, only PPNA1, at 40 µmol/l, was able to completely eradicate a high inoculum of MRSA CY11 (105 CFU/ml) (in 6 hours). Our anti-rpoA-TAT construct was able to achieve the same effect, however at a much lower concentration (16 µmol/l).
This study confirms rpoA is a viable drug target for developing novel antibacterial agents for treatment of MRSA infections. The PNA-TAT construct we designed successfully inhibits MRSA growth in pure culture, cell culture, and in vivo in a C. elegans whole animal model. The PNA-TAT construct does not bind to HSA indicating this agent has potential for use in treating systemic MRSA infections. Further work confirming the anti-rpoA PNA-TAT construct's activity in other animal models of MRSA infection (to examine toxicity to host tissues and identify an appropriate route of administration) is an important next step to further examine the potential of PNA-TAT as a novel antimicrobial agent.
Materials and Methods
Chemicals, reagents, and kits. Mueller-Hinton broth II, Dulbecco's modified Eagle's medium, Dulbecco's phosphate buffered saline, chloroform, isopropanol, agarose, ethidium bromide, Tris-Borate-ethylenediaminetetraacetate buffer, free water and primers were purchased commercially from Sigma-Aldrich (St. Louis, MO). Trypticase soy broth (TSB) and trypticase soy agar (TSA) were purchased from BD/Difco (Sparks, Maryland) and mannitol salt agar was purchased from Hardy Diagnostics (Santa Maria, California). Vancomycin hydrochloride was purchased from Gold Biotechnology (St. Louis, MO). Fetal bovine serum was purchased from Life Technologies (Grand Island, NY). Recombinant lysostaphin was purchased from AMBI Products LLC (Lawrence, NY). TRIzol Max Bacterial RNA Isolation Kit, SYBR Green PCR Master Mix, SuperScript II Reverse Transcriptase and 1 kb plus DNA ladder were purchased from Invitrogen (Carlsbad, CA). Turbo DNA-free Kit and DEPC-treated water were purchased from Ambion (Foster city, CA). RNAprotect Bacteria Reagent (QIAGEN, Valencia, CA) and random hexamers (Applied Biosystems, Carlsbad, CA) were also purchased from commercial vendors.
Bacterial strains and C. elegans. Bacterial strains used in this study are presented in Table 1. The temperature-sensitive sterile mutant strain C. elegans AU37 (sek-1(km4); glp-4(bn2) I) was used as a whole animal model for testing the PNA's impact against MRSA infection in vivo. Worms were grown on nematode growth media plates cultivated with Escherichia coli OP50. For infection worms were maintained on TSA agar plates seeded with MRSA Sanger 252 as described earlier54
Cell penetrating peptide and PNA. The HIV-1 TAT CPP (GRKKKRRQRRRYK) was synthesized and purified by GenScript (Piscataway, NJ). A PNA was designed to be complementary to a conserved region of the rpoA gene (including the start codon) present in 12 different Staphylococcus species. The PNA-TAT (GRKKKRRQRRRYK-O-tttctatcattt-NH2) was synthesized and purified by PNA Bio (Thousand Oaks, CA) and was conjugated to the HIV-1 TAT using manual coupling chemistry.
In vitro antimicrobial activity of CPP and PNA. The MICs of the anti-rpoA PNA-TAT construct, free PNA and the TAT CPP were determined against multiple Staphylococcus species. Bacteria (ranging from 2 × 105 to 3.15 × 105 CFU/ml) were cultured in Mueller-Hinton broth II and a modified version of the broth microdilution method was used to determine the MIC.55 Briefly, low-binding clear microcentrifuge tubes (USA Scientific, Ocala, FL) were used instead of 96-well plates. Bacteria were incubated with each agent for 16 hours at 37 °C before the numbers of viable bacteria were counted by serial dilution and plating on TSA plates. The MIC was scored as the lowest concentration where no turbidity was observed in the microcentrifuge tubes.
Time-kill assay. MRSA Sanger 252 in late logarithmic growth phase was diluted to ~2.75 × 105 CFU/ml and incubated with 16 μmol/l (2 × MIC) of PNA-TAT (in triplicate) at 37°C for 24 hours and samples were collected every 2 hours.56,57 Linezolid and vancomycin were used as controls at the same concentration (16 μmol/l). Samples were serially diluted and plated onto TSA plates. Plates were then incubated at 37°C for 16 hours before viable CFU were determined.
Effect of PNA on MRSA gene expression. To quantify the expression of rpoA and the subsequent effect on expression of toxin genes including, hla toxin and PVL, total RNA was extracted using the TRIzol Max Bacterial RNA Isolation Kit, according to the manufacturer's protocol, with few modifications. Briefly, MRSA USA300 was cultured for 3 hours until reaching an optical density (OD600) of 0.24. A small aliquot (20 µl) was then centrifuged at 6,000×g for 2 minutes. The supernatant was discarded and the pellet was resuspended in 50 µl fresh trypticase soy broth. PNA-TAT was subsequently added at the following concentrations 0.5 × MIC (8 µmol/l), 1 × MIC (16 µmol/l) and 2 × MIC (32 µmol/l). Water served as a negative control. The samples were incubated in a 37°C shaking incubator for 16 hours.
After immediate stabilization of RNA in all samples by RNAprotect Bacteria Reagent and subsequent lysis of bacteria using 100 µg/ml recombinant lysostaphin in Tris EDTA (TE) buffer (1mol/l Tris pH 8, 0.5 mol/l ethylenediaminetetraacetate pH 8, dH2O), total RNA was extracted from the treated and untreated samples using the TRIzol Max Bacterial RNA Isolation Kit. The phase separation step, using chloroform, was repeated twice to minimize the carryover of phenol and guanidine isothiocyonate. To remove genomic DNA contamination, the RNA samples were treated with Turbo DNAse, according to the manufacturer's protocol. The absence of genomic DNA was confirmed using conventional PCR and running samples on a 1% agarose gel. RNA quantity and quality were determined using an Epoch Microplate Spectrophotometer (Bioteck Instruments, Winooski, Vermont).
For first strand cDNA synthesis, 150 nanograms of Turbo DNAse treated RNA was reverse transcribed using random hexamers and SuperScript II Reverse Transcriptase (according to the manufacturer's protocol). Specific primers for MRSA300 16S rRNA, rpoA, hla, and PVL genes were manually designed from GenBank sequences of S. aureus subsp. aureus USA300_FPR3757 (GenBank: CP000255.1) and purchased from Sigma-Aldrich (Supplementary Table S2). The specificity of the primers was confirmed using Basic Local Alignment Search Tool and conventional PCR using the genomic DNA of MRSA USA300.
Relative quantification for all cDNA samples was carried out, in triplicate, using an ABi 7300 Real-Time PCR System (Applied Biosystems) with the following conditions; 95 ºC for 10 minutes as an initial step for DNA polymerase activation (1 cycle) and 95ºC for 15 seconds, 58ºC for 30 seconds and 72ºC for 45 seconds for melting, annealing, and extension, respectively (40 cycles for each). 16S rRNA was used as the internal reference gene.12 Real-Time quantitative reverse-transcriptase PCR results were analyzed by using the 2(-Delta Delta C(T)) method.58
Cell culture infection assay. To determine the ability of PNA-TAT to inhibit growth of MRSA Sanger 252 present inside macrophages, a modified version of a cell culture infection assay was performed, as described elsewhere.9,59,60 Briefly, J774 cells in 96-well plates were infected with MRSA Sanger 252 at a 1:10 multiplicity of infection (MOI) for 45 minutes and treated subsequently with recombinant lysostaphin to kill extracellular bacteria. PNA-TAT at a final concentration of 8 or 16 μmol/l was added (in triplicate) to the infected J774 cells (maintained with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) and the cells were incubated for 4 hours at 37°C with 5% CO2. Vancomycin was used as a control. After incubation the cells were washed three times with phosphate buffered saline, lysed using 0.1% Triton X-100 and the intracellular bacteria were serially diluted and plated on TSA plates. Plates were incubated at 37°C for 16 hours before viable CFU were counted.
In vitro cytotoxicity analysis of rpoA-TAT against J774 cells. rpoA-TAT PNA was assayed (at concentrations of 4, 8, and 16 µmol/l) against murine macrophage (J774) cells to determine the potential toxic effect to mammalian cells in vitro.61 Briefly, cells were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum at 37 °C with CO2 (5%). Control cells received sterile water alone at a concentration equal to that in drug-treated cell samples. The cells were incubated with the PNA (in triplicate) in a 96-well plate at 37 ºC with CO2 (5%) for 4 hours prior to addition of the assay reagent MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (Promega, Madison, WI, USA) for 4 hours. Absorbance readings (at OD490) were taken using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA). The quantity of viable cells after treatment was expressed as a percentage of the viability of water-treated control cells (average of triplicate wells ± SD). The toxicity data was analyzed via Student t-test, (P < 0.05), utilizing GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA).
PNA efficacy in infected C. elegans animal model. To assess the efficacy of PNA-TAT in vivo, C. elegans were infected with MRSA Sanger 252. Briefly, worms were grown for 5 days at 15°C (permitting worms to lay eggs) on nematode growth media agar plates seeded with a lawn of E. coli OP50. The eggs were harvested by bleaching62 and maintained for 24 hours at room temperature with gentle agitation for hatching. Hatched larvae were transferred to a new nematode growth media plate seeded with E. coli OP50 and were kept at room temperature for 5 days until worms reached the adult stage of growth. Adult worms were collected and washed three times with M9 media in a 1:10 ratio to get rid of E. coli before transfer to TSA agar plates seeded with a lawn of MRSA Sanger 252 for infection.63 After 2 hours of infection, worms were collected and washed with M9 buffer five times before incubation with PNA-TAT. Worms were transferred to low-binding microcentrifuge tubes (10 worms per tube). PNA-TAT and vancomycin were added to the tubes (in triplicate) to achieve a final concentration equivalent to 1 × MIC, 2 × MIC, or 3 × MIC. Sterile water served as a negative control. After treatment for 18 hours, worms were washed five times with M9 buffer. Worms were examined microscopically to examine morphological changes and viability. Worms were lysed in microcentrifuge tubes containing 200 mg of 1.0-mm silicon carbide particles (Biospec Products, Bartlesville, OK) that were vortexed for one minute. Samples were serially diluted and plated onto mannitol salt agar to select for MRSA. Plates were incubated at 37°C for 18–20 hours before viable CFU was determined.
Assessment of PNA-TAT binding to HAS. The impact of protein binding on the antimicrobial effect of PNA-TAT was evaluated using HSA (Sigma-Aldrich). Briefly, PNA-TAT, at 4 × MIC (32 µmol/l), was added to diluted overnight cultures (~5 × 105 CFU/ml) of MRSA Sanger 252, in triplicate, using Mueller-Hinton broth II in the presence or absence of 4% HSA (in low adhesion tubes). Vancomycin (4 × MIC) and daptomycin (4 × MIC) served as negative and positive controls, respectively.64 The media for testing daptomycin was supplemented with 50 µg/ml CaCl2. Sterile water and 4% HSA were used as negative controls. After 12 hours incubation of the tubes with shaking at 37ºC, the samples were serially diluted in sterile water and 5 µl drops were plated onto TSA plates. The agar plates were incubated at 37 ºC for 24 hours before visual scoring of the bacteria.
Statistical analysis. Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). Statistical significance was determined using the two-tailed Student's t-test. P ≤ 0.05 was considered significant. Data are presented as mean ± SD.
SUPPLEMENTARY MATERIAL Figure S1. In vitro cytotoxicity analysis of rpoA-TAT against J774 cells. Table S1. Sequence alignment of rpoA gene among Staphylococcus species. Table S2. Primers used in this study.
Acknowledgments
The authors thank the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program supported under NIAID/NIH Contract # HHSN272200700055C for providing the MRSA strains used in this study.
Supplementary Material
References
- Lazarevic, V, Beaume, M, Corvaglia, A, Hernandez, D, Schrenzel, J and François, P (2011). Epidemiology and virulence insights from MRSA and MSSA genome analysis. Future Microbiol 6: 513–532. [DOI] [PubMed] [Google Scholar]
- Foster, TJ (2005). Immune evasion by staphylococci. Nat Rev Microbiol 3: 948–958. [DOI] [PubMed] [Google Scholar]
- Lin, YC and Peterson, ML (2010). New insights into the prevention of staphylococcal infections and toxic shock syndrome. Expert Rev Clin Pharmacol 3: 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel, J, Maciag-Gudowska, A, Mikolajczyk, T, Bzowska, M, Sturdevant, DE, Whitney, AR et al. (2009). Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS One 4: e5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelbaum, PC (2006). The emergence of vancomycin-intermediate and vancomycin-resistant Staphylococcus aureus. Clin Microbiol Infect 12 (Suppl 1): 16–23. [DOI] [PubMed] [Google Scholar]
- Nielsen, PE (2004). PNA Technology. Mol Biotechnol 26: 233–248. [DOI] [PubMed] [Google Scholar]
- Bai, H, Zhou, Y, Hou, Z, Xue, X, Meng, J and Luo, X (2011). Targeting bacterial RNA polymerase: promises for future antisense antibiotics development. Infect Disord Drug Targets 11: 175–187. [DOI] [PubMed] [Google Scholar]
- Soofi, MA and Seleem, MN (2012). Targeting essential genes in Salmonella enterica serovar typhimurium with antisense peptide nucleic acid. Antimicrob Agents Chemother 56: 6407–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alajlouni, RA and Seleem, MN (2013). Targeting listeria monocytogenes rpoA and rpoD genes using peptide nucleic acids. Nucleic Acid Ther 23: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, H, You, Y, Yan, H, Meng, J, Xue, X, Hou, Z et al. (2012). Antisense inhibition of gene expression and growth in gram-negative bacteria by cell-penetrating peptide conjugates of peptide nucleic acids targeted to rpoD gene. Biomaterials 33: 659–667. [DOI] [PubMed] [Google Scholar]
- Rajasekaran, P, Alexander, JC, Seleem, MN, Jain, N, Sriranganathan, N, Wattam, AR et al. (2013). Peptide nucleic acids inhibit growth of Brucella suis in pure culture and in infected murine macrophages. Int J Antimicrob Agents 41: 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, H Sang, G, You,Y, Xue, X, Zhou, Y, Hou, Zet al. (2012). Targeting RNA polymerase primary sigma(70) as a therapeutic strategy against methicillin-resistant Staphylococcus aureus by antisense peptide nucleic acid. PLoS One 7: 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abushahba, MF, Mohammad, H, Thangamani, S, Hussein, AA and Seleem, MN (2016). Impact of different cell penetrating peptides on the efficacy of antisense therapeutics for targeting intracellular pathogens. Sci Rep 6: 20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi, T and Nielsen, PE (2014). Cellular delivery of peptide nucleic acids (PNAs). Methods Mol Biol 1050: 193–205. [DOI] [PubMed] [Google Scholar]
- Patenge, N, Pappesch, R, Krawack, F, Walda, C, Mraheil, MA, Jacob, A et al. (2013). Inhibition of growth and gene expression by PNA-peptide conjugates in Streptococcus pyogenes. Mol Ther Nucleic Acids 2: e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryselius, R, Aswasti, SK, Rajarao, GK, Nielsen, PE and Good, L (2003). The translation start codon region is sensitive to antisense PNA inhibition in Escherichia coli. Oligonucleotides 13: 427–433. [DOI] [PubMed] [Google Scholar]
- Rasmussen, LC, Sperling-Petersen, HU and Mortensen, KK (2007). Hitting bacteria at the heart of the central dogma: sequence-specific inhibition. Microb Cell Fact 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, MD, Humphrey, BJ, Wang, YF, Kourbatova, EV, Ray, SM and Blumberg, HM (2006). Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann Intern Med 144: 309–317. [DOI] [PubMed] [Google Scholar]
- Moy, TI, Ball, AR, Anklesaria, Z, Casadei, G, Lewis, K and Ausubel, FM (2006). Identification of novel antimicrobials using a live-animal infection model. Proc Natl Acad Sci USA 103: 10414–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen, H, Bojer, MS, Marinus, MG, Xu, T, Struve, C, Krogfelt, KA et al. (2013). The alkaloid compound harmane increases the lifespan of Caenorhabditis elegans during bacterial infection, by modulating the nematode's innate immune response. PLoS One 8: e60519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otter, JA and French, GL (2010). Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Europe. Lancet Infect Dis 10: 227–239. [DOI] [PubMed] [Google Scholar]
- Gillet, Y, Issartel, B, Vanhems, P, Fournet, JC, Lina, G, Bes, M et al. (2002). Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 359: 753–759. [DOI] [PubMed] [Google Scholar]
- Bocchini, CE, Hulten, KG, Mason, EO Jr, Gonzalez, BE, Hammerman, WA and Kaplan, SL (2006). Panton-Valentine leukocidin genes are associated with enhanced inflammatory response and local disease in acute hematogenous Staphylococcus aureus osteomyelitis in children. Pediatr 117: 433–440. [DOI] [PubMed] [Google Scholar]
- David, MZ and Daum, RS (2010). Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23: 616–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ağca, H, Topaç, T, Ozmerdiven, GE, Celebi, S, Köksal, N, Hacımustafaoğlu, M et al. (2014). Investigation of methicillin resistant Staphylococcus aureus in neonatal intensive care unit. Int J Clin Exp Med 7: 2209–2213. [PMC free article] [PubMed] [Google Scholar]
- Regev-Yochay, G, Rubinstein, E, Barzilai, A, Carmeli, Y, Kuint, J, Etienne, J et al. (2005). Methicillin-resistant Staphylococcus aureus in neonatal intensive care unit. Emerg Infect Dis 11: 453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis, C, Hachem, R, Raad, II, Perego, CA, Dvorak, T, Hulten, KG et al. (2011). Outbreak of community-acquired methicillin-resistant Staphylococcus aureus skin infections among health care workers in a cancer center. Am J Infect Control 39: 112–117. [DOI] [PubMed] [Google Scholar]
- Wilson, P, Andrews, JA, Charlesworth, R, Walesby, R, Singer, M, Farrell, DJ et al. (2003). Linezolid resistance in clinical isolates of Staphylococcus aureus. J Antimicrob Chemother 51: 186–188. [DOI] [PubMed] [Google Scholar]
- Hiramatsu, K (2001). Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis 1: 147–155. [DOI] [PubMed] [Google Scholar]
- Gordon, RJ and Lowy, FD (2008). Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 46 (Suppl 5): S350–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep, BA, Afasizheva, A, Le, HN, Kajikawa, O, Matute-Bello, G, Tkaczyk, C et al. (2013). Effects of linezolid on suppressing in vivo production of staphylococcal toxins and improving survival outcomes in a rabbit model of methicillin-resistant Staphylococcus aureus necrotizing pneumonia. J Infect Dis 208: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead, AL and Li, B (2011). Nanomedicine as an emerging approach against intracellular pathogens. Int J Nanomedicine 6: 3281–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones, E, Colino, CI and Lanao, JM (2008). Delivery systems to increase the selectivity of antibiotics in phagocytic cells. J Control Release 125: 210–227. [DOI] [PubMed] [Google Scholar]
- Moore, CL, Hingwe, A, Donabedian, SM, Perri, MB, Davis, SL, Haque, NZ et al. (2009). Comparative evaluation of epidemiology and outcomes of methicillin-resistant Staphylococcus aureus (MRSA) USA300 infections causing community- and healthcare-associated infections. Int J Antimicrob Agents 34: 148–155. [DOI] [PubMed] [Google Scholar]
- Soong, G, Paulino, F, Wachtel, S, Parker, D, Wickersham, M, Zhang, D et al. (2015). Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. MBio 6:e00289–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire, S, Kosowska-Shick, K, Appelbaum, PC, Glupczynski, Y, Van Bambeke, F and Tulkens, PM (2011). Activity of moxifloxacin against intracellular community-acquired methicillin-resistant Staphylococcus aureus: comparison with clindamycin, linezolid and co-trimoxazole and attempt at defining an intracellular susceptibility breakpoint. J Antimicrob Chemother 66: 596–607. [DOI] [PubMed] [Google Scholar]
- Cruciani, M, Gatti, G, Lazzarini, L, Furlan, G, Broccali, G, Malena, M et al. (1996). Penetration of vancomycin into human lung tissue. J Antimicrob Chemother 38: 865–869. [DOI] [PubMed] [Google Scholar]
- Van Bambeke, F, Barcia-Macay, M, Lemaire, S and Tulkens, PM (2006). Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives. Curr Opin Drug Discov Devel 9: 218–230. [PubMed] [Google Scholar]
- Anastassopoulou, CG, Fuchs, BB and Mylonakis, E (2011). Caenorhabditis elegans-based model systems for antifungal drug discovery. Curr Pharm Des 17: 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewbank, JJ and Zugasti, O (2011). C. elegans: model host and tool for antimicrobial drug discovery. Dis Model Mech 4: 300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochansky, CJ, McMasters, DR, Lu, P, Koeplinger, KA, Kerr, HH, Shou, M et al. (2008). Impact of pH on plasma protein binding in equilibrium dialysis. Mol Pharm 5: 438–448. [DOI] [PubMed] [Google Scholar]
- Lee, BL, Sachdeva, M and Chambers, HF (1991). Effect of protein binding of daptomycin on MIC and antibacterial activity. Antimicrob Agents Chemother 35: 2505–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, DA, Di, L and Kerns, EH (2010). The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9: 929–939. [DOI] [PubMed] [Google Scholar]
- Rippa, V, Cirulli, C, Di Palo, B, Doti, N, Amoresano, A and Duilio, A (2010). The ribosomal protein L2 interacts with the RNA polymerase alpha subunit and acts as a transcription modulator in Escherichia coli. J Bacteriol 192: 1882–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, H, Sang, G, You, Y, Xue, X, Zhou, Y, Hou, Z et al. (2012). Targeting RNA polymerase primary σ70 as a therapeutic strategy against methicillin-resistant Staphylococcus aureus by antisense peptide nucleic acid. PLoS One 7: e29886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson, M, Nielsen, A, Kjærulff, L, Gotfredsen, CH, Wietz, M, Ingmer, H et al. (2011). Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Mar Drugs 9: 2537–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick, RP, Ross, HF, Projan, SJ, Kornblum, J, Kreiswirth, B and Moghazeh, S (1993). Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J 12: 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korem, M, Gov, Y, Kiran, MD and Balaban, N (2005). Transcriptional profiling of target of RNAIII-activating protein, a master regulator of staphylococcal virulence. Infect Immun 73: 6220–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabelskaya, S, Bordeau, V and Felden, B (2014). Dual RNA regulatory control of a Staphylococcus aureus virulence factor. Nucleic Acids Res 42: 4847–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfeldt, E, Taylor, D, von Gabain, A and Arvidson, S (1995). Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J 14: 4569–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, WC, Coyle, BJ and Williams, P (2004). Virulence regulation and quorum sensing in staphylococcal infections: competitive AgrC antagonists as quorum sensing inhibitors. J Med Chem 47: 4633–4641. [DOI] [PubMed] [Google Scholar]
- Nekhotiaeva, N, Awasthi, SK, Nielsen, PE and Good, L (2004). Inhibition of Staphylococcus aureus gene expression and growth using antisense peptide nucleic acids. Mol Ther 10: 652–659. [DOI] [PubMed] [Google Scholar]
- Liang, S, He, Y, Xia, Y, Wang, H, Wang, L, Gao, R et al. (2015). Inhibiting the growth of methicillin-resistant Staphylococcus aureus in vitro with antisense peptide nucleic acid conjugates targeting the ftsZ gene. Int J Infect Dis 30: 1–6. [DOI] [PubMed] [Google Scholar]
- Thomsen, LE, Slutz, SS, Tan, MW and Ingmer, H (2006). Caenorhabditis elegans is a model host for Listeria monocytogenes. Appl Environ Microbiol 72: 1700–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne, PA (2007). Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M7-A7. Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. Nineteenth informational supplement M100-S19. Wayne, PA: Clinical and Laboratory Standards Institute; 2009. [Google Scholar]
- Mohamed, MF, Hamed, MI, Panitch, A and Seleem, MN (2014). Targeting methicillin-resistant Staphylococcus aureus with short salt-resistant synthetic peptides. Antimicrob Agents Chemother 58: 4113–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, MF, Hammac, GK, Guptill, L and Seleem, MN (2014). Antibacterial activity of novel cationic peptides against clinical isolates of multi-drug resistant Staphylococcus pseudintermedius from infected dogs. PLoS One 9: e116259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, KJ and Schmittgen, TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Thangamani, S, Mohammad, H, Abushahba, MF, Sobreira, TJ, Hedrick, VE, Paul, LN et al. (2016). Antibacterial activity and mechanism of action of auranofin against multi-drug resistant bacterial pathogens. Sci Rep 6: 22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangamani, S, Younis, W and Seleem, MN (2015). Repurposing clinical molecule ebselen to combat drug resistant pathogens. PLoS One 10: e0133877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad, H, Reddy, PV, Monteleone, D, Mayhoub, AS, Cushman, M and Seleem, MN (2015). Synthesis and antibacterial evaluation of a novel series of synthetic phenylthiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA). Eur J Med Chem 94: 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, JR and Ausubel, FM (2008). Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. Methods Mol Biol 415: 403–427. [DOI] [PubMed] [Google Scholar]
- Stiernagle, T. (2006). Maintenance of C. elegans WormBook : the online review of C. elegans biology. pp. 1-11. [DOI] [PMC free article] [PubMed]
- McKay, GA, Beaulieu, S, Sarmiento, I, Arhin, FF, Parr, TR Jr and Moeck, G (2009). Impact of human serum albumin on oritavancin in vitro activity against enterococci. Antimicrob Agents Chemother 53: 2687–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.