

Abstract
The complement system is an integral component of the humoral immune system, and describes a cascade of interacting proteins responsible for the opsonization and lysis of foreign pathogens, in addition to the recruitment of immune cells. However, complement activation is also implicated in the progression and complication of immune dysfunctions such as sepsis. Microparticle (MP) biomaterials capable of tuning the local magnitude of serum complement activation could improve complement-mediated cytotoxicity to serum-resistant bacteria or calm an overactive immune response during sepsis. We demonstrate that model Fc-functionalized microparticles can be designed to either enhance or diminish the local cytotoxic effect of complement activation in human serum. The particles were formed with either the antibody Fc domains oriented outward from the particle surface or randomly adsorbed in a non-oriented fashion. In the oriented Fc form, complement products were directly sequestered to the particle surface, including C5a, a potent anaphylatoxin that, when elevated, is associated with poor sepsis prognosis. The oriented particle also lowered the cytotoxicity of serum and thus decreased the antibiotic effect when compared to serum alone. Conversely, the non-oriented microparticles were found to sequester similar levels of C5a, but much lower levels of iC3b and TCC on the microparticle surface, thereby increasing the amount of the soluble terminal complement complex. In addition, the non-oriented microparticles extend the distance over which TCC forms and enhance serum cytotoxicity to bacteria. Together, these two types of complement-modulating particles provide the first biomaterial that can functionally modify the range of complement activation at sites distant from the particle surface. Thus, biomaterials that exploit Fc presentation provide new possibilities to functionally modulate complement activation to achieve a desired clinical result.
Introduction
The humoral immune response consists of immunoglobulins and complement proteins that opsonize and inactivate invading pathogens, recruit phagocytic cells through chemotactic mediators, and initiate adaptive immune responses.1-3 A central mechanism for recognizing threats in the body is opsonization—the coating of invading pathogens with antibodies, complement proteins, or other molecules to mark them as foreign. The crystallizable fragment (Fc) of each immunoglobulin class is a key effector of post-pathogen opsonization that initiates recognition by immune cells, namely neutrophils and macrophages, and activation of the complement cascade, which is directly cytotoxic to invading pathogens.4-7
In the classical complement or antibody-mediated pathway, activation begins after the binding of the protein C1q to closely apposed Fc regions of a single IgM or multiple IgG antibodies. The subsequent enzymatic cascade results in the formation of the terminal complement complex (TCC), also known as the membrane attack complex, consisting of proteins C5b-C9.8-10 TCC directly lyses pathogens through membrane insertion and formation of a pore.11 The complement cascade also generates a series of protein cleavage products that act as potent anaphylatoxins, including C3a and C5a.12 The other complement activation pathways, the alternative and lectin-mediated pathways, differ in their activation methods but converge at the essential complement opsonin C3b.
Due to the importance of the signal amplification and control points present in the cascade, complement activation is highly susceptible to subtle variations in initiation conditions.13 The use of particles decorated with Fc or other molecular regulators or prompters of complement as a biomaterial can be highly effective in modulating the cascade through the adjustment of particular design parameters, including the initiator molecule spacing and valency.14, 15 Since Fc valency and orientation have been demonstrated to regulate complement activation in vitro 16-18 we hypothesize that Fc-conjugated biomaterials can also modulate the molecular and functional effects of complement, including cytotoxicity and further downstream immune responses. However, much of the work related to Fc-coated microparticles applied to immunomodulation concerns the activation of macrophages through phagocytosis and subsequent cytokine release.19-21 In addition, an incomplete understanding of the biophysical effects of complement activation by Fc-coated biomaterials, including the physical distance over which activated complement products can impact targets, limits the rational design of these biomaterial approaches.
In contrast to biological functionalization, the effects of material chemistry on complement activation has been more widely studied, including the influence of biomaterial surface coating on innate immune responses such as inflammation 22-25 and the modification of complement responses.26 Modifications to particle surface chemistry can also incite and alter the alternative pathway of complement activation and deposition, affecting both adaptive and innate immunity.27
In this study, we demonstrate that Fc microparticles can prompt functional changes to the magnitude and effective range of complement-mediated effects. Using one form of Fc particles displaying non-oriented IgG, we increase serum cytotoxicity towards bacteria by increasing the local magnitude and physical range of activated complement products that culminate in increased TCC deposition upon nearby bacteria. In demonstrating this effect, we performed one of the first quantitative studies of the range of complement-mediated cytotoxicity. In the other form of Fc particles, Fc regions of IgG molecules are oriented outward upon binding immobilized antigen. This mode of display results in post-activation complement products that are instead tightly adsorbed onto the microparticle surface. This sequestration decreases serum bacterial cytotoxicity, likely because it substantially decreases serum C5a anaphylatoxin levels. A biomaterial that can lower the serum levels of C5a is of substantial interest to lessen the clinical effects of sepsis 28 and other autoimmune complications.29 Taken together, through biophysical variation of molecular initiators of complement displayed by the Fc microparticles, we can tune their functional effects to be either potent immune activators (non-oriented Fc particles) or immune suppressors (oriented Fc particles).
Materials and Methods
Microparticle Functionalization
To create microparticles with oriented Fc, carboxylated polystyrene microparticles 1 μm in diameter (Bangs Laboratories, Fisher, IN) were first washed (10 min, 2000×g) in PBS to remove any buffers or preservatives. Next the particles were incubated in a saturating solution of bovine serum albumin (BSA, Sigma Aldrich, St. Louis, MO). Although unsuitable for in vivo use as they are non-degradable, these polystyrene microparticles were chosen as the base of our model system as they are highly uniform, have a systematic means of determining protein coverage as determined by the manufacturer, and we have previously utilized them as complement modulating platforms.14, 15 Despite the negative charges of both the BSA and the carboxylated surface, with electrostatic repulsion predominating at physiological pH (binding occurs in PBS), passive adsorption and ionic interactions have been reported (Bangs Labs Tech Note 204).30 In addition, it has been demonstrated that for extensively carboxylated (>2 COOH/nm2) polystyrene microspheres, hydrogen bonding with BSA prevails over hydrophobic interactions.31 This is likely occurring here, as we have determined from estimates provided by the manufacturer that the carboxyl density may be as high as 4/nm2 on average. Rabbit polyclonal anti-BSA IgG antibody (Abcam, Cambridge, MA) was then added in various molar ratios of antibody to the immobilized BSA antigen (2:1, 1:5, and BSA-only), as described previously.15 In the case of oriented MP roughly 86 μg of BSA was bound to the surface, and in both oriented and non-oriented 2:1 MP roughly 76 μg of IgG was bound to the surface, as we have determined previously.15 Non-oriented Fc particles were formed by incubation of anti-BSA IgG antibody without a previous BSA saturation. The saturating amount of IgG was found from the MP manufacturer's documentation (Bangs Labs Tech Note 205) and confirmed by verifying a maximal fluorescence after dilutions of antibody. The omission of BSA randomizes the proportion of exposed Fc domains on the microparticle surface and reduces fluorescence when labeled with tagged secondary antibodies. Flow cytometry was performed after primary antibody incubation with a fluorescent secondary antibody (donkey polyclonal anti-rabbit IgG – DyLight® 650, Abcam, Cambridge, MA). Flow cytometry (BD Accuri C6, BD Biosciences, San Jose, CA) was used to confirm the presence of the primary antibody on the microparticle surface and from fluorescent particle counts the concentration of microparticles was determined.
Validation of Fc Orientation Control in Microparticle Formation
To demonstrate that the surface-bound IgG was in fact oriented or non-oriented with respect to the bead surface, atomic force microscopy (AFM) imaging of the particle surfaces were conducted. Glass microscope slides were cleaned and 5 μL of 1.8 mg/mL Cell-Tak (Corning Life Sciences, Tewksbury, MA) in 5% acetic acid was mixed into a 60 μL PBS droplet and allowed to incubate for 1 hr. After several washes in PBS, 30 μL of microparticles from either condition were added and incubated 45 min to ensure attachment. Following another wash cycle, images of the surface of both particle types were collected using an MFP-3D Bio AFM (Asylum Research, Goleta, CA) paired to a MSCT cantilever E (Bruker Instruments, Billerica, MA). A scan area of 1 μm2 was collected at a scan rate of 0.2 Hz, with a resolution of 512 scan lines per image. To remove the microparticle curvature, the images were flattened and poorly resolved regions at the edge of the image were omitted from the image analysis. Igor Pro 6 (Wavemetrics, Lake Oswego, OR) was used for image analysis.
To further demonstrate the dissimilar availability of Fc regions due to differing orientation of the surface-bound IgG between the particle types, AFM binding studies were performed (See Fig S3). Carboxylated polystyrene particles 4 μm in diameter (Bangs Labs) were carefully glued to MSCT C cantilevers via micromanipulation stage and allowed to dry overnight. The cantilevers to be functionalized with oriented Fc microparticles were cleaned for five minutes in ethanol, dried with nitrogen gas, immersed in 2 mg/mL BSA for 30 minutes, washed in PBS, immersed in 0.02 mg/mL anti-BSA IgG for 30 minutes, and finally washed in PBS. For cantilevers with non-oriented microparticles, the same process was followed except for the BSA functionalization step. At all times, the cantilevers were immersed in liquid to avoid drying of the surface-attached molecules.
Cleaned glass slides were immersed in 60 μL of 0.15 mg/mL Cell-Tak in 5% acetic acid for 1 hour, washed in PBS, and then immersed in 1 mg/mL BSA for 30 minutes. Excess BSA was removed by washing 20 times with 500 mL PBS.
The cantilever was placed into the AFM and brought into contact with the BSA-functionalized surface at an approach velocity of 100 nm/s. Upon contact, the cantilever was allowed to deflect 1 nm vertically, which is sufficiently small to avoid damaging the immobilized molecules, before retracting at 100 nm/s. We assume that the number of molecular bonds that formed was a function of both contact time and contact area and thus the velocity and contact deflection parameters were held fixed across all experimental groups to minimize variation in adhesion within the groups and isolate the role of antibody orientation on adhesion. The blocking assay was performed by perfusing the 50 μL fluid cell with 30 μL of 1 mg/mL BSA to a final concentration of 0.38 mg/mL, and incubating for 30 minutes before collecting force curves. The thermal calibration method was used to calibrate the cantilever spring constant.32 The adhesive work was calculated by integrating the force-distance retraction data using a trapezoidal approximation. All data analysis was performed in Igor Pro 6.
ELISAs for Complement Activation and Deposition
Activation products initiated by the oriented and non-oriented particles were analyzed by enzyme-linked immunosorbent assay (ELISA) on the microparticle surfaces as well as within soluble serum fractions (Fig 2D). Approximately 20 million microparticles in 86 μL of microparticle solution was added to 14 μL of normal human serum (Life Technologies, Carlsbad, CA) and incubated for an hour at 37 °C to initiate complement activation. To understand the abundance of complement mediators free in solution versus deposited upon microparticle surfaces, the incubated solution was centrifuged at 3000 G for 10 minutes and the supernatant was decanted. Both the remaining microparticle pellet and supernatant were diluted separately 1:200 in PBS and placed in the microassay wells (MP washed 1X in PBS). Heat aggregated gamma globulin (HAGG) was used as a positive control complement activator for both the microparticle-containing serum samples and the naive serum samples. MicroVue CH50 Eq ELISA kits were used (Quidel, San Diego, CA) to assess the levels of the terminal complement complex (TCC) on the microparticle surface and in serum. The kit quantifies the amount of total TCC, expressed in CH50 unit equivalents per milliliter. The CH50 unit corresponds to the volume of test serum required for 50% lysis in sheep erythrocytes exposed to rabbit IgM, and is a standard means to assess complement fixation. Given that TCC experiences rapid hydrolytic degradation in serum33, the determination of complement intermediate products in solution and on the particle surface was supplemented with a Microvue iC3b ELISA (Quidel, San Diego, CA), performed using the same procedures detailed above. A C5a ELISA (Quidel, San Diego, CA) was performed to determine the amount of C5a, a potent anaphylatoxin and phagocyte chemoattractant.12 To simulate the serum of a septic patient, 100 μL solution of normal human serum was spiked with 250,000 DH5α E. coli. As before, the microparticles, bacteria, and serum were combined and incubated at 37 °C for 1 hour, after which the solution was spun down for 10 minutes at 3000 G. The supernatant was aspirated to separate the microparticle-containing fraction from the serum fraction.
Fig 2.
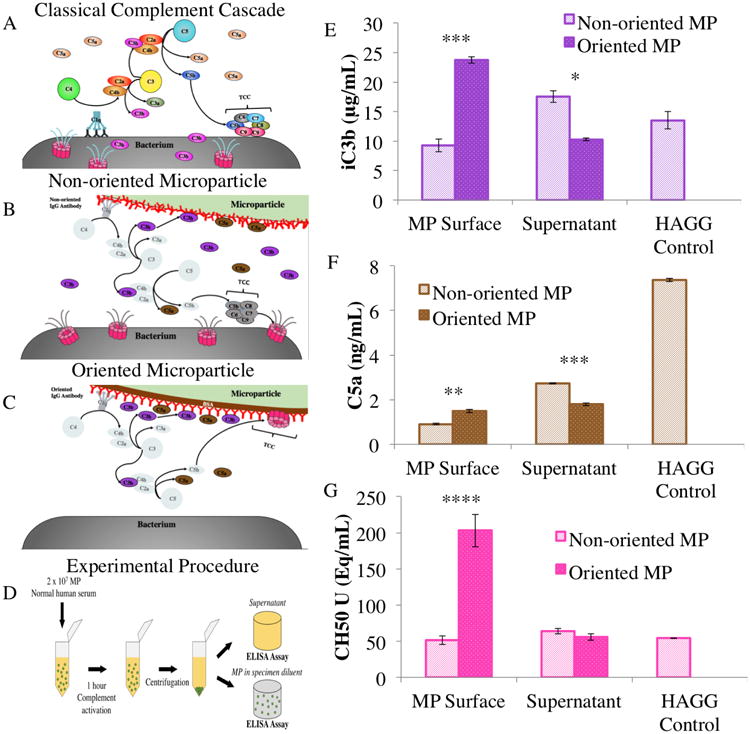
General classical complement cascade (A) The classical complement protein activation pathway. Models of complement activation and deposition for non-oriented (B) and oriented (C) microparticles C1q molecules bind to the Fc region of IgG antibodies to begin the classical complement pathway. Oriented microparticles have high levels of adsorbed iC3b and TCC while non-oriented microparticles activate and release high levels of iC3b into the serum. Low levels of TCC are seen on the non-oriented microparticle surface. Schematic of ELISA microparticle/serum separation procedure (D) The combined solution was centrifuged at 3000 G for 10 minutes, and the supernatant was separated and tested independently of the microparticles, which were reconstituted in specimen diluent (reagent included with kit). Quantification of complement protein activation and deposition for oriented and non-oriented microparticles (E-G) ELISAs were performed to quantify the levels of iC3b, C5a, and TCC in units of CH50 in whole solution, in the supernatant, and on the surface of microparticles. For each, 1 μm oriented and non-oriented microparticles with Fc densities of 2:1 were tested. Statistics were calculated from four replicates. * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001; the stars correlate to statistical difference between corresponding oriented and non-oriented measurements (i.e. oriented serum vs. non-oriented serum).
Effect of Fc Microparticles on E. coli Viability
Approximately 500,000 DH5α Escherichia coli bacteria in 10 μL of Luria-Bertani (LB) broth were added to human serum with and without the oriented and non-oriented Fc microparticles to determine their effects on bacterial survival. These E. coli solution aliquots were mixed with 30 μL of microparticles in PBS at various concentrations to test several microparticle-to-bacteria ratios. To this, lysogeny broth (LB, ThermoFisher Scientific, Waltham, MA) and a volume of normal human serum (Pierce, ThermoFisher Scientific, Waltham, MA) were added such that the serum constituted either 1, 10, or 55% of the mixture by volume, with each tube at a total volume of 125 μL. Conditions using 1% serum were necessary to ensure sensitivity after plating, as more concentrated serum rendered the non-oriented MP too cytotoxic to allow for manual colony counting. Control experiments included non-functionalized microparticles and bacteria in serum, bacteria in broth only, and bacteria with isopropyl alcohol (Sigma Aldrich, St. Louis, MO). Other controls evaluated were non-oriented microparticles only, oriented microparticles only, serum only, and broth only to verify that results observed were not due to contamination. Tubes were first incubated for 4 hours at 37 °C, then diluted 10 fold 9 times to provide a range of bacteria sufficient for manual colony counting. Dilute or original bacterial sample (10 μL) was added to 100 μL of broth, transferred onto agar/LB broth plates in four separate 10 μL droplets, streaked, and incubated at 37 °C for 16 hours. Any colonies formed were then photographed and colony-forming units were counted visually (See S1 Fig). Bacterial lawns were assigned a value of 160 colonies, which estimates a minimum number of colonies that would fit in the plate area. Cytotoxicity for each experimental condition was calculated by determining the fraction of apparent sample bacterial colonies when compared to the bacteria only control.
Confocal Microscopy of TCC Deposition and Live/Dead Staining
To correlate dead bacteria to TCC (Fig 6C), both oriented and non-oriented Fc microparticles were added to 500,000 bacteria in a 100:1 microparticle-to-bacterium ratio in a 1% serum solution diluted in LB broth. The solutions were centrifuged at 3000 G for 10 minutes, after which the supernatant was removed. The primary rabbit anti-TCC antibody (anti-C5b-9, Abcam, Cambridge, MA) was added at 1:100 to the pellet to a total volume of 100 μL and incubated at 4°C for 45 minutes. The sample was centrifuged again and a secondary goat anti-rabbit Alexa Fluor® 532 antibody (ThermoFisher Scientific, Waltham, MA) was added at 1:250 (v/v) to the pellet to a total volume of 100 μL and incubated at 4°C for 45 minutes. The solution was again centrifuged, and reconstituted to 100 μL in 10% serum. A live/dead bacterial stain was also added using the LIVE/DEAD BacLight Bacterial Viability Kit (ThermoFisher Scientific, Waltham, MA) at 3 μL per sample. The solutions were then immediately deposited on glass slides in 5 μL droplets and incubated for 30 minutes to give time to bind non-specifically. The identification of live and dead bacteria was determined by confocal microscopy (Zeiss Elyra PS.1 / LSM 780, Zeiss, Oberkochen, Germany).
Fig 6.
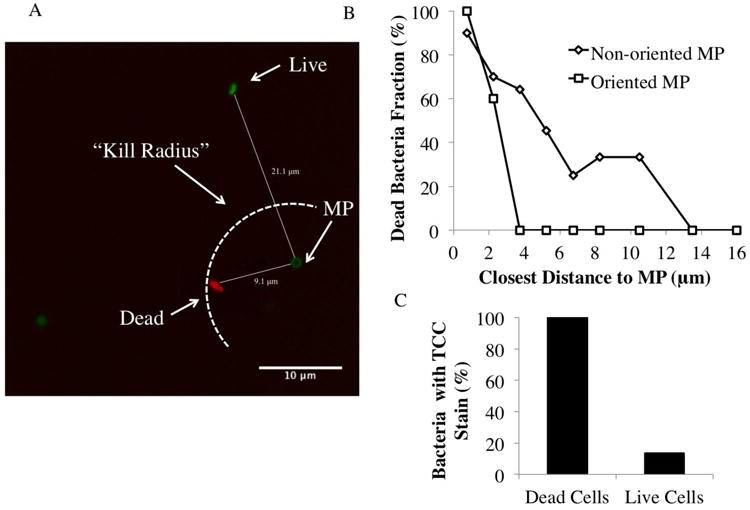
Confocal imaging of microparticle/microparticle interaction (A). The image shows a red dead stain, green live stain, and dark green MP stain. DH5α E. Coli and 1 μm non-oriented microparticles (2:1 Fc density) were imaged. The arc indicates a theoretical “kill radius” inside which complement-mediated lysis is greatest. Relating bacterial death and distance to closest microparticle (B) This plot shows the percentage of bacteria that are dead at various distances from individual microparticles, comparing the non-oriented MP (triangles) and oriented MP (squares). Two way ANOVA was performed and showed a significant difference between non-oriented and oriented effect on serum cytotoxicity (overall p-value (prob>F)=0.0012, and p-value(prob>t) = 0.0463 for effect of oriented vs. non-oriented). Accuracy of TCC stain (C) The bar chart shows the percentage of dead cells linked to the TCC stain, and the percentage of live cells paired to a TCC stain.
To attain the relationship between distance from MP and number of dead bacteria (Fig 6A and 6B), MP and bacteria in the same numbers as mentioned in the previous paragraph were allowed to evaporate on a glass slide. Then, the same live/dead stain was added along with a 10% serum solution; samples were then immediately imaged. MP were discernible via their circular shape and darker color (from adsorbing excess live stain).
Statistical Analysis
Statistical analysis of the results was performed in JMP®, Version 12 (SAS Institute Inc., Cary, NC, 1989-2007). Comparisons were evaluated with ANOVA followed by Tukey-Kramer honest significant difference (HSD) post-analysis.
Results and Discussion
Confirming Control over Fc Orientation and Measuring Binding Force Curves
Available Fc on the particle surface was quantified in order to validate the control over Fc orientation. It was observed that, for equal amounts of IgG, the non-oriented MP showed lower amounts of available Fc when compared to its oriented MP counterpart; the high-density non-oriented MP displayed amounts of Fc similar to low density oriented MP. In addition, AFM imaging of IgG-coated MPs displays nanoscale features of the surface consistent with antibody coating.34, 35 The oriented particle surface featured smaller variation in topography height, consistent with a more uniform presentation of the molecule surface. In contrast, the non-oriented Fc particles showed much greater variation in surface topography, consistent with more randomness to the conjugation.
To characterize the availability of the antigen binding regions of the immobilized IgG, and thus the comparative orientation of the Fc region of the molecule, AFM adhesion measurements were conducted between the two particle types and an immobilized BSA antigen surface (see Fig 1C-D). The non-oriented antibody-functionalized microparticles resulted in an order of magnitude higher adhesive work (87,050±43,900 pN-nm) than the oriented antibody-coated microparticles (7600±8300 pN-nm), with a statistical significance of p < 0.0001 (see Fig 1F). This work did not diminish over repeated adhesion events (see Fig S4), indicating that the molecules were stable and did not detach or denature during the experiment. Furthermore, the adhesion forces were largest for the non-oriented MPs as we expect due to greater availability of Fab and there decreased availability of the Fc region. The addition of soluble BSA into the fluid cell reduced the adhesion of the oriented MP interactions, indicating the blocking of specific bond formation (see Fig S5).
Fig 1.
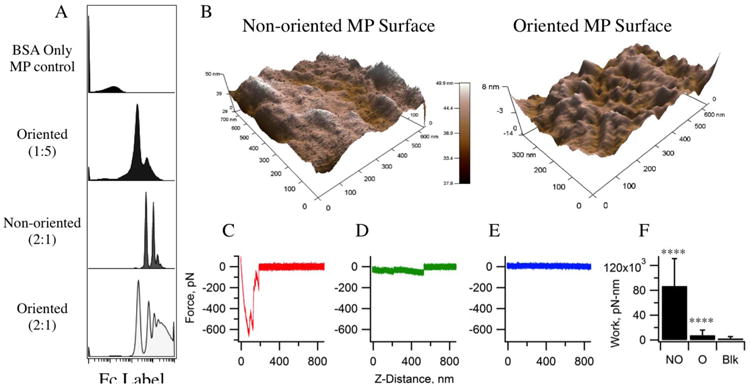
Understanding the amount of available Fc on microparticle surfaces (A) Mean fluorescence intensity was plotted to show the proportions of MP with available Fc. This confirms the decreased amount of available Fc when comparing non-oriented MP to oriented MP. Surface analysis of MP measuring physical effects of oriented vs. non-oriented Fc to confirm orientation control (B) 3-D AFM images of non-oriented and oriented 1 μm MPs. The non-oriented Fc surface shows higher roughness and variability in physical heights; the standard deviation of these heights is 1.8 nm, while that of the oriented Fc is only 1.5 nm. Average peak heights of features for oriented and non-oriented, respectively, are 11.3 +/- 1.2 nm and 10.7 +/- 1.8 nm. The widths were 101.1 +/- 28.5 nm and 197.2 +/- 67.5 nm (p-value < 0.01), respectively. Representative AFM binding force curves for the (C) non-oriented, (D) oriented, and (E) BSA-blocked oriented interactions. (F) The adhesive work performed by the molecular bonds for each interaction, where non-oriented is “NO”, oriented is “O”, and BSA-blocking of O is “Blk”. The work done by the non-oriented antibody-functionalized MP and oriented antibody-coated MP differed significantly; * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001.
Oriented and Non-oriented Fc Microparticles Sequester Different Amounts of Complement Proteins iC3b, C5a, and TCC
To delineate between complement activation by Fc microparticles with different antibody presentations, ELISAs were conducted to test for the presence of three essential complement proteins or protein complexes: iC3b (the bound, inactivated form of the C3b opsonin), C5a, and TCC (Fig 2E to 2G). Oriented Fc microparticles had relatively high levels of iC3b and TCC sequestered on the microparticle surfaces, as illustrated in Fig 2C. The non-oriented Fc microparticles showed lower levels of these proteins adsorbed to the surface, as shown in Fig 2B. Both the non-oriented and the oriented Fc particles bound comparable amounts of C5a. The supernatant serum collected showed low levels of all complement products for oriented Fc microparticles compared to the positive HAGG control. Non-oriented Fc microparticles in contrast showed higher levels of iC3b and TCC products in solution compared to the HAGG control.
Complement Toxicity to E. coli in Serum Can Be Tuned By Fc Microparticle Design
We hypothesized that the different Fc functionalization schemes, which resulted in different molecular activation of complement, could also result in functional differences in complement activation. To test this hypothesis we examined the impact of Fc particles on serum-mediated bacterial cytotoxicity. E. coli were incubated in diluted serum with the addition of oriented Fc microparticles or non-oriented Fc microparticles. Oriented Fc microparticles were found to substantially inhibit the serum cytotoxicity to the bacteria (Fig 3A). Conversely, the addition of non-oriented Fc microparticles increased the cytotoxicity of serum. The modulation of serum cytotoxicity is consistent with the changes in measured serum levels of C3b (complement cascade common pathway initiation) and the sequestration of TCC (cytolytic activity) upon the particle surfaces with the addition of the oriented Fc and non-oriented Fc microparticles. Serum levels of TCC are difficult to quantify as the complex is readily hydrolyzed when unbound 33, yet we hypothesize that non-oriented Fc microparticles activate the cascade in such a way that TCC formation upon the E. coli membrane is favored, resulting in cell death, as illustrated in Fig 2B. This cytotoxic effect was exhibited at a range of serum levels from physiological (55%) to diluted serum concentrations as low as 0.014% by volume (See Supplemental Fig 3). As previously stated, 1% serum was used for experiments as it was sufficiently dilute to ensure countable colony formation when exposed to the non-oriented Fc microparticles. The addition of the non-oriented Fc microparticles resulted in a significantly lower number of colony forming units than serum alone, thus supporting their use as a cytotoxic biomaterial. In general, a negative correlation was found between the amount of TCC on the microparticle surface and the cytotoxicity of that treatment (Fig 3B).
Fig 3.
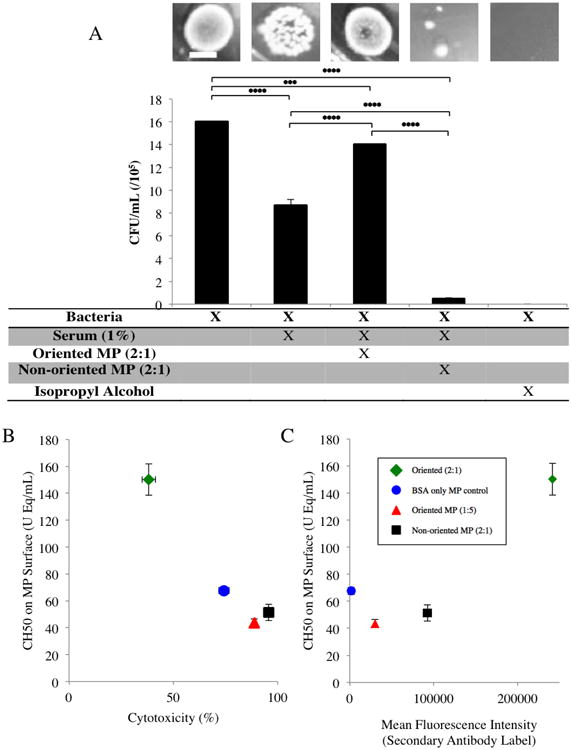
Scale Bar = 5 mm The viability of E. coli after treatment with normal human serum and in combination with oriented and non-oriented microparticles (A) The addition of 1% serum resulted in significantly lower colony formation than bacteria alone. When 1 μm 2:1 oriented microparticles were added at a 200:1 ratio with 1% serum, E. coli viability was significantly increased in comparison to treatment with serum alone. The 1 μm 2:1 non-oriented microparticles were also added at a 200:1 ratio with 1% serum and resulted in a significant increase in bacterial death. The CH50 values presented (correlating to TCC formation) are those measured on the surface of the microparticle. * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001. Relating TCC binding on microparticle surface to cytotoxicity (B) Higher levels of TCC present on the microparticle surface correlate with lower cytotoxicity. Relating TCC binding to Fc availability (C) Flow cytometry data in which an anti-Fc fluorescent secondary antibody (DyLight® 650) was used to quantify Fc density for each given particle. X and Y error bars are present in all data points.
The oriented Fc microparticles were coated with immobilized BSA such that the Fc region of each bound antibody projects outward. We observed moderate complement binding to the BSA on particles (Fig 3B), consistent with previous reports from our laboratory15 as well as others.36, 37 To characterize the influence of the BSA coating in the cytotoxicity of the oriented microparticles, we tested particles with BSA-only and those of low Fc density (1:5 molar ratio of Fc to immobilized BSA) when cultured with E. coli. BSA only control microparticles show low levels of complement activation and sequestration, but not enough to account for the difference observed between non-oriented and oriented Fc designs (Fig 3B). Oriented Fc microparticles with low Fc density resulted in substantially increased cytotoxicity compared to the 2:1 oriented and on par with the 2:1 non-oriented Fc microparticles. The 1:5 oriented microparticles were slightly less cytotoxic than 2:1 non-oriented microparticles and had similarly low levels of TCC on the particle surface, suggesting that low Fc density contributes to the activation of complement with low levels of TCC sequestration. Together these data suggest that random Fc orientation and low Fc density both increase the functional activation of complement without significant binding to the particle surface. We quantified available Fc on the particle surface using a secondary antibody label, specific to the rabbit heavy chain, of which the Fc fragment is composed.38 Particles with a higher Fc fluorescence intensity correlated with greater levels of TCC binding (Fig 3C), suggesting that the increase in available Fc is responsible for greater affinity to complement proteins. The proportion of particles with high Fc fluorescence intensity is greater for oriented MP with more extensive Fc coverage than both non-oriented and low Fc density oriented (Fig 1A), suggesting that the extent of bound secondary label corresponds to the density of available Fc.
The polystyrene microparticles are meant to demonstrate a proof-of-concept of how Fc-functionalized biomaterials can be designed to modulate complement activation. Given that polystyrene is not degradable under physiological conditions, future work will utilize Fc- decorated decomposable polymers, such as chitosan and PLGA, that display a high density of functional groups for derivatization and/or C3b binding, namely hydroxyl39, 40 amine39, 41, and carboxyl groups42. These particles will also be of smaller size, as we have previously demonstrated that smaller particles (0.5 μm) functionalized in a similar manner activate complement more extensively than 1 μm particles.15 Ultimately we envision these particles for integration into a dialysis-type machine, in which complement mediators are sequestered. It is well-known that hemodialysis and hemodialysis filters in current use activate complement, leading to chronic inflammation that may result in worsened morbidity and mortality.43, 44 We are also currently investigating complement activation prompted by engineered virus-like particles, which are of a more therapeutically-viable scale (∼ 30 nm in diameter). At this size, the EPR effect can be exploited45 and the particles can be carried by lymphatic conduits.46
Complement Toxicity to E. coli in Serum Can Be Tuned By Fc Microparticle Number
The effects of Fc microparticle numbers were evaluated over a range of particle-to-bacterium ratios (Fig 4A). Various numbers of Fc microparticles were cultured with E. coli with constant diluted serum volume and bacteria number. At the 1:1 microparticle to bacterium ratio for both oriented and non-oriented particles, the Fc microparticles did not affect bacterial cytotoxicity compared to serum alone. Higher numbers of Fc microparticles altered the cytotoxic impact of the serum. The non-oriented particles showed maximal increase of serum cytotoxicity at the 100:1 condition, though this was reduced as we increased the microparticles per bacterium ratio. In contrast, the oriented Fc particles increased complement sequestration with increasing particle number, monotonically reducing serum cytotoxicity.
Fig 4.
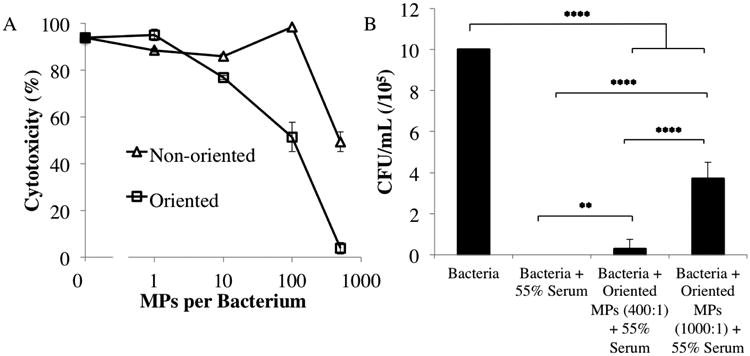
The relationship between cytoprotection and microparticle-to-cell ratio for oriented microparticles (A) The 1 μm 2:1 oriented microparticles were added to approximately 500,000 E. coli in 1% serum (of total volume) at various microparticle-to-cell ratios. The 125 μL total volume included: 500,000 E. coli (10 μL in broth), microparticle solution at the specified ratio (30 μL in PBS), serum (1% of total volume), and broth. Each subsequent dilution is 10× less concentrated than the last. Two way ANOVA was performed and showed significant difference between non-oriented and oriented effects on serum cytotoxicity (overall p-value(prob>F)=0.0017, and p-value(prob>t) = 0.0377 for effect of oriented vs. non-oriented). The effect of oriented microparticles on E. coli viability at physiological levels of normal human serum (B) Serum was added to 55% of the total volume. The 1 μm 2:1 oriented microparticles significantly increased cell viability in the presence of serum at both 400:1 and 1000:1 microparticle-to-cell ratios. * p<0.05, ** p<0.01, *** p<0.001, and **** p<0.0001.
To demonstrate the functional effects of oriented Fc microparticles at physiological serum concentrations, oriented Fc microparticles and bacteria were incubated with 55% serum. Serum alone at this concentration is completely cytotoxic to bacteria (Fig 4B). However, for conditions to which microparticles were added (400:1 and 1000:1 microparticle-to-bacteria ratio) cytotoxicity was decreased and a significant number of colonies were counted when plated. The results show that the oriented Fc microparticles protect bacteria from complement cytolysis even in very hostile conditions.
C5a Binds Non-specifically to Both Oriented and Non-oriented Fc Microparticles
To determine the effect of particle Fc configuration in reducing the C5a level available, C5a ELISAs were conducted on serum simulating infection. Previous work established an average baseline C5a concentration in the serum of a healthy individual as approximately 7 ng/mL 47, which was confirmed by our results (Fig 5B). The addition of 250,000 bacteria to 100 μL serum solution causes a noticeable increase in C5a concentration. Notably, the addition of either non-oriented or oriented Fc microparticles reduces the serum C5a concentrations to levels below the normal serum concentration (Fig 5B). We also examined the impact of particle number on serum C5a level, and found that as microparticle number increased, the fraction of C5a removed from serum also increased (Fig 5C).
Fig 5.
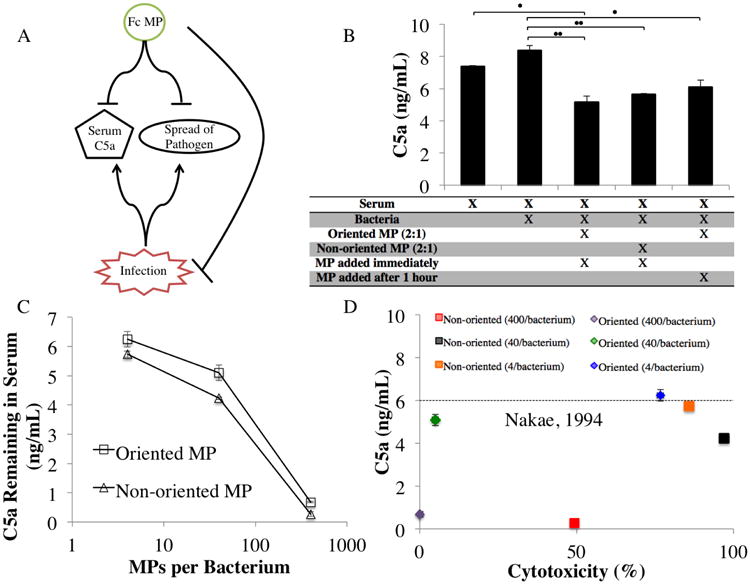
Graphic depicting concept for ideal sepsis treatment (A) This flow diagram illustrates the concept that an ideal treatment would halt spread of the pathogen while also mitigating serum C5a levels. Comparison of microparticle configurations in reducing C5a serum levels (B) The addition of 250,000 DH5a E. coli bacteria to a 100 μL solution of 14% serum showed an increase in C5a from 7 to 8.6 ng/mL. The subsequent addition of either oriented microparticles or non-oriented microparticles showed a significant reduction in serum C5a levels. Analyzing the effect of microparticle number on percent C5a reduction (C) Different microparticle-to-bacterium ratios were tested at increasing orders of magnitude. A positive correlation between microparticle number and C5a reduction in serum was observed. Note that the non-oriented Fc microparticles reduced C5a concentrations equivalently to the oriented microparticles at the particle-to-cell ratios tested (overall p-value (prob>F)=0.0343, and p-value (prob>t) = 0.0117 for effect of oriented vs. non-oriented). Examining the relationship between C5a serum levels and cytotoxicity in a microparticle design that may promote survival in patients with sepsis (D) The survival level of C5a binding is referenced from Nakae, 1994 47, which corresponds to serum C5a levels well-correlated to survival among sepsis patients.
We compared the cytotoxicity and serum C5a levels of several Fc microparticle types (Fig 5D). The results suggest that particle Fc configuration can be used as a means both to maximize bacterial cytotoxicity and to achieve non-specific C5a sequestration. We note that Fc microparticles with non-oriented Fc arrangement both increase bacterial cytotoxicity while lowering C5a in serum. As such, they warrant further investigation as a possible approach to combat bacterial-induced immune dysfunction as seen in sepsis, a systemic inflammatory response to excessive pathogen colonization that can result in multiple organ failure.48, 49 Utilizing the non-oriented Fc microparticles, immune signaling can be quieted while the underlying infection is targeted. A dashed line is included to indicate C5a levels above which a poorer prognosis is expected.47 The particles that achieve high bacterial cytotoxicity with high C5a sequestration represent a theoretical ideal for the treatment of immune overreactions such as sepsis (Fig 5A).
The modulation of functional complement responses via Fc microparticles is promising and calls for further examination into applications of complement modulation for novel antibiotics and as a countermeasure to sepsis and autoimmune disease. The appearance of high levels of serum anaphylatoxins, including C3a, C4a, and C5a 50, indicate a loss of control of complement activation and a dysregulation of homeostatic processes.47, 51 As such, a biomaterial approach using Fc microparticles to remove the aberrant factors may provide a pathway to regain regulation and mitigate the exaggerated pro-inflammatory pathways of worsening sepsis.28
Other work has demonstrated that limiting C5a levels may improve outcomes in sepsis, as septic mice treated with C5a antibodies showed significant increases in survival.52, 53 Given that the non-oriented particles can both sequester C5a and increase cytotoxicity, these particles may be relevant to effectively counter the underlying infection due to its combinatorial approach.49
Confocal Microscopy Confirms that TCC is Associated with Bacterial Cytotoxicity
We performed multicolor confocal imaging studies of bacteria and Fc microparticles of both non-oriented and oriented forms. Live bacteria were stained green, dead bacteria stained red, and TCC inserted in the bacterial membrane and on the microparticles themselves stained yellow (Fig 6A). Bacterial cytotoxicity was found to increase with proximity to Fc microparticles (Fig 6B), indicating that complement-mediated cytotoxicity exhibited a defined cytotoxic radius from the physical location of activation. Moreover, this cytotoxic radius of the particles was dependent upon the Fc orientation. TCC staining was found to preferentially localize with the dead bacteria stain, consistent with TCC-mediated bacterial cytotoxicity (Fig 6C). Analysis of the images indicates that the distance from the nearest Fc particle corresponding to 50% cytotoxicity was 3 μm for oriented Fc microparticles and increased to 6 μm for non-oriented Fc microparticles. Thus, we demonstrate that cell death is TCC-mediated and that the non-oriented Fc particles extend the effective range of functional complement activation.
The results of complement activation from the two Fc microparticle designs suggest that complement activation can be tuned in magnitude and physical distance to systematically enhance cytotoxicity and diminish anaphylatoxins in serum. Figs 1C and 1D summarize two possible models of the complement responses that arise from the two forms of Fc microparticles. The observed extended radius of complement products resulting from non-oriented Fc functionalization may stem from a variety of factors. Randomized Fc orientation, for example, may allow for faster and more probable release of complement products resulting from the decreased accessibility of Fc. This explanation is consistent with the high cytotoxicity also observed for low-density oriented Fc microparticles (Fig 3B). The different effects exhibited by the different orientations of Fc linked to microparticles suggest that multiple biophysical parameters can tune the functional responses of complement. This potential to tune the complement response is supported by observations that C1q can bind single IgG molecules, but multivalent C1q-Fc binding increases the binding constant three orders of magnitude 16, 54 and allows for subsequent interaction with the C1r-C1s proenzyme complex.54, 55 It is possible that the Fab portions of immobilized antibody may contribute to activate C3 via the alternative pathway, affecting the response observed for the non-oriented microparticles.56
There have been few direct studies of the physical factors concerning spatiotemporal activation of complement. When activated, C3b exposes a thioester bond 57 that can covalently attach to acceptor hydroxyl or amine groups on proteins, glycoproteins, carbohydrates, and phospholipid bacterial surfaces to initiate TCC formation and immune cytolysis.58 Using particles as the site of complement activation to affect bacteria distant from the particle would seem to be a limited approach due to the short half-life of the thioester bond of activated C3b. In studies of the binding of C3 to trypsin-conjugated agarose, Sim et al. found a lower bound for the thioester half-life of 60 μs before hydrolysis and inactivation of ‘nascent’ C3b 59, though no maximum half-life was established. Assuming a diffusion constant of 4.5 × 10-7 cm2/s 60 three dimensional diffusional motion for 60 μs produces an effective radius of ∼0.19 μm. Targets beyond this distance would seem to require a longer lifetime of the thioester bond, or a transport mechanism in addition to diffusion, e.g. natural convectional flows due to static thermal gradients. In another study using electron microscopy, the site of C3 binding was shown to be topographically distinct from the site of C3 activation.61 Together these studies support a complement activation model in which a transport mechanism can create a reaction zone distinct from the activation site.
As the magnitude and effective cytotoxic radius of complement is expanded, non-target host cells may experience increased susceptibility. Red blood cells are well-known to be lysed by increased complement activation.62, 63 Under normal conditions, complement is stringently regulated to avoid complement activation against host tissues. We direct the reader to a comprehensive review on the subject.64, 65 Host cells both display surface glycoproteins and/or are bound by soluble mediators that prevent inappropriate terminal complement activation. The host cell surface glycoprotein CD59 impedes the formation of TCC (consisting of proteins C5b, C6, C7, C8, and C9) by blocking the polymerization of C9 to the other constituents.66 Surface-bound complement receptor 1 expressed predominantly by erythrocytes, leukocytes, and monocytes and the more universal CD55 (decay-accelerating factor) compete with the soluble mediator factor B of the alternative pathway for surface-bound C3b, inhibiting the formation of the alternative pathway C3 convertase.67 CD55 also binds surface-bound C4b; this prevents the cleavage of C2 by C4b, blocking the formation of the classical C3 convertase.68 CD46, another widely-expressed surface-bound glycoprotein, also binds C3b, and serves as a cofactor of factor I, which inactivates bound C3b to iC3b.67, 69 In this way, formation of both the alternative C3 convertase and the subsequent C5 convertase are hindered. Finally, circulating factor H selectively binds to vertebrate cells via recognition of their sialic acid residues, where it both acts as a cofactor for factor I and competes with factor B to obstruct alternative C3 convertase formation.67 Although complement dysregulation is a hallmark of a number of pathologies, these are usually accompanied by deficient or defective complement regulators.70 Excess complement activation may result in off-target tissue injury via both direct lysis and the release of proteolytic enzymes from mesenchymal cells stimulated by TNF-α produced by upstream C3a/C5a activity.71 Future work in animal models will be undertaken to determine the most effective, tolerable dosing such that a complement-based therapy may be realized. In addition, targeting the complement activation to specific bacteria types may improve future selectivity in the antibiotic effect.72
Conclusions
Herein we have demonstrated that microparticles differentially functionalized with Fc prompt opposite complement-mediated outcomes. Those with oriented Fc molecules bind several key complement proteins: iC3b, C5a, and TCC. This binding results in an increased cytoprotective effect on E. coli, even in the presence of normally cytotoxic, physiological serum levels. By contrast, microparticles presenting non-oriented antibody molecules prompt increased generation of iC3b and TCC while still binding comparable levels of C5a, resulting in an increased cytolytic effect on E. coli compared to serum alone. Although the oriented microparticles are more apt to sequester C5a, both modalities display this capability. It is important to note that these data support a congruency between the Fc orientation/binding studies (AFM and Flow Cytometry) and the functional (ELISA and Cytotoxicity) assays. Overall, these data suggest that the classical complement pathway can be selectively modulated by Fc presentation and orientation.
Supplementary Material
Acknowledgments
The authors thank the Georgia Research Alliance through the Immunoengineering Seed Grant for support of this research as well as the NSF DMR division (1507238). The authors would also like to thank the Petit Scholars program for support of BAH. Research reported in this presentation was supported by the National Institutes of Health under 5T32EB006343T32 (GT BioMAT training grant) at the Georgia Institute of Technology (MCB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DH5α bacteria were provided courtesy of Caitlin Austin. The authors are grateful to Matt Devlin and Michael Harrington for their assistance in preliminary experiments.
References
- 1.Heinrich V, Lee CY. J Cell Sci. 2011;124:3041–3051. doi: 10.1242/jcs.086413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkin J, Cohen B. Lancet. 2001;357:1777–1789. doi: 10.1016/S0140-6736(00)04904-7. [DOI] [PubMed] [Google Scholar]
- 3.Tan TT, Coussens LM. Curr Opin Immunol. 2007;19:209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neill CP, Lee LK, Swartz MA, Hubbell JA. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 5.Moghimi SM, Andersen AJ, Ahmadvand D, Wibroe PP, Andresen TL, Hunter AC. Adv Drug Deliv Rev. 2011;63:1000–1007. doi: 10.1016/j.addr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Montdargent B, Labarre D, Jozefowicz M. J Biomater Sci Polym Ed. 1991;2:25–35. doi: 10.1163/156856291x00034. [DOI] [PubMed] [Google Scholar]
- 7.Szebeni J, Muggia F, Gabizon A, Barenholz Y. Adv Drug Deliv Rev. 2011;63:1020–1030. doi: 10.1016/j.addr.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 8.Porter RR, Reid KB. Advances in protein chemistry. 1979;33:1–71. doi: 10.1016/s0065-3233(08)60458-1. [DOI] [PubMed] [Google Scholar]
- 9.Schumaker VN, Zavodszky P, Poon PH. Annu Rev Immunol. 1987;5:21–42. doi: 10.1146/annurev.iy.05.040187.000321. [DOI] [PubMed] [Google Scholar]
- 10.Duncan AR, Winter G. Nature. 1988;332:738–740. doi: 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- 11.Janeway CA, Jr, Travers P, Walport M. Immunobiology: The Immune System in Health and Disease. Garland Science; New York: 2001. [Google Scholar]
- 12.Shin HS, Snyderman R, Friedman E, Mellors A, Mayer MM. Science. 1968;162:361–363. doi: 10.1126/science.162.3851.361. [DOI] [PubMed] [Google Scholar]
- 13.Lachmann PJ, Hughes-Jones NC. Springer Seminars in Immunopathology. 7:143–162. doi: 10.1007/BF01893018. [DOI] [PubMed] [Google Scholar]
- 14.Pacheco P, White D, Sulchek T. PLoS One. 2013;8 doi: 10.1371/journal.pone.0060989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pacheco P, Le B, White D, Sulchek T. Nano LIFE. 2013;3 doi: 10.1142/S1793984413410018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJR, van de Winkel JGJ, Wilson IA, Koster AJ, Taylor RP, Saphire EO, Burton DR, Schuurman J, Gros P, Parren PWHI. Science (New York, NY) 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes-Jones NC, Gorick BD, Howard JC, Feinstein A. European journal of immunology. 1985;15:976–980. doi: 10.1002/eji.1830151003. [DOI] [PubMed] [Google Scholar]
- 18.Cragg MS, Morgan SM, Chan HT, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ. Blood. 2003;101:1045–1052. doi: 10.1182/blood-2002-06-1761. [DOI] [PubMed] [Google Scholar]
- 19.Ahsan F, Rivas IP, Khan MA, Torres Suárez AI. Journal of Controlled Release. 2002;79:29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 20.Champion JA, Walker A, Mitragotri S. Pharmaceutical Research. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallo P, Gonçalves R, Mosser DM. Immunology Letters. 2010;133:70–77. doi: 10.1016/j.imlet.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Molecular Immunology. 2007;44:82–94. doi: 10.1016/j.molimm.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 23.Tang L, Liu L, Elwing HB. Journal of Biomedical Materials Research. 1998;41:333–340. doi: 10.1002/(sici)1097-4636(199808)41:2<333::aid-jbm19>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 24.Mosqueira VCF, Legrand P, Gulik A, Bourdon O, Gref R, Labarre D, Barratt G. Biomaterials. 2001;22:2967–2979. doi: 10.1016/s0142-9612(01)00043-6. [DOI] [PubMed] [Google Scholar]
- 25.Kazatchkine MD, Carreno MP. Biomaterials. 1988;9:30–35. doi: 10.1016/0142-9612(88)90066-x. [DOI] [PubMed] [Google Scholar]
- 26.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Nat Biotech. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 27.Thomas SN, van der Vlies AJ, O'Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Biomaterials. 2011;32:2194–2203. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 28.Ward PA. Nature Reviews Immunology. 2004;4:133–142. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 29.Kohl J. Immunol Res. 2006;34:157–176. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 30.Saha B, Saikia J, Das G. RSC Advances. 2013;3:7867–7879. [Google Scholar]
- 31.Yoon JY, Park HY, Kim JH, Kim WS. Journal of Colloid and Interface Science. 1996;177:613–620. [Google Scholar]
- 32.Haider A, Potter D, Sulchek TA. Proceedings of the National Academy of Sciences. 2016 doi: 10.1073/pnas.1608792113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lachmann PJ, Thompson RA. The Journal of experimental medicine. 1970;131:643–657. doi: 10.1084/jem.131.4.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iijima M, Somiya M, Yoshimoto N, Niimi T, Kuroda S. Scientific reports. 2012;2:790. doi: 10.1038/srep00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouerghi O, Touhami A, Othmane A, Ben Ouada H, Martelet C, Fretigny C, Jaffrezic-Renault N. Sensors and Actuators B: Chemical. 2002;84:167–175. [Google Scholar]
- 36.Andersson J, Ekdahl KN, Lambris JD, Nilsson B. Biomaterials. 2005;26:1477–1485. doi: 10.1016/j.biomaterials.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Savay S, Szebeni J, Baranyi L, Alving CR. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2002;1559:79–86. doi: 10.1016/s0005-2736(01)00408-4. [DOI] [PubMed] [Google Scholar]
- 38.Goat anti-Rabbit IgG (H+L) Secondary Antibody, DyLight 650 conjugate. 2016 https://www.thermofisher.com/antibody/product/Goat-anti-Rabbit-IgG-H-L-Secondary-Antibody-Polyclonal/84546.
- 39.Law SK, Dodds AW. Protein Science: A Publication of the Protein Society. 1997;6:263–274. doi: 10.1002/pro.5560060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Nature biotechnology. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Yin Y, Wang L, Zhang W, Chen X, Yang X, Xu J, Ma G. Biomacromolecules. 2013;14:3321–3328. doi: 10.1021/bm400930k. [DOI] [PubMed] [Google Scholar]
- 42.Thomas SN, van der Vlies AJ, O'Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Biomaterials. 2011;32:2194–2203. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 43.Ekdahl KN, Lambris JD, Elwing H, Ricklin D, Nilsson PH, Teramura Y, Nicholls IA, Nilsson B. Advanced drug delivery reviews. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reis ES, DeAngelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD. Immunobiology. 2015;220:476–482. doi: 10.1016/j.imbio.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren Y, Mu Y, Jiang L, Yu H, Yang S, Zhang Y, Wang J, Zhang H, Sun H, Xiao C, Peng H, Zhou Y, Lu W. International Journal of Pharmaceutics. 2016;502:249–257. doi: 10.1016/j.ijpharm.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 46.Cubas R, Zhang S, Kwon S, Sevick-Muraca EM, Li M, Chen C, Yao Q. Journal of immunotherapy (Hagerstown, Md: 1997) 2009;32:118–128. doi: 10.1097/CJI.0b013e31818f13c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakae H, Endo S, Inada K, Takakuwa T, Kasai T, Yoshida M. Research communications in chemical pathology and pharmacology. 1994;84:189–195. [PubMed] [Google Scholar]
- 48.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD. Nature immunology. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charchaflieh J, Wei J, Labaze G, Hou YJ, Babarsh B, Stutz H, Lee H, Worah S, Zhang M. Hindawi Publishing Corporation. 2012;2012:8. doi: 10.1155/2012/407324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gennaro R, Simonic T, Negri A, Mottola C, Secchi C, Ronchi S, Romeo D. European Journal of Biochemistry. 1986;155:77–86. doi: 10.1111/j.1432-1033.1986.tb09460.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakae H, Endo S, Inada K, Yoshida M. Surgery today. 1996;26:225–229. doi: 10.1007/BF00311579. [DOI] [PubMed] [Google Scholar]
- 52.Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, Markiewski MM, Mastellos D, Strey CW, Pierson CL, Lambris JD, Zetoune FS, Ward PA. The Journal of Clinical Investigation. 110:101–108. doi: 10.1172/JCI15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 54.Feinstein A, Richardson N, Taussig MI. Immunology today. 1986;7:169–174. doi: 10.1016/0167-5699(86)90168-4. [DOI] [PubMed] [Google Scholar]
- 55.Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Trends in Immunology. 2004;25:368–373. doi: 10.1016/j.it.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 56.Antón LC, Ruiz S, Barrio E, Marqués G, Sánchez A, Vivanco F. European Journal of Immunology. 1994;24:599–604. doi: 10.1002/eji.1830240316. [DOI] [PubMed] [Google Scholar]
- 57.Abdul Ajees A, Gunasekaran K, Volanakis JE, Narayana SVL, Kotwal GJ, Krishna Murthy HM. Nature. 2006;444:221–225. doi: 10.1038/nature05258. [DOI] [PubMed] [Google Scholar]
- 58.Müller-Eberhard HJ, Dalmasso AP, Calcott MA. The Journal of Experimental Medicine. 1966;123:33–54. doi: 10.1084/jem.123.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sim RB, Twose TM, Paterson DS, Sim E. Biochemical Journal. 1981;193:115–127. doi: 10.1042/bj1930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tack BF, Prahl JW. Biochemistry. 1976;15:4513–4521. doi: 10.1021/bi00665a028. [DOI] [PubMed] [Google Scholar]
- 61.Mardiney MR, Müller-Eberhard HJ, Feldman JD. The American Journal of Pathology. 1968;53:253–261. [PMC free article] [PubMed] [Google Scholar]
- 62.Packman CH, Rosenfeld SI, Jenkins DE, Thiem PA, Leddy JP. Journal of Clinical Investigation. 1979;64:428–433. doi: 10.1172/JCI109479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Girard-Pierce KR, Stowell SR, Smith NH, Arthur CM, Sullivan HC, Hendrickson JE, Zimring JC. Blood. 2013;122:1793–1801. doi: 10.1182/blood-2013-06-508952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim DD, Song WC. Clinical immunology (Orlando, Fla) 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 65.Zipfel PF, Skerka C. Nature reviews Immunology. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 66.Maio M, Brasoveanu LI, Coral S, Sigalotti L, Lamaj E, Gasparollo A, Visintin A, Altomonte M, Fonsatti E. International journal of oncology. 1998;13:305–318. doi: 10.3892/ijo.13.2.305. [DOI] [PubMed] [Google Scholar]
- 67.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Frontiers in Immunology. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lublin DM, Atkinson JP. Annual Review of Immunology. 1989;7:35–58. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- 69.Riley-Vargas RC, Gill DB, Kemper C, Liszewski MK, Atkinson JP. Trends in Immunology. 2004;25:496–503. doi: 10.1016/j.it.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 70.Markiewski MM, Lambris JD. The American Journal of Pathology. 2007;171:715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beutler B. Journal of investigative medicine: the official publication of the American Federation for Clinical Research. 1995;43:227–235. [PubMed] [Google Scholar]
- 72.Tang JL, Schoenwald K, Potter D, White D, Sulchek T. Langmuir: the ACS journal of surfaces and colloids. 2012;28:10033–10039. doi: 10.1021/la3010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.