

Abstract
To understand how cells form tissues, we need to understand how the tyrosine kinases are involved in controlling cell mechanics, whether they act directly as parts of mechanosensing machines or indirectly. Cells test the critical parameter of matrix rigidity by locally contracting (“pinching”) matrices and measuring forces, and the depletion of contractile units causes transformation. We report here that knocking down the receptor tyrosine kinases (RTKs), AXL, and ROR2, alters rigidity sensing and increases the magnitude or duration of local contraction events, respectively. Phospho-AXL and ROR2 localize to contraction units and bind major contractile components, tropomyosin 2.1 (AXL), myosin IIA (AXL), and filamin A (ROR2). At a molecular level, phosphorylated AXL localizes to active myosin filaments and phosphorylates tropomyosin at a tyrosine critical for adhesion formation. ROR2 binding of ligand is unnecessary, but binding filamin A helps function. Thus, AXL and ROR2 alter rigidity sensing and consequently morphogenic processes by directly controlling local mechanosensory contractions without ligands.
Keywords: Rigidity sensing, nano pillars, protein tyrosine kinases, AXL, ROR2, mechanotransduction
Graphical abstract
There is considerable evidence from genetic studies that tissue morphogenesis, cell migration, and polarization are controlled by receptor tyrosine kinase (RTK) activity in normal and disease states.1,2 Many neoplastically transformed cells can ignore the softness of their microenvironment,3 and RTK alterations may play a significant role in these defects. However, the links between the mechanosensory systems and the RTKs are still unknown.
The ability of cells to exert contractile forces on the matrixadhesion sites was recognized in pioneering work4 that triggered the development of various approaches that made it possible to quantitatively assess these forces.5–10 The recent development of submicrometer elastomeric pillar arrays enabled measurement of the local contractile activity at the periphery of spreading cells with high spatial resolution and linked those contractions to the rigidity sensing of the matrix.11,12
In particular, neighboring pillars located close to the cell leading edge often bend toward each other, revealing previously overlooked “pinching” activity.11 It is hypothesized that local contractile units (CUs), resembling muscle sarcomeres, mediate these contractions.11,13,14 Indeed, microscopy reveals that α-actinin is localized close to the pillars’ tips, while myosin-II appears in between the neighboring pillars, in agreement with a predicted sarcomere-like organization.12 This raises the question of whether such “sarcomere-like” local pinching activity during the initial spreading stage is related to a cell adhesion-dependent mechanosensing process.
In experiments with cells lacking the receptor-like protein tyrosine phosphatase alpha (RPTPα), local contractions did not form,11 and cells were unable to sense substrate rigidity.15 A screen of the effects of depletion of each of the 85 human RTKs on rigidity sensing identified about 16 RTKs that were needed for normal rigidity sensing.16 This raised the possibility that the tyrosine kinases might be involved in the pinching process. We have focused on two, AXL and ROR2, since cells lacking these kinases developed elongated adhesions and stress fibers on both rigid (2 MPa) and soft (5 kPa) surfaces.16 In addition, alterations of AXL are often involved in neoplastic transformation, invasion, and metastasis.17–19 On the other hand, ROR2, a Wnt5A receptor in the noncanonical Wnt signaling pathway, is required for limb formation and establishment of planar cell polarity in embryos.20,21 Plus, it is involved in the epithelial-to-mesenchymal transition, invasion, and metastasis in neoplastic development.20,22
To further elucidate the relationship between local contractility and adhesion-mediated mechanosensing, we examine here the local pinching activity of cells lacking two receptor tyrosine kinases, AXL and ROR2.16 Previous studies indicate that the overall cell contractility response to substrate rigidity is not affected by these knockdowns: both AXL and ROR2 knockdown cells exert greater global traction (measured as “net contractile moment”6) on stiff than on soft substrates, like control cells.16 Surprisingly, AXL or ROR2 depletion alters the local mechanical steps in rigidity sensing by increasing either magnitude or duration of elementary contractile events at the periphery of early spreading cells, indicating that these kinases regulate local rather than global contractions.
From several studies of cell spreading on the submicrometer pillar arrays, a working model of rigidity sensing has emerged.11–13 Basically, sarcomere-like contractile units contract to a constant displacement of ~120 nm for ~30 s, and if contractile force exceeds a threshold of ∼25 pN, then nascent matrix adhesions will transition to mature adhesions.11–14,23 In a similar way, we sense the rigidity of a material by pinching it to roughly measure the force needed to produce a pinch of constant displacement. Our finding that rigidity-related kinases affect the mechanical parameters of the pinching process and modify sarcomere components strongly supports this working model.
Fibroblasts Develop Standard Pinches for Rigidity Sensing
Human foreskin fibroblasts (HFFs) spread normally on the tops of cylindrical fibronectin-coated PDMS pillars (500 nm diameter and 1 μm center-to-center) as on fibronectin-coated glass (ref 11 and Figure 1A). The local forces exerted by HFF cells on the pillars at the early stage of spreading (10–30 min after plating) were analyzed by examining the dynamics of pillar deflections for pillars covered during cell spreading (Figure 1B). Three different heights of the pillars (750, 1500, and 2000 nm) were used, and the effective stiffness of the pillar substrates were calculated24 to be 125, 16, and 7 kPa, respectively. In agreement with previous studies,11,12 the displacements of the neighboring pillars were often directed toward each other (Figure 1A) and highly correlated in time (Figure 1B), indicating that a single contractile unit bent both pillars.
Figure 1.
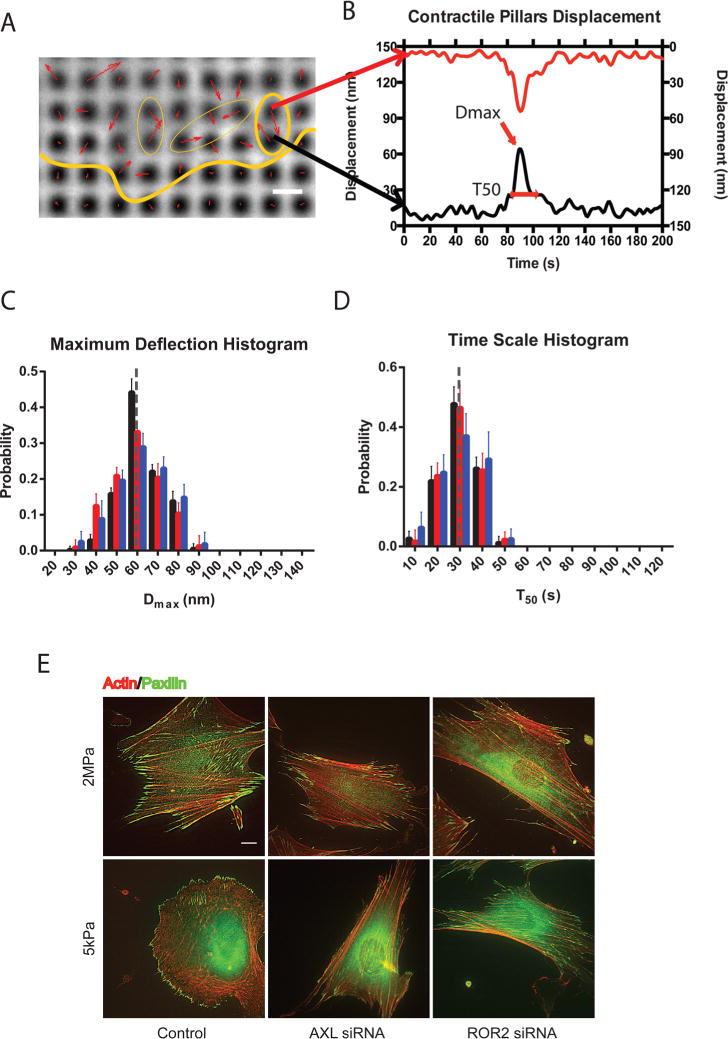
HFF cells pull to a maximum displacement of 60 nm over a 30 s period regardless of the pillar rigidity. (A) Displacements (red arrows) of pillars that show several contractile pairs (marked with yellow ovals) at the periphery of a spreading HFF cell (cell edge marked with yellow line; scale bar, 1 μm). (B) Displacements as a function of time of two opposing pillars showing peak displacement (Dmax) and time of contraction above half-maximal displacement (T50). (C) Histogram plots of Dmax for normal HFF cells spreading on pillars of different heights. Black represents 750 nm high pillars, red for 1500 nm high pillars, and blue for 2000 nm high pillars. Error bars represent SDs of the fractions from analyses of 10 different cells in 3 independent experiments. The gray line highlights the histogram peak at Dmax = 60 nm. (D) Distributions of T50 for normal HFF cells spreading on pillars of different heights. Gray line highlights the histogram peak at T50 = 30 s. (E) Staining for actin (red) and paxillin (green) in control cells (left column) and cells with AXL (middle column) or ROR2 (right column) knockdown on hard (upper panel) and soft (lower panel) PDMS surfaces after 6 h plating.
For further analysis, each pillar from a contractile pair was characterized by two values: the magnitude of the maximal peak deflection during the period of observation, Dmax, and the duration of the maximal peak of deflection, T50 (the full width at half-maximum value, fwhm) (see Figure 1B and ref 11). We computed the distribution and median values of these parameters for each cell separately. In the final statistics, we characterized each type of treatment by pooling the median data obtained for the similarly treated cells.
We found that the displacements of local paired contractions of control HFF cells were similar (Dmax around 60 nm for a single pillar) irrespective of the pillars’ height (Figure 1C), indicating that cells exerted higher forces on shorter, more rigid pillars. The average deflection time was also similar (T50 around 30 s) for the three heights of pillars (Figure 1D). The noise levels for reference pillars far away from the cells were Dmax ≈ 13 nm and T50 ≈ 5 s, which were significantly lower than for cellular pulls (Supplementary Figure 1A, B, and C). Thus, paired pillars were contracted toward each other by an average of 60 nm for a deflection time of 30 s, and this was consistent with previously published studies on mouse embryo fibroblasts.11,12
We analyzed the effect of knockdown of two protein tyrosine kinases, AXL and ROR2, on the local contractility assessed by pillar deflections. These kinases were shown to be involved in adhesion-mediated mechanosensing.16 On the soft substrate (Young modulus E = 5 kPa), control HFF cells were rounder (lower aspect ratio) and had a reduced focal adhesion area, as compared to the rigid substrate (E ≥ 100 kPa).16 In contrast, the AXL- or ROR2-depleted cells had similar shapes, aspect ratios, and focal adhesion areas on the stiff and soft substrates (Figure 1E, Supplementary Figure 1D, and ref 16). Thus, the depleted cells were able to respond to soft surfaces as if they were rigid.
Knockdown of AXL and ROR2 Alters Rigidity Sensing Displacement and Time Scale, Respectively
After AXL siRNA transfection, cells had significantly reduced AXL protein levels (Figure 2A) and produced significantly greater maximum deflections (Dmax) (Figure 2B and C). However, the duration of deflection (T50) was the same as control cells on both stiff and soft pillars (compare the histograms in Figure 1C,D with those in Figure 2C and D, respectively, and Supplementary Figure 2). The expression of GFP-fused mouse AXL, which was not targeted by the siRNA, rescued the normal contractions (Figures 2E and F). This indicated that the change in cell contractility was not a result of off-target effects of the siRNA. Thus, AXL depletion caused a significant increase in Dmax but no change in the duration of deflection.
Figure 2.
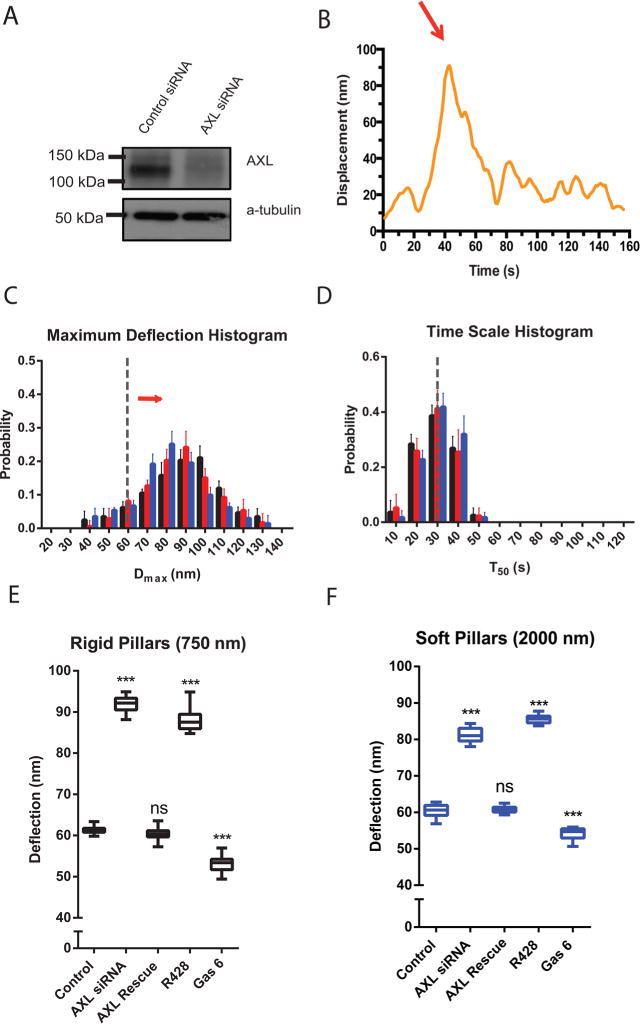
Knockdown of AXL increases the displacement. (A) AXL level is knocked down in Western blots of control siRNA vs AXL siRNA transfected cells. (B) Displacement vs time of a single pillar in a contractile unit under an AXL knockdown cell. (C) Histograms of Dmax values for AXL knockdown cells spreading on pillars of different heights. Black represents 750 nm high pillars, red for 1500 nm high pillars, and blue for 2000 nm high pillars. Error bars denote SDs of the percentages from 10 different cells in 3 independent experiments. The gray line highlights the control histogram peak at Dmax = 60 nm. The red arrow indicates the shifting of AXL knockdown histogram distribution. (D) Histograms of T50 values for AXL knockdown cells spreading on pillars of different heights. (E and F) Box-and-whisker plots of median values of Dmax for similarly treated cells on rigid pillars (E) and soft pillars (F) (n = 10 in each case; ***p < 0.001).
We next tested if AXL kinase activity was involved in regulating the pillar deflection using the AXL chemical inhibitor R42825 and the AXL-ligand, Gas 6, known to activate AXL in several cell types.26 The R428 inhibitor caused a significant increase in Dmax (Figure 2E and F), similar to the AXL knockdown. Stimulation with Gas 6 before and during cell spreading reduced the Dmax values (Figure 2E and F) and broadened the Dmax distribution. Thus, inhibiting AXL activity caused an increase in deflection of both soft and rigid pillars, while activation of AXL by Gas 6 caused a slight decrease.
ROR2 depletion by siRNA also altered the local contractile activity (Figure 3A and B). ROR2 knockdown cells exhibited a normal maximal pillar deflection (Dmax) (Figure 3C and Supplementary Figure 3). However, the duration of the deflection (T50) was significantly increased on both rigid and soft pillars, (∼50 s, as compared to ∼30 s in control cells) (Figure 3C–F). This difference was not due to siRNA off-target effects, since transfection of ROR2 depleted cells with a GFP-fused mouse ROR2 construct restored the normal T50 value (Figure 3E and F). Thus, ROR2 depletion increased the duration of local cell contractions.
Figure 3.
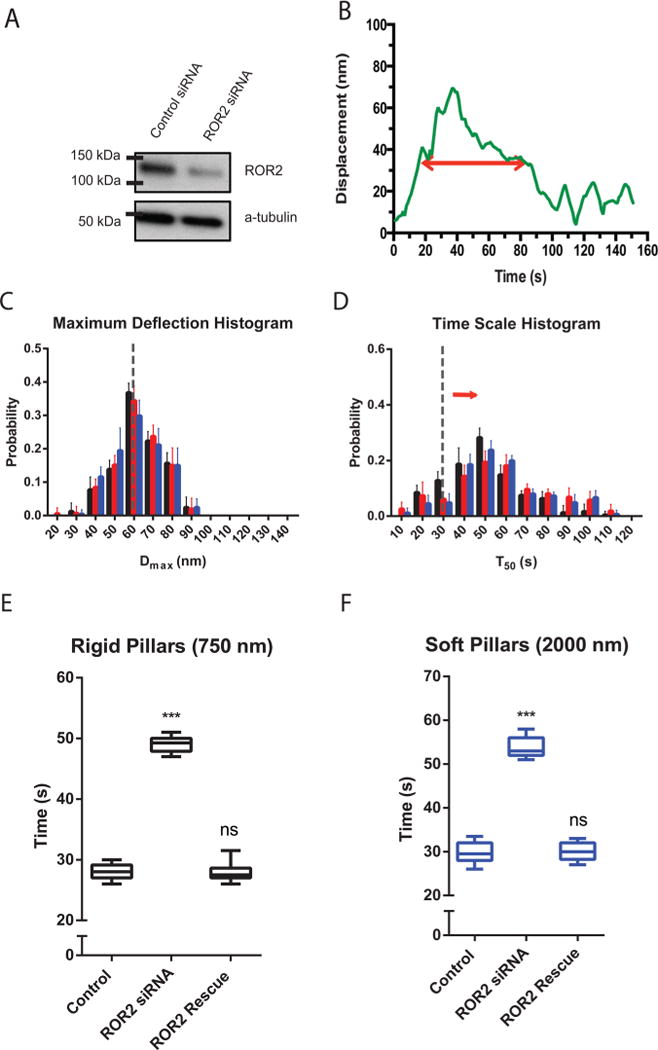
ROR2 silencing increases the duration of contractions, not length. (A) ROR2 level is knocked down in Western blot of ROR2 siRNA vs control siRNA transfected cells. (B) Typical displacement vs time of a single pillar under a ROR2 knockdown cell. (C) Histogram plots of Dmax values for ROR2 knockdown cells spreading on pillars of three different heights. (D) Histogram plots of T50 values for ROR2 knockdown cells spreading on pillars of three different heights. Black represents 750 nm high pillars, red for 1500 nm high pillars, and blue for 2000 nm high pillars. Error bars denote SDs of the percentages from 10 different cells in 3 independent experiments. (E and F) Box-and-whisker plots of median values of T50 distributions for similarly treated cells on rigid pillars (E) and soft pillars (F) (n = 10 in each case; ***p < 0.001).
Depletion of AXL by 70% using siRNA in HFF cells did not affect endogenous ROR2 expression level significantly (Supplementary Figure 4). This suggested that these two kinases did not have any functional interaction with each other.
AXL Regulated Rigidity Sensing Threshold
Previous investigations12 indicated that rigidity sensing occurred early in the contraction process, when there was a 1–2 s pause at roughly 25 pN of force on the pillars that corresponded with a dramatic increase in the accumulation of α-actinin at the pillars. To more carefully examine contractile pillar displacement, we also tracked pillar movements using continuous bright field imaging at an imaging rate of 100 Hz.12 A higher accuracy (<1 nm) was obtained by filtering pillar displacement data with a 15-point median filter (Figure 4A and B gray line). After a step-detection algorithm detected discrete elementary steps (Figure 4A and B black line), we observed on rigid substrates (8.4 pN/nm) that the average number of steps before the first pause was 4, whereas on the soft substrate (1.6 pN/nm) this number increased to 11 with control cells (Figure 4B and Supplementary Figure 5A), consistent with previous observations.12 Surprisingly, after AXL depletion, the force generated before the pause was higher because of more steps on both stiff and soft pillars, as compared with the control group (Figure 4C and Supplementary Figure 5B). AXL knockdown cells made 11 steps before the first pause on stiff pillars (Figure 4E) and 19 steps on soft pillars (Figure 4F). At the same time, ROR2 silencing did not cause any change in the step number before the first pause on either soft or stiff pillars (Figure 4D–F and Supplementary Figure 5C). Thus, the early rigidity sensing pause threshold was increased by AXL depletion consistent with the increased magnitude of contraction.
Figure 4.
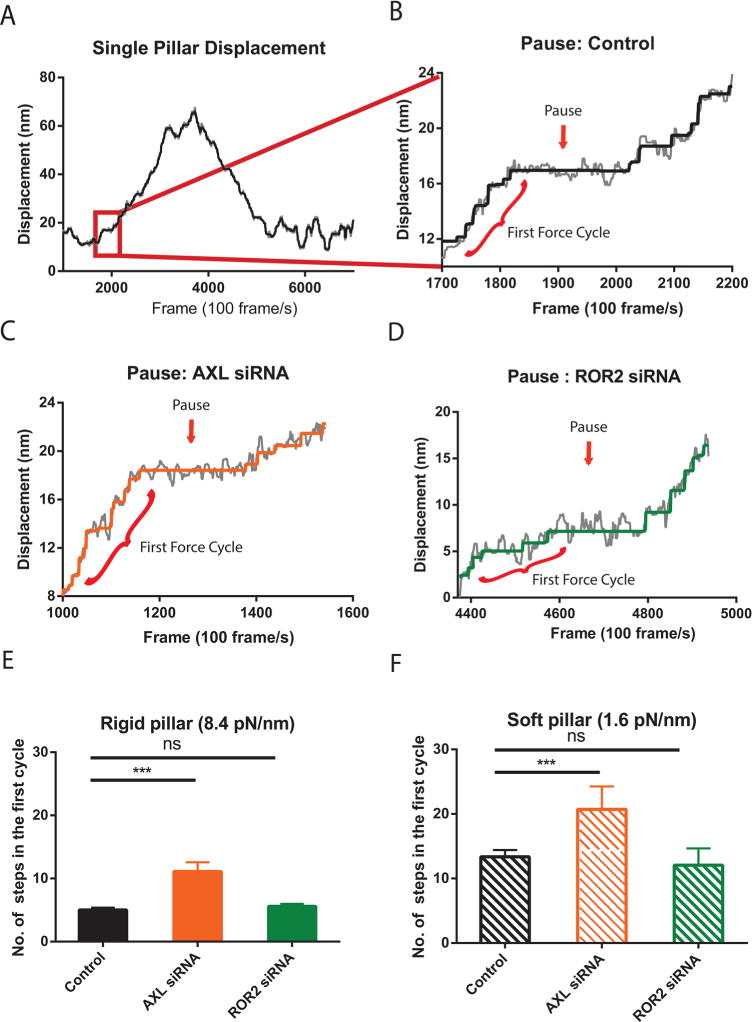
Depletion of AXL raises the force threshold for rigidity sensing. (A) Typical single pillar displacement in a contractile pair. The gray line is the median filtered data. The black line is the step-filtered curve. (B) A zoom-in graph of a typical pause during very early stage of pillar displacement. The red line highlighted the first force cycle. (C) A typical pause example of a single pillar under a AXL knockdown cell spreading on a stiff pillar surface. (D) A typical pause example of a single pillar under a ROR2 knockdown cell spreading on a stiff surface. (E and F) Bar plot of average ± SEM number of steps during first force cycle on stiff (E) and soft (F) pillar surface (n = 20 in each case; ***p < 0.001).
AXL and ROR2 Bind Different Components of Rigidity Sensing Complexes
Previous studies reported that local contraction units (CUs) had a sarcomere-like structure, with α-actinin concentrated around the edges of the pillars and myosin II located in between the contracted pillars.11,12 To investigate whether AXL and ROR2 were associated with the CUs, we analyzed if they colocalized with α-actinin after 15 min spreading on the pillars. Immunofluorescence revealed that phospho-AXL staining indeed overlapped with α-actinin-staining around the pillars at the cell periphery (Figure 5A and Supplementary Figure 6A). Staining of ROR2 also demonstrated some colocalization with α-actinin (Figure 5E and Supplementary Figure 6B), even though the ROR2-staining was also prominent in the central part of the cell. Thus, it appeared that AXL, and to some extent ROR2, was associated with local contraction units of spreading cells.
Figure 5.
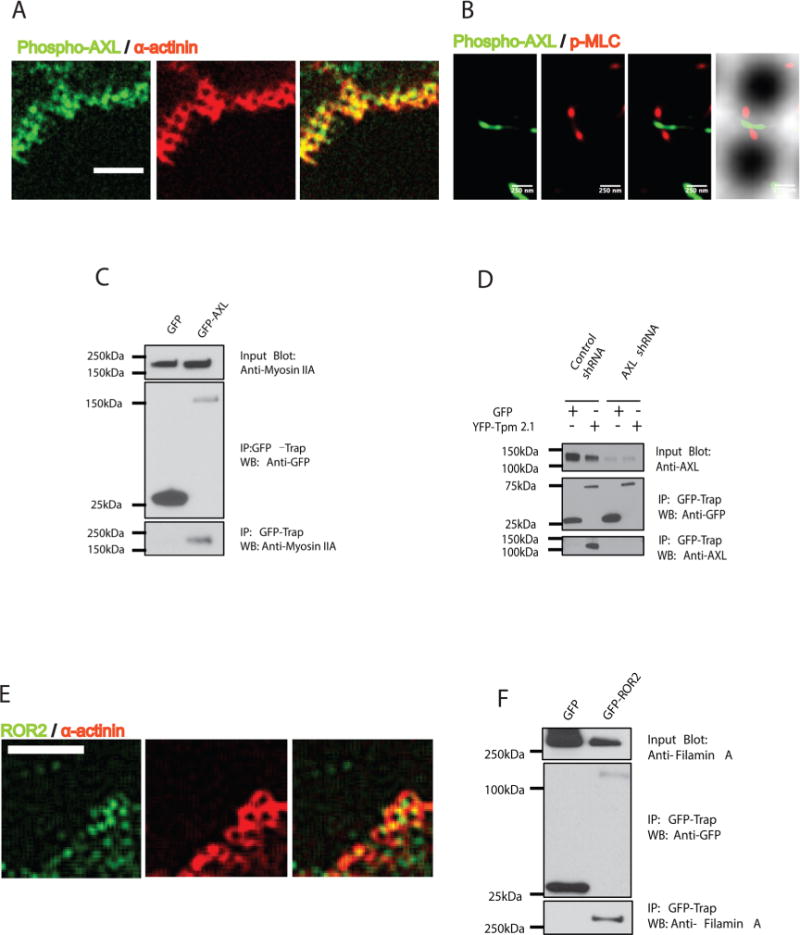
AXL and ROR2 associate with components of contraction units. (A) Phospho-AXL (green) colocalized around pillars together with α-actinin (red) at the cell periphery during initial spreading (15 min; scale bar 5 μm). (B) Phospho-AXL (green) overlaps p-MLC (red) in super-resolution images (3B analysis) in contractile regions at cell edges. (C) GFP-AXL transfected HFF cell lysates were immunoprecipitated with GFP nanotrap beads, followed by immunoblotting with anti-Myosin IIA antibody. (D) YFP-Tpm 2.1 transfected normal HFF cell or AXL knockdown cell lysates were immunoprecipitated with GFP nanotrap beads, followed by immunoblotting with anti-AXL antibody. (E) ROR2 (green) overlaps with α-actinin around the pillars at the cell periphery during initial spreading time (15 min; scale bar 5 μm). (F) GFP-ROR2 transfected HFF cell lysate was immunoprecipitated with GFP nanotrap beads, followed by immunoblotting with antifilamin A antibody.
To identify binding partners of AXL and ROR2 in the local contraction units, we performed a series of coimmunoprecipitation experiments. After GFP nanotrap beads pulled down GFP or GFP-fused proteins from lysates of transfected HFF cells, we probed for proteins of interest with specific antibodies. GFP-AXL, but not GFP alone, pulled down the endogenous myosin IIA heavy chain in HFF cells (Figure 5C). As a further test for this interaction, we immunoprecipitated AXL with GFP-myosin IIA (Supplementary Figure 7A). Previously, an interaction of AXL with the heavy chain of myosin IIB was also detected.27 In addition, YFP-Tpm 2.1 pulled down endogenous AXL from the lysate of control, but not from AXL-knockdown HFF cells (Figure 5D). In parallel experiments, interactions of ROR2 with either myosin-II or Tpm 2.1 were not detected (Supplementary Figure 7C and D). Instead, in a similar assay, ROR2 bound with filamin A (in agreement with previous publications28), while AXL did not (Figure 5F and Supplementary Figure 7B). Neither AXL nor ROR2 interacted with nonmuscle α-actinins 1 or 4 (Supplementary Figure 7E and F), indicating that they were not generally associated with actin-binding proteins. Altogether, these results showed that AXL and ROR2 specifically interacted with components of the local contraction unit complex.
Using Bayesian localization superresolution microscopy,29 we found a colocalization of phospho-AXL with active myosin II visualized by an antibody to phosphorylated myosin light chain (phospho-MLC). Phospho-AXL staining overlapped with ∼60% of myosin-II mini-filaments at the cell periphery (Figure 5B and Supplementary Figure 6C and D). These observations indicated that tyrosine kinases could locally modify the mechanosensory contractile units.
AXL Phosphorylates Tropomyosin 2.1 in Early Spreading
Many anchorage-independent cancer cells, for example, MDA-MB-231 cells, exhibited a very low expression level of Tpm 2.1.30,31 In previous pillar measurements,12 the MDA-MB-231 cell line was a good model to examine Tpm 2.1 roles in early rigidity sensing activity. To further investigate the relationship between AXL and Tpm 2.1, we transfected YFP-Tpm 2.1 or GFP in MDA-MB-231 and used GFP nanotrap beads to pull down proteins from cell lysates. YFP-Tpm 2.1 tyrosine phosphorylation in MDA-MB-231 cells was inhibited by the AXL inhibitor, R428, during initial spreading (Figure 6A and B). As a control, cells transfected with GFP-myosin IIA were treated similarly, and the level of p-Tyr reactivity was not inhibited by R428 (Supplementary Figure 8). This indicated that Tpm 2.1 was a target of AXL tyrosine phosphorylation during early spreading. Further, as previously reported,12 MDA-MB-231 cells produced a larger Dmax value (84 nm) compared with normal fibroblasts, and the density of contractile pairs was about fifty-fold lower than after expression of YFP-Tpm 2.1 (Supplementary Figure 9A). In addition to restoring the density of contractile pairs, YFP-Tpm 2.1 transfection restored normal contraction displacements in MDA-MB-231 cells (51 nm) (Figure 6C black groups). Interestingly, inhibition of AXL by R428 caused a dramatic increase in Dmax versus controls in YFP-Tpm 2.1 transfected groups (an increment of 47 nm) but not in nontransfected groups (an increment of 5 nm) (Figure 6F and Supplementary Figure 9B). Thus, Tpm 2.1 was phosphorylated by AXL and AXL kinase activity was needed for normal contractility in the initial spreading stage of MDA-MB-231 cells expressing Tpm 2.1.
Figure 6.
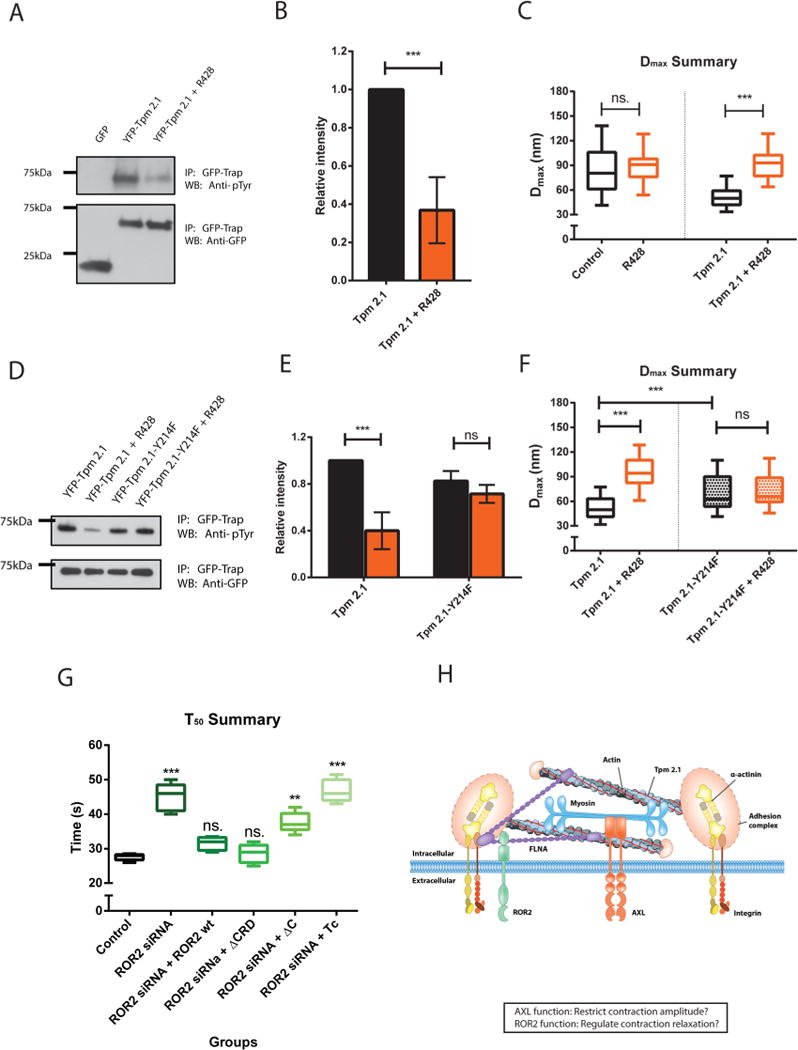
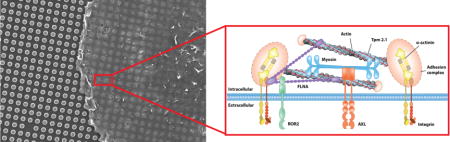
Tpm 2.1 is a downstream target of AXL during early spreading. The ROR2 ligand binding domain is not necessary for sarcomere pinching activity regulation. (A) GFP or YFP-Tpm 2.1 transfected MDA-MB-231 cells lysates were immunoprecipitated with GFP nanotrap beads, followed immunoblotting with anti-phosphotyrosine antibody under control or R428 treated conditions. (B) Relative intensity analysis of the blot. Black represents the control group, orange for R428 treated group; (n = 3 in each case; ***p < 0.001. (C) Box-and-whisker plots of median values of Dmax distributions for nontransfected or YFP-Tpm 2.1 transfected cells under control (black) or R428 (orange) treatment on 750 nm high pillars. (D) YFP-Tpm 2.1 or YFP-Tpm 2.1-Y214F transfected MDA-MB-231 cells lysates were immunoprecipitated with GFP nanotrap beads, followed by immunoblotting with antiiphosphotyrosine antibody under control or R428 treated conditions. (E) Relative intensity analysis of the blot (black for control group; orange for R428 treated group). (F) Box-and-whisker plots of median values of Dmax distributions for YFP-Tpm 2.1 or YFP-Tpm 2.1-Y214F transfected MDA-MB-231 cells under control (black) or R428 (orange) treatment on 750 nm high pillars. (G) Box-and-whisker plots of median values of T50 distributions for ROR2 silenced cells rescued with different mutants on 750 nm high pillars (***p < 0.001; **p < 0.01). (H) Cartoon model of the structure of a typical contraction unit with the relevant molecular components.
To better understand the function of Tpm 2.1 tyrosine phosphorylation in rigidity sensing, we mutated all of the possible tyrosine phosphorylation sites within Tpm 2.1. After reintroducing different tyrosine mutants of Tpm 2.1 in MDA-MB-231 cells, the tyrosine phosphorylation of only the Y214F mutant was unaffected by R428 inhibition (Figure 6D and E). In addition, different from Tpm 2.1 transfected groups, Dmax values of Tpm 2.1 -Y214F transfected MDA-MB-231 cells did not change significantly after R428 treatment (70–72 nm with R428, Figure 6F). (Compare the black box with the orange box on the left or right side, respectively). Finally, Tpm 2.1-Y214F did not restore normal adhesions as Tpm 2.1 did on fibronectin-coated glass (Supplementary Figure 10A, B, and C), whereas the phosphorylation-mimicking mutant of Tpm plasmid, Tpm 2.1-Y214E, partially restored the size of adhesions in MDA-MB231 cells (Supplementary Figure 10D). Thus, Tpm 2.1-Y214F was not able to rebuild normal contractility or adhesions, and it was not phosphorylated by AXL.
To determine which domain of ROR2 was required for its function in regulating contraction unit time scale, we examined three different ROR2 mutants that were reported before;28 (1) ΔCRD lacking the Wnt-5a binding site, (2) ΔC lacking the filamin A binding site PRD domain, and (3) Tc lacking both the kinase domain and PRD domain in ROR2 silenced HFF cells. ROR2-ΔCRD transfection restored the normal T50 value of 30 s as did ROR2-wt (wild type) transfection of ROR2 silenced HFF cells. On the other hand, ROR2-ΔC transfection gave a larger T50 value (37 s). In addition, the ROR2-Tc transfection showed no change in T50 value compared with ROR2 depleted cells (Figure 6G). Thus, the ROR2 kinase and filamin A binding domains but not ligand binding domain were important for its function in sarcomere unit regulation in early rigidity sensing.
In this study, we find that local contractions for matrix rigidity sensing are controlled by the RTKs, AXL and ROR2, in the absence of ligands. The alterations induced by knockdowns of AXL and ROR2 in local contractile activities are different. AXL knockdown significantly increases the amplitude of pillar deflections Dmax as well as the number of steps before a pause to reinforce the adhesions but does not affect the deflection time T50. In contrast, ROR2 knockdown does not affect Dmax or steps before pausing but significantly increases T50. These alterations in the contractile activity correlate with localization of these kinases to peripheral sarcomeric structures (more prominent for AXL), and physical association of AXL and ROR2 with specific sarcomeric proteins (myosin II and tropomyosin-2.1 for AXL and filamin A for ROR2). The interaction of AXL with endogenous myosin-IIA is supported both biochemically and by super-resolution imaging. The association with Tpm 2.1 is especially interesting because alterations in both AXL18,19 and Tpm 2.130,31 are related to disruption of adhesion-dependent signaling in the course of neoplastic transformation. Further, since the pause in the contraction correlates with reinforcement of the adhesion, the increase in the number of steps and consequently in the force before pausing could explain why softer surfaces are perceived as rigid in the AXL knockdown cells. Although the exact role of Tpm 2.1 in the control of the length of contractions is not clear, there is a definite correlation between the phosphorylation of Tpm 2.1 on tyrosine 214 by AXL and the regulation of contraction length. In addition, mutation of tyrosine 214 to phenylalanine causes dramatic changes in the adhesion size. Thus, we suggest that mechanosensing of rigidity by the contractile units involves a mechanical feedback between the AXL kinase, myosin II filaments and Tpm 2.1.
In the case of ROR2, its binding partner, filamin A, also has mechanosensory functions32,33 and has been linked to the relaxation of contractions.34 We understand less about the relaxation process than contraction; however, the rate of relaxation appears remarkably similar to the contraction rate. Filamin A helps to organize contractile networks in the cytoplasm and to link the microtubule-rich endoplasm with the peripheral ectoplasm. As a cross-linker in filament networks, filamin A could be an important site for regulation of the network tension. Thus, ROR2 could also cause a secondary relaxation of the local contractions through an indirect effect on contractile units or on signals from the network that would inhibit myosin and produce relaxation.
Even though the effects of AXL and ROR2 knockdown on force generation are different, these knockdowns produce similar effects on the focal adhesion response to substrate rigidity. Since AXL-knockdown cells generate larger pillar deflections than control cells, they apply greater force on the nascent integrin adhesion; similarly, ROR2-knockdown cells generate longer deflections and therefore exert force on nascent adhesions for longer periods of time than control cells. Thus, we suggest that, even on soft substrates, the integral of contractile force over time for these knockdown cells is higher than threshold values needed to activate growth and maturation of the nascent adhesions.
In conclusion, (i) there is a pronounced effect of AXL and ROR2 knockdowns on local contractile activity; (ii) these kinases colocalize with local contractile units; (iii) they associate with mechanosensory sarcomeric proteins, and (iv) AXL kinase phosphorylation of Tpm 2.1 on tyrosine 214 is needed for activity. Thus, we suggest that these kinases are directly involved in controlling the contractile pinching activity at the cell periphery. Further, localization near the contractile units plays a decisive role in the regulation of adhesion-dependent mechanosensing by these protein tyrosine kinases. This is important for cell viability since the local contraction units are needed for cell apoptosis on soft surfaces and the depletion of the contraction units by either Tpm 2.1 or α-actinin 4 depletion causes growth on soft surfaces (transformation).12,23 One surprising aspect of RTK involvement in contractility is that neither the ligand of AXL nor ROR2 is present in the assay of the control cell contractions, indicating that the kinase roles in mechanosensing are independent of ligand. Further, the inhibition of AXL kinase in the absence of ligand decreases Tpm 2.1 phosphorylation, and the ligand-binding domain of ROR2 is not needed to restore normal activity. This is consistent with reports of EGF receptor activity being involved in cell mechanosensing in the absence of ligand.35 Obviously, other potential regulatory factors, such as other RTKs,16 phosphatases,1,36 or chemical species37 could affect similar or other characteristics of the sarcomeric contractile units. Further definition of how RTKs function in mechanosensing is important for understanding how mechanical factors interplay with cell biochemical pathways to control organ form and mechanics.
Supplementary Material
Acknowledgments
We thank all of the members of the Sheetz lab and Bershadsky lab for their kind help. We thank Y. Minami, M. Nishita, Q. Lu, and P. Gunning for providing plasmids, Shuamin Liu for useful Matlab scripts writing instructions; B. Zhang for helping preparing the illustrations and Z. Chen for providing MDAMB-231 cells. This research was supported by funding to the MBI, National University of Singapore. H.W. was supported by a Marie Curie International Outgoing Fellowship within the Seventh European Commission Framework Programme (PIOF-GA-2012-332045). A.D.B. holds the Joseph Moss Professorial Chair in Biomedical Research at the Weizmann Institute and is a Senior Principal Research Scientist at the National University of Singapore. M.P.S. is supported by NIH grants, NUS grants, and Mechanobiology Institute, National University of Singapore.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.6b02995.
Materials and methods and supplementary figures (PDF)
Author Contributions
B.Y., A.D.B., and M.P.S. designed the research; B.Y. performed the experiments and analyzed the data. Z.Z.L. contributed technical assistance with coimmunoprecipitation experiment. H.W. contributed to high frequency video microscopy, 3B super-resolution, and data analysis. F.H.M. contributed to pillar mold fabrication and SEM imaging. B.Y., A.D.B., and M.P.S. wrote the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Giannone G, Sheetz MP. Trends Cell Biol. 2006;16(4):213–23. doi: 10.1016/j.tcb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. Curr Opin Cell Biol. 2009;21(2):140–6. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butcher DT, Alliston T, Weaver VM. Nat Rev Cancer. 2009;9(2):108–22. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris AK, Wild P, Stopak D. Science. 1980;208(4440):177–9. doi: 10.1126/science.6987736. [DOI] [PubMed] [Google Scholar]
- 5.Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Nat Cell Biol. 2001;3(5):466–72. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 6.Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ. Am J Physiol Cell Physiol. 2002;282(3):C595–605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- 7.Dembo M, Wang YL. Biophys J. 1999;76(4):2307–16. doi: 10.1016/S0006-3495(99)77386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galbraith CG, Sheetz MP. Proc Natl Acad Sci U S A. 1997;94(17):9114–8. doi: 10.1073/pnas.94.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J, Leonard M, Oliver T, Ishihara A, Jacobson K. J Cell Biol. 1994;127(6):1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Proc Natl Acad Sci U S A. 2003;100(4):1484–9. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghassemi S, Meacci G, Liu S, Gondarenko AA, Mathur A, Roca-Cusachs P, Sheetz MP, Hone J. Proc Natl Acad Sci U S A. 2012;109(14):5328–33. doi: 10.1073/pnas.1119886109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolfenson H, Meacci G, Liu S, Stachowiak MR, Iskratsch T, Ghassemi S, Roca-Cusachs P, O’Shaughnessy B, Hone J, Sheetz MP. Nat Cell Biol. 2015;18:33. doi: 10.1038/ncb3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iskratsch T, Wolfenson H, Sheetz MP. Nat Rev Mol Cell Biol. 2014;15(12):825–33. doi: 10.1038/nrm3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfenson H, Iskratsch T, Sheetz MP. Biophys J. 2014;107(11):2508–14. doi: 10.1016/j.bpj.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang G, Huang AH, Cai Y, Tanase M, Sheetz MP. Biophys J. 2006;90(5):1804–9. doi: 10.1529/biophysj.105.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prager-Khoutorsky M, Lichtenstein A, Krishnan R, Rajendran K, Mayo A, Kam Z, Geiger B, Bershadsky AD. Nat Cell Biol. 2011;13(12):1457–65. doi: 10.1038/ncb2370. [DOI] [PubMed] [Google Scholar]
- 17.Lemke G. Cold Spring Harbor Perspect Biol. 2013;5(11):a009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paccez JD, Vogelsang M, Parker MI, Zerbini LF. Int J Cancer. 2014;134(5):1024–33. doi: 10.1002/ijc.28246. [DOI] [PubMed] [Google Scholar]
- 19.Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. Oncotarget. 2014;5(20):9546–63. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Endo M, Nishita M, Fujii M, Minami Y. Int Rev Cell Mol Biol. 2015;314:117–48. doi: 10.1016/bs.ircmb.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Gao B, Song H, Bishop K, Elliot G, Garrett L, English MA, Andre P, Robinson J, Sood R, Minami Y, Economides AN, Yang Y. Dev Cell. 2011;20(2):163–76. doi: 10.1016/j.devcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren D, Minami Y, Nishita M. Genes Cells. 2011;16(3):304–15. doi: 10.1111/j.1365-2443.2011.01487.x. [DOI] [PubMed] [Google Scholar]
- 23.Meacci G, Wolfenson H, Liu S, Stachowiak MR, Iskratsch T, Mathur A, Ghassemi S, Gauthier N, Tabdanov E, Lohner J, Gondarenko A, Chander AC, Roca-Cusachs P, O’Shaughnessy B, Hone J, Sheetz MP. Mol Biol Cell. 2016 doi: 10.1091/mbc.E16-02-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghibaudo M, Saez A, Trichet L, Xayaphoummine A, Browaeys J, Silberzan P, Buguin A, Ladoux B. Soft Matter. 2008;4(9):1836–1843. [Google Scholar]
- 25.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, Zhang J, Ding P, Apatira A, Chua J, Brandt R, Pine P, Goff D, Singh R, Payan DG, Hitoshi Y. Cancer Res. 2010;70(4):1544–54. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 26.Stitt TN, Conn G, Goret M, Lai C, Bruno J, Radzlejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al. Cell. 1995;80(4):661–70. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 27.Cavet ME, Smolock EM, Menon P, Konishi A, Korshunov VA, Berk BC. Hypertension. 2010;56(1):105–11. doi: 10.1161/HYPERTENSIONAHA.109.144642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishita M, Yoo SK, Nomachi A, Kani S, Sougawa N, Ohta Y, Takada S, Kikuchi A, Minami Y. J Cell Biol. 2006;175(4):555–62. doi: 10.1083/jcb.200607127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox S, Rosten E, Monypenny J, Jovanovic-Talisman T, Burnette DT, Lippincott-Schwartz J, Jones GE, Heintzmann R. Nat Methods. 2011;9(2):195–200. doi: 10.1038/nmeth.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bharadwaj S, Thanawala R, Bon G, Falcioni R, Prasad GL. Oncogene. 2005;24(56):8291–303. doi: 10.1038/sj.onc.1208993. [DOI] [PubMed] [Google Scholar]
- 31.Raval GN, Bharadwaj S, Levine EA, Willingham MC, Geary RL, Kute T, Prasad GL. Oncogene. 2003;22(40):6194–203. doi: 10.1038/sj.onc.1206719. [DOI] [PubMed] [Google Scholar]
- 32.Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP. Nature. 2011;478(7368):260–3. doi: 10.1038/nature10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rognoni L, Stigler J, Pelz B, Ylanne J, Rief M. Proc Natl Acad Sci U S A. 2012;109(48):19679–84. doi: 10.1073/pnas.1211274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lynch CD, Gauthier NC, Biais N, Lazar AM, Roca-Cusachs P, Yu CH, Sheetz MP. Mol Biol Cell. 2011;22(8):1263–73. doi: 10.1091/mbc.E10-08-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Umesh V, Rape AD, Ulrich TA, Kumar S. PLoS One. 2014;9(7):e101771. doi: 10.1371/journal.pone.0101771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hafizi S, Alindri F, Karlsson R, Dahlback B. Biochem Biophys Res Commun. 2002;299(5):793–800. doi: 10.1016/s0006-291x(02)02718-3. [DOI] [PubMed] [Google Scholar]
- 37.Huang JS, Cho CY, Hong CC, Yan MD, Hsieh MC, Lay JD, Lai GM, Cheng AL, Chuang SE. Free Radical Biol Med. 2013;65:1246–56. doi: 10.1016/j.freeradbiomed.2013.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.