

Abstract
Mitophagy is an essential process for mitochondrial quality control and turnover. It is activated by two distinct pathways, one dependent on ubiquitin and the other dependent on receptors including FUNDC1. It is not clear whether these pathways coordinate to mediate mitophagy in response to stresses, or how mitophagy receptors sense stress signals to activate mitophagy. We find that the mitochondrial E3 ligase MARCH5, but not Parkin, plays a role in regulating hypoxia‐induced mitophagy by ubiquitylating and degrading FUNDC1. MARCH5 directly interacts with FUNDC1 to mediate its ubiquitylation at lysine 119 for subsequent degradation. Degradation of FUNDC1 by MARCH5 expression desensitizes mitochondria to hypoxia‐induced mitophagy, whereas knockdown of endogenous MARCH5 significantly inhibits FUNDC1 degradation and enhances mitochondrial sensitivity toward mitophagy‐inducing stresses. Our findings reveal a feedback regulatory mechanism to control the protein levels of a mitochondrial receptor to fine‐tune mitochondrial quality.
Keywords: MARCH5, mitochondrial autophagy, mitophagy receptor, ubiquitylation
Subject Categories: Autophagy & Cell Death; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Mitochondria govern numerous cell activities, including oxidative phosphorylation 1, 2, 3, energy production 4, 5, apoptosis 6, 7, 8, inflammation responses 9, 10, 11, and aging 12, 13. They are also the primary source of reactive oxygen species (ROS), which contribute to oxidative stress and mitochondrial dysfunction 14, 15. To protect against mitochondrial damage, cells have evolved well‐coordinated quality control mechanisms that maintain overall mitochondrial health. This is achieved through continuous cycles of mitochondrial fission/fusion, and mitochondrial turnover, including balanced mitochondrial biogenesis and mitochondrial autophagy (mitophagy). A number of molecules have been identified that mediate the mitochondrial fission/fusion cycle 16, 17, 18, mitochondrial biogenesis 19, 20, 21, and mitophagy 22, 23, 24. A finely tuned regulatory network orchestrates these processes in response to both cellular and environmental cues. Stringent control of the levels of regulatory proteins (by transcriptional or translational regulation) and of post‐translational modifications or cleavage events will determine mitochondrial behaviors and functions 25, 26, 27, 28, 29. Dysregulation of these processes impairs mitochondrial quality, which is related to the pathogenesis of many aging‐related diseases such as diabetes 30, 31, neurodegenerative disorders 32, 33, and cancer 34, 35.
Mitophagy is a cellular process that selectively removes damaged or superfluous mitochondria. It may comprise an initial “decision‐making” phase and subsequent engagement of the autophagy machinery to engulf and eliminate mitochondria. It is proposed that the damaged or malfunctioning mitochondria may send out signals to activate the cellular “policing” system, leading to subsequent autophagic removal 14, 36, 37. In response to acute stress, the PINK1‐Parkin pathway mediates mitophagy to eliminate those mitochondria which have lost membrane potential. Parkin is an E3 ligase, and mutations in the gene encoding Parkin usually cause Parkinson's disease associated with mitochondrial function deficiency 38, 39. The loss of mitochondrial membrane potential results in the accumulation of PINK1 at the surface of mitochondria. Activated PINK1 phosphorylates Parkin and ubiquitin to recruit Parkin, which ubiquitylates many mitochondrial outer membrane proteins including Tom20, Mfn1, and Mfn2 40, 41. PINK1 is also able to recruit autophagy receptors such as OPTN and NDP52 to mediate the engulfment of mitochondria. Recent evidence shows that several mitophagy receptors are involved in mediating mitophagy. ATG32, the first reported mitophagy receptor in yeast, mediates aggregation of ATG8 (LC3 in homolog), which results in the recruitment of vesicles that engulf damaged mitochondria 42, 43, 44. In mammals, Nix/BNIP3L‐mediated mitophagy plays a crucial role in erythroid cell maturation 45, 46. We recently discovered a new mitochondrial receptor, FUNDC1, which mediates hypoxia‐induced mitophagy 47. In normal situations, FUNDC1 is phosphorylated by Src kinase and CK2. In response to hypoxia, dephosphorylation of FUNDC1 by PGAM5 enhances its interaction with LC3, which induces the formation of isolation membranes (precursors of autophagic vesicles) to engulf damaged mitochondria 28. It is less understood how mitophagy receptors sense stress signals to activate mitophagy and how mitophagy is orchestrated with other mitochondrial events.
MARCH5 (also named as MITOL) is a mitochondrially localized RING‐finger E3 ligase that is involved in mitochondrial dynamics by ubiquitylating Fis1 48, Mfn1 49, and Mfn2 50. Recently, MARCH5 was found to play a role in ubiquitin‐mediated degradation of MiD49 and recruitment of Drp1 51. Furthermore, MARCH5 is also involved in apoptosis regulation 49, 51 and the maintenance of the stemness of mouse ES cells 52. In this study, we reveal that the mitophagy receptor FUNDC1 is a new substrate of MARCH5, and we suggest that under hypoxic conditions, the MARCH5/FUNDC1 axis is involved in sensing the stress signal and fine‐tuning the mitophagy response.
Results
Hypoxia induces FUNDC1 degradation independent of Parkin
We have previously shown that genetic manipulation of FUNDC1 expression strongly affects mitophagy in response to mitochondrial stresses, suggesting that the protein level of this mitophagy receptor is closely associated with mitophagy activities. In order to gain insights into how mitophagy is regulated in response to mitochondrial stresses, we sought to address the molecular regulation of FUNDC1 protein homeostasis. In response to hypoxia, FUNDC1 was degraded and this degradation could be largely inhibited by MG132, a specific proteasome inhibitor (Fig 1A). We noticed that the FUNDC1 protein level was reduced as early as 3 h, which preceded the degradation of other mitochondrial proteins such as Tim23 and Tom20 (Fig 1A and B). Chloroquine, an autophagic flux inhibitor, also prevented FUNDC1 degradation, and MG132 and chloroquine together completely prevented FUNDC1 degradation under hypoxic conditions (Fig 1C). Similarly, MG132 and chloroquine were able to prevent FUNDC1 degradation induced by cycloheximide (CHX) treatment (Appendix Fig S1). These data suggest that FUNDC1 protein levels are regulated in both a proteasomal‐ and autophagy‐dependent manner under hypoxic stress. Importantly, FUNDC1 was degraded before it was dephosphorylated (Fig 1D and E). Dephosphorylation of FUNDC1 is considered as a biomarker of mitophagy induction 28, 47; thus, FUNDC1 was degraded before hypoxia‐induced mitophagy occurred.
Figure 1. Hypoxic stress induces Parkin‐independent degradation of FUNDC1 via ubiquitylation.
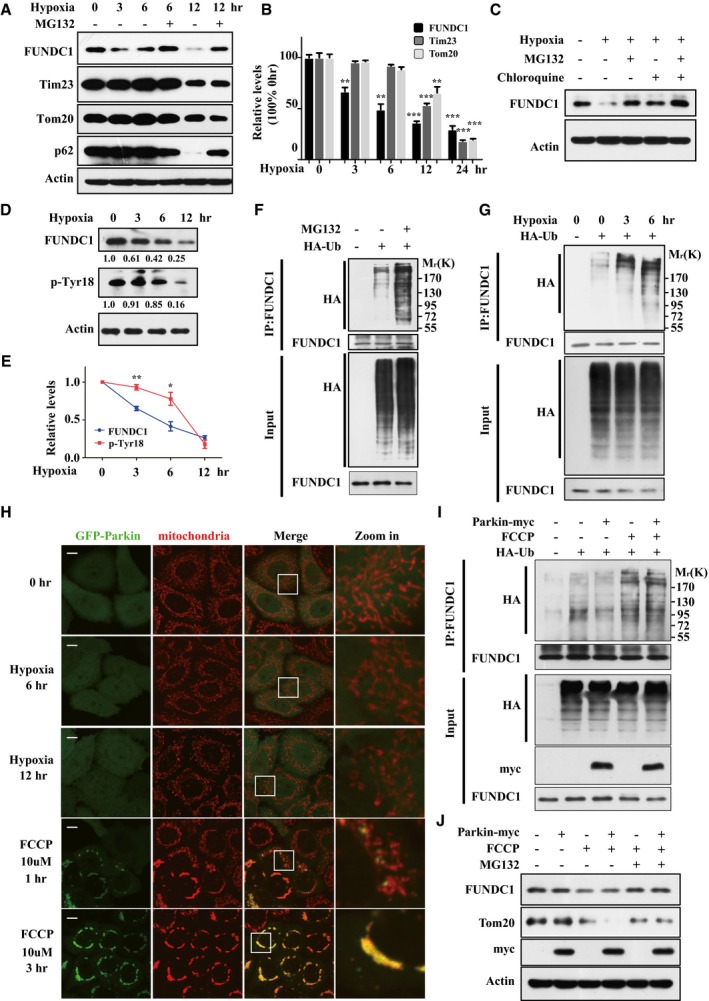
- HeLa cells were exposed to 1% O2 for the indicated time, with or without MG132 (10 μM), and then FUNDC1, Tim23, and Tom20 proteins were detected by Western blotting.
- Quantification of mitochondrial membrane proteins as indicated in (A) (mean ± SEM from three independent experiments, Student's t‐test, **P < 0.01, ***P < 0.001; asterisks indicate a significant difference between 0 h and indicated hypoxic condition to the respective color‐coded proteins).
- HeLa cells were exposed to 1% O2 for 12 h, with MG132 (10 μM) or chloroquine (10 μM), and then FUNDC1 protein levels were detected by Western blotting.
- HeLa cells were exposed to 1% O2 for the indicated time, and then the levels of FUNDC1 and Tyr18‐phosphorylated FUNDC1 were analyzed using antibodies against FUNDC1 and p‐Tyr18 FUNDC1, respectively.
- Quantification of FUNDC1 and p‐Tyr18 FUNDC1 as indicated in (D) (mean ± SEM from three independent experiments, Student's t‐test, *P < 0.05, **P < 0.01).
- HeLa cells were transfected with HA‐Ub for 24 h and then treated with or without MG132 (5 μM) for 6 h. Ubiquitylation assays were performed as described in Materials and Methods. The ubiquitylation level of FUNDC1 was detected using an anti‐HA antibody.
- HeLa cells were transfected with HA‐Ub for 24 h, then exposed to 1% O2 for the indicated time. Ubiquitylation assays were performed as described in Materials and Methods. The ubiquitylation level of FUNDC1 was detected using an anti‐HA antibody.
- HeLa cells stably expressing GFP‐Parkin were exposed to 1% O2 or treated with 10 μM FCCP for the indicated time. Cells were then fixed and immunostained with an antibody against the mitochondrial protein cytochrome c (Cyto C, red), and examined by immunofluorescence microscopy to detect mitochondrial translocation of GFP‐Parkin. Scale bar, 5 μm.
- HeLa cells were transfected with Parkin‐myc or the empty myc‐vector together with HA‐Ub for 24 h, and then treated with 10 μM FCCP. Ubiquitylation assays were performed as described in Materials and Methods, and the ubiquitylation level of FUNDC1 was detected by Western blotting using an anti‐HA antibody.
- HeLa cells were transfected with Parkin‐myc or the empty myc‐vector for 24 h, and then FCCP (10 μM) and MG132 (10 μM) were added 4 h before harvesting. Levels of FUNDC1 and Parkin‐myc were detected by Western blotting using anti‐FUNDC1 and anti‐myc antibodies, respectively.
Source data are available online for this figure.
We first investigated whether FUNDC1 is ubiquitylated for degradation during hypoxia‐induced mitophagy. Indeed, FUNDC1 exhibited ubiquitylation (Fig 1F), which was significantly enhanced in a time‐dependent manner in cells exposed to hypoxia (Fig 1G). It is well characterized that Parkin, an E3 ligase, plays crucial roles in mitophagy; therefore, we asked whether Parkin mediates FUNDC1 ubiquitylation and degradation. Unlike FCCP treatment, hypoxia did not induce translocation of GFP‐Parkin to mitochondria (Fig 1H). Overexpression of Parkin failed to enhance FUNDC1 ubiquitylation (Fig 1I) or degradation (Fig 1J) in HeLa cells. As a control, FCCP induced mitochondrial translocation of Parkin and degradation of Tom20 (Fig 1J). These observations revealed that FUNDC1 can undergo ubiquitin‐mediated degradation in a Parkin‐independent manner upon hypoxic treatment.
Mitochondrial E3 ligase MARCH5 is responsible for FUNDC1 ubiquitylation under hypoxic conditions
We next wished to identify the E3 ligase that mediates ubiquitylation and degradation of FUNDC1. We screened several E3 ligases that were previously reported to affect mitochondrial behaviors or show mitochondrial localization, and found that MARCH5, an E3 ligase located on the mitochondrial outer membrane, induced drastic FUNDC1 degradation (Fig EV1A). It has been reported that MARCH5 regulates mitochondrial dynamics by ubiquitylating Mfn1, Mfn2, Fis1, and MiD49 48, 49, 50. Ectopic expression of MARCH5, but not Parkin, dramatically increased the level of ubiquitylation of FUNDC1 (Figs 2A and EV1B). Conversely, knockdown of MARCH5 markedly decreased FUNDC1 ubiquitylation and degradation as revealed by Western blotting, and this decrease was rescued by shRNA‐resistant MARCH5 (MARCH5‐Res), but not the MARCH5 C65S,C68S mutant (MARCH5‐Res‐CS), which lacks E3 ligase activity (Fig 2B). In vitro ubiquitylation assays further revealed that FUNDC1 was ubiquitylated by MARCH5, but not by two E3 ligase‐dead mutants, CS or H43W (Fig 2C).
Figure EV1. Identification of MARCH5 as the E3 ligase for FUNDC1 degradation.
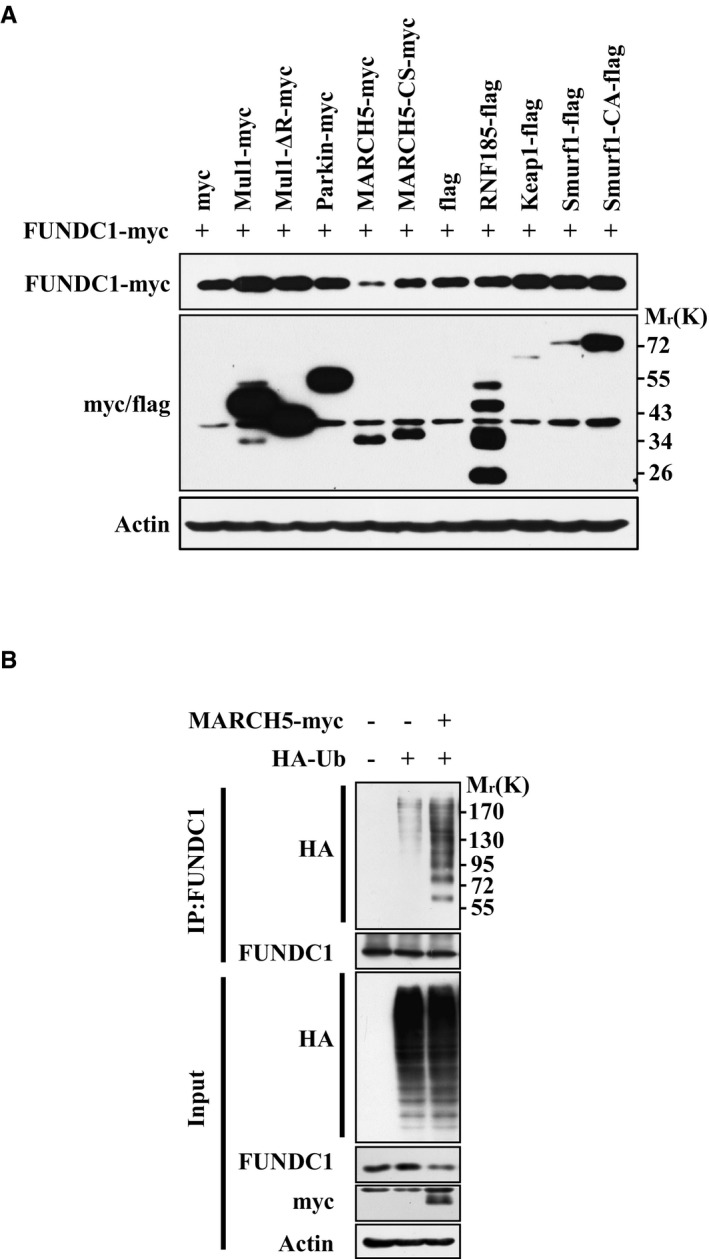
- FUNDC1‐knockdown HeLa cells were transfected with FUNDC1‐myc for 24 h, and then with plasmids expressing the indicated mitochondria‐associated E3 ligases. FUNDC1‐myc protein level was detected by Western blotting.
- HeLa cells were transfected with MARCH5‐myc or the empty myc‐vector together with HA‐Ub for 24 h. Ubiquitylation assays were performed as described in Materials and Methods, and ubiquitylated FUNDC1 was detected using an anti‐HA antibody. FUNDC1 and MARCH5‐myc expression was detected by Western blotting.
Source data are available online for this figure.
Figure 2. MARCH5 is responsible for FUNDC1 ubiquitylation.
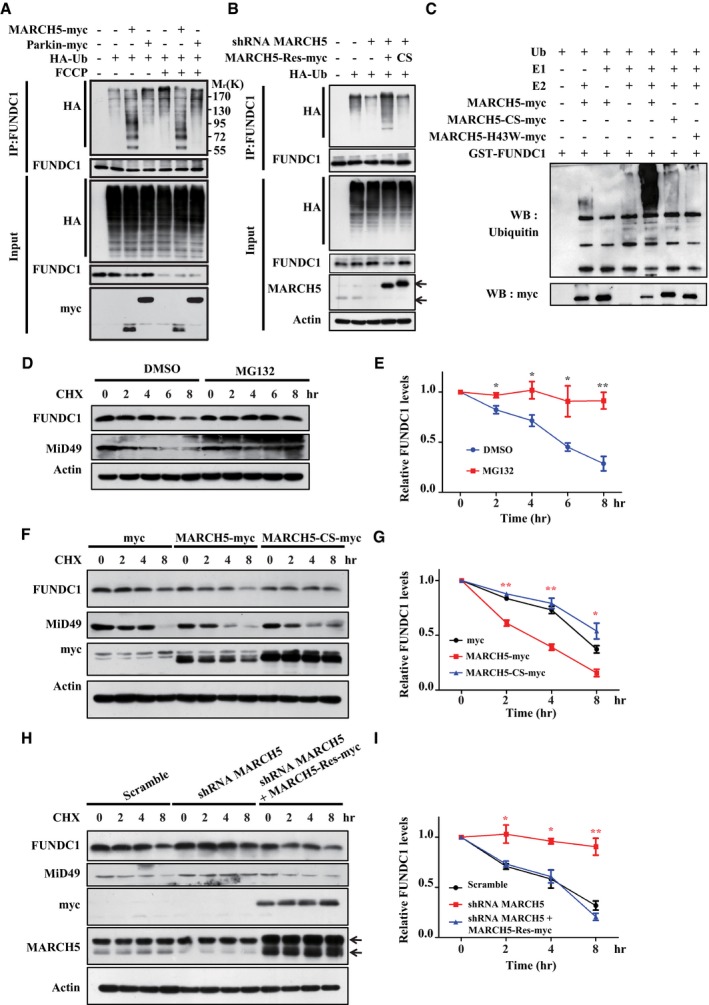
- HeLa cells expressing HA‐Ub were transfected with MARCH5‐myc, Parkin‐myc, or the empty myc‐vector and then treated with FCCP (10 μM) for 3 h. Ubiquitylation assays were performed as described in Materials and Methods. The ubiquitylation level of FUNDC1 was detected using an anti‐HA antibody.
- MARCH5‐knockdown cells were transfected with MARCH5‐Res‐myc, MARCH5‐Res‐CS‐myc, or the empty myc‐vector together with HA‐Ub for 24 h. Ubiquitylation assays were performed as described in Materials and Methods. The ubiquitylation level of FUNDC1 was detected using an anti‐HA antibody. The levels of FUNDC1 and MARCH5 proteins were detected by Western blotting using anti‐FUNDC1 and anti‐MARCH5 antibodies, respectively. Black arrows indicate endogenous and exogenous MARCH5.
- In vitro ubiquitylation assays were performed as described in Materials and Methods. The ubiquitylated form of FUNDC1 was detected by immunoblotting using an anti‐ubiquitin antibody.
- HeLa cells were treated with CHX (10 μM) for the indicated time, with or without MG132, and then subjected to Western blotting analysis of FUNDC1 and MiD49.
- Quantification of FUNDC1 protein level in (D) (mean ± SEM from three independent experiments, Student's t‐test, *P < 0.05, **P < 0.01).
- HeLa cells were transfected with MARCH5‐myc, MARCH5‐CS‐myc, or the empty myc‐vector for 24 h. CHX (10 μM) was added for the indicated time, and the cell lysates were subjected to Western blotting analysis of FUNDC1 and MiD49.
- Quantification of FUNDC1 protein levels in (F) (mean ± SEM from three independent experiments, Student's t‐test, *P < 0.05, **P < 0.01).
- MARCH5‐knockdown cells were transfected with MARCH5‐Rescue‐myc or the empty myc‐vector for 24 h. CHX (10 μM) was added for the indicated time, and the cell lysates were subjected to Western blotting analysis of FUNDC1, MiD49, and MARCH5. Black arrows indicate endogenous and exogenous MARCH5.
- Quantification of FUNDC1 protein levels in (H) (mean ± SEM from three independent experiments, Student's t‐test, *P < 0.05, **P < 0.01).
Source data are available online for this figure.
To further demonstrate that the ubiquitylation mediated by MARCH5 promotes degradation of FUNDC1, we performed CHX chase assays. In the presence of MG132, the degradation of FUNDC1 was greatly reduced (Fig 2D and E). Ectopic expression of wild‐type MARCH5, but not the CS mutant, promoted FUNDC1 degradation when HeLa cells were treated with CHX (Fig 2F and G). The degradation of FUNDC1 was significantly suppressed when MARCH5 was knocked down, and re‐introduction of shRNA‐resistant MARCH5 increased FUNDC1 degradation (Fig 2H and I). Similar patterns were observed for MiD49, a previously reported MARCH5 substrate (Fig 2D, F and H).
MARCH5 interacts with FUNDC1
To further confirm that FUNDC1 is a novel substrate of MARCH5, we carried out co‐immunoprecipitation analysis and found that FUNDC1 interacted with MARCH5 (Fig 3A and B). Moreover, purified glutathione S‐transferase (GST)‐fused MARCH5 was able to pull down FUNDC1 from HeLa cell lysates (Fig 3C), suggesting a possible direct interaction between MARCH5 and its substrate FUNDC1. We further determined the interacting domains using truncation and/or deletion analysis. Immunoprecipitation analyses revealed that MARCH5 interacted with FUNDC1 through amino acids 140–228, which comprise the second cytosolic domain of MARCH5 (Fig 3D). Similarly, FUNDC1 interacted with MARCH5 through amino acids 94–110, a motif that is also exposed to the cytosol (Fig 3E).
Figure 3. MARCH5 interacts with FUNDC1.
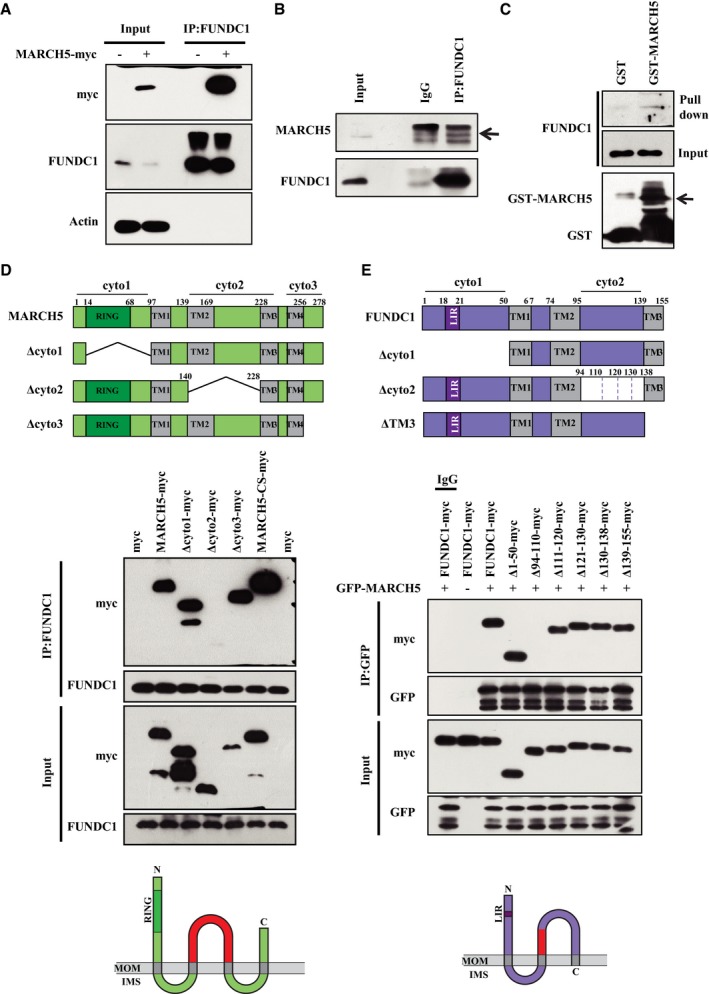
- MARCH5‐myc was transfected into HeLa cells for 24 h, and then immunoprecipitation assays were performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated endogenous FUNDC1 and MARCH5‐myc proteins were detected by Western blotting with anti‐FUNDC1 and anti‐myc antibodies, respectively.
- Immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated endogenous MARCH5 was detected by Western blotting with an anti‐MARCH5 antibody. The black arrow indicates endogenous MARCH5.
- Purified GST and GST‐tagged MARCH5 proteins were used for GST affinity isolation of endogenous FUNDC1, and blotted with an anti‐FUNDC1 antibody. The black arrow indicates the purified GST‐MARCH5.
- Truncated forms of MARCH5‐myc were constructed based on its functional domains. HeLa cells were co‐transfected with full‐length or truncated forms of MARCH5‐myc, and immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated MARCH5 and FUNDC1 were detected by Western blotting with anti‐myc and anti‐FUNDC1 antibodies, respectively. The red area of the schematic diagram (bottom) indicates the domain of MARCH5 that interacts with FUNDC1.
- Truncated forms of FUNDC1‐myc were constructed based on its functional domains. GFP‐MARCH5 was co‐transfected with full‐length or truncated forms of FUNDC1‐myc, and immunoprecipitation was performed with an anti‐GFP antibody. Co‐immunoprecipitated MARCH5 and FUNDC1 were detected by Western blotting with anti‐GFP and anti‐myc antibodies, respectively. The red area of the schematic diagram (bottom) indicates the domain of FUNDC1 that interacts with MARCH5.
Source data are available online for this figure.
MARCH5 mediates FUNDC1 ubiquitylation at lysine 119
We next sought to identify the site of MARCH5‐mediated ubiquitylation within FUNDC1. To do this, we individually mutated the 11 lysine (K) residues of FUNDC1 to arginine (R) (Fig 4A). Wild‐type FUNDC1 and the individual K‐to‐R mutants were co‐expressed with HA‐Ub in FUNDC1‐knockdown HeLa cells. Compared to wild‐type FUNDC1, the ubiquitylation level was strongly decreased in the K119R mutant (Fig 4B), suggesting that K119 of FUNDC1 is the potential ubiquitylation site. To confirm this finding, we performed in vitro ubiquitylation assays and showed that purified GST‐FUNDC1‐K119R was hardly ubiquitylated by MARCH5‐myc isolated from HeLa cell lysates (Fig 4C).
Figure 4. MARCH5 promotes FUNDC1 degradation by ubiquitylation at lysine 119.
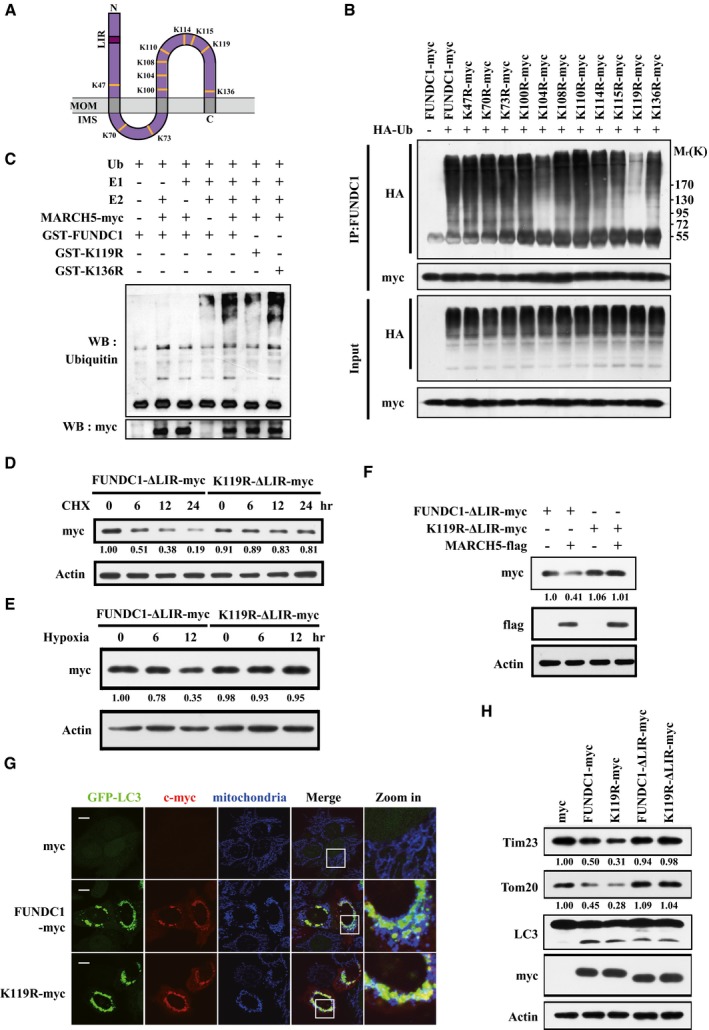
- Schematic representation of the FUNDC1 protein in the mitochondrial outer membrane showing the LIR domain and 11 lysine residues.
- FUNDC1‐knockdown cells were transfected with myc‐tagged wild‐type FUNDC1 or the different lysine‐to‐arginine mutants together with HA‐Ub. Ubiquitylation assays were performed as described in Materials and Methods, and ubiquitylated FUNDC1 was detected using an anti‐HA antibody.
- In vitro ubiquitylation assays were performed as described in Materials and Methods, and the ubiquitylated form of FUNDC1 was detected by immunoblotting using an anti‐ubiquitin antibody.
- FUNDC1‐knockdown cells expressing FUNDC1‐∆LIR‐myc or K119R‐∆LIR‐myc were treated with 10 μM CHX for the indicated time, and then FUNDC1‐myc protein levels were detected by Western blotting.
- FUNDC1‐knockdown cells expressing FUNDC1‐∆LIR‐myc or K119R‐∆LIR‐myc were exposed to 1% O2 for the indicated time, and then FUNDC1‐myc protein levels were detected by Western blotting.
- FUNDC1‐knockdown cells expressing FUNDC1‐∆LIR‐myc or K119R‐∆LIR‐myc were transfected with MARCH5‐flag for 24 h. FUNDC1‐myc and MARCH5‐flag proteins were detected by Western blotting.
- FUNDC1‐knockdown cells expressing GFP‐LC3 were co‐transfected with FUNDC1‐myc or K119R‐myc (red, anti‐myc) for 24 h. The K119R mutant of FUNDC1 increased the co‐localization between GFP‐LC3 dots and mitochondria (blue, anti‐Tom20) when compared with wild‐type FUNDC1. Scale bar, 5 μm.
- FUNDC1‐knockdown HeLa cells were transfected with plasmids encoding FUNDC1‐myc or the indicated mutants for 24 h. Tim23, Tom20, LC3, and FUNDC1‐myc proteins were detected by Western blotting.
Source data are available online for this figure.
We next investigated whether K119 ubiquitylation is responsible for FUNDC1 stability. We constructed deletion mutants of FUNDC1 (FUNDC1‐∆LIR‐myc) and FUNDC1‐K119R (K119R‐∆LIR‐myc) which lack the LIR motif and therefore cannot be degraded by mitophagy 47. Mutation of K119 to R strongly inhibited FUNDC1 degradation in CHX chase assays (Fig 4D and Appendix Fig S2A) or in response to hypoxic treatment (Fig 4E and Appendix Fig S2B). Degradation of the FUNDC1 K119R‐∆LIR‐myc mutant, but not FUNDC1‐∆LIR‐myc, was blocked by co‐expression of MARCH5‐flag (Fig 4F). Collectively, our data demonstrated that K119 ubiquitylation of FUNDC1 is responsible for its MARCH5‐mediated degradation.
We wished to understand whether degradation of FUNDC1 triggered by ubiquitylation at K119 has a functional consequence on mitophagy. We expressed wild‐type FUNDC1 and the FUNDC1‐K119R mutant in FUNDC1‐knockdown HeLa cells stably expressing GFP‐LC3 and observed the formation GFP‐LC3 puncta, which are indicators of mitophagosomes. We found that the FUNDC1‐K119R mutant induced more GFP‐LC3 puncta that co‐localized with mitochondria (marked by Tom20 antibody) (Fig 4G and Appendix Fig S3A). Western blotting analyses further confirmed that ectopic expression of FUNDC1‐K119R induced a greater reduction in the mitochondrial proteins Tim23 and Tom20 than wild‐type FUNDC1 (Fig 4H and Appendix Fig S3B). Consistent with the previous report, deletion of the LIR prevented mitophagosome formation and mitochondrial marker protein degradation 47.
The MARCH5/FUNDC1 axis fine‐tunes hypoxia‐induced mitophagy
We next addressed the biological significance of the MARCH5/FUNDC1 axis in hypoxia‐induced mitophagy. Knockdown of MARCH5 induced a significant increase in the co‐localization between GFP‐LC3 puncta and mitochondria compared to the scramble control (Fig 5A and B). Re‐introduction of shRNA‐resistant MARCH5‐myc, but not MARCH5‐CS‐myc or MARCH5‐∆cyto2‐myc (which does not interact with FUNDC1), in MARCH5‐knockdown cells was able to reduce mitophagosome formation (Fig 5A and B). Consistent with this, Western blotting results showed that knockdown of MARCH5 prevented FUNDC1 degradation and induced a marked reduction in mitochondrial proteins (Tim23 and Tom20) in response to hypoxia, and these changes were rescued by re‐introduction of shRNA‐resistant MARCH5, but not the E3 ligase‐dead mutant MARCH5‐CS (Fig 5C). To demonstrate the indispensability of FUNDC1 for MARCH5‐regulated mitophagy, we knocked down FUNDC1 in MARCH5 RNAi cells. Compared with the effect in MARCH5 RNAi cells, the hypoxia‐induced mitophagy process was completely blocked in FUNDC1 and MARCH5 double‐knockdown cells (Fig 5D). The co‐localization between GFP‐LC3 puncta and mitochondria was significantly reduced (Fig 5E and F). These results demonstrated that MARCH5 fine‐tunes hypoxia‐induced mitophagy by regulating the ubiquitin‐mediated degradation of FUNDC1.
Figure 5. MARCH5 ubiquitylates FUNDC1 to fine‐tune hypoxia‐induced mitophagy.
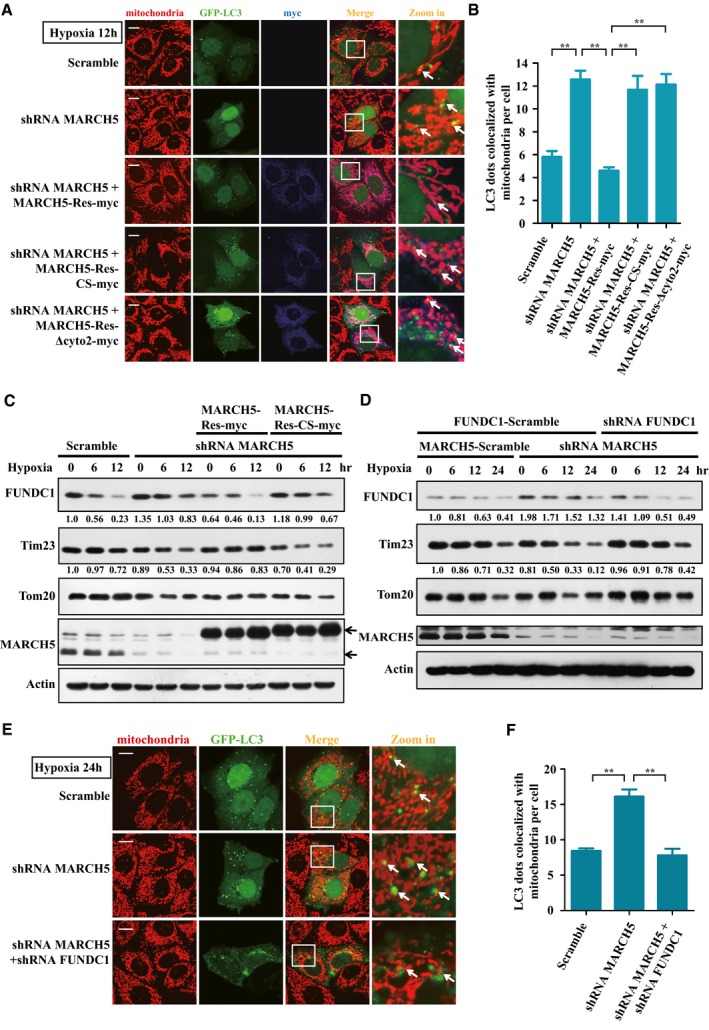
- HeLa cells stably expressing MARCH5 shRNA were co‐transfected with GFP‐LC3 and MARCH5‐Rescue‐myc, the E3 activity‐dead mutant (MARCH5‐Res‐CS‐myc), or the interaction domain mutant (MARCH5‐Res‐∆cyto2‐myc) at a ratio of 5:1 for 24 h, and were then exposed to 1% O2 for 12 h. The expression of MARCH5‐myc (blue, anti‐myc) and the co‐localization of GFP‐LC3 dots with mitochondria (red, anti‐Tom20) were detected. Scale bar, 5 μm. White arrows indicate mitochondria co‐localized with GFP‐LC3.
- Quantification of the number of GFP‐LC3 puncta co‐localized with mitochondria per cell in (A) (mean ± SEM from three independent experiments, Student's t‐test, **P < 0.01).
- HeLa cells stably expressing MARCH5 shRNA were transfected with MARCH5‐Rescue‐myc or E3 activity‐dead mutant for 24 h, and then exposed to 1% O2 for the indicated time. FUNDC1, Tim23, Tom20, and MARCH5 protein levels were detected by Western blotting.
- HeLa cells stably expressing MARCH5 shRNA were transfected with FUNDC1 shRNA or FUNDC1 scramble plasmids for 24 h, and then exposed to 1% O2 for the indicated time. FUNDC1, Tim23, Tom20, and MARCH5 protein levels were detected by Western blotting analysis.
- HeLa cells stably expressing MARCH5 shRNA were co‐transfected with GFP‐LC3 and FUNDC1 shRNA or FUNDC1 scramble plasmids at a ratio of 5:1 for 24 h, and then exposed to 1% O2 for another 24 h. Co‐localization between GFP‐LC3 dots and mitochondria (red, anti‐Tom20) was detected (white arrows). Scale bar, 5 μm.
- Quantification of the number of GFP‐LC3 puncta co‐localized with mitochondria per cell in (E) (mean ± SEM from three independent experiments, Student's t‐test, **P < 0.01).
Source data are available online for this figure.
Hypoxic stress reduces MARCH5 homo‐oligomerization and enhances its interaction with FUNDC1
We next addressed how MARCH5 senses the hypoxic signal to promote FUNDC1 degradation and fine‐tune mitophagy. We observed that hypoxia progressively enhanced the interaction between MARCH5 and FUNDC1 (Fig 6A), whereas the self‐interaction of MARCH5 was significantly decreased (Fig 6B and C). MARCH5 is able to form dimers or oligomers, which mediate degradation of dysfunctional mutant MARCH5 proteins to maintain mitochondrial quality 53. Indeed, cross‐linking analysis showed that MARCH5 homo‐oligomers were reduced under hypoxic conditions (Fig EV2). A previous study showed that the MARCH5 G103/107L mutant (GL) fails to form oligomers 53. We thus constructed the MARCH5 GL mutant and co‐transfected it with wild‐type MARCH5. We observed that the GL mutant markedly reduced MARCH5 oligomerization while increasing the interaction with endogenous FUNDC1 in the presence of hypoxia (Fig 6D and E). Importantly, the MARCH5 GL mutant promoted more degradation of FUNDC1 than wild‐type MARCH5 (Fig 6E), indicating that the interaction between FUNDC1 and MARCH5 is important for FUNDC1 degradation. We believe that the oligomeric switch of MARCH5 is important for degradation of FUNDC1; however, exact nature of this complex (oligomeric) and its interaction needs to be explored further. Taken together, our data demonstrated that hypoxic stress triggers disassembly of MARCH5 homo‐oligomers and simultaneously facilitates its interaction with FUNDC1 in response to initial mitochondrial insults before the execution of mitophagy.
Figure 6. Hypoxic stress triggers disassembly of MARCH5 homo‐oligomers and enhances the interaction of MARCH5 with FUNDC1.
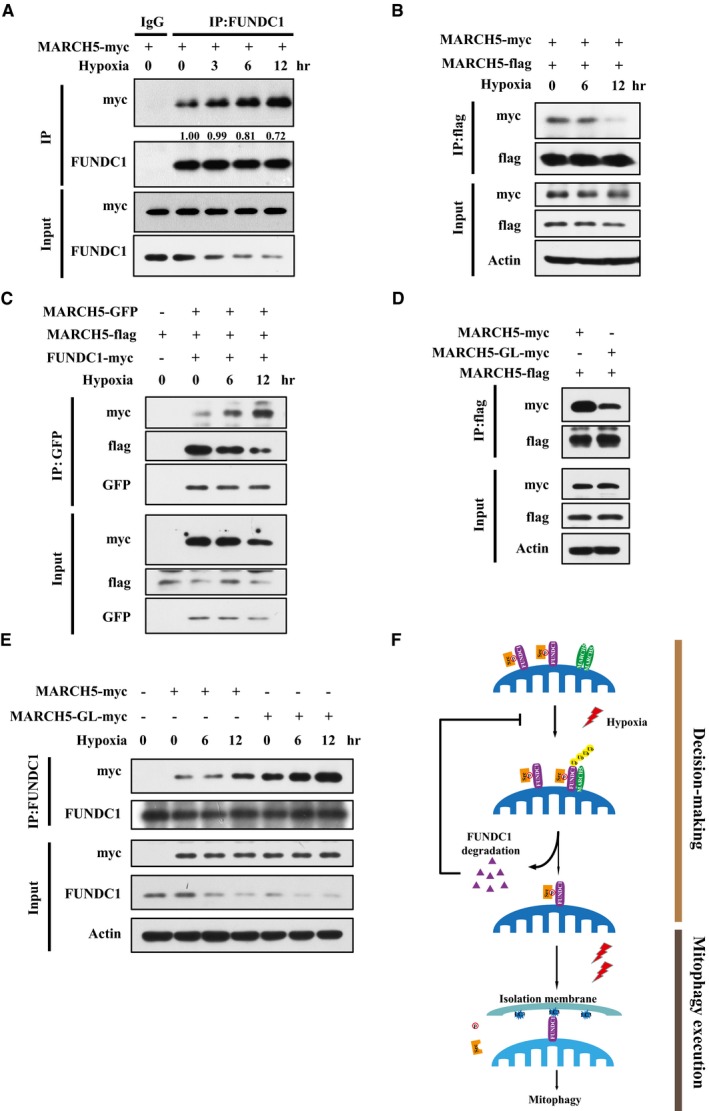
- HeLa cells were transfected with MARCH5‐myc for 24 h, and then exposed to 1% O2 for the indicated time. Immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated MARCH5 and FUNDC1 were detected by Western blotting with anti‐myc and anti‐FUNDC1 antibodies, respectively.
- HeLa cells were co‐transfected with MARCH5‐myc and MARCH5‐flag for 24 h, and then exposed to 1% O2 for the indicated time. Immunoprecipitation was performed with an anti‐flag antibody. Co‐immunoprecipitated MARCH5‐myc and MARCH5‐flag were detected by Western blotting with anti‐myc and anti‐flag antibodies, respectively.
- HeLa cells were transfected with MARCH5‐GFP and MARCH5‐flag together with FUNDC1‐myc for 24 h, and then exposed to 1% O2 for the indicated time. Immunoprecipitation was performed with an anti‐GFP antibody. Co‐immunoprecipitated MARCH5‐GFP, MARCH5‐flag, and FUNDC1 were detected by Western blotting with anti‐GFP, anti‐flag, and anti‐FUNDC1 antibodies, respectively.
- HeLa cells expressing MARCH5‐flag were transfected with MARCH5‐myc or the MARCH5‐G103/107L‐myc mutant (MARCH5‐GL‐myc) for 24 h, and immunoprecipitation was performed with an anti‐flag antibody. Co‐immunoprecipitated MARCH5‐myc and MARCH5‐flag were detected by Western blotting with anti‐myc and anti‐flag antibodies, respectively.
- HeLa cells were transfected with MARCH5‐myc or the MARCH5‐GL‐myc mutant for 24 h, and then exposed to 1% O2 for the indicated time. Immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated MARCH5‐myc and FUNDC1 were detected through Western blotting with anti‐myc and anti‐FUNDC1 antibodies, respectively.
- A hypothetical model of the MARCH5/FUNDC1 axis in response to hypoxic stress. In the “decision‐making” phase, the initial hypoxic stress induces disassembly of MARCH5 and increases the interaction between MARCH5 and FUNDC1, which leads to FUNDC1 ubiquitylation and degradation. FUNDC1 degradation limits the extent of mitophagic degradation of mitochondria. During the “mitophagy execution” phase, prolonged hypoxic stress decreases the interaction between Src kinase and FUNDC1, which leads to FUNDC1 dephosphorylation. Dephosphorylated FUNDC1 interacts with LC3, thus recruiting LC3‐bound isolation membranes to damaged mitochondria, and eventually leading to mitophagy.
Source data are available online for this figure.
Figure EV2. Hypoxic stress promotes disassembly of MARCH5 homo‐oligomer.
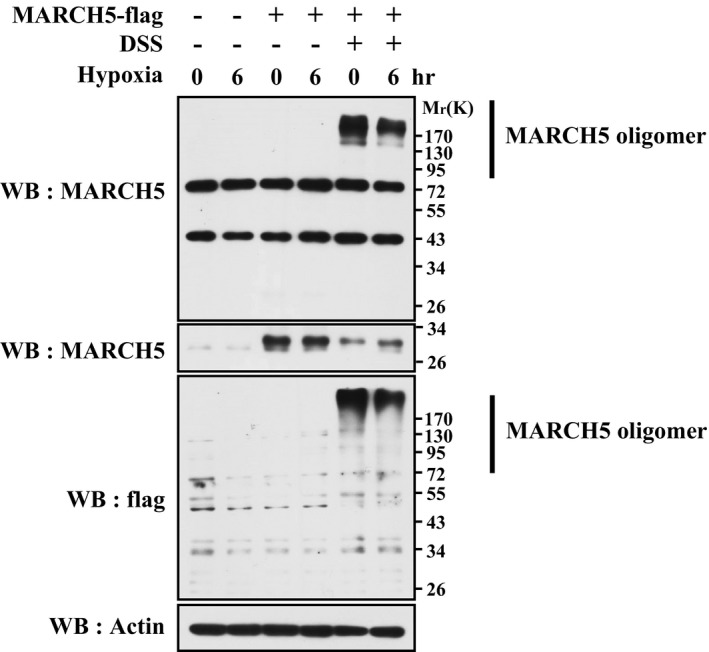
HeLa cells expressing MARCH5‐flag were exposed to 1% O2 for the indicated time, and then isolated cells were treated with DSS (1 mM) for 10 min. MARCH5 polymerization was detected by Western blotting analysis with anti‐flag and anti‐MARCH5 antibodies. Source data are available online for this figure.
Discussion
Both ubiquitin‐ and receptor‐dependent mitophagy pathways have been described, and it has been speculated that there is cross‐talk between these two pathways. We demonstrated that FUNDC1 is a new substrate of the mitochondrial E3 ligase MARCH5 and that MARCH5‐mediated ubiquitylation and degradation of FUNDC1 is one of the early events in response to hypoxia. We show that MARCH5 mediates ubiquitylation of FUNDC1 at K119 to promote its proteasome‐dependent degradation under hypoxic stress. Our results suggest a new regulatory mechanism of FUNDC1‐mediated mitophagy, which is distinct from the Parkin‐mediated pathway. Indeed, hypoxia does not induce translocation of Parkin to mitochondria (Fig 1H), and FUNDC1 overexpression induces mitophagy in Parkin‐ or PINK1‐deficient cells (Y. Zhu, J. Han, unpublished data). It is somewhat surprising that Parkin, as a potent E3 ligase, does not mediate ubiquitylation or degradation of FUNDC1, although a previous report showed that FUNDC1 is modified by ubiquitin when Parkin is highly expressed 54. Our data thus support the idea that distinct pathways exist to mediate mitophagy in response to different stresses 55.
We reveal a MARCH5/FUNDC1 axis as a novel mechanism by which cells can sense hypoxic stress and activate mitophagy. It is paradoxical that, in response to hypoxia, MARCH5 promotes the degradation of FUNDC1, thereby reducing mitophagic activity (Fig 5). Unlike Parkin‐regulated mitophagy that ubiquitylates a large amount of mitochondrial outer membrane proteins and then recruits autophagy adaptor p62 and isolation membrane, MARCH5 fine‐tunes mitophagy by specifically ubiquitylating and degrading receptor protein FUNDC1 in response to hypoxic stress. Regulation of the MARCH5/FUNDC1 axis desensitizes mitochondrial degradation and avoids improper clearness of undamaged mitochondria. Our findings uncover the distinct roles of E3 ligases in different mitophagy pathways. This axis may serve as a negative feedback mechanism to protect mitochondria from removal by mitophagy. Such a mechanism gives the cell extra time to “make a critical decision” about the fate of mitochondria, which is a matter of life and death for the cell. We have previously shown that reversible phosphorylation of FUNDC1, in particular at Tyr18, serves as a master switch for the activation of mitophagy 47. Initially, hypoxic stress leads to disassembly of MARCH5 homo‐oligomers and increases the interaction between MARCH5 and FUNDC1. Ubiquitylation and degradation of FUNDC1 desensitizes mitochondria to avoid unnecessary removal of mitochondria via mitophagy. However, prolonged and/or severe hypoxic stress leads to dephosphorylation of FUNDC1 which significantly enhances its interaction with LC3, thus activating mitophagy to remove damaged or unwanted mitochondria (Fig 6F). Src kinase directly phosphorylates FUNDC1 at Tyr18 to avoid premature activation of FUNDC1 dephosphorylation and mitophagy 47. Mitophagic stresses are able to decrease the interaction between Src and FUNDC1 28. We expressed Src‐GFP in HeLa cells and carried out co‐immunoprecipitation assays in cells exposed to hypoxia for different times and found that the interaction between FUNDC1 and Src‐GFP was reduced for up to 12 h (Fig EV3A), whereas FUNDC1 degradation occurred as early as 3 h (Fig 1A and D). These results are consistent with the suggestion that ubiquitin‐mediated FUNDC1 degradation occurs prior to its activation by dephosphorylation. Moreover, the phosphorylation status of FUNDC1 does not affect its degradation by MARCH5 (Fig EV3B). Our model contrasts with acute mitochondrial stress, which may activate PINK1/Parkin for quick removal of damaged mitochondria. Also, the MARCH5/FUNDC1 axis has a unique role in fine‐tuning hypoxia‐induced mitophagy, as MARCH5 failed to regulate the level of Nix/BNIP3L (Fig EV4).
Figure EV3. The interaction between FUNDC1 and Src is significantly decreased upon prolonged hypoxic stress.
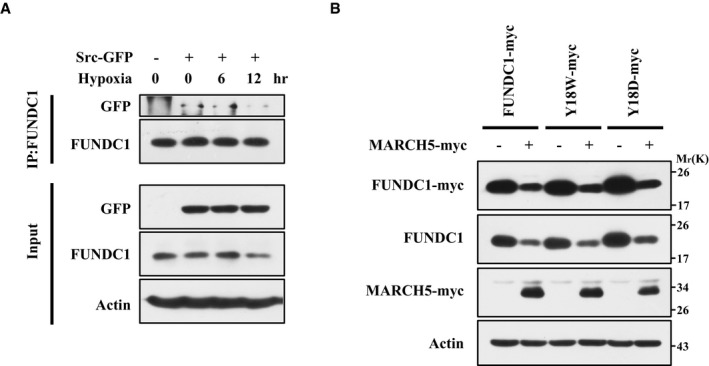
- HeLa cells were transfected with Src‐GFP for 24 h and then exposed to 1% O2 for the indicated time. Immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated Src‐GFP and FUNDC1 were detected by Western blotting with anti‐GFP and anti‐FUNDC1 antibodies, respectively.
- FUNDC1‐knockdown HeLa cells were transfected with FUNDC1‐myc, FUNDC1‐Y18W‐myc, and FUNDC1‐Y18D‐myc together with MARCH5‐myc for 24 h, and then FUNDC1‐myc protein level was detected by Western blotting.
Source data are available online for this figure.
Figure EV4. MARCH5 is not responsible for Nix/BNIP3L degradation.
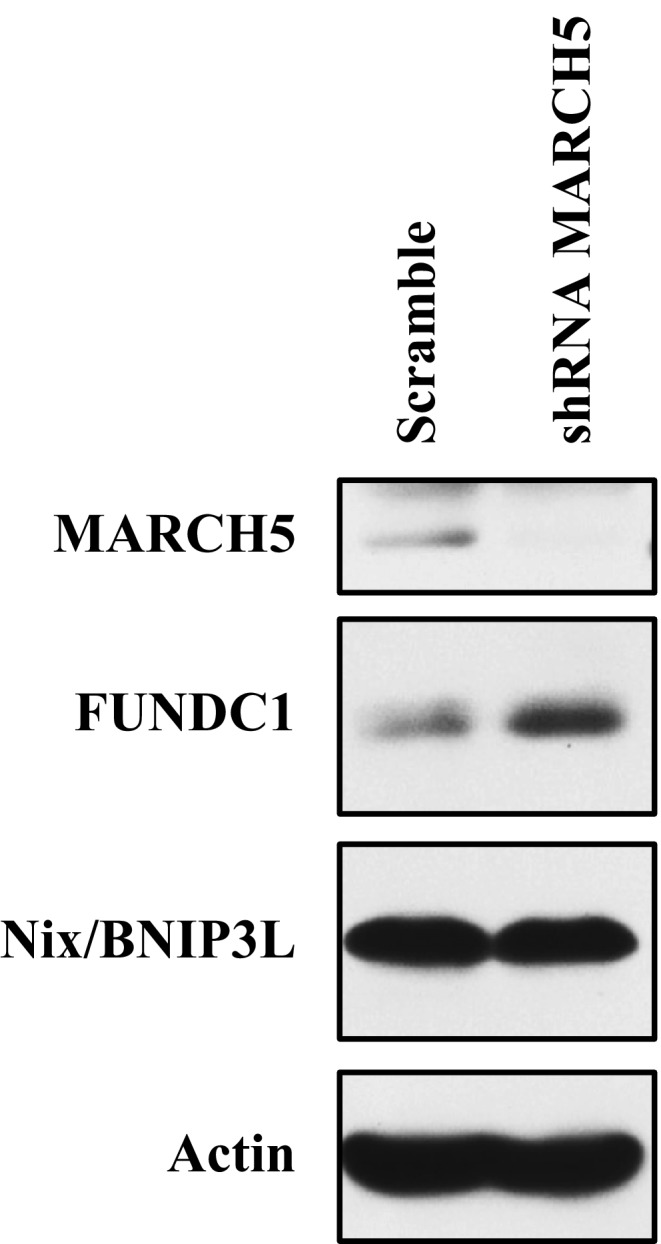
HeLa cells stably expressing MARCH5 shRNA or scramble plasmid were harvested and lysed for Western blotting. MARCH5, FUNDC1, and Nix/BNIP3L were detected using anti‐MARCH5, anti‐FUNDC1, and anti‐Nix/BNIP3L antibodies, respectively. Source data are available online for this figure.
It remains to be determined how the MARCH5/FUNDC1 axis is activated by hypoxia. Previous research suggested that hypoxic stress induces changes in the status of the redox system and subsequent ROS generation 56. We observed that mito‐TEMPO, a specific mito‐ROS scavenging agent, blocked the interaction between wild‐type/mutant MARCH5 and FUNDC1 while also inhibiting FUNDC1 degradation (Fig EV5). These observations led us to suggest that hypoxia‐induced ROS generation serves as the initial signal to activate MARCH5/FUNDC1 to fine‐tune the graded response to hypoxic stress. Our findings thus identify MARCH5/FUNDC1 as a critical module for sensing oxidative stress signals and for subsequently adjusting mitochondrial behaviors in response to complex environmental cues.
Figure EV5. Hypoxia‐induced ROS generation promotes MARCH5–FUNDC1 interaction.
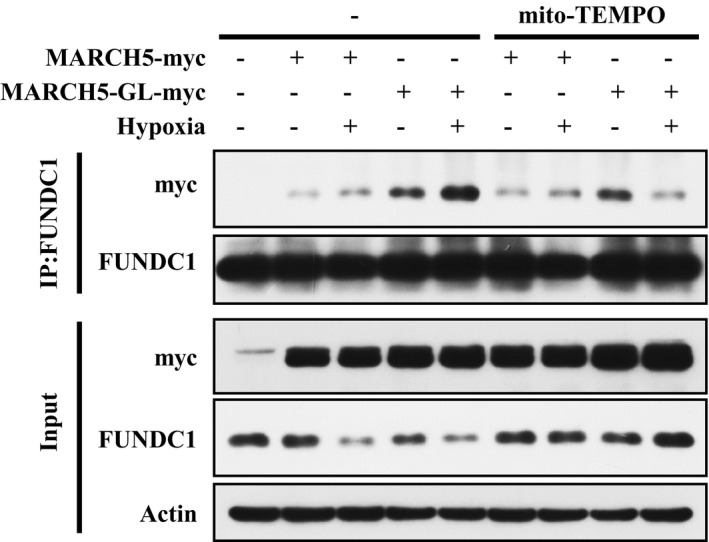
HeLa cells were transfected with MARCH5‐myc or the MARCH5‐GL‐myc mutant for 24 h and then exposed to 1% O2 for 12 h, with or without mito‐TEMPO (10 μM). Immunoprecipitation was performed with an anti‐FUNDC1 antibody. Co‐immunoprecipitated MARCH5‐myc and FUNDC1 were detected by Western blotting with anti‐myc and anti‐FUNDC1 antibodies, respectively. Source data are available online for this figure.
Previous studies have shown that MARCH5 is a key regulator of mitochondrial dynamics, as it is able to ubiquitylate Fis1, Mfn1, Mfn2, and MiD49. In particular, MARCH5 RNAi increases MiD49 expression, thereby enhancing Drp1 recruitment, which triggers mitochondrial fission 51. We recently reported that FUNDC1 is also involved in regulation of mitochondrial dynamics by coordinating the interaction with OPA1 and Drp1 57. We also observed that the level of mitochondrial fragmentation under hypoxic conditions was higher in MARCH5 siRNA HeLa cells than in MARCH5‐scramble cells (Z. Chen, unpublished data). These results indicate that intimate connections exist between the mitochondrial fission/fusion cycle and mitophagy, and these processes are coordinated at the protein level by MARCH5.
Materials and Methods
Cell culture and transfection
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin–streptomycin and maintained in 5% CO2 at 37°C. For hypoxia treatment, cells were transferred to a special hypoxia chamber (Billups‐Rothenberg) with a pre‐analyzed gas mixture containing 1% O2, 5% CO2, and 95% N2. For transfection, plasmids were transfected into cells with MegaTran 1.0 (OriGene) according to the manufacturer's instructions.
Expression constructs
The expression plasmids for human MARCH5, FUNDC1, Mulan, Parkin, RNF185, Keap1, and Smurf1 were generated by amplifying the corresponding cDNA by PCR and cloning it into pcDNA4‐TO‐myc‐his‐B, pFLAG‐CMV4, or pEGFP‐C1 expression vectors. The recombinant GST fusion proteins GST‐MARCH5 and GST‐FUNDC1 were generated by cloning the corresponding cDNA into the pGEX‐4T‐1 vector. The site‐specific mutants and deletion mutants were generated according to the manufacturer's (Stratagene, 200521) instructions.
RNA interference assay
We are grateful to Mian Wu (University of Science and Technology of China, Anhui, China) for the MARCH5 shRNA. The targeted sequence in MARCH5 for RNA interference (5′‐GCCAATCCTTTAGCAGATCA‐3′) was cloned into the pLKO.1‐puro vector; the target FUNDC1 sequence for RNA interference (5′‐GCAGCACCTGAAATCAACA‐3′) was cloned into the pSilencer2.1‐neo vector. HeLa cells were transfected with the corresponding vectors and selected with puromycin or neomycin to establish stable knockdown cell lines.
SDS–PAGE and Western blotting
Cells were harvested and lysed in lysis buffer (137 mM NaCl, 20 mM Tris, pH 7.4, 2 mM EDTA, 1% NP‐40, 10% glycerol, 2.5 mM sodium pyrophosphate, 1 mM Na3VO4, and protease inhibitors). Equivalent protein quantities were subjected to SDS–PAGE and transferred to nitrocellulose membranes. The membranes were blocked in 5% non‐fat milk or 5% BSA for 1 h at room temperature, and then probed with the indicated primary antibodies, followed by the appropriate HRP‐conjugated anti‐mouse/rabbit secondary antibodies (KPL). Immunoreactive bands were visualized with a chemiluminescence kit (Pierce). The following antibodies were used: antibodies against Tim23 (1:2,000, BD Biosciences), Tom20 (1:1,000, BD Biosciences), HA (1:1,000, Sigma), actin (1:10,000, Sigma), myc (1:2,000 Sigma), ubiquitin (1:1,000, Cell Signaling), MiD49 (1:500, Sigma), flag (1:2,000 Sigma), GFP (1:1,000, Invitrogen), GST (1:2,000, Santa Cruz), and LC3 (1:1,000 Sigma). Polyclonal antibodies against FUNDC1 (1:1,000) and p‐FUNDC1 (Tyr18) (1:1,000) were generated by immunizing rabbits with purified FUNDC1 peptides or phosphopeptides, and then affinity‐purified (Abgent). The antibody against MARCH5 was produced by immunizing mice with the purified 1–72aa truncation of MARCH5. The ImageJ program was used for densitometric analyses of Western blots, and the quantification results were normalized to the loading control.
Immunoprecipitation
Cells were collected and lysed with 0.6 ml of lysis buffer plus protease inhibitors (Roche Applied Science) for 40 min on ice. After centrifugation at 12,000 g for 15 min, the lysates were immunoprecipitated with 1 μg specific antibody overnight at 4°C, and 30 μl protein A agarose beads (Invitrogen) or protein G agarose beads (Santa Cruz) were washed and then added for an additional 3 h. Thereafter, the precipitants were washed four times with lysis buffer, and the immune complexes were boiled with loading buffer for 3 min and analyzed by SDS–PAGE and Western blotting.
GST pulldown
All GST‐tagged proteins were expressed in Escherichia coli Rosetta (DE3). GST fusion proteins were purified on glutathione–Sepharose 4 Fast Flow beads (GE Health Science). For GST pulldown, 4 μg of GST‐MARCH5 protein was incubated with cell lysates for 2 h at 4°C and then washed five times with 1 ml PBS buffer. The precipitate complex was boiled with loading buffer for 3 min at 95°C and subjected to SDS–PAGE and Western blotting. The nitrocellulose membrane was stained with Ponceau S and then incubated with anti‐FUNDC1 antibody.
Immunofluorescence microscopy
HeLa cells were transfected with the indicated plasmids or subjected to hypoxia, and then fixed with 3.7% formaldehyde in DMEM for 15 min at 37°C. The fixed cells were permeabilized with 0.2% Triton X‐100 (Sigma) for 15 min on ice, blocked and incubated with the indicated primary antibodies at room temperature for 1 h, then incubated with fluorescence‐conjugated secondary antibodies at room temperature for 40 min. Cell images were captured with an LSM 510 Zeiss confocal microscope (Carl Zeiss Jena, Germany). The following fluorescent primary antibodies were used: antibodies against cytochrome c (1:200, BD Biosciences), Tom20 (1:100, BD Biosciences), and myc (1:200, Sigma). The following fluorescent secondary antibodies were used: goat anti‐mouse IgG FITC (1:300, Invitrogen), goat anti‐mouse IgG Cy3 (1:300, Invitrogen), goat anti‐mouse Cy5 (1:300, Invitrogen), goat anti‐rabbit IgG Cy3 (1:300, Invitrogen), and goat anti‐rabbit Cy5 (1:300, Invitrogen).
In vivo ubiquitylation assay
Cells were transiently transfected with plasmids expressing HA‐Ub for 24 h, with or without myc‐tag vectors. Cells were exposed to hypoxia or treated with FCCP, together with MG132 (Sigma) before collection. Cells were washed with PBS and then lysed in 200 μl denaturing buffer (150 mM Tris–HCl, pH 7.4, 1% SDS) by boiling for 10 min. The lysates were made up to 1 ml with lysis buffer and immunoprecipitated using 1 μg anti‐FUNDC1 antibody, then subjected to Western blotting with anti‐HA or anti‐FUNDC1 to detect FUNDC1 ubiquitylation.
In vitro ubiquitylation assay
In vitro ubiquitylation assays were carried out in ubiquitylation buffer (50 mM Tris, pH 7.4, 5 mM MgCl2, 2 mM dithiothreitol) with human recombinant E1 (100 ng, Abcam), human recombinant E2 UbcH5c (200 ng, Upstate), and ubiquitin (10 μg, Boston Biochem). MARCH5 or MARCH5 mutants were immunopurified from cell lysates of cells expressing MARCH5‐myc or its mutants; 2 μg GST‐FUNDC1 and GST‐FUNDC1 lysine mutants were expressed and purified from E. coli Rosetta (DE3). Ubiquitylation reaction mixtures (30 μl volume) were incubated for 2 h at 30°C and then boiled with loading buffer for 5 min and analyzed by Western blotting using anti‐ubiquitin and anti‐myc antibodies.
Cycloheximide chase assay
The stability of endogenous FUNDC1 was determined by CHX chase assays. Scramble or MARCH5‐knockdown HeLa cells were seeded in six‐well plates (Nest), or cells were transiently transfected with MARCH5‐myc, MARCH5‐CS‐myc mutant, or MARCH5‐Rescue‐myc‐vector for 24 h. Cells were treated with CHX (100 μg/ml) for the indicated time and then collected and lysed by Western blotting.
Statistical analysis
All data were obtained from at least three independent experiments and expressed as means ± SEM. Statistical analyses were performed using the Student's two‐tailed t‐test, with P‐values < 0.05 considered significant. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the corresponding controls were indicated. All statistical calculations were performed with GraphPad Prism software.
Author contributions
QuC, ZC, and LL designed the experiments. ZC and QuC wrote the manuscript. ZC performed most of the experiments with the help of QiC, YW, YL, HW, WZ, SAS, SS and YZ. XW and JW provided technical support for the project. All authors provided intellectual input and read the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We would like to thank Mian Wu from University of Science and Technology of China for MARCH5 shRNA and control plasmids. This research was supported by the 973 program projects (2015CB856303; 2016YFA0500201) from the Ministry of Science and Technology (MOST) and Natural Science Foundation of China (31671446; 31520103904; 31471306).
EMBO Reports (2017) 18: 495–509
References
- 1. Hatefi Y (1985) The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem 54: 1015–1069 [DOI] [PubMed] [Google Scholar]
- 2. Smeitink J, van den Heuvel L, DiMauro S (2001) The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2: 342–352 [DOI] [PubMed] [Google Scholar]
- 3. Blumberger J (2015) Recent advances in the theory and molecular simulation of biological electron transfer reactions. Chem Rev 115: 11191–11238 [DOI] [PubMed] [Google Scholar]
- 4. Chandel NS (2015) Evolution of mitochondria as signaling organelles. Cell Metab 22: 204–206 [DOI] [PubMed] [Google Scholar]
- 5. Stock D, Leslie AG, Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286: 1700–1705 [DOI] [PubMed] [Google Scholar]
- 6. Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309–1312 [DOI] [PubMed] [Google Scholar]
- 7. Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell‐free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157 [DOI] [PubMed] [Google Scholar]
- 8. Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933 [PubMed] [Google Scholar]
- 9. Chen LT, Hu MM, Xu ZS, Liu Y, Shu HB (2016) MSX1 modulates RLR‐mediated innate antiviral signaling by facilitating assembly of TBK1‐associated complexes. J Immunol 197: 199–207 [DOI] [PubMed] [Google Scholar]
- 10. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, Shu HB (2009) The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30: 397–407 [DOI] [PubMed] [Google Scholar]
- 11. Picard M, McManus MJ, Gray JD, Nasca C, Moffat C, Kopinski PK, Seifert EL, McEwen BS, Wallace DC (2015) Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc Natl Acad Sci USA 112: E6614–E6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jensen MB, Jasper H (2014) Mitochondrial proteostasis in the control of aging and longevity. Cell Metab 20: 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finkel T (2015) The metabolic regulation of aging. Nat Med 21: 1416–1423 [DOI] [PubMed] [Google Scholar]
- 14. Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiol Rev 94: 909–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shadel GS, Horvath TL (2015) Mitochondrial ROS signaling in organismal homeostasis. Cell 163: 560–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22: 79–99 [DOI] [PubMed] [Google Scholar]
- 17. Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11: 872–884 [DOI] [PubMed] [Google Scholar]
- 18. Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287 [DOI] [PubMed] [Google Scholar]
- 19. Doerks T, Copley RR, Schultz J, Ponting CP, Bork P (2002) Systematic identification of novel protein domain families associated with nuclear functions. Genome Res 12: 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryan MT, Hoogenraad NJ (2007) Mitochondrial‐nuclear communications. Annu Rev Biochem 76: 701–722 [DOI] [PubMed] [Google Scholar]
- 21. Scarpulla RC (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: 611–638 [DOI] [PubMed] [Google Scholar]
- 22. Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu L, Sakakibara K, Chen Q, Okamoto K (2014) Receptor‐mediated mitophagy in yeast and mammalian systems. Cell Res 24: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durcan TM, Fon EA (2015) The three ‘P's of mitophagy: PARKIN, PINK1, and post‐translational modifications. Genes Dev 29: 989–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li W, Zhang X, Zhuang H, Chen HG, Chen Y, Tian W, Wu W, Li Y, Wang S, Zhang L et al (2014) MicroRNA‐137 is a novel hypoxia‐responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem 289: 10691–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- 28. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C et al (2014) A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor‐mediated mitophagy. Mol Cell 54: 362–377 [DOI] [PubMed] [Google Scholar]
- 29. Guo C, Hildick KL, Luo J, Dearden L, Wilkinson KA, Henley JM (2013) SENP3‐mediated deSUMOylation of dynamin‐related protein 1 promotes cell death following ischaemia. EMBO J 32: 1514–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maechler P, Wollheim CB (2001) Mitochondrial function in normal and diabetic beta‐cells. Nature 414: 807–812 [DOI] [PubMed] [Google Scholar]
- 31. Patti ME, Corvera S (2010) The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 31: 364–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schon EA, Przedborski S (2011) Mitochondria: the next (neurode)generation. Neuron 70: 1033–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quiros PM, Langer T, Lopez‐Otin C (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16: 345–359 [DOI] [PubMed] [Google Scholar]
- 34. Fulda S, Galluzzi L, Kroemer G (2010) Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 9: 447–464 [DOI] [PubMed] [Google Scholar]
- 35. Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and cancer. Cell 166: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA (2002) Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal 4: 769–781 [DOI] [PubMed] [Google Scholar]
- 37. Court FA, Coleman MP (2012) Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci 35: 364–372 [DOI] [PubMed] [Google Scholar]
- 38. Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev 91: 1161–1218 [DOI] [PubMed] [Google Scholar]
- 39. Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshii SR, Kishi C, Ishihara N, Mizushima N (2011) Parkin mediates proteasome‐dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D (2011) Phosphorylation of serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 22: 3206–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17: 98–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Okamoto K, Kondo‐Okamoto N, Ohsumi Y (2009) Mitochondria‐anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17: 87–97 [DOI] [PubMed] [Google Scholar]
- 45. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A et al (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- 48. Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura‐Hoshino M, Sada K, Hotta H, Yamamura H et al (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25: 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park YY, Nguyen OT, Kang H, Cho H (2014) MARCH5‐mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5: e1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N et al (2013) MITOL regulates endoplasmic reticulum‐mitochondria contacts via Mitofusin2. Mol Cell 51: 20–34 [DOI] [PubMed] [Google Scholar]
- 51. Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C et al (2016) Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress‐induced apoptosis through regulation of MiD49 protein. Mol Biol Cell 27: 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gu H, Li Q, Huang S, Lu W, Cheng F, Gao P, Wang C, Miao L, Mei Y, Wu M (2015) Mitochondrial E3 ligase March5 maintains stemness of mouse ES cells via suppression of ERK signalling. Nat Commun 6: 7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim SH, Park YY, Yoo YS, Cho H (2016) Self‐clearance mechanism of mitochondrial E3 ligase MARCH5 contributes to mitochondria quality control. FEBS J 283: 294–304 [DOI] [PubMed] [Google Scholar]
- 54. Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wei H, Liu L, Chen Q (2015) Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta 1853: 2784–2790 [DOI] [PubMed] [Google Scholar]
- 56. Waypa GB, Smith KA, Schumacker PT (2016) O2 sensing, mitochondria and ROS signaling: the fog is lifting. Mol Aspects Med 47–48: 76–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L et al (2016) Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12: 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6