

Abstract
Defects in mitochondrial respiratory chain complex IV (CIV) frequently cause encephalocardiomyopathies. Human CIV assembly involves 14 subunits of dual genetic origin and multiple nucleus‐encoded ancillary factors. Biogenesis of the mitochondrion‐encoded copper/heme‐containing COX1 subunit initiates the CIV assembly process. Here, we show that the intermembrane space twin CX 9C protein CMC1 forms an early CIV assembly intermediate with COX1 and two assembly factors, the cardiomyopathy proteins COA3 and COX14. A TALEN‐mediated CMC1 knockout HEK293T cell line displayed normal COX1 synthesis but decreased CIV activity owing to the instability of newly synthetized COX1. We demonstrate that CMC1 stabilizes a COX1‐COA3‐COX14 complex before the incorporation of COX4 and COX5a subunits. Additionally, we show that CMC1 acts independently of CIV assembly factors relevant to COX1 metallation (COX10, COX11, and SURF1) or late stability (MITRAC7). Furthermore, whereas human COX14 and COA3 have been proposed to affect COX1 mRNA translation, our data indicate that CMC1 regulates turnover of newly synthesized COX1 prior to and during COX1 maturation, without affecting the rate of COX1 synthesis.
Keywords: CMC1, complex IV, COX1, cytochrome c oxidase, mitochondrial respiratory chain
Subject Categories: Membrane & Intracellular Transport, Physiology, Protein Biosynthesis & Quality Control
Introduction
Mitochondrial respiratory chain complex IV (CIV) is a heme a/a 3‐copper terminal cytochrome c oxidase that reduces oxygen to water. CIV also contributes to the creation of the proton gradient across the inner membrane that drives ATP synthesis through oxidative phosphorylation (OXPHOS). CIV deficiencies in humans severely affect cellular aerobic energy production and therefore are a common cause of encephalo‐ and cardiomyopathies 1.
Human CIV contains 14 subunits. The three catalytic subunits (COX1, COX2, and COX3) are encoded in the mitochondrial genome. The remaining subunits (COX4, COX5a, COX5b, COX7a, COX6c, COX7c, COX6b, COX6a, COX7b, COX8, NDUFA4), some of which have tissue‐specific isoforms, are nucleus‐encoded, synthesized in the cytoplasm, and imported into mitochondria. The CIV redox centers are located in subunits COX1 and COX2. COX1 harbors a heme a center and a binuclear heme a 3‐copper (CuB) center, and COX2 contains a dinuclear copper center (CuA). Formation of the redox centers is not only essential for CIV function but also for complex assembly. In fact, CIV biogenesis involves first the maturation of COX1 and COX2 and then the ordered assembly of all other subunits around COX1. During the process, COX4 and COX5a are added to a first subcomplex formed only by COX1 and several COX1‐specific chaperones. Then, matured COX2 is incorporated, and the remaining subunits are subsequently added to yield the CIV monomer 2, 3.
Complex IV biogenesis is extensively regulated and requires the assistance of numerous conserved assembly factors interceding at all stages of the process 4, 5, 6. Studies in the yeast Saccharomyces cerevisiae have shown that the two major targets for regulation are COX1 synthesis and incorporation of metal prosthetic groups. In S. cerevisiae, COX1 mRNA translation is regulated by heme B availability 7, and heme A biosynthesis is regulated by both an early CIV assembly intermediate 8 and COX1‐dependent oligomerization of the heme O synthase COX10 9. Furthermore, Cox1 synthesis is coordinated with its assembly into CIV by the existence of a negative feedback loop 10, 11.
In S. cerevisiae, Mss51 is a COX1 mRNA‐specific translation activator that also interacts with Cox1 protein and Cox1‐specific chaperones Cox14 and Coa3 in a stable complex 11, 12, 13. Only when Cox1 proceeds in the assembly pathway, Mss51 is released and becomes available for new rounds of Cox1 synthesis 11. This system serves in part to prevent the accumulation of partially matured or unassembled Cox1, which can be deleterious 14. In humans, COX1 synthesis is specifically activated by the late‐onset Leigh's syndrome protein TACO1 15, which does not interact with COX1. However, although a functional human homolog of yeast Mss51 has not been identified, mitochondrial cardiomyopathy proteins COA3 (also called MITRAC12, COX25, or CCDC56) and COX14 (or C12orf62) are both COX1 chaperones that interact with newly synthesized COX1 and drive it through the entire CIV assembly process 16, 17, 18, 19. Human CIV assembly intermediates containing newly synthesized COX1 and imported respiratory chain subunits have been termed MITRAC complexes 19. Intriguingly, it has been proposed that MITRAC complexes, and specifically the COX1‐COX14‐COA3 complex, could regulate COX1 synthesis in humans as it occurs in S. cerevisiae 16, 19, 20. SURF1, another COX1 assembly factor 21, 22 shown to be involved in heme a center formation in bacteria 23 but never in mitochondria, is also part of these MITRAC complexes 19. More recent work has identified a new MITRAC component, termed MITRAC7, which affects the biogenesis pathway by stabilizing newly synthesized COX1 in assembly intermediates that contain COX14 and COA3 and also subunits COX4 and COX6c. These observations have suggested the existence of a quality control checkpoint during CIV assembly that regulates COX1 turnover 20.
The list of CIV assembly factors includes several members of the twin CX9C motif family of proteins, which contain two structural disulfide bonds and are located in the intermembrane space 24, 25, 26. The roles of these proteins in the CIV assembly process remain in most cases enigmatic. Only COX17, a CIV‐specific copper chaperone, has a recognized function in copper delivery to metallochaperones COX11 and SCO1/2 for further delivery to COX1 or COX2, respectively 27, 28, 29, 30, 31, 32, 33. Other twin CX9C proteins known to be critical for CIV assembly/stability/function in S. cerevisiae and/or human cells include the mitochondrial cardiomyopathy proteins PET191 and COA6 34, 35, 36 as well as Cmc1, Cmc2, Cox19, and Cox23 37, 38, 39, 40. Saccharomyces cerevisiae and human COA6 interact with SCO1/SCO2 to somehow facilitate the copper‐dependent biogenesis of COX2 35, 41, 42, and S. cerevisiae Cox19 interacts with Cox11 probably to promote copper transfer to Cox1 43. Regarding Cmc1, Cmc2, Pet191, and Cox23, although indirect links to copper homeostasis or delivery have been reported, their functions remain largely unknown 37, 39, 40, 44. As an interesting observation, human CMC1 was co‐purified with COX1 19.
Here, we have used extensive genetics and biochemical analyses to gain insight into the role of human CMC1 in CIV assembly. Our data demonstrate that CMC1 acts in the intermembrane space to stabilize the COX1‐COX14‐COA3 complex prior to the incorporation of subunits COX4 and COX5a. Additionally, this COX1‐COX14‐COA3‐CMC1 complex is independent of COX10, COX11, MITRAC7, and SURF1, hinting that CMC1 probably maintains COX1 in a maturation‐competent state before insertion of its prosthetic groups. Finally, we show that COX1‐COX14‐COA3‐CMC1 complex is stable and COX1 synthesis is normal when CIV assembly is stalled at COX2 or COX3 biogenesis steps, discarding any COX1 mRNA translational regulation in these conditions.
Results
TALEN‐mediated generation of CMC1 knockout (KO) lines in HEK293T cells
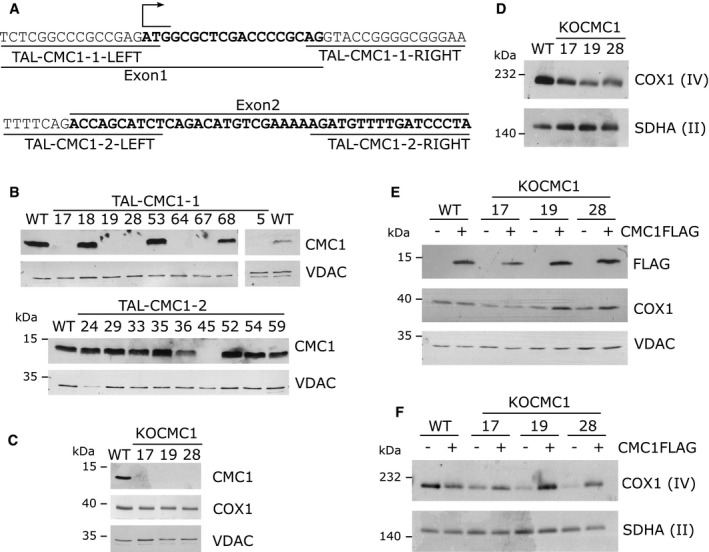
To analyze the function of human CMC1 in mitochondria, we used the transcription activator‐like effector nuclease (TALEN) technology 24, 25 to create stable CMC1 knockout lines in HEK293T (human embryonic kidney) cells. Cellectis Bioresearch (Paris, France) designed and generated TALEN binding pairs to disrupt CMC1 expression. The constructs target DNA regions either within the first exon of CMC1 directly downstream the start codon or at the beginning of the second exon (Fig 1A). When both TALENs bind CMC1 DNA, the endonuclease induces a double‐strand break, which is mutagen prone. We co‐transfected HEK293T cells with either the TALEN pair #1 or #2, and subsequently, single cells were isolated by fluorescence‐activated cell sorting. After growth, 55 clones from TALEN pair #1 and 66 clones from TALEN pair #2 transfections were screened for CMC1 steady‐state levels by immunoblotting. CMC1 levels were decreased in seven clones from TALEN pair #1 and two clones from TALEN pair #2 (Fig 1B). CMC1 gene was sequenced in seven clones. One clone with 50% residual CMC1 level (#2–36) was found to be heterozygous, whereas the clones without detectable CMC1 protein had each two mutant alleles predicted to lead to protein absence (Table 1). These knockout clones are henceforth referred as KO‐CMC1.
Figure 1. TALEN‐mediated generation of KO‐CMC1 clones in HEK293T.
-
ASchematic representation of the first and second exons of the CMC1 locus and the sequences recognition sites of the two TALEN pairs.
-
B, CImmunoblot analysis of the steady‐state levels of (B) CMC1 in HEK293T (WT) and TALEN‐transfected HEK293T cell lines, or (C) COX1 and CMC1 in three KO‐CMC1 clones. VDAC was used as a loading control.
-
DSteady‐state levels of CIV extracted with lauryl maltoside in three KO‐CMC1 clones analyzed by BN–PAGE and detected by immunoblotting with an anti‐COX1 antibody. Complex II (SDHA) serves as a loading control.
-
EImmunoblot analysis in whole‐cell extracts of COX1 and CMC1‐FLAG steady‐state levels in KO‐CMC1 cells carrying an empty plasmid or stably expressing CMC1‐FLAG compared with the parental line (WT).
-
FSteady‐state levels of CIV in three KO‐CMC1 clones carrying an empty plasmid or stably expressing CMC1‐FLAG analyzed as in panel (D).
Source data are available online for this figure.
Table 1.
CMC1 alleles in TAL‐CMC1 clones
| TAL‐ clones | CMC1 genotype | DNA | Protein |
|---|---|---|---|
| TAL1‐05 | C. Hetero | c.8_17del/c.(−22)_19del | p.Leu3 fs/np |
| TAL1‐17 | C. Hetero | c.10_11del/c.12_15insACGA | p.Asp4 fs/p.Asp4 fs |
| TAL1‐19 | C. Hetero | c.(12_19+1)del/c.(6_19+3)del | p.Pro5 fs/p.Leu3 fs |
| TAL1‐28 | Mut. Homo | c.10_14del | p.Asp4 fs |
| TAL1‐64 | C. Hetero | c.14C>G 15_18del/c.(−5_19+1)del | p.Pro5 fs/np |
| TAL1‐67 | C. Hetero | c.(14_19+5)del/c.9C>T (11_19+2)del | p.Pro5 fs/p.Pro5 fs |
| TAL2‐36 | Hetero | WT/c.40G>T 41–45del | WT/p.Glu14Ter |
The DNA numbering refers to the coding sequence (c.) and the protein (p.) number to the predicted full polypeptide 67. C: compound; Mut: mutant; Hetero: heterozygous; Homo: homozygous; del: deletion; +: position in introns; −: position before ATG; >: substitution; ins: insertion; fs: frameshift; np: no protein synthesized (start codon deleted); Ter: stop codon; WT: wild type.
Complex CIV steady‐state levels are decreased in KO‐CMC1 cell lines
Cmc1 was first described in S. cerevisiae as required for full expression of CIV 26, although its specific role remains unknown. The protein is not essential for CIV assembly since a strain carrying a null cmc1 allele retains 40% of CIV activity 26. This observation suggested that Cmc1 could have a redundant function or that it enhances the efficiency of a CIV assembly step.
To evaluate the requirement of CMC1 for CIV assembly in human cells, we analyzed three KO‐CMC1 cell lines for steady‐state levels of COX1 subunit by SDS–PAGE (Fig 1C) and of CIV complex by BN–PAGE (Fig 1D) followed by immunoblotting. In the three clones tested, COX1 and CIV steady‐state levels were decreased (Fig 1C and D). This CIV assembly/stability defect was fully restored by stable expression of C‐terminal FLAG‐tagged CMC1 (Fig 1E and F), eliminating the possibility that our results were attributable to off‐target effects that arose during the gene disruption procedure. Our data indicate that, as in S. cerevisiae, human CMC1 is not essential but required to stabilize CIV or enhance its formation rate in HEK293T cells. Given the consistency among the phenotypes of the three KO‐CMC1 clones analyzed, we chose one of them (TAL1‐19) for all subsequent studies.
CMC1 participates exclusively in the biogenesis of CIV
The KO‐CMC1 cell line had a basal respiratory rate lowered to 70% of the parental line (Fig 2A). This phenotype was a direct consequence of an isolated CIV deficiency (Fig 2B) because the steady‐state levels of subunits from OXPHOS complexes I, II, III, and V, as the fully assembled complexes, were unchanged in KO‐CMC1 cells (Fig 2C and D). The fact that a ~45% decrease in CIV activity limits cell respiration to ~70% supports the notion that there is a low‐reserve cytochrome c oxidase capacity in cultured HEK293T cells, in agreement with previous cyanide titration experiments that had shown that in several cell lines, the CIV activity capacity is in low excess (16–40%) with respect to that required to support the endogenous respiration rate 27. As a note, although CMC1‐FLAG is overexpressed in our transfected cell lines (Fig 2D), CIV levels and activity are not reproducibly different than in the parental HEK293T cell line (Fig 2B–D).
Figure 2. CMC1 is required for cytochrome c oxidase biogenesis or stability.
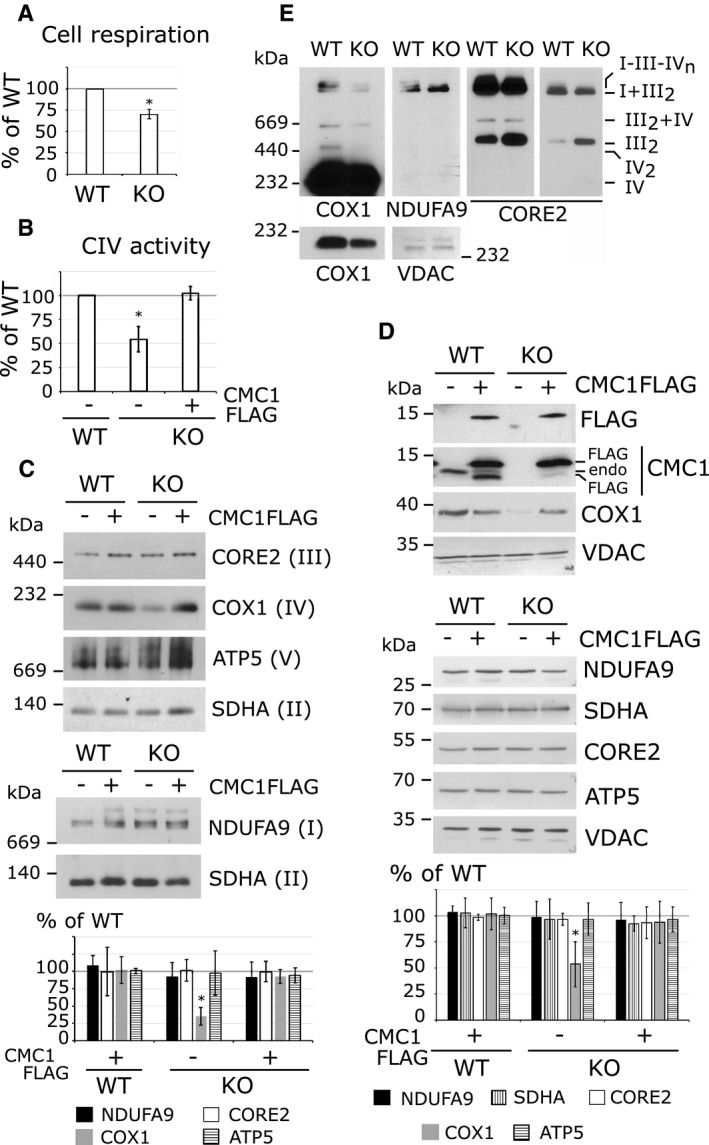
- Endogenous cell respiration rate expressed as percentage of the HEK293T control cell line. The bars represent average ± SD. n = 3; t‐test: *P = 0.00066.
- Cytochrome c oxidase activity in HEK293T (WT) and KO‐CMC1 cell lines carrying an empty plasmid or stably expressing CMC1‐FLAG. The values are expressed as percentage of the control and normalized by citrate synthase activity. The bars represent average ± SD. n = 3, t‐test: *P = 0.004 for KO, P = 0.6 for KO + CMC1‐FLAG.
- Steady‐state levels of OXPHOS complexes extracted with lauryl maltoside in HEK293T and KO‐CMC1 cell lines carrying an empty plasmid or stably expressing CMC1‐FLAG, analyzed by BN–PAGE and detected by immunoblotting with the indicated antibodies. The digitalized signal was quantified using ImageJ program, normalized by SDHA and expressed as percentage of the HEK293T control. The bars represent average ± SD. n = 3 – 10, t‐test: *P = 5 × 10−11 for CIV in KO‐CMC1.
- Immunoblot analysis of CMC1 and OXPHOS complex subunits in HEK293T and KO‐CMC1 cell lines carrying an empty plasmid or stably expressing CMC1‐FLAG. NDUFA9 is a subunit of complex I, SDHA of CII, CORE2 or CIII and ATP5 of the F1Fo‐ATP synthase. Endo: endogenous. The signal was normalized by VDAC and expressed as percentage of the control. The bars represent average ± SD. n = 4, t‐test: *P = 0.0004 for COX1 in KO‐CMC1.
- Steady‐state levels of respiratory chain complexes and supercomplexes extracted with digitonin from HEK293T and KO‐CMC1 cell lines, and analyzed by BN–PAGE. The panels show immunoblots probed with the indicated antibodies. Two different exposure times are presented for COX1 and CORE2. VDAC serves as a loading control.
Source data are available online for this figure.
We also investigated the effect of knocking out CMC1 on the ability of respiratory complexes to form supercomplexes, by analyzing digitonized whole‐cell extracts using BN–PAGE. The KO‐CMC1 cell line showed normal levels of I + III2 complexes but supercomplexes containing CIV (III2 + IV and I + III2 + IVn) were decreased or too scarce to be detected, whereas the CIII dimer levels were increased (Fig 2E). In summary, biochemical analyses of KO‐CMC1 cells indicate that CMC1 functions to promote CIV assembly or stability without affecting other respiratory chain enzyme complexes.
CMC1 is not required for COX1 synthesis but some COX1 post‐translational events
A characteristic of mammalian CIV assembly mutants is the frequent accumulation of assembly intermediates, which are informative regarding the assembly step that could be impaired. Here, BN–PAGE analysis of KO‐CMC1 cells disclosed no accumulation of subassemblies containing COX1 when extracted with lauryl maltoside (Fig 3A) and a mild accumulation when extracted with digitonin (Fig 3B). In lauryl maltoside extraction conditions, accumulation of CIV intermediates, containing COX1 alone or assembled with COX4 and COX5a, has been reported in fibroblasts from patients carrying mutations in assembly factors such as the COX2 metallochaperone SCO1, the COX1 biogenetic factor SURF1, or the COX2 chaperone COX20 28, 29. On the contrary, COX1‐containing intermediates are absent or not highly accumulated in fibroblasts from patients carrying mutations in COX1 biogenetic factors such as the translational activator TACO1 15, the heme O synthase COX10 28, or the COX1 chaperones COA3 and COX14 16, 17. Here, consistently with the BN–PAGE data, the steady‐state levels of all CIV subunits tested are decreased in the absence of CMC1 (Fig 3C). The decrease is more pronounced (20–30%) for late assembled subunits like COX2, COX3 and COX6b, than for the earlier assembled proteins COX1 and COX4 (70–50%). In HEK293T cells, when CIV assembly stalls at the level of COX2 biogenesis, the accumulation of COX1 and COX4 in subcomplexes allows the maintenance of normal steady‐state levels of these two proteins, like in engineered KO‐COX20 HEK293T mutant cell lines 30. Therefore, our observations are compatible with a role of CMC1 in some early event during COX1 biogenesis.
Figure 3. CMC1 is not required for COX1 synthesis but some COX1 post‐translational events.
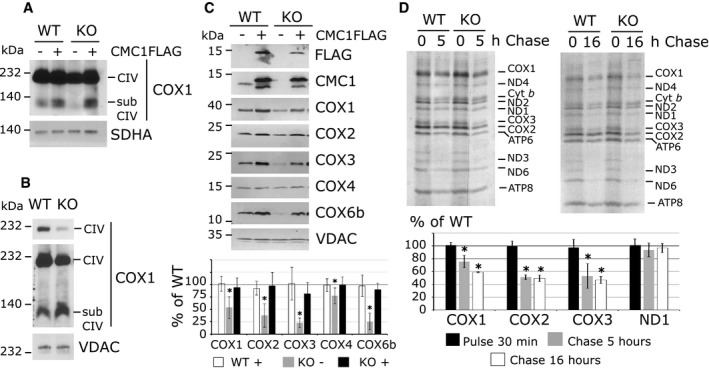
-
A, BAnalysis of COX1‐containing complexes in cell lines depleted for CMC1 and expressing CMC1‐FLAG or an empty plasmid. Cells were extracted by lauryl maltoside (A) or digitonin (B), separated by BN–PAGE and analyzed by immunoblotting. subCIV: subcomplexes IV. In (B), two different expositions of COX1 are represented.
-
CImmunoblot analysis of the steady‐state levels of CIV subunits in mitochondria isolated from HEK293T and KO‐CMC1 cells carrying an empty plasmid or stably expressing CMC1‐FLAG. In the graph, the digitalized signal was normalized by VDAC signal and expressed as percentage of the control. The bars represent average ± SD. n = 4–6, t‐test: P is statistically different in KO‐CMC1 cells for COX1 *P = 0.0004, COX2 *P = 0.002, COX3 *P = 5 × 10−6, COX4 *P = 0.007 and COX6b *P = 0.0001.
-
DMitochondrial translation products were pulse‐labeled in HEK293T and KO‐CMC1 cells with [35S]‐methionine for 30 min in the presence of emetine to inhibit cytosolic translation. In pulse experiments, the cells were then washed and incubated with fresh complete culture media for 5 or 16 h. The radiolabeled mitochondrial proteins were separated by SDS–PAGE and visualized by autoradiography. In the graph, the radiolabeled signals were quantified as explained in Fig 2C, normalized by Cyt b and expressed as % of wild type (WT). The bars represent average ± SD. n = 2 – 4; t‐test: in reading order for 5‐h chase and 16‐h chase P = 0.0005, 2 × 10−5, 1 × 10−5, 0.0004, 0.001, 0.001, 0.3, 0.6.
Source data are available online for this figure.
We then tested whether CMC1 could play a role in COX1 mRNA translation. Pulse [35S]‐methionine labeling showed that COX1 and all other mtDNA‐encoded proteins were synthesized at normal rates in the KO‐CMC1 cell line (Fig 3D). However, COX1, COX2, and COX3 were unstable in KO‐CMC1 cells following 5‐ or 16‐h chase (Fig 3D). Similar quantification results were obtained when the COX1 radiosignal was normalized either by the signal of cytochrome b or ND1 (Fig 3D). In conclusion, CMC1 is not required for COX1 synthesis but is required for some COX1 post‐translational events such as stability, maturation, or assembly.
CMC1 is a membrane‐bound protein that interacts with newly synthesized COX1
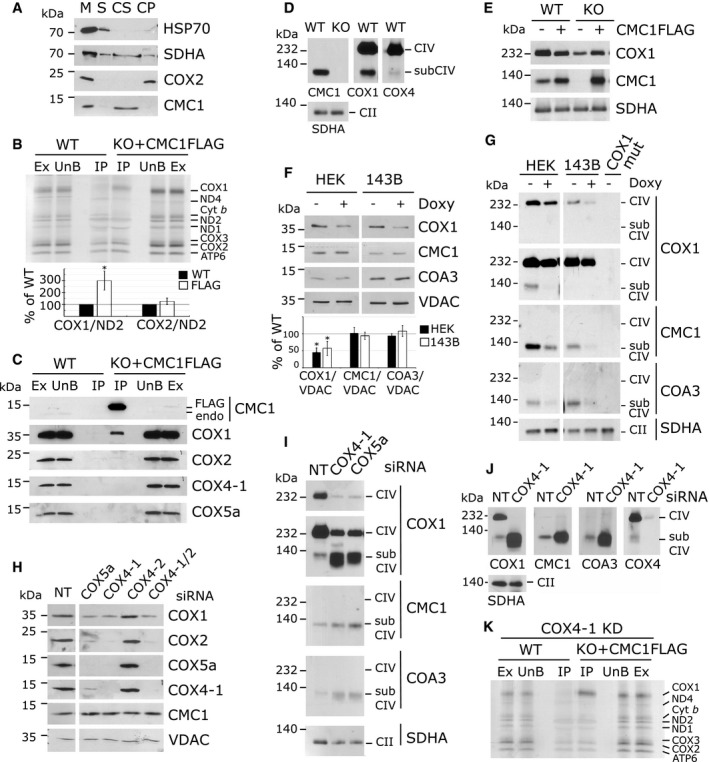
Saccharomyces cerevisiae Cmc1 is imported into the mitochondrial IMS by the Mia40/Erv1 oxidative folding pathway 31 and binds loosely to the inner membrane 26. Assays based on mild sonication of HEK293T purified mitochondria followed by extraction with alkaline carbonate disclosed that human CMC1 is also an extrinsic membrane‐bound protein (Fig 4A).
Figure 4. CMC1 interacts with newly synthesized COX1 in a stable complex prior the incorporation of COX4 and COX5a.
-
ATest of CMC1 solubility by mild sonication and alkaline carbonate extraction. Mitochondria (M) isolated from HEK293T cells were sonicated, and the soluble (S) and membrane‐bound fractions were separated by centrifugation. The pellet was subsequently extracted with alkaline sodium carbonate and fractionated into supernatant (CS) and pellet (CP). The different fractions were analyzed by immunoblotting using antibodies that recognized CMC1 and the controls COX2 (membrane protein), SDHA (loosely bound to the inner membrane), and HSP70 (soluble protein).
-
BMitochondrial translation products were pulse‐labeled in HEK293T (WT) and KO‐CMC1 + CMC1‐FLAG cells for 30 min. CMC1‐FLAG was immunoprecipitated using anti‐FLAG beads. IP is 15× enriched. Ex: extract/input; UnB: unbound/flow through; IP: immunoprecipitation. In the graph, COX1 and COX2 were quantified, normalized by ND2 and expressed relatively to the control. The bars represent average ± SD. n = 3; t‐test: *P = 0.008 for COX1, P = 0.17 for COX2.
-
CImmunoprecipitation of CMC1‐FLAG from a KO‐CMC1 + CMC1‐FLAG stable cell line using anti‐FLAG‐conjugated beads. Samples were separated by SDS–PAGE and immunoblotted with antibodies. HEK293T (WT) extracts were used as a negative control. IP is 40× enriched. Endo: endogenous.
-
D, EBN–PAGE analysis of HEK293T (WT), KO‐CMC1, and KO‐CMC1 + CMC1‐FLAG cells extracted with lauryl maltoside. The blots were probed with CMC1, COX1, COX4‐1, and SDHA antibodies.
-
FSDS–PAGE and immunoblot analysis of HEK293T and 143B cells treated 24 h with doxycycline. COX1, CMC1, and COA3 were quantified, normalized by VDAC and expressed relatively to the untreated sample. The bars represent average ± SD. n = 4 or 5, t‐test: from left to right: P = 0.00001; 0.006; 0.8; 0.3; 0.08; and 0.3.
-
GBN–PAGE analysis of cell extracts prepared as in (D and E) from HEK293T, 143B, or 143B‐COX1 mutant cells, following 24‐h inhibition of mitochondrial protein synthesis with doxycycline or left untreated. Two different exposure times are presented for COX1.
-
HSDS–PAGE and immunoblot analysis in HEK293T cells transfected for 3 days with non‐targeting (NT), COX4‐1, COX4‐2, COX4‐1 and COX4‐2 together or COX5a siRNAs.
-
I, JBN–PAGE and immunoblot analysis of CIV subassemblies (subCIV) in cellular extracts from HEK293T cells transfected with non‐targeting (NT), COX4‐1 or COX5a siRNAs for 3 (I) or 6 days (J). Two different exposure times are presented for COX1 (I).
-
KAs (B) but after 24 h of COX4‐1 knockdown by siRNA.
Source data are available online for this figure.
The studies presented in the previous section suggested a role for CMC1 in some early event of COX1 biogenesis. This possibility is also supported by a previous study that identified CMC1 as a COX1 interactor in human cells 19, although the meaning of this interaction remained unexplored. To address whether CMC1 acts as a COX1 chaperone, we then investigated possible physical interactions of CMC1 with newly synthesized COX1 or any other mitochondrion‐synthesized proteins. For this purpose, we labeled these proteins with [35S]‐methionine in KO‐CMC1 cells stably expressing CMC1‐FLAG, and adsorbed a cell extract onto anti‐FLAG‐agarose beads. In the [35S]‐methionine‐labeled proteins recovered from the beads, COX1 was selectively enriched when compared with the parental line (Fig 4B). CMC1‐FLAG was even able to co‐immunoprecipitate a small fraction of the total COX1 in extracts from unlabeled cells (Fig 4C), presumably corresponding to COX1 in assembly intermediate/s since, as discussed in the next sections, CMC1 signal does not overlap with fully assembled CIV in BN–PAGE assays. In conclusion, the stabilizing effect of CMC1 on CIV subunits is due to a direct interaction of CMC1 with newly synthesized COX1.
CMC1 forms a stable complex with COX1 in wild‐type mitochondria before the incorporation of COX4 and COX5a
To better understand the function of CMC1 in COX1 biogenesis, we analyzed its native molecular mass. BN–PAGE analysis of wild‐type whole‐cell extracts using an anti‐CMC1 antibody detected a complex migrating as ~120 kDa (Fig 4D), CMC1 being 13 kDa and COX1 40 kDa. This CMC1 complex co‐migrates with some COX1‐ and COX4‐containing subcomplexes (Fig 4D). Similar results were obtained when BN–PAGE analyses were performed in extracts from KO‐CMC1 cells expressing CMC1‐FLAG (Fig 4E).
To determine whether the stable ~120‐kDa CMC1 complex contains COX1, COX4 (14 kDa), and COX5a (13 kDa), we used two approaches. First, we tested whether the CMC1 complex could be detected in the absence of these CIV subunits. Removal of newly synthesized COX1 by inhibition of mitochondrial protein synthesis with doxycycline for 24 h prevented the formation of the CMC1 complex in two different human cell lines (Fig 4G) whereas CMC1 steady‐state levels remained unchanged (Fig 4F). The CMC1 complex was also undetectable in a 100% COX1 mutant cybrid cell line (Fig 4G), further indicating that this complex contains newly synthesized COX1.
To test the potential presence of COX4 and COX5a in the CMC1 complex, we performed siRNA‐mediated silencing of the two corresponding genes (Fig 4H). COX4 exists in two tissue‐specific oxygen‐regulated isoforms, COX4‐1 is expressed in normoxia and COX4‐2 in hypoxia 32. Although COX4‐1 should be the predominant isoform present in HEK293T cells under our growth conditions, we tested the effect of silencing each isoform. As expected, in COX4‐1 knockdown, CIV is highly decreased as visualized by COX2 steady‐state levels whereas knockdown of COX4‐2 has no effect (Fig 4H) and could not be detected with a specific antibody. Additionally, COX4‐2 is undetectable in HEK293T cells using a specific antibody.
When COX4‐1 or COX5a are silenced, the absence of each protein led to their reciprocal instability (Fig 4H), suggesting they may form a pre‐assembly module as it has been observed in S. cerevisiae 33 and human cells 34. However, the CMC1 complex was still present and accumulated in the absence of COX4‐1 and COX5a (Fig 4I and J). Furthermore, in the absence of COX4‐1, the immunoprecipitation of newly synthesized COX1 by CMC1‐FLAG was enhanced compared to the wild type (Fig 4K). Therefore, COX4 and COX5a are not part of the CMC1 complex, which is consistent with the fact that these two proteins were not co‐immunoprecipitated with CMC1‐FLAG (Fig 4C). These data also show that, in the wild type, at least two co‐migrating COX1‐containing subassemblies are detected by BN–PAGE, one containing CMC1 but not COX4 and COX5 and another without CMC1 but with COX4 and COX5.
In conclusion, CMC1 forms an early CIV assembly intermediate with newly synthesized COX1 before the incorporation of COX4 and COX5a.
COA3 and COX14 are components of the CMC1 complex
In an effort to define the components of the CMC1 complex, we tested its dependence on two known COX1 chaperones, COA3 (14 kDa) and COX14 (7 kDa), which interact with newly synthesized COX1 16, 18, 19. We approached this question by siRNA‐mediated knockdown of COA3 or COX14 followed by analysis of the CMC1 complex. Silencing of COA3 was highly effective (Fig 5A). The efficiency of COX14 silencing could not be directly estimated since we did not find any optimal anti‐COX14 antibody. However, we followed COX1 steady‐state levels as a surrogate and found them highly decreased after 7 days of COX14 silencing, as expected (Fig 5A). Silencing COX14 or COA3 resulted in decreased CMC1 steady‐state levels after 7 days of knockdown but not after 3 days (Fig 5A). However, the CMC1 complex was undetectable at both knockdown times (Fig 5B) despite that COX1 synthesis, analyzed by short pulses of 10 or 30 min, was essentially unaffected (Fig 5C). COA3 (13 kDa) and COX14 (7 kDa) are probably components of the CMC1 complex with COX1 (40 kDa) and CMC1 (13 kDa).
Figure 5. CMC1 cooperates with COA3 and COX14 in the biogenesis of COX1.
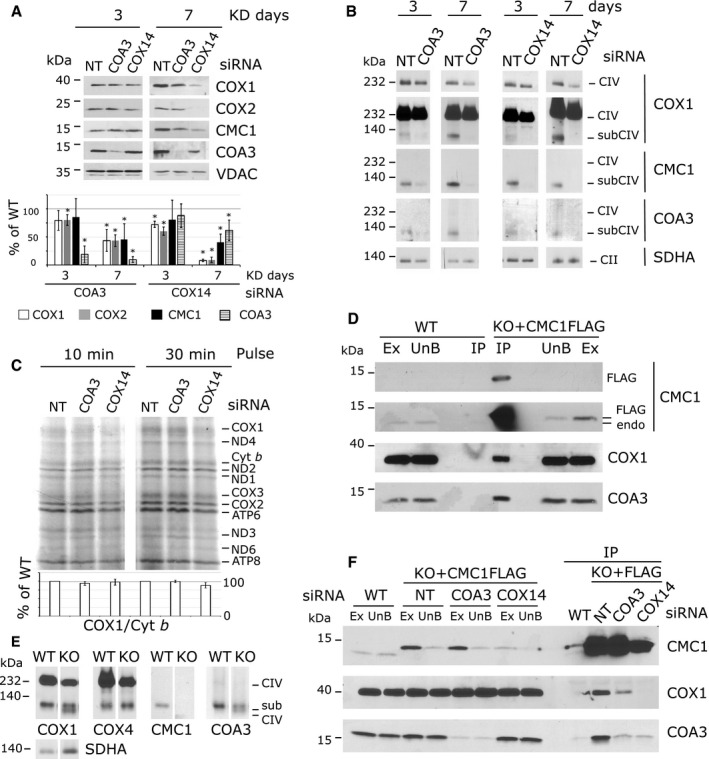
- SDS–PAGE and immunoblot analysis in HEK293T cells after 3 and 7 days of transfection with by non‐targeting (NT), COX14, or COA3 siRNA to knock down their expression. The signal was normalized by VDAC and expressed as percentage of the non‐targeted control. The bars represent average ± SD. n = 3 – 6, t‐test: in reading order P = 0.06; 0.007; 0.9; 4 × 10−5; 0.008; 0.0007; 0.03; 7 × 10−6; 2 × 10−7; 4 × 10−7; 0.2; 0.2; 2 × 10−7; 5 × 10−6; 0.002; 0.02.
- BN–PAGE and immunoblot analysis of CMC1, COA3, and COX1 in the same samples as (A). Two different expositions are presented for COX1.
- Mitochondrial protein synthesis after a 10‐ or 30‐min pulse with [35S]‐methionine in cells knocked down for 3 days. In the lower panel, the radiolabeled signal was quantified as in Fig 3 and expressed as percentage of the control. The bars represent average ± SD. n = 3, t‐test: P = 0.15; 0.7; 0.8; 0.07 in reading order.
- Immunoprecipitation of CMC1‐FLAG in extracts prepared from KO‐CMC1 + CMC1‐FLAG and HEK293T cells using anti‐FLAG‐conjugated agarose beads. Samples were separated by SDS–PAGE and immunoblotted with anti‐FLAG, CMC1, COX1, and COA3 antibodies. Ex: extract; UnB: unbound; IP: immunoprecipitation; endo: endogenous. IP is 40× enriched.
- BN–PAGE and immunoblot analysis of CMC1, COX4, COA3, and COX1 complexes in HEK293T (WT) and KO‐CMC1 cells.
- Immunoprecipitation of CMC1‐FLAG with FLAG‐conjugated beads from a KO‐CMC1 + CMC1‐FLAG stable cell line after 5 days of transfection with non‐targeting (NT), COX14 or COA3 siRNA to knock down their expression. Samples were separated by SDS–PAGE and immunoblotted with antibodies against CMC1, COX1, and COA3. IP is 40× enriched.
Source data are available online for this figure.
A COA3 antibody had revealed a band co‐migrating with CMC1 on BN–PAGE that was fully dependent on COX1 synthesis (Fig 4G) but independent of the presence of COX4 or COX5 (Fig 4I and J). BN–PAGE analysis in wild‐type cells further favors the existence of a complex that contains COX1, COA3, and CMC1 (Fig 5E). In the absence of CMC1, COA3 still interacts with COX1 in two slightly different migrating complexes (Fig 5E) that support, albeit inefficiently, some biogenesis of COX1 and fully assembled CIV (35% CIV in KO‐CMC1 cells). In support of this view, only the larger of these two complexes contains COX4 (Fig 5E). Furthermore, COA3 and COX1 co‐immunoprecipitated with CMC1‐FLAG in control cells (Fig 5D) but not in COX14‐ or COA3‐silenced cells (Fig 5F). Therefore, we conclude that CMC1 teams up with COA3 and COX14 to provide stability to newly synthesized COX1.
CMC1‐COA3‐COX1 complex is independent of MITRAC7, SURF1, COX10, and COX11
To further analyze the components of the CMC1 complex and to define the COX1 biogenetic step that coincides with the release of CMC1, we tested the dependence of this complex on four additional known COX1 biogenetic factors, SURF1, MITRAC7, COX11, and COX10.
We tested the occurrence of the CMC1 complex in fibroblasts from a Leigh's syndrome patient carrying a mutation in SURF1 35 (Fig 6A), a COX1 assembly factor 21, 22, shown to be involved in heme a center formation in bacteria 23. CMC1 complex was formed in mitochondria from SURF1‐deficient fibroblasts (Fig 6A); therefore, SURF1 is not part of this complex neither it affects its formation or stability.
Figure 6. Formation of the CMC1‐COX1‐COX14‐COA3 complex is independent of MITRAC7, SURF1, COX10, and COX11.
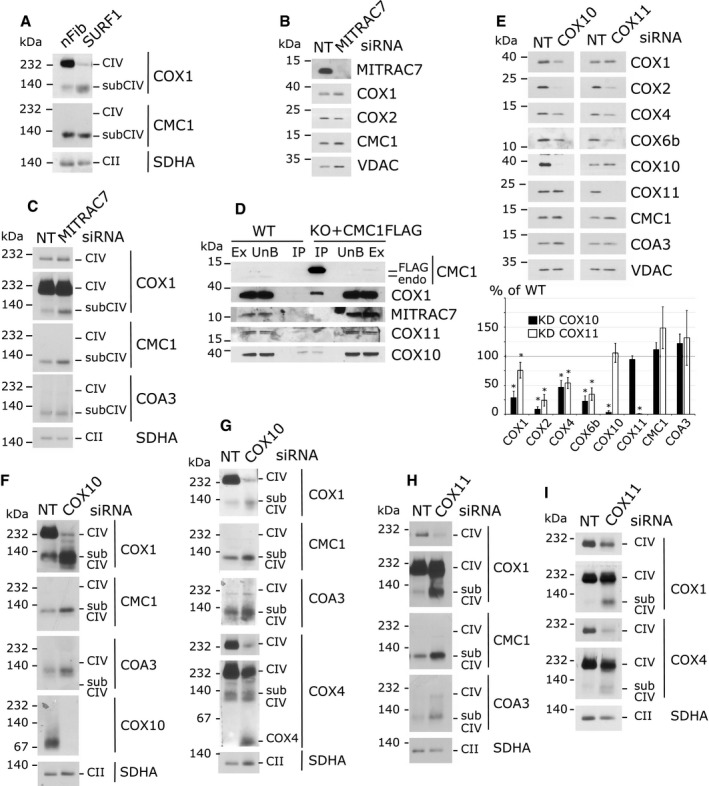
-
ABN‐PAGE and immunoblot analysis of CMC1 and COX1 complexes in control human neonatal fibroblasts (nFib) and SURF1 mutant fibroblast cell extracts.
-
B, CSDS–PAGE (B) or BN–PAGE (C) and immunoblot analysis of the steady‐state levels of the indicated proteins (B) or protein complexes (C) in HEK293T cells transfected for 7 days with non‐targeting (NT) or MITRAC7 siRNAs. Two different exposure times are presented for COX1(C).
-
DImmunoprecipitation of CMC1‐FLAG from a KO‐CMC1 + CMC1‐FLAG stable cell line using anti‐FLAG‐conjugated beads. Samples were separated by SDS–PAGE and immunoblotted with antibodies against CMC1, COX1, MITRAC7, COX10, and COX11. HEK293T (WT) mitochondrial extracts were used as a negative control. Ex: extract; UnB: unbound; IP: immunoprecipitation; endo: endogenous. IP is 40× enriched.
-
ESDS–PAGE and immunoblot analysis of the steady‐state levels of the indicated proteins in HEK293T cells transfected for 7 days with non‐targeting (NT), COX10 or COX11 siRNAs. In the lower panel, the signal was normalized by VDAC and expressed as percentage of the control. The bars represent average ± SD. n = 3 or 4; t‐test: in reading order P = 0.0003, 0.03, 2 × 10−6, 0.0002, 0.002, 0.001, 0.0002, 0.0005, 2 × 10−7, 0.6, 0.2, 5 × 10−10, 0.06, 0.07, 0.06, 0.3.
-
F–IBN–PAGE and immunoblot analysis of the steady‐state levels of the indicated complexes in HEK293T cells transfected for 5 (F), 8 (G), or 7 days (H, I) with non‐targeting (NT), COX10 or COX11 siRNAs. Two different exposure times are presented for COX4 (G, I) and COX1 (H, I).
Source data are available online for this figure.
Furthermore, we analyzed HEK293T cells siRNA‐silenced for MITRAC7 (Fig 6B and C). MITRAC7 interacts with COX1 in a subassembly complex containing COX4 and participates to the biogenesis of CIV 20. Silencing of MITRAC7 leads to a 50% decrease in CIV activity 20. In our experiment, MITRAC7 was well silenced by siRNA (Fig 6B), but no obvious defect in CIV assembly was observed arguing for a non‐essential or mild role of this factor in CIV biogenesis. Nevertheless, MITRAC7 has been clearly shown to interact with an assembly complex containing COX1 and COX4 36, and its absence does not affect the formation of the CMC1 complex nor its size on BN–PAGE (Fig 6C). MITRAC7 is not co‐immunoprecipitated with CMC1‐FLAG (Fig 6D), further demonstrating its absence from the CMC1 complex.
The heme O synthase COX10 is essential for heme A synthesis and COX1 maturation 37. It is interesting to notice that COX10 was detected on BN–PAGE as a ~80‐kDa band, a size that could correspond to a homodimer (Fig 6F). Cox10 oligomerization has been described in S. cerevisiae, where the active state of Cox10 appears to be a homo‐oligomeric complex of ~300 kDa, whose formation is dependent on the newly synthesized Cox1 and the presence of an early Cox1 assembly intermediate 9, 38. In COX10‐silenced cells, CIV subunits steady‐state levels were decreased as expected (Fig 6E), and CIV assembly was almost completely blocked (Fig 6F and G). In COX10 knockdown cells, the CMC1/COA3/COX1 complex was still formed and accumulated compared to the control (Fig 6F and G), whereas some COX4‐containing complex running at the same level on BN–PAGE was still detected (Fig 6G). Additionally, a portion of COX4, probably forming a heterodimer with COX5a, migrates to the bottom of the gel (Fig 6G). In conclusion, when heme A is unavailable, COX1 assembly and maturation stall at the CMC1/COA3/COX14 complex level; however, some biogenesis continue with the addition of COX4 in either aberrant or unstable complexes as they are not accumulated and do not lead to CIV fully assembled complex.
In yeast S. cerevisiae, Cox11 is essential for CIV assembly and was shown to receive copper from Cox17 39, 40. In Rhodobacter sphaeroides, Cox11 was shown to insert copper into COX1 41. In human, although the involvement of COX11 in CIV assembly has not been previously shown, the protein and its copper‐binding motif are well conserved. When we tested CIV assembly in COX11‐silenced cells, the steady‐state levels of COX2 and COX6b were found particularly decreased (around 25% of the non‐targeted control, Fig 6E), whereas COX1 and COX4 are less affected (75% and 50% decreased, Fig 6E). These phenotypes correlate with the BN–PAGE analysis where CIV is decreased, and CIV subcomplexes containing COX1 and COX4 are accumulated (Fig 6H and I). Therefore, we assume that the COX11 function in copper insertion into COX1 is largely conserved form bacteria to human. When copper insertion into COX1 is impaired, both the CMC1/COA3/COX1 complex and some COX4 containing subcomplexes (probably at least the COX14/COX1/COA3/COX4 complex) accumulate (Fig 6H and I). These data suggest that copper insertion into COX1 is not an absolute requirement for COX4 interaction with COX1. Thus, copper insertion into CIV assembly line could occur after COX4 incorporation. It is also possible that in human cells, as seen in R. sphaeroides 41, heme A could be inserted into COX1 in the absence of copper and form a relatively stable complex, that in human mitochondria would be an off‐pathway complex that could interact with COX4 but would be unable to progress toward holo‐CIV assembly.
Our results indicate that the CMC1 complex is formed in mitochondria from SURF1‐deficient fibroblasts (Fig 6A) as well as in MITRAC7‐, COX11‐, or COX10‐silenced cells (Fig 6C and F–I). Consistently, MITRAC7, COX11, or COX10 did not co‐purify with CMC1‐FLAG (Fig 6D). Hence, we concluded that these proteins are neither components of the CMC1 complex nor influence its formation. Additionally, our data indicate that the COX1 fraction present in the CMC1 complex is probably in an immature state, lacking heme and/or copper prosthetic groups.
COX1 synthesis and formation of the CMC1‐COX1‐COA3‐COX14 complex are not impaired by downstream defects in CIV biogenesis
In S. cerevisiae, Cox1 synthesis is tightly regulated by the status of CIV assembly 10, 11. The COX1 mRNA‐specific translation activator Mss51 is involved in coordinating Cox1 synthesis with CIV assembly through its interaction with newly synthesized Cox1 and several assembly factors including Cox14 and Coa3 10, 11, 12, 13. The net result of this regulation in most S. cerevisiae mutants stalled in CIV assembly, including cox2 and cox3 mutants, is the marked downregulation of Cox1 synthesis 11. Intriguingly, although the key element of this regulatory system, Mss51, is not functionally conserved in humans, it has been proposed that the COX1‐COX14‐COA3 complex could also regulate COX1 synthesis in humans as it occurs in S. cerevisiae 16, 19, 20.
Here, we have shown that depletion of COX10 (Fig 6F) or early‐assembled CIV subunits such as COX4 or COX5a induces the accumulation of the CMC1‐COX1‐COA3‐COX14 complex (Fig 4I and J). Now, we tested whether both COX1 synthesis and the formation/stability of the CMC1‐COX1‐COA3‐COX14 complex are affected when CIV biogenesis is stalled at a step downstream the formation of the COX1‐COX4‐COX5a intermediate. For this purpose, we used cybrid cell lines carrying mitochondrial DNA homoplasmic for mutant COX1 (as a control), COX2, or COX3.
As expected, in the COX1 mutant control cybrid, COX1 was not detected, COX2 and COX3 were normally synthesized, and the CMC1 complex was undetectable (Figs 7A and E, and 4G). Furthermore, the absence of COX1 rendered COA3 and CMC1 partially unstable as observed in COX1 mutant cells (Fig 7B) or in cells where COX1 mRNA translation was inhibited for 6 days (Fig 7F). In HEK293T cells, depletion of COX14 or COA3 led to CMC1 instability (Fig 5A). Also, the stability of COA3 was dependent on COX14 (Fig 5A) as reported in patient fibroblasts 17. Therefore, in the absence of one of its components, the turnover of the other partners in the CMC1‐COX1‐COA3‐COX14 complex is accelerated.
Figure 7. COX1 synthesis and formation of the CMC1‐COX1‐COA3‐COX14 complex are not impaired by downstream defects in CIV biogenesis.
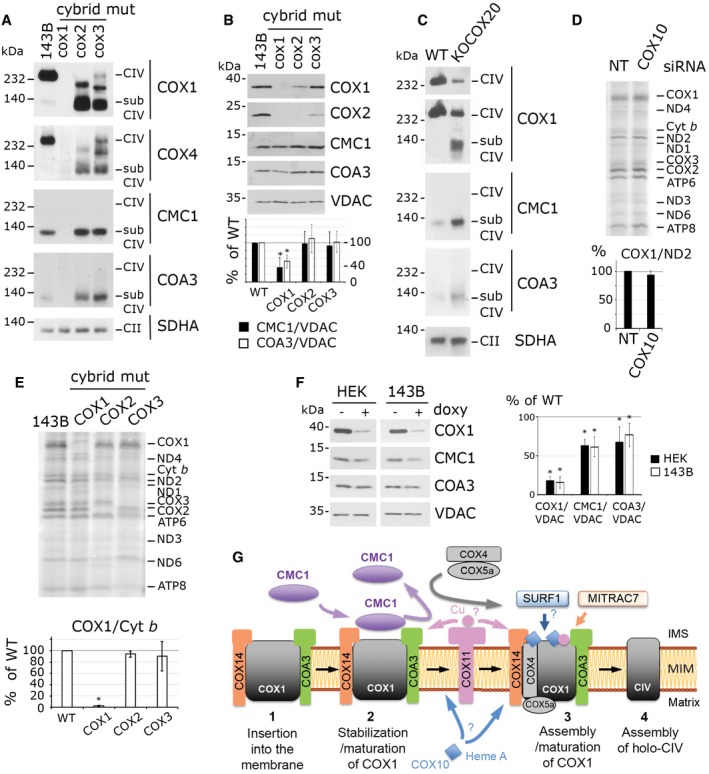
-
A, BBN–PAGE (A) and SDS–PAGE (B) and immunoblot analysis of CMC1, COA3, COX1, and COX4 complexes and proteins, respectively, in COX1, COX2, and COX3 homoplasmic mutant cells and the parental 143B cell line. CMC1 and COA3 signals were normalized by VDAC and expressed as percentage of the control cell line. CMC1 and COA3 amounts are statistically different from the wild type in COX1 cybrids. The bars represent average ± SD. n = 7; t‐test: *P = 0.000005 for CMC1 and *P = 0.00002 for COA3.
-
CBN–PAGE and immunoblot analysis of CMC1, COA3, and COX1 complexes in KO‐COX20 mutant cells and the parental HEK293T cell line. The two upper panels are two different levels of exposure.
-
DMitochondrial protein synthesis after a 30‐min pulse with [35S]‐methionine in COX10‐depleted cells. Quantification of COX1, normalized by ND2, is expressed as percentage of the non‐targeted siRNA‐transfected HEK293T cells. The bars represent average ± SD. n = 3, t‐test: P = 0.19.
-
EMitochondrial protein synthesis after a 20‐min pulse with [35S]‐methionine in the same cell lines as in panel (A). Quantification of COX1 was normalized by Cyt b and expressed as percentage of the control 143B line. The bars represent average ± SD. n = 2; t‐test: *P = 0.00001 in COX1, P = 0.26 in COX2 and P = 0.65 in COX3 cybrids.
-
FSDS–PAGE and immunoblot analysis of CMC1, COA3, and COX1 in 143B or HEK293T doxycycline‐treated cells for 6 days. Quantification was measured, normalized by VDAC and expressed as percentage of untreated cells. The bars represent average ± SD. n = 4 or 5; t‐test: in order in the graph, *P = 7 × 10−6, 6 × 10−9, 0.0001, 0.0001, 0.015, 0.008.
-
GModel depicting the role of CMC1 in controlling post‐translational events in COX1 biogenesis. According to our model, newly synthesized COX1 would first bind COX14 and COA3, followed by CMC1. CMC1 would promote COX1 stability during or before COX1 maturation and would be released from the growing COX1 complex before the incorporation of COX4‐1 and COX5a.
Source data are available online for this figure.
Importantly, COX1 synthesis in COX10‐depleted cells (Fig 7D) or in the absence of COX2 or COX3 was as in the parental cell line (Fig 7E). Similar results were obtained in HEK293T cells KO for COX20, which codes for a COX2 assembly chaperone 30. Furthermore, the absence of COX2 or COX3 in cybrid cell lines, or of COX20 in HEK293T cells not only did not impair formation of the CMC1 complex but also stimulated its accumulation (Fig 7A and C), whereas CMC1 and COA3 steady‐state levels remained stable (Fig 7B).
In conclusion, contrary to what occurs in yeast S. cerevisiae mitochondria, human COX1 synthesis is not downregulated when CIV biogenesis is stalled at the COX2 or COX3 incorporation steps in cultured cell lines. Additionally, the early CMC1‐COA3‐COX14‐COX1 assembly intermediate is stable or accumulated in all the CIV mutant cell lines tested with the exception of those involving one of the components of this complex. Taken together, the data presented in this section argue against a COX1 synthesis regulatory checkpoint in human mitochondria, and rather support enhanced COX1 turnover when its progression in the assembly process is blocked.
Discussion
Biogenesis of respiratory chain complex IV involves the coordinated expression of the nuclear and mitochondrial genetic systems, the stoichiometric accumulation of the 14 CIV subunits, and the incorporation of metal prosthetic groups into COX1 and COX2, two of the mitochondrial DNA‐encoded catalytic core subunits 4. CIV‐specific biogenetic factors assist every step of the process 4, 6. Mutations in CIV subunits and assembly factors are the most frequent cause of mitochondrial encephalocardiomyopathies in humans, highlighting their biomedical relevance 5, 42. From a biological perspective, understanding the intricate CIV assembly process is a fascinating task. Here, we have identified CMC1 as a COX1 chaperone that, although not essential, markedly enhances the stability of newly synthesized COX1 prior to its assembly with other CIV subunits.
Following the model presented in Fig 7G, in this study we show that: 1) CMC1 stimulates CIV biogenesis by stabilizing newly synthesized COX1 in an early CIV assembly complex containing the chaperones COA3 and COX14; 2) the release of CMC1 from this COX1 assembly intermediate occurs before the incorporation of CIV subunits COX4 and COX5a to the growing subassembly; 3) the CMC1 complex accumulates in the absence of COX1 metallation (heme A or copper, COX10 and COX11 knockdown); and 4) the CMC1 complex accumulates in several mutants affecting later steps of CIV assembly like in COX2 or COX3 mutant cybrids, COX4‐ or COX5a‐silenced cells, or in knockout of the COX2 chaperone COX20, whereas the synthesis of COX1 remains unaffected in these mutants.
COX1 is a highly hydrophobic protein that spans 12 transmembrane domains in the inner membrane, connected by short hydrophilic loops that protrude either in the IMS or the matrix. In S. cerevisiae, COX1 is co‐translationally inserted into the membrane by the Oxa1 insertase machinery 43, whereas human OXA1L is apparently not required for CIV biogenesis 44. Two conserved COX1 chaperones, the cardiomyopathy proteins COX14 and COA3, are single transmembrane proteins with a hydrophilic C‐terminus in the IMS 11, 13, 45. They interact with COX1 and probably direct its insertion into the inner membrane. Certainly, they promote the stability of newly synthesized COX1 since in their absence COX1 is rapidly degraded 16, 17, 18. Beyond their role in early folding and stability, COX14 and COA3 also chaperone COX1 in later final steps of CIV holoenzyme assembly 18, 19. On the contrary, CMC1 acts on a very specific segment of the COX1 biogenetic process.
Our data clearly demonstrate that CMC1 interacts with newly synthesized COX1 in the COX1‐COX14‐COA3 complex and is released from this complex most likely when COX1 acquires its prosthetic groups or interacts with subunits COX4‐1 and COX5a (see the model in Fig 7G). The CMC1 complex forms in the absence of COX4‐1 or COX5a and in the absence of assembly factors SURF1 and MITRAC7, known to act on the COX4‐containing complex 20, 21, 46. The CMC1 complex also forms in the absence of COX10 or COX11, indicating that COX1 in the CMC1 complex is not necessarily metallated, a possibility supported by the observation in S. cerevisiae that the fraction of Cox1 present in the Cox1‐Mss51‐Cox14‐Coa3 complex does not contain either heme A or copper 7, 47. The ~120‐kDa CMC1 complex contains at least CMC1 (12 kDa), COA3 (12 kDa), COX1 (40 kDa), and COX14 (7 kDa) which do not add up to 120 kDa. Several possibilities could count for the discrepancy. The stoichiometry of these proteins in the complex could differ from 1:1:1:1, additional unidentified proteins could be present, or the migration on BN–PAGE of this particularly hydrophobic protein complex could differ from its real molecular weight. The clarification of this point warrants future investigations.
CMC1 is an IMS twin Cx9C protein. First discovered in S. cerevisiae, it was not shown to interact with Cox1. CMC1 was hypothesized to be involved in the CIV copper transfer pathway based on its similarity with the copper chaperone COX17 and the partial suppression of the CIV assembly defect in a cmc1 null yeast by exogenous copper 26. However, CMC1 lacks the COX17 CC‐copper‐binding motif 48, and in our hands, copper supplementation to the media did not rescue the CIV assembly defect of human KO‐CMC1 cells. It remains plausible, however, that CMC1 chaperoning of COX1 might be required for efficient COX1 maturation, an event that could coincide with the release of CMC1 from the CMC1‐COX1‐COA3‐COX14 complex, as explained earlier. It is tempting to speculate that CMC1 could promote conformational changes on COX1 or stabilize it, to render it competent for incorporation of its prosthetic groups. Future studies to identify CMC1 residues involved in its interaction with COX1 are warranted. In support of this folding modulator possibility, several twin Cx9C proteins have been shown to function as such. Three examples include the yeast CIV assembly factor Cox19 that interacts with Cox11 to promote copper transfer 49, Mdm35 that forms a complex with the phosphatidic acid‐binding protein Ups1 to maintain it in a lipid transfer‐competent state 50, 51, and Mia40 that in addition to its role as oxidoreductase, acts as a chaperone for IMS proteins 52. We conclude that CMC1 is a COX1‐specific chaperone that promotes COX1 stability presumably by supporting the membrane insertion and folding of the COX1 transmembrane domains and/or rendering the protein in a competent state for maturation or further assembly with additional CIV subunits.
In S. cerevisiae, COX1 synthesis is coordinated with CIV biogenesis via several regulatory mechanisms 4. Relevant to our work, the Cox1‐Cox14‐Coa3 complex traps the COX1 mRNA‐specific translational activator Mss51, whose release is coupled with Cox1 maturation or assembly with other CIV subunits. By this mechanism, Cox1 synthesis is attenuated to only approximately 10% of wild type in most CIV assembly mutants 11. A putative human homolog of Mss51 has been recently described in humans, but it does not participate in COX1 biogenesis 53. Additionally, TACO1, the single mammalian‐specific COX1 mRNA translational activator identified, binds to the COX1 mRNA and associates with the mitoribosome 54 but is not a COX1 chaperone 15. However, based on the observation of a mildly attenuated COX1 synthesis in [35S]‐methionine pulses in fibroblasts from COX14‐deficient patients or in HEK293T cells treated with COX14 or COA3 siRNA, it has been proposed that the COX1‐COX14‐COA3 complex could regulate COX1 synthesis in humans as it occurs in S. cerevisiae 16, 19, 20. When we performed similar short pulse‐labeling assays in COX14‐ or COA3‐silenced HEK293T cells, we did not observe a dramatic decrease in COX1 synthesis in our experimental conditions, whereas the CMC1‐COX1‐COA3‐COX14 complex was severely decreased. We interpret these results as a sign of extremely fast degradation of newly synthesized COX1 rather than a feedback loop decreasing COX1 synthesis. As an alternative explanation, if COX1 membrane insertion were co‐translational, a failure of this process in the absence of COX14 or COA3 could lead to protein misfolding and, potentially, mitoribosome stalling and premature translation termination. However, this would be very different from the translational regulatory systems identified in the yeast S. cerevisiae 55. Furthermore, our data show that KO‐CMC1 HEK293T cells, as well as COX2 or COX3 homoplasmic mutant cybrids and COX10‐silenced HEK293T cells (Fig 7), exhibit normal COX1 synthesis, clearly demonstrating that contrary to S. cerevisiae, human COX1 synthesis is not downregulated in a variety of CIV assembly mutants.
Why the translational regulatory system would not have been conserved along evolution up to humans? S. cerevisiae mitochondrial messenger RNAs, including COX1, have long 5′UTR where translation activators bind, whereas those in human mitochondrial mRNAs are extremely short. Therefore, the translation machinery in human mitochondria has probably evolved alternate mechanisms to recognize mRNAs, initiate protein synthesis, and eventually regulate translation. As a case in point, Mss51 and TACO1 are functionally restricted to yeast or humans, respectively. Beyond the differences in COX1 expression, in both yeast and human cells, the concerted stoichiometric accumulation of CIV subunits is largely regulated by posttranslational degradation of unassembled components, the pathway that seems prevalent in human cells. We speculate that the development of feedback translational regulatory loops in S. cerevisiae mitochondria to tightly control the expression of key catalytic subunits, such as COX1 in CIV or cyt b in CIII 55, may relate to its facultative aerobe/anaerobe status. Saccharomyces cerevisiae can undergo fast switches from fermentation to respiration depending on environmental conditions and carbon source availability. In support of this view, even in the obligatory aerobe yeast Schizosaccharomyces pombe, Mss51 as well as the cyt b translational activators Cbp3 and Cbp6 are not required for the translation of mitochondrial mRNAs, but fulfill post‐translational functions, thus probably accounting for their conservation in S. pombe 56. Saccharomyces cerevisiae might have recruited these proteins for translational activation as a later adaptation, to facilitate the coupling of mitochondrial translation and respiratory complex assembly in response to its specific energetic needs, which differ from those of S. pombe and those of human cells and tissues.
Materials and Methods
Human cell lines
Human neonatal fibroblasts (nFib, CCD‐10645 k), HEK293T embryonic kidney cells (CRL‐3216), and 143B osteosarcoma cells (CRL‐8303) were obtained from ATCC. A patient SURF1‐deficient fibroblast line that carries a homozygous frameshift mutation that retained ~20% of residual CIV activity was previously reported 35. Fibroblast cell lines were immortalized by human papillomavirus E6/E7 57. All cybrid cell lines were constructed using the osteosarcoma 143B TK−206 rho zero cell line and enucleated control fibroblasts as described 58. COX1 mutant cybrid cells carry a homoplasmic G6930A mitochondrial mutation that leads to a stop codon and a truncated version of COX1 missing 33% of its sequence 59. COX2 mutant cybrid cells carry a homoplasmic G7896A mutation that leads to stop codon 60. COX3 cybrids carry a homoplasmic frameshift mutation due to the insertion of an extra C at nucleotide position 9537 that leads to the absence of COX3 protein 61. KO‐COX20 was constructed in HEK293T using the TALEN technology 30.
Cells were grown in high‐glucose DMEM media with 110 mg/l sodium pyruvate, 50 mg/l uridine, and 10% FBS at 37°C under 5% CO2. Cells were transfected with Lipofectamine 2000 (Invitrogen), mixed with plasmids in OPTIMEM‐I media (Gibco). Stable lines were established by transfection of HEK293T or KO‐CMC1 cell lines with pIRESpuro2 empty vector or pIRESpuro2 containing CMC1‐FLAG. Two days after transfection, the media was supplemented with 2.5 μg/ml puromycin and drug selection was maintained for at least 1 month.
Plasmids
Two pairs of TALEN plasmids designed to KO CMC1 were ordered at Cellectis (Paris, France). The left and right TALENs for pair #1 were designed to bind the TCTCGGCCCGCCGAGAT and AGGTACCGGGGCGGGAA DNA sequences, respectively, at the CMC1 locus. TALENs for pair #2 were designed to bind TTTTCAGACCAGCATCT and AGATGTTTGATCCCTA DNA sequences.
CMC1‐FLAG was amplified by PCR from HEK293T cDNA with the primers 5′‐ccggGCTAGCatggcgctcgaccccgcagacc‐3′ and 5′‐gcgcGGATCCttacttgtcgtcatcgtctttgtagtccatgcttgttggaagcttctg‐3′. CMC1FLAG was cloned under the control of a CMV promoter in the pIRESpuro2 plasmid using NheI and BamHI sites.
siRNA (stealth RNAi™ siRNA duplex oligoribonucleotides)
Non‐targeting siRNA (UGGUUUACAUGUCGACUAA) was ordered from Thermo Scientific. COA3 (UCGGGAGAAGCUGACACCCGAGCAA), COX14 (GACAUUGGCUAUAAGACCUUCUCUA), COX4‐1 (GGCUACCAGGGUAUUUAGCCUAGUU), COX4‐2 (GAGAUGAACCGUCG CUCCAAUGAGU), COX5a (GGCUAUCCAGUCAGUUCGCUGCUAU), and MITRAC7 (GCGCCGCCUUCUAUCCCAUCUACU) siRNAs were obtained from Invitrogen. COX11 (GCUUUAAUGCAGAUGUGCAtt) and COX10 (GGUGCCAUUUGACUCAAACtt) siRNAs were ordered at Ambion‐Life Technologies.
Antibodies
The antibodies used in this study are listed in Table 2. Secondary antibodies coupled with horseradish peroxidase were obtained from Santa Cruz Biotechnology. To eliminate interference of the light and heavy chains of the immunoprecipitation antibody, we used secondary mouse TrueBlot (Rockland) antibody.
Table 2.
Antibodies used in this study
| Against | Company/ref | Used and tested in this manuscript |
|---|---|---|
| COX1 | Abcam Ab14705 | No signal on Western in respective COX1, COX2, or COX3 mutant cybrids |
| COX2 | Abcam Ab110258 | |
| COX3 | Abcam Ab110259 | |
| COX4‐1 | Abcam Ab14744 | Signal on immunoblot highly decreased in respective siRNA knockdown or knockout cells |
| COX4‐2 | Abcam Ab70112 | |
| COX5a | Sigma HPA027525 | |
| CMC1 | Sigma HPA043333 | |
| COA3 MITRAC12 | Sigma HPA031966 | |
| COX10 | Sigma HPA032005 | |
| COX11 | Sigma HPA044020 | |
|
MITRAC7 C2ORF52 |
Sigma HPA016552 | |
| COX6b | Abcam Ab110266 | Immunoblot signal is decreased in several mutants as expected |
| FLAG | Sigma F3165 | Detects the tagged FLAG protein |
| SDHA | Abcam Ab14715 | Detect a band of the expected size on SDS–PAGE/ and BN–PAGE/immunoblot |
| NDUFA9 | Abcam Ab14713 | |
|
UQCRC2 CORE2 |
Abcam Ab14745 | |
| ATP5 | Abcam Ab14748 | |
| VDAC | Abcam Ab14734 |
siRNA transfection
HEK293T cells grown on a 6‐well plate at 10% confluency were transfected with the indicated Stealth RNAi duplexes (at 10 nM) or a scrambled control [BLOCK‐iT Alexa Fluor (Invitrogen)] using 5 μl of Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's specifications.
Whole‐cell extracts and mitochondrial isolation
Whole‐cell extracts were obtained by solubilization in RIPA buffer (25 mM Tris–HCl pH 7.6; 150 mM NaCl; 1% NP‐40; 1% sodium deoxycholate; and 0.1% SDS) with 1 mM PMSF and 1× mammalian protease inhibitor cocktail (Sigma). Extracts were cleared by 5‐min centrifugation at 10,000 g at 4°C.
Mitochondria from human cells were isolated from at least ten 80% confluent 175‐cm2 flasks as described previously 30, 62.
BN–PAGE analysis
Cells (2.5 × 106) were incubated in 270 μl of PBS with 2 mg/ml digitonin (Sigma) at 4°C for 10 min. Cells were centrifuged at 10,000 g for 5 min at 4°C and washed two times in PBS. Pellets were solubilized in 100 μl of 1.5 M aminocaproic acid, 50 mM Bis–Tris pH 7.0, and 2 μl of 20% lauryl maltoside (for monomeric complexes analysis) or 10 μl of 10% digitonin (for supercomplexes analysis). After spinning at 22,000 g for 30 min at 4°C, 10 μl of 750 mM aminocaproic acid, 50 mM Bis–Tris pH 7.0, 0.5 mM, EDTA, and 5% Serva blue G was added to the clarified extracts. For each sample, extracts from ~2.5 to 5 × 105 cells were loaded on a linear 3–12% or 4–16% gradient gel.
Immunoblotting
Protein concentration was measured with the Folin phenol reagent 63. 20–60 μg of mitochondrial proteins or 40–60 μg of whole‐cell protein extract was separated by SDS–PAGE in the Laemmli buffer system 64. After transfer, nitrocellulose membranes were decorated with antibodies followed by a second reaction with anti‐mouse or anti‐rabbit IgG conjugated to horseradish peroxidase. Chemiluminescence was used for the final detection.
Mitochondrial protein synthesis
Mitochondrial protein synthesis was determined by pulse‐labeling 80% confluent cells as described by 65 with some adaptations. Human HEK293T cells were grown in a six‐well plate pre‐coated for 1 h at room temperature with 0.1 mg/ml collagen in PBS to increase cell adherence. Cells were washed twice with PBS and incubated 20 min in DMEM without methionine. Then, the media was supplemented with 100 μg/ml emetine for 10 min to inhibit cytoplasmic protein synthesis. 100 μCi of [35S]‐methionine was added to the media for the indicated pulse time. For pulse samples, after incubation cells were washed one time with PBS and collected. For chase samples, cells were washed twice with PBS and incubated in complete media for the indicated chase time. Cells were then harvested by trypsinization, and whole‐cell extracts were prepared by solubilization in RIPA buffer. One hundred microgram was loaded on a 17.5% SDS–PAGE with a 5% stacking part. After running, the gel was transferred to a nitrocellulose membrane and exposed to a film.
Immunoprecipitation
For immunoprecipitation of FLAG‐tagged proteins, cells were grown to 80% confluency and harvested by trypsinization. 20 × 106 cells were incubated in 1,600 μl PBS and 560 μl digitonin (Sigma) at 8 mg/ml for 10 min at 4°C. The membrane part was recovered by centrifugation at 10,000 g for 5 min at 4°C. This pellet was washed twice with 1 ml PBS and then extracted in 800 μl 1.5 M aminocaproic acid, 50 mM Bis–Tris pH 7, 0.4% lauryl maltoside, 1 mM PMSF, and 8 μl of a protease inhibitor cocktail (Sigma, P8340). After 30‐min centrifugation at 22,000 g, the extract (Ex) was incubated with 50 μl anti‐FLAG‐conjugated agarose beads (anti‐DYKDDDDK beads, Clontech) previously washed in PBS and aminocaproic acid buffer. Following 4‐h incubation at 4°C on an orbital shaker, the unbound material (UnB) was recovered and the beads were washed five times in 500 μl 1.5 M aminocaproic acid, 50 mM Bis–Tris pH 7, 0.05% lauryl maltoside. The beads were then supplemented with 50 μl of Laemmli buffer 2× and boiled 5 min to release the bound material. Representative amounts of all fractions were loaded on an SDS–PAGE gel in the following proportions; Ex (1×), UnB (1×), and IP (40×).
For co‐immunoprecipitation studies of CMC1‐FLAG with newly synthesized mitochondrial translation products, cells were pulse radiolabeled with [35S]‐methionine for 30 min. The same protocol than above was applied on 11 × 106 cells and using 40 μl FLAG beads. IP sample is 15× enriched compared to the extract.
Determination of cellular respiration and mitochondrial enzyme activities
Basal endogenous cell respiration was measured using an XFp Extracellular Flux Analyzers (Seahorse Bioscience) according to the manufacturer specification in 1 mM pyruvate, 2 mM glutamine, 5 mM glucose XF Base medium. Values were normalized by total cell number. CIV complex and TCA enzyme citrate synthase activities were determined spectrophotometrically in freeze‐thawed cells as described previously 66.
Mitochondrial protein solubility assay
Two hundred microgram of mitochondria in 200 μl of STE buffer (0.6 M sorbitol, 20 mM Tris pH 7.5, 1 mM EDTA, 1 mM PMSF) was sonicated for 3 s at intensity 2 using a Virtis Virsonic 100 ultrasonic cell disrupter and then centrifuged at 35,000 g for 15 min at 4°C. The supernatant containing the soluble proteins (S) was removed. The pellet was then resuspended in 100 μl of 200 mM Na2CO3 pH 11.5 to extract extrinsic proteins, incubated 30 min on ice and centrifuged at 35,000 g for 15 min at 4°C. The pellet was extracted a second time in the same conditions. The 200 μl of supernatant after alkaline carbonate extraction represents the extrinsic proteins loosely associated with membranes (CS). The pellet, containing intrinsic transmembrane proteins, was resuspended in 200 μl STE (CP). Equivalent samples from the different fractions were analyzed by immunoblotting.
Statistical analysis—quantification of Western signal
The Western signals were quantified using ImageJ. All data are presented as average ± SD of the percentage of control. Values were analyzed for statistical significance by Student's t‐test. *: P < 0.05 was considered significant. n is the number of independent repetition realized. The actual n and P‐values for each comparison are listed in the figure legends.
Author contributions
MB and AB designed the experiments. MB performed the experiments. MB and AB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Dr. Flavia Fontanesi for critical reading of the manuscript. We thank Dr. Giovanni Manfredi, Dr. Massimo Zeviani, Dr. Valeria Tiranti, J.A. Enriquez, and Dr. Agnes Rotig for providing cell lines. We thank Dr. Carlos Moraes and Dr. Claudia Pereira for their technical aid. This research was supported by NIGMS‐RO1 grants GM071775, GM105781, and GM112179 (to AB), NIGMS‐R35 MIRA grant R35GM118141 (to AB), Muscular Dystrophy Association Research Grant MDA‐381828 (to AB), and an American Heart Association postdoctoral fellowship (to MB).
EMBO Reports (2017) 18: 477–494
References
- 1. Ghezzi D, Zeviani M (2012) Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv Exp Med Biol 748: 65–106 [DOI] [PubMed] [Google Scholar]
- 2. Fornuskova D, Stiburek L, Wenchich L, Vinsova K, Hansikova H, Zeman J (2010) Novel insights into the assembly and function of human nuclear‐encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem J 428: 363–374 [DOI] [PubMed] [Google Scholar]
- 3. Nijtmans LG, Taanman JW, Muijsers AO, Speijer D, Van den Bogert C (1998) Assembly of cytochrome‐c oxidase in cultured human cells. Eur J Biochem 254: 389–394 [DOI] [PubMed] [Google Scholar]
- 4. Soto IC, Fontanesi F, Liu J, Barrientos A (2012) Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim Biophys Acta 1817: 883–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rak M, Benit P, Chretien D, Bouchereau J, Schiff M, El‐Khoury R, Tzagoloff A, Rustin P (2016) Mitochondrial cytochrome c oxidase deficiency. Clin Sci (Lond) 130: 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dennerlein S, Rehling P (2015) Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci 128: 833–837 [DOI] [PubMed] [Google Scholar]
- 7. Soto IC, Fontanesi F, Myers RS, Hamel P, Barrientos A (2012) A heme‐sensing mechanism in the translational regulation of mitochondrial cytochrome c oxidase biogenesis. Cell Metab 16: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barros MH, Tzagoloff A (2002) Regulation of the heme A biosynthetic pathway in Saccharomyces cerevisiae . FEBS Lett 516: 119–123 [DOI] [PubMed] [Google Scholar]
- 9. Khalimonchuk O, Kim H, Watts T, Perez‐Martinez X, Winge DR (2012) Oligomerization of heme o synthase in cytochrome oxidase biogenesis is mediated by cytochrome oxidase assembly factor Coa2. J Biol Chem 287: 26715–26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perez‐Martinez X, Butler CA, Shingu‐Vazquez M, Fox TD (2009) Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol Biol Cell 20: 4371–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barrientos A, Zambrano A, Tzagoloff A (2004) Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae . EMBO J 23: 3472–3482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fontanesi F, Soto IC, Horn D, Barrientos A (2010) Mss51 and Ssc1 facilitate translational regulation of cytochrome c oxidase biogenesis. Mol Cell Biol 30: 245–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mick DU, Vukotic M, Piechura H, Meyer HE, Warscheid B, Deckers M, Rehling P (2010) Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J Cell Biol 191: 141–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khalimonchuk O, Bird A, Winge DR (2007) Evidence for a pro‐oxidant intermediate in the assembly of cytochrome oxidase. J Biol Chem 282: 17442–17449 [DOI] [PubMed] [Google Scholar]
- 15. Weraarpachai W, Antonicka H, Sasarman F, Seeger J, Schrank B, Kolesar JE, Lochmuller H, Chevrette M, Kaufman BA, Horvath R et al (2009) Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late‐onset Leigh syndrome. Nat Genet 41: 833–837 [DOI] [PubMed] [Google Scholar]
- 16. Weraarpachai W, Sasarman F, Nishimura T, Antonicka H, Aure K, Rotig A, Lombes A, Shoubridge EA (2012) Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet 90: 142–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ostergaard E, Weraarpachai W, Ravn K, Born AP, Jonson L, Duno M, Wibrand F, Shoubridge EA, Vissing J (2015) Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J Med Genet 52: 203–207 [DOI] [PubMed] [Google Scholar]
- 18. Clemente P, Peralta S, Cruz‐Bermudez A, Echevarria L, Fontanesi F, Barrientos A, Fernandez‐Moreno MA, Garesse R (2013) hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J Biol Chem 288: 8321–8331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mick DU, Dennerlein S, Wiese H, Reinhold R, Pacheu‐Grau D, Lorenzi I, Sasarman F, Weraarpachai W, Shoubridge EA, Warscheid B et al (2012) MITRAC links mitochondrial protein translocation to respiratory‐chain assembly and translational regulation. Cell 151: 1528–1541 [DOI] [PubMed] [Google Scholar]
- 20. Dennerlein S, Oeljeklaus S, Jans D, Hellwig C, Bareth B, Jakobs S, Deckers M, Warscheid B, Rehling P (2015) MITRAC7 acts as a COX1‐specific chaperone and reveals a checkpoint during cytochrome c oxidase assembly. Cell Rep 12: 1644–1655 [DOI] [PubMed] [Google Scholar]
- 21. Yao J, Shoubridge EA (1999) Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum Mol Genet 8: 2541–2549 [DOI] [PubMed] [Google Scholar]
- 22. Nijtmans LG, Artal Sanz M, Bucko M, Farhoud MH, Feenstra M, Hakkaart GA, Zeviani M, Grivell LA (2001) Shy1p occurs in a high molecular weight complex and is required for efficient assembly of cytochrome c oxidase in yeast. FEBS Lett 498: 46–51 [DOI] [PubMed] [Google Scholar]
- 23. Bundschuh FA, Hannappel A, Anderka O, Ludwig B (2009) Surf1, associated with Leigh syndrome in humans, is a heme‐binding protein in bacterial oxidase biogenesis. J Biol Chem 284: 25735–25741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (2010) Targeting DNA double‐strand breaks with TAL effector nucleases. Genetics 186: 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li T, Huang S, Zhao X, Wright DA, Carpenter S, Spalding MH, Weeks DP, Yang B (2011) Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res 39: 6315–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Horn D, Al‐Ali H, Barrientos A (2008) Cmc1p is a conserved mitochondrial twin Cx9C protein involved in cytochrome c oxidase biogenesis. Mol Cell Biol 28: 4354–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Villani G, Greco M, Papa S, Attardi G (1998) Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J Biol Chem 273: 31829–31836 [DOI] [PubMed] [Google Scholar]
- 28. Williams SL, Valnot I, Rustin P, Taanman JW (2004) Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1 . J Biol Chem 279: 7462–7469 [DOI] [PubMed] [Google Scholar]
- 29. Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, van den Brand MA, Hendriks‐Franssen MG, Gilissen C, Veltman JA et al (2013) A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet 22: 656–667 [DOI] [PubMed] [Google Scholar]
- 30. Bourens M, Boulet A, Leary SC, Barrientos A (2014) Human COX20 cooperates with SCO1 and SCO2 to mature COX2 and promote the assembly of cytochrome c oxidase. Hum Mol Genet 23: 2901–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bourens M, Dabir DV, Tienson HL, Sorokina I, Koehler CM, Barrientos A (2012) Role of twin Cys‐Xaa9‐Cys motif cysteines in mitochondrial import of the cytochrome c oxidase biogenesis factor Cmc1. J Biol Chem 287: 31258–31269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL (2007) HIF‐1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129: 111–122 [DOI] [PubMed] [Google Scholar]
- 33. Glerum DM, Tzagoloff A (1997) Submitochondrial distributions and stabilities of subunits 4, 5, and 6 of yeast cytochrome oxidase in assembly defective mutants. FEBS Lett 412: 410–414 [DOI] [PubMed] [Google Scholar]
- 34. Stiburek L, Vesela K, Hansikova H, Pecina P, Tesarova M, Cerna L, Houstek J, Zeman J (2005) Tissue‐specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1 . Biochem J 392: 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Von Kleist‐Retzow JC, Yao J, Taanman JW, Chantrel K, Chretien D, Cormier‐Daire V, Rotig A, Munnich A, Rustin P, Shoubridge EA (2001) Mutations in SURF1 are not specifically associated with Leigh syndrome. J Med Genet 38: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Markov DA, Savkina M, Anikin M, Del Campo M, Ecker K, Lambowitz AM, De Gnore JP, McAllister WT (2009) Identification of proteins associated with the yeast mitochondrial RNA polymerase by tandem affinity purification. Yeast 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Glerum DM, Tzagoloff A (1994) Isolation of a human cDNA for heme A: farnesyltransferase by functional complementation of a yeast cox10 mutant. Proc Natl Acad Sci USA 91: 8452–8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bestwick M, Khalimonchuk O, Pierrel F, Winge DR (2010) The role of Coa2 in hemylation of yeast Cox1 revealed by its genetic interaction with Cox10. Mol Cell Biol 30: 172–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR (2004) Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome c oxidase. J Biol Chem 279: 35334–35340 [DOI] [PubMed] [Google Scholar]
- 40. Carr HS, George GN, Winge DR (2002) Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)‐binding protein. J Biol Chem 277: 31237–31242 [DOI] [PubMed] [Google Scholar]
- 41. Hiser L, Di Valentin M, Hamer AG, Hosler JP (2000) Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J Biol Chem 275: 619–623 [DOI] [PubMed] [Google Scholar]
- 42. Fontanesi F, Barrientos A (2014) Mitochondrial cytochrome c oxidase assembly in health and human diseases. In Mitochondrial Disorders Caused by Nuclear Genes, pp. 239–259. New York, NY: Springer; [Google Scholar]
- 43. Hell K, Neupert W, Stuart RA (2001) Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J 20: 1281–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stiburek L, Fornuskova D, Wenchich L, Pejznochova M, Hansikova H, Zeman J (2007) Knockdown of human Oxa1l impairs the biogenesis of F1Fo‐ATP synthase and NADH:ubiquinone oxidoreductase. J Mol Biol 374: 506–516 [DOI] [PubMed] [Google Scholar]
- 45. Fontanesi F, Clemente P, Barrientos A (2011) Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae . J Biol Chem 286: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tiranti V, Galimberti C, Nijtmans L, Bovolenta S, Perini MP, Zeviani M (1999) Characterization of SURF‐1 expression and Surf‐1p function in normal and disease conditions. Hum Mol Genet 8: 2533–2540 [DOI] [PubMed] [Google Scholar]
- 47. Khalimonchuk O, Bestwick M, Meunier B, Watts TC, Winge DR (2010) Formation of the redox cofactor centers during Cox1 maturation in yeast cytochrome oxidase. Mol Cell Biol 30: 1004–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Banci L, Bertini I, Ciofi‐Baffoni S, Janicka A, Martinelli M, Kozlowski H, Palumaa P (2008) A structural‐dynamical characterization of human Cox17. J Biol Chem 283: 7912–7920 [DOI] [PubMed] [Google Scholar]
- 49. Bode M, Woellhaf MW, Bohnert M, van der Laan M, Sommer F, Jung M, Zimmermann R, Schroda M, Herrmann JM (2015) Redox‐regulated dynamic interplay between Cox19 and the copper‐binding protein Cox11 in the intermembrane space of mitochondria facilitates biogenesis of cytochrome c oxidase. Mol Biol Cell 26: 2385–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Potting C, Wilmes C, Engmann T, Osman C, Langer T (2010) Regulation of mitochondrial phospholipids by Ups1/PRELI‐like proteins depends on proteolysis and Mdm35. EMBO J 29: 2888–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tamura Y, Iijima M, Sesaki H (2010) Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J 29: 2875–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weckbecker D, Longen S, Riemer J, Herrmann JM (2012) Atp23 biogenesis reveals a chaperone‐like folding activity of Mia40 in the IMS of mitochondria. EMBO J 31: 4348–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moyer AL, Wagner KR (2015) Mammalian Mss51 is a skeletal muscle‐specific gene modulating cellular metabolism. J Neuromuscul Dis 2: 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richman TR, Spahr H, Ermer JA, Davies SM, Viola HM, Bates KA, Papadimitriou J, Hool LC, Rodger J, Larsson NG et al (2016) Loss of the RNA‐binding protein TACO1 causes late‐onset mitochondrial dysfunction in mice. Nat Commun 7: 11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fontanesi F (2013) Mechanisms of mitochondrial translational regulation. IUBMB Life 65: 397–408 [DOI] [PubMed] [Google Scholar]
- 56. Kuhl I, Fox TD, Bonnefoy N (2012) Schizosaccharomyces pombe homologs of the Saccharomyces cerevisiae mitochondrial proteins Cbp6 and Mss51 function at a post‐translational step of respiratory complex biogenesis. Mitochondrion 12: 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lochmuller H, Johns T, Shoubridge EA (1999) Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp Cell Res 248: 186–193 [DOI] [PubMed] [Google Scholar]
- 58. King MP, Attadi G (1996) Mitochondria‐mediated transformation of human rho (0) cells. Methods Enzymol 264: 313 [DOI] [PubMed] [Google Scholar]
- 59. D'Aurelio M, Pallotti F, Barrientos A, Gajewski CD, Kwong JQ, Bruno C, Beal MF, Manfredi G (2001) In vivo regulation of oxidative phosphorylation in cells harboring a stop‐codon mutation in mitochondrial DNA‐encoded cytochrome c oxidase subunit I. J Biol Chem 276: 46925–46932 [DOI] [PubMed] [Google Scholar]
- 60. Campos Y, Garcia‐Redondo A, Fernandez‐Moreno MA, Martinez‐Pardo M, Goda G, Rubio JC, Martin MA, del Hoyo P, Cabello A, Bornstein B et al (2001) Early‐onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA cytochrome c oxidase II gene. Ann Neurol 50: 409–413 [DOI] [PubMed] [Google Scholar]
- 61. Tiranti V, Corona P, Greco M, Taanman JW, Carrara F, Lamantea E, Nijtmans L, Uziel G, Zeviani M (2000) A novel frameshift mutation of the mtDNA COIII gene leads to impaired assembly of cytochrome c oxidase in a patient affected by Leigh‐like syndrome. Hum Mol Genet 9: 2733–2742 [DOI] [PubMed] [Google Scholar]
- 62. Fernandez‐Vizarra E, Ferrin G, Perez‐Martos A, Fernandez‐Silva P, Zeviani M, Enriquez JA (2010) Isolation of mitochondria for biogenetical studies: an update. Mitochondrion 10: 253–262 [DOI] [PubMed] [Google Scholar]
- 63. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275 [PubMed] [Google Scholar]
- 64. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 65. Leary SC, Sasarman F (2009) Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol Biol 554: 143–162 [DOI] [PubMed] [Google Scholar]
- 66. Barrientos A, Fontanesi F, Díaz F (2009) Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet 63: 19.3.1‐19.3.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15: 7–12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7