

Abstract
High blood pressure (BP) has been identified as a major risk factor for cardiovascular complications. Although two-way association between BP and hypertensive complications makes hypertension a near-ideal biomarker, BP as “the cause” for the complications of HT per se still needs more evidence. Another entirely possible hemodynamic candidate for causing hypertensive cardiovascular adverse events can be flow or its iterations, which might have escaped the attention because of its perfect correlation with pressure and harder technical measurement. In this article, we analyze the evidence in hand to compare flow- and pressure-related phenomena to delineate which of the two is the dominant mediator of complications related to hypertension and should be the target for therapy. A “flow-” rather than a “pressure-” based factor, as the causative or major driving mediator of common hypertensive complications, may change our understanding of hypertension pathophysiology.
Keywords: Atherosclerosis, cardiovascular outcomes, coronary heart disease, hypertension, stroke
Introduction
High blood pressure (BP) has been identified as a major risk factor for cardiovascular (CV) diseases. Data from observational studies have indicated that the risk from coronary artery disease and stroke increases progressively from 115/75 mm Hg upward, and for every 20/10 mm Hg increase in BP, there is a doubling of mortality from both (1). Also, it has been convincingly shown that lowering high BP with any drug significantly reduces adverse events (2). This two-way association makes hypertension a type 2 biomarker, which is defined as a surrogate end point that is expected to predict clinical benefit (or harm or lack of benefit or harm) on the basis of epidemiological, therapeutic, pathophysiological, or other scientific evidence (3). However, targeting high BP may not be only clear and simple answer to the hypertension (HT) problem, since BP as “the cause” for the complications of HT per se still needs more evidence.
Essentially, rather than being “a cause” by its nature, BP is “the result” of two interacting processes (4): the flow from the heart into the arteries and the resistance against this flow, which can be expressed in mean terms as follows:
Pressure = flow x resistance
Given that resistance is a localized anatomical element, which one of the other two physiologic elements exerts its effects on target organs and results in adverse events is questionable.
From a physiological standpoint, the flow-part, not the pressure-part, of this relationship seems to be more critical. The autoregulation systems of the target organs aim to keep the organ flow constant by changing vascular resistance via manipulating vascular diameters (5). While keeping organ flow constant in a certain pressure range, however, adjustment of vascular diameters also changes flow velocity and profile. Increased velocity increases shear stress, but after a certain threshold this may result in a tendency to turbulence and low shear stress in some vascular segments (Fig. 1). Hence, autoregulation preserves perfusion level in expense of disturbed flow-vascular wall relationship, which may culminate in arteriosclerosis and atherosclerosis (5). Furthermore, after structural adaptive response of the arteries to this disturbance occurred, it may still not be eliminated and even may propagate itself. Although a detailed discussion of the effects of flow and its iterations on vascular wall is beyond the scope of this paper, it needs to be stressed that accumulated bulk of evidence from hypertension research, which is focused on pressure and its manipulation, may in fact be related with these flow-related phenomena. A flow-related real causative factor may have been hiding for decades behind the perfect correlation between flow and pressure and the convenience of pressure measurement.
Figure 1.
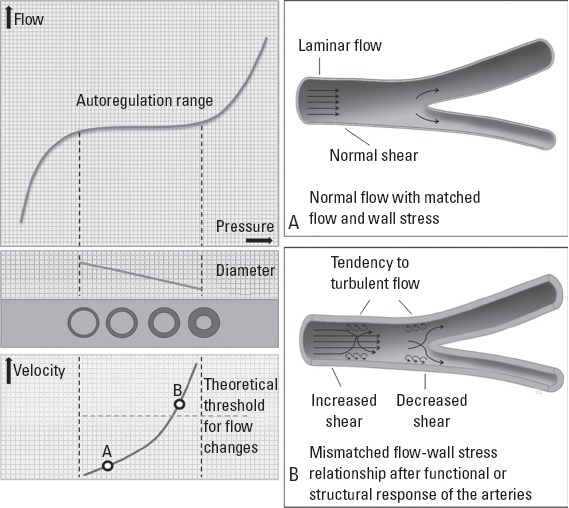
Since autoregulation maintains the flow in a certain pressure range, but not other flow-related parameters (e.g., flow velocity, flow profile, and shear forces), flow, and wall stress may be mismatched after a critical threshold has exceeded (left panel). After a certain amount of decrease in luminal cross-sectional area (critical limit separates A from B), which may require a certain amount of structural change, faster flowing blood may create a tendency to turbulence and low shear stress at certain vascular segments, such as bifurcations, bends, and kinks (right panel, B). After protective changes to avoid hyperperfusion induced structural changes, flow-wall stress mismatch sustains in face of normalized or even reduced flow
If a “flow-” rather than a “pressure-” based factor is the causative or major driving mediator of common “hypertensive” complications, future approach to HT and its diagnosis, risk stratification, and treatment may take a different path. In this article, we are going to assess the evidence on hand to elucidate if HT and antihypertensive treatment exert most of their effects through flow rather than pressure. In this regard, we are going to examine the effects of high BP on CV outcomes in situations where (1) pressure changes significantly while flow remains constant, (2) pressure does not change significantly compared to control, while flow may be the dominant changing factor and (3) other supporting observations (Table 1).
Table 1.
Evidence supporting flow against pressure as the causative factor of common hypertensive complications
| Against pressure as the dominant factor | For flow as the dominant factor | |
|---|---|---|
| Pressure changes whereas organ flow is constant | ||
| Hydrostatic pressure | ||
| Effects of gravity | Involvement of the brain (the lowest pressure) | Involvement of organs with higher flow |
| Positional change | No effect of HSP | Target organ flow is unchanged |
| No gravity | No effect of HSP | Target organ flow is unchanged |
| High BP in physiologic conditions | ||
| Exercise | No effect of exercise induced BP increase | Target organ flow is unchanged |
| Comparative physiology | No effect of HT in the giraffe | Target organ flow is unchanged (?) |
| Pressure is constant whereas organ flow may be changing | ||
| Hemodynamic studies | Vascular changes predating increase in BP | Increased flow as the possible earliest finding |
| Residual risk | Arbitrary definition of HT | |
| Resistant hypertension | Natural BP > treated BP for the same level | |
| Significant residual risk in treated HT | ||
| Resistance to treatment > achieved BP level | ||
| Other | ||
| Type of damage | Less directly pressure-related complications | Dominance of endothelial complications |
BP - blood pressure; HSP - hydrostatic pressure; >, better or more important
Pressure being the dominant changing factor Evidence from hydrostatic pressure-related phenomena
The effects of hydrostatic pressure on CV system are generally overlooked, since we deliberately endeavor to eliminate its effects on our measurements by making these measurements at level of the heart. However, according to Bernoulli’s principle hydrostatic pressure has to decrease or increase as the fluid vertically gains or loses potential energy, respectively (6). Therefore, in the upright position, arterial and venous distending pressures are ~40 mm Hg lower in head and ~90 mm Hg higher in lower extremity, whereas arteriovenous pressure gradient driving flow is not much affected because both arterial and venous pressures change in the same direction and magnitude. This difference provides an excellent opportunity for studying differential effects of pressure and flow, as discussed below.
Differential effects of gravity on hydrostatic pressure vs. flow
Although comparing different organs may be subject to misjudgments because of their inherent structural differences and the dissimilarity of exposed pulsatile stress (7), intriguing observational evidence comes forth when one considers differential effects of gravity on distending and perfusion pressures: (1) one of the foremost targets of high BP, the brain, is the organ with the lowest BP in whole body, (2) target organ involvement in HT does not fit into craniocaudally-increasing pattern of hydrostatic pressure, (3) hypertensive complications tend to cluster in organs with higher flow, such as brain, heart, and kidneys, rather than the organs with vascular beds exposed to higher pressures.
Positional changes in a gravitational field
Among organs targeted by HT, the brain is an outstanding example for studying differential effects of gravity on pressure and flow, since its position changes continuously during the day with regard to hydrostatic reference point (i.e., the heart).
Humans spend about one-third of their whole day in supine position for sleeping, which causes an approximately 40 mm Hg increase in cerebral systolic and diastolic BPs, but no change in perfusion pressure. Yet, neither supine sleeping position nor sleeping duration seems to have any effect on hypertensive cerebrovascular damage. On the contrary, studies exploring circadian rhythmicity of CV events provide solid proof by consistently showing that supine hours showed not only lack of any evidence of increased CV event rate but also the lowest incidence in whole day (8–10). Also, the circadian rhythm of myocardial infarction shows a very similar pattern to circadian rhythm of stroke (11), which strongly argues against any genuine effect of hydrostatic pressure, since the heart is a gravity-independent organ in contrast to the brain.
Temporal coincidence of CV events with 24-h variation in BP, which shows a sharp rise near awakening and the highest value around midmorning (12), led some authors to speculate a direct effect of increased BP on vessels or atherosclerotic lesions (8–10). But in reality, it is the flow that increases under the sway of neurohormonal arousal, cerebral arterial pressure generally decreases upon waking up, as the magnitude of morning BP surge is generally less than the decrease in hydrostatic pressure (13). Lastly, sleeping duration has an inverse correlation with incidence of CV events, (14) although the mechanism is certainly more complex.
Absence of a gravitational field
Although being exposed to microgravity during long space travels causes many important changes in CV system, the most dominant effect is an approximately 40 mm Hg higher BP, which would have been resulted in a 16-times increase in stroke rates, but this has never been reported despite a total of nearly 128 person-years of manned spaceflight (15). This can be explained by the fact that perfusion pressure remains constant as both arterial and venous pressures increase due to loss of gravity (16, 17). On the other hand, increased distending pressure results in filtration of vascular fluids into the brain parenchyma causing intracranial HT. Accordingly, radiological studies showed that astronauts show evidence of intracranial HT such as globe flattening, optic disc edema, but not lesions specific to arterial HT, such as white matter lesions or lacunar infarcts (18).
High BP in physiologic conditions
Exercise
Both healthy individuals and patients with cardiovascular disease exhibit systolic BPs over 180 mm Hg at peak exercise, which is definitely in the hypertensive range according to resting standards. However, this physiologic increase in BP during exercise generally does not lead to acute adverse events (19). Although well-controlled longitudinal studies seeking hypertensive complications related to exercise are not taken in healthy individuals; professional athletes, who are usually exposed to exercise-induced BP increases, virtually never experience increased renal or cerebral hypertensive complications. Conversely, they enjoy a favorable CV risk profile, less incident HT, and longer life expectancy (20). The distinctive feature of physiological increase in BP during exercise is that the blood flow through target organs of HT is not increased out of proportion to demand, since blood flow is directed to working muscle.
Comparative physiology
Many mammals share similar BP values with humans, but the giraffe (Giraffa camelopardalis) is a unique mammal, which has an exceptionally high mean blood pressure of about 250 mm Hg, as its head is 250–300 centimeters above its heart, and its heart must overcome this huge hydrostatic pressure difference. Interestingly, this extraordinarily high BP does not culminate in severe vascular lesions, nor does it lead to stroke, heart, or kidney failure, despite the fact that usual life expectancy is about 20–25 years in the wild and even longer in zoos. Despite some anatomical differences, including thicker left ventricular and arterial walls, no signs of degenerative vascular or cardiac lesions were found in older giraffes. Although not specifically addressed, it seems excessively high BP and resulting hypertrophic vascular remodeling do not culminate in accelerated vascular damage when flow or flow-related parameters of vital organs kept within normal ranges (21–23).
Taken together, abovementioned evidence support that when target organ flow is kept in physiologic limits, increased BP does not result in any increase in CV event rate.
Pressure not being the dominant changing factor Hemodynamic studies on HT
In the starting or preceding phase of essential HT, most invasive studies in younger individuals have demonstrated a near-normal systemic vascular resistance, but increased cardiac output compared with age-matched controls (24–29). Also, microvascular changes begin to appear in this hyperdynamic phase, even before BP exceeds the diagnostic threshold for HT. Analysis of retinal microcirculation is a powerful tool in this regard, not only because it is the only place where vascular structure can be observed in vivo, but also it shows a strong association with the vascular status in other target organs (30–32). Several longitudinal studies have shown that signs of retinopathy can already be observed in young individuals before the occurrence of HT (33–35). These observations suggest that for a given BP within normal or pre-hypertensive limits, only patients with increased flow develop clinical hypertension and its consequences.
Chronic vasoconstriction in face of high BP (or perhaps high flow) to maintain normal blood flow induces hypertrophic remodeling of arteries, which results in reduction in luminal diameter and further increases in vascular resistance (36, 37). Indeed, age-related increases in BP in hypertensive subjects is associated with a progressive increase in vascular resistance, which at the age of 50 may be almost twice the value seen at the age of 20 (24). As a result, cardiac output and specific organ flow decrease as vascular resistance increases in sustained HT, sometimes without any further change in BP (24, 36, 37). It should be underlined that despite the flow itself is decreased, flow-related wall stress still remains increased after structural changes takes place, as discussed above (5). In this manner, essential HT may have close similarities to pulmonary HT from left-to-right shunts, in which initial insult is flow-related, and this insult continues to exert its effects even after flow itself decreases (37).
Residual risk
Another testimony to BP not being the primary offender is the fact that for the same BP level, treated and naturally-occurring BP have different prognostic meanings (38). Ironically, antihypertensive drug use seems to be a risk factor in HT, as it is accepted as a parameter in several risk assessment models (39–41). Despite achieving the same BP, why treatment always leaves a residual risk behind is explained by already occurred structural changes before intervention or an autoregulation system set to a higher BP range (42, 43). But these explanations are far from being convincing, because residual risk is observed in a wide array of patients (1) with different baseline HT levels and durations, (2) with different baseline risk levels, (3) with controlled HT for long durations and (4) taking different number and classes of drugs (38, 44). On the other hand, such a residual risk does not occur with lipid-lowering therapy (43).
Even the definition of HT contradicts with the explanation of residual risk. A steady gradient between BP levels and CV continues down to BPs that is well below the average for the population, which shows no separate subcategories such as with and without HT (1). Therefore, hypertension can be defined pragmatically as the level of BP at which treatment is worthwhile. Moreover, it seems some comorbidities and aging may shift this “HT-threshold” upward, contrary to the expectation of lower BP target would provide greater benefit for higher-risk patients (45). This pragmatic definition is essentially another reflection of residual risk, which indicates that residual risk is 100% at “HT-threshold”. However, explaining residual risk with already occurred damage or a shift in autoregulation range creates a curious paradox since its most extreme form (100% residual risk) is found in patients labeled as normal.
Although complete elimination of residual risk may require targeting other parameters beyond mean office BP, such as central BP, BP variability, and 24-h ambulatory BP, an alternative explanation is the pathophysiologic difference between naturally occurring and treated BP may be related with the other hemodynamic parameter, the flow. Reducing central reservoir pressure may influence specific organ flow parameters to a limited extent and may explain the residual risk left behind. This can also explain why ACEI/ARBs show BP-independent effects on renal outcomes as they target specific organ hemodynamics besides their systemic antihypertensive effects (45).
Resistance to therapy
Some clinical trials (46–49) have identified a subgroup of patients who are at a high risk for subsequent adverse outcomes, whose BP is less responsive to antihypertensive treatment, and in whom further lowering of BP with additional drugs is not necessarily accompanied by an outcome reduction. For example, in a recent study, a 22% increase in stroke risk was observed in patients with stage-I HT for every additional class of medication taken compared with those with untreated stage-I HT (46). Authors rightly entitled this study as “Is the blood pressure control for stroke prevention the correct goal?” A possible explanation is that despite additional BP-lowering agents, compensatory mechanisms maintaining critical organ flow may resist against lowering of BP, and at the same time, this critical flow level sustains the effects of a flow-related insulting factor.
In conclusion, abovementioned indirect evidence suggest that in situations where BP differs little with comparator and CV events are still increased, the flow, or one of its iterations, may be the possible culprit.
Other observations
The usual type of hypertensive damage also gives clues about the real inciting mechanism. Blood vessels are exposed to two kinds of mechanical forces; namely shear stress and cyclic strain of the vascular wall, which is mainly determined by cyclic change of BP. Although not mutually exclusive, shear stress affects predominantly endothelial cells, whereas BP changes influence largely medial structures of the arterial wall (50). Many of major CV events related to HT are atherosclerotic and thrombotic in nature rather than having a connection with the force exerted by BP on arterial wall. For example, HT usually does not cause bursting of arteries by high BP, as many patients and sometimes clinicians think, cerebral complications of HT are mostly ischemic due to atherosclerosis and arteriolosclerosis rather than being hemorrhagic. Similarly, many of the antihypertensive trials, if not all, targeted to achieve a reduction in myocardial infarction (44, 45). Given that both atherosclerosis and atherothrombosis are endothelium-related disorders with a well-known connection with flow-mediated hemodynamic forces (e.g., shear stress), it may be another indication that flow or its iterations, rather than BP, are the risk factor for these CV events.
Limitations, unresolved problems, and future directions
Our proposition that an association exists between flow-mediated perturbations and vascular damage is not new. For many years, HT has been regarded as a risk factor for atherosclerotic CV disease more than being a disease on its own, which acknowledges that it uses intermediaries to cause its adverse effects. Moreover, close link between atherosclerotic events and flow-related phenomena is known for a long time. Nevertheless, our perception of HT has always been pressure-based; the diagnosis, risk stratification and treatment targets are all in pressure domain (45). To our knowledge, a flow-related factor has never been proposed as the causative factor for HT complications. This perspective change may lead to important differences in understanding, definition, diagnosis, and treatment of HT. A new flow-related-parameter-based definition; (1) may extend the definition of HT and recognize patients-at-risk in “pre-hypertensive” range earlier; (2) may fine-tune pressure-based thresholds used for diagnosing, stratification of risk and defining treatment targets in HT, especially in patients with high risk and comorbidities; (3) may address residual risk, the Achilles’ heel of HT management, more accurately; (4) may lead to development of therapies targeting resistance or flow without changing BP. The development of such preventive approaches may differ organ to organ, and, more importantly, flow modulation may go far beyond arterial system and venous manipulations can be of value in this regard.
However, discrediting pressure as the dominant factor may not automatically elevate flow or its iterations to its place for all clinical scenarios. Some hypertensive complications can be directly attributed to pressure, such as left ventricular hypertrophy, vessel aneurysm formation, and rupture. Flow-related factors may require pressure related changes in the first place for exerting their deleterious effects on the arteries. For example, a change in flow profile (from laminar to turbulent) may require a certain degree of luminal narrowing due to structural remodeling arteries induced by pressure load (Fig. 1). Also, pulse pressure may still have a specific risk by exerting tensile stress on arteries, but it should be reevaluated with the studies designed to differentiate its effects from the effects of pulsatile flow. Most importantly, which of the flow-related parameters are responsible for CV effects and whether guiding treatment with manipulation of this parameter would be successful need to be clarified.
Conclusion
Although never specifically addressed, indirect evidence in hand supports that pressure per se may not be the causative factor for the majority of HT-associated complications and flow-based hemodynamic factors may be the principal mediator of HT-associated damage. This understanding may change the way we approach HT. Future risk prediction for specific outcomes might be scrutinized via examining flow profiles in the organs under investigation and possible modifications of these parameters may significantly contribute to HT management. Most important of all, treatments targeting flow-related phenomena and their effects may go far beyond current understanding. These areas need to be explored with the light of this new perspective.
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – All authors; Design – All authors; Supervision – All authors; Analysis and/or interpretation – All authors; Writing – All authors; Critical review – All authors.

From Prof. Dr. Arif Akşit’s collections
References
- 1.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality:a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–13. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 2.Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease:metaanalysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009:338–b1665. doi: 10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasan RS. Biomarkers of cardiovascular disease:molecular basis and practical considerations. Circulation. 2006;113:2335–62. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 4.Levy M. The cardiac and vascular factors that determine systemic blood flow. Circ Res. 1979;44:739–47. doi: 10.1161/01.res.44.6.739. [DOI] [PubMed] [Google Scholar]
- 5.Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282:2035–42. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 6.Hicks JW, Badeer HS. Gravity and the circulation: “open” vs. “closed” systems. Am J Physiol. 1992;262:R725–32. doi: 10.1152/ajpregu.1992.262.5.R725. [DOI] [PubMed] [Google Scholar]
- 7.O'Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney:cause and logic of therapy. Hypertension. 2005;46:200–4. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- 8.Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med. 1985;313:1315–22. doi: 10.1056/NEJM198511213132103. [DOI] [PubMed] [Google Scholar]
- 9.Sloan MA, Price TR, Foulkes MA, Marler JR, Mohr JP, Hier DB, et al. Circadian rhythmicity of stroke onset. Intracerebral and subarachnoid hemorrhage. Stroke. 1992;23:1420–6. doi: 10.1161/01.str.23.10.1420. [DOI] [PubMed] [Google Scholar]
- 10.Elliott WJ. Circadian variation in the timing of stroke onset:a meta-analysis. Stroke. 1998;29:992–6. doi: 10.1161/01.str.29.5.992. [DOI] [PubMed] [Google Scholar]
- 11.Reavey M, Saner H, Paccaud F, Marques-Vidal P. Exploring the periodicity of cardiovascular events in Switzerland:variation in deaths and hospitalizations across seasons, day of the week and hour of the day. Int J Cardiol. 2013;168:2195–200. doi: 10.1016/j.ijcard.2013.01.224. [DOI] [PubMed] [Google Scholar]
- 12.Millar-Craig MW, Bishop CN, Raftery EB. Circadian variation of blood-pressure. Lancet. 1978;1:795–7. doi: 10.1016/s0140-6736(78)92998-7. [DOI] [PubMed] [Google Scholar]
- 13.Sheppard JP, Hodgkinson J, Riley R, Martin U, Bayliss S, McManus RJ. Prognostic significance of the morning blood pressure surge in clinical practice:a systematic review. Am J Hypertens. 2015;28:30–41. doi: 10.1093/ajh/hpu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gangwisch JE. A review of evidence for the link between sleep duration and hypertension. Am J Hypertens. 2014;27:1235–42. doi: 10.1093/ajh/hpu071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.List of spaceflight records. [last accessed on 20.04.2016]. https://en.wikipedia.org/wiki/List_of_spaceflight_records#cite_note-Spacefacts_listflights-33 .
- 16.Arbeille P, Fomina G, Roumy J, Alferova I, Tobal N, Herault S. Adaptation of the left heart, cerebral and femoral arteries, and jugular and femoral veins during short- and long-term head-down tilt and space flights. Eur J Appl Physiol. 2001;86:157–68. doi: 10.1007/s004210100473. [DOI] [PubMed] [Google Scholar]
- 17.Herault S, Fomina G, Alferova I, Kotovskaya A, Poliakov V, Arbeille P. Cardiac, arterial and venous adaptation to weightlessness during 6-month MIR spaceflights with and without thigh cuffs. Eur J Appl Physiol. 2000;81:384–90. doi: 10.1007/s004210050058. [DOI] [PubMed] [Google Scholar]
- 18.Kramer LA, Sargsyan AE, Hasan KM, Polk JD, Hamilton DR. Orbital and intracranial effects of microgravity:findings at 3-T MR imaging. Radiology. 2012;263:819–27. doi: 10.1148/radiol.12111986. [DOI] [PubMed] [Google Scholar]
- 19.Aslanger E, Assous B, Bihry N, Beauvais F, Logeart D, Cohen-Solal A. Association between baseline cardiovascular mechanics and exercise capacity in patients with coronary artery disease. Anatolian J Cardiol. 2015 Nov 18; doi: 10.5152/AnatolJCardiol.2015.6471. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma S, Merghani A, Mont L. Exercise and the heart:the good, the bad, and the ugly. Eur Heart J. 2015;36:1445–53. doi: 10.1093/eurheartj/ehv090. [DOI] [PubMed] [Google Scholar]
- 21.Goetz RH, Warren JV, Gauer OH, Patterson JL, Jr, Doyle JT, Keen EN, et al. Circulation of the giraffe. Circ Res. 1960;8:1049–58. doi: 10.1161/01.res.8.5.1049. [DOI] [PubMed] [Google Scholar]
- 22.Zhang QG. Hypertension and counter-hypertension mechanisms in giraffes. Cardiovasc Hematol Disord Drug Targets. 2006;6:63–7. doi: 10.2174/187152906776092640. [DOI] [PubMed] [Google Scholar]
- 23.Munis JR, Lozada LJ. Giraffes, siphons, and Starling resistors:cerebral perfusion pressure revisited. J Neurosurg Anesthesiol. 2000;12:290–6. doi: 10.1097/00008506-200007000-00029. [DOI] [PubMed] [Google Scholar]
- 24.Lund-Johansen P. Hemodynamics in early essential hypertension. Acta Med Scand. 1967;181(Suppl 482):1–101. [Google Scholar]
- 25.Frohlich ED, Tarazi RC, Dustan HP. Re-examination of the hemodynamics of hypertension. Am J Med Sci. 1969;257:9–23. doi: 10.1097/00000441-196901000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Julius S, Pascual AV, Sannerstedt R, Mitchell C. Relationship between cardiac output and peripheral resistance in borderline hypertension. Circulation. 1971;43:382–90. doi: 10.1161/01.cir.43.3.382. [DOI] [PubMed] [Google Scholar]
- 27.Jern S. Psychological and hemodynamic factors in borderline hypertension. Acta Med Scand Suppl. 1982;662:1–55. [PubMed] [Google Scholar]
- 28.Andersson OK, Sannerstedt R, Beckman M. Essential hypertension-implications for pathogenesis from repeated hemodynamic investigations in young men with elevated blood pressure. J Hypertens. 1982;1(Suppl 2):91–3. [PubMed] [Google Scholar]
- 29.Witt N, Wong TY, Hughes AD, Chaturvedi N, Klein BE, Evans R, et al. Abnormalities of retinal microvascular structure and risk of mortality from ischemic heart disease and stroke. Hypertension. 2006;47:975–81. doi: 10.1161/01.HYP.0000216717.72048.6c. [DOI] [PubMed] [Google Scholar]
- 30.Sairenchi T, Iso H, Yamagishi K, Irie F, Okubo Y, Gunji J, et al. Mild retinopathy is a risk factor for cardiovascular mortality in Japanese with and without hypertension:the Ibaraki prefectural health study. Circulation. 2011;124:2502–11. doi: 10.1161/CIRCULATIONAHA.111.049965. [DOI] [PubMed] [Google Scholar]
- 31.Sabanayagam C, Tal ES, Shankar A, Lee J, Sun C, Wong TY. Retinal arteriolar narrowing increases the likelihood of chronic kidney disease in hypertension. J Hypertens. 2009;27:2209–17. doi: 10.1097/HJH.0b013e328330141d. [DOI] [PubMed] [Google Scholar]
- 32.Wong TY, Mitchell P. Hypertensive retinopathy. N Engl J Med. 2004;351:2310–7. doi: 10.1056/NEJMra032865. [DOI] [PubMed] [Google Scholar]
- 33.Wong TY, Klein R, Sharrett AR, Duncan BB, Douper DJ, Klein BE, et al. Atherosclerosis risk in communities study. Retinal arteriolar diameter and risk for hypertension. Ann Intern Med. 2004;140:248–55. doi: 10.7326/0003-4819-140-4-200402170-00006. [DOI] [PubMed] [Google Scholar]
- 34.Ikram MK, Witteman JC, Vingerlimg JR, Breteler MM, Hofman A, De Jong PT. Retinal vessel diameters and risk of hypertension:the Rotterdam study. Hypertension. 2006;47:189–94. doi: 10.1161/01.HYP.0000199104.61945.33. [DOI] [PubMed] [Google Scholar]
- 35.Izzard AS, Rizzoni D, Agabiti-Rosei E, Heagerty AM. Small artery structure and hypertension:adaptive changes and target organ damage. J Hypertens. 2005;23:247–50. doi: 10.1097/00004872-200502000-00002. [DOI] [PubMed] [Google Scholar]
- 36.Laurent S, Boutouytrie P. The structural factor of hypertension:large and small artery alterations. Circ Res. 2015;116:1007–21. doi: 10.1161/CIRCRESAHA.116.303596. [DOI] [PubMed] [Google Scholar]
- 37.Dickinson MG, Bartelds B, Borgdorff MA, Berger RM. The role of disturbed blood flow in the development of pulmonary arterial hypertension:lessons from preclinical animal models. Am J Physiol Lung Cell Mol Physiol. 2013;305:L1–14. doi: 10.1152/ajplung.00031.2013. [DOI] [PubMed] [Google Scholar]
- 38.Zanchetti A. Bottom blood pressure or bottom cardiovascular risk? How far can cardiovascular risk be reduced? J Hypertens. 2009;27:1509–20. doi: 10.1097/HJH.0b013e32832e9500. [DOI] [PubMed] [Google Scholar]
- 39.NCEP. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 40.Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, May M, Brindle P. Derivation and validation of QRISK, a new cardiovascular disease risk score for the United Kingdom:prospective open cohort study. BMJ. 2007;335:136–47. doi: 10.1136/bmj.39261.471806.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zethelius B, Berglund L, Sundström J, Ingelsson E, Basu S, Larsson A, et al. Use of multiple biomarkers to improve the prediction of death from cardiovascular causes. N Engl J Med 200. 358:2107–16. doi: 10.1056/NEJMoa0707064. [DOI] [PubMed] [Google Scholar]
- 42.Zanchetti A. Blood pressure targets of antihypertensive treatment:up and down the J-shaped curve. Eur Heart J. 2010;31:2837–40. doi: 10.1093/eurheartj/ehq281. [DOI] [PubMed] [Google Scholar]
- 43.Blacher J, Evans A, Arveiler D, Amouyel P, Ferrières J, Bingham A, et al. PRIME Study Group. Residual cardiovascular risk in treated hypertension and hyperlipidaemia:the PRIME Study. J Hum Hypertens. 2010;24:19–26. doi: 10.1038/jhh.2009.34. [DOI] [PubMed] [Google Scholar]
- 44.Andersson OK, Almgren T, Persson B, Samualsson O, Hedner T, Wilhemsen L. Survival in treated hypertension:follow up study after two decades. BMJ. 1998;317:167–71. doi: 10.1136/bmj.317.7152.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults:report from the panel members appointed to the Eighth Joint National Committee (JNC 8) JAMA. 2014;311:507–20. doi: 10.1001/jama.2013.284427. [DOI] [PubMed] [Google Scholar]
- 46.Howard G, Banach M, Cushman M, Goff DC, Howard VJ, Lackland DT, et al. Is blood pressure control for stroke prevention the correct goal? The lost opportunity of preventing hypertension. Stroke. 2015;46:1595–600. doi: 10.1161/STROKEAHA.115.009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Zhang X, Liu L, Wang Y, Tang X, Zanchetti A FEVER Study Group. Higher cardiovascular risk and impaired benefit of antihypertensive treatment in hypertensive patients requiring additional drugs on top of randomized therapy:is adding drugs always beneficial? J Hypertens. 2012;30:2202–12. doi: 10.1097/HJH.0b013e3283582eec. [DOI] [PubMed] [Google Scholar]
- 48.Lithell H, Hansson L, Skoog I, Elmfeldt D, Hofman A, Olofsson B, et al. SCOPE Study Group. SCOPE Study Group. The Study on COgnition and Prognosis in the Elderly (SCOPE);outcomes in patients not receiving add-on therapy after randomization. J Hypertens. 2004;22:1605–12. doi: 10.1097/01.hjh.0000133730.47372.4c. [DOI] [PubMed] [Google Scholar]
- 49.Weber MA, Julius S, Kjeldsen SE, Jia Y, Brunner HR, Zappe DH, et al. Cardiovascular outcomes in hypertensive patients:comparing single-agent therapy with combination therapy. J Hypertens. 2012;30:2213–22. doi: 10.1097/HJH.0b013e3283582ed6. [DOI] [PubMed] [Google Scholar]
- 50.Safar ME, Blacher J, Jankowski P. Arterial stiffness, pulse pressure, and cardiovascular disease—Is it possible to break the vicious circle? Atherosclerosis. 2011;218:263–71. doi: 10.1016/j.atherosclerosis.2011.04.039. [DOI] [PubMed] [Google Scholar]