

Abstract
Objective:
Germline mutations in the bone morphogenetic protein receptor type-2 (BMPR2) gene are considered to be a major risk factor for pulmonary arterial hypertension (PAH). BMPR2 mutations have been reported in 10%–20% of idiopathic PAH and in 80% of familial PAH cases. The aim of this study was to evaluate the frequency of mutations in the serine/threonine kinase domain of the BMPR2 gene in a group of patients from a single PAH referral center in Turkey.
Methods:
This cross-sectional study used a DNA-sequencing method to investigate BMPR2 mutations in the serine-threonine-kinase domain in 43 patients diagnosed with PAH [8 with idiopathic PAH and 35 with congenital heart disease (CHD)] from a single PAH referral center. Patients were included if they had a hemodynamically measured mean pulmonary arterial pressure of >25 mm Hg with a mean pulmonary capillary wedge pressure of ≤15 mm Hg. Patients with severe left heart disease and/or pulmonary disease that could cause pulmonary hypertension were excluded. Associations between categoric variables were determined using the chi-square test. Differences between idiopathic and CHD-associated PAH groups were compared with the unpaired Student’s t-test for continuous variables.
Results:
We detected a missense mutation, [p.C347Y (c.1040G>A)], in one patient with idiopathic PAH in exon 8 of the BMPR2 gene. The mutation was detected in a 27-year-old female with a remarkable family history for PAH. She had a favorable response to endothelin receptor antagonists. No mutations were detected in the exons 5–11 of the BMPR2 gene in the PAH-CHD group.
Conclusion:
A missense mutation was detected in only one of the eight patients with idiopathic PAH. The BMPR2 missense mutation rate of 12.5% in this cohort of Turkish patients with idiopathic PAH was similar to that seen in European registries. The index patient was a young female with a family history remarkable for PAH; she had a good long-term response to PAH-specific treatment, probably due to the early initiation of the treatment. Genetic screening of families affected by PAH might have great value in identifying the disease at an early stage.
Keywords: DNA sequencing, BMPR2 mutation, pulmonary arterial hypertension
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by increased pulmonary arterial resistance, leading to right heart failure with high mortality (1, 2). Idiopathic PAH is histologically characterized by endothelial and smooth muscle cell proliferation, medial hypertrophy, inflammation, and thrombosis, particularly in distal pulmonary arteries (1-7).
The presence of germline mutations in patients with idiopathic PAH is referred to as heritable PAH (3, 4, 8-11). Mutations in the transforming growth factor-β (TGF-β) receptor family, including the BMPR2, SMAD9, ACVRL1 (formerly ALK1), and endoglin genes, have been identified in patients with PAH and PAH associated with hereditary hemorrhagic telangiectasia (2, 10, 12-17).
Mutations in the bone morphogenic protein receptor type 2 (BMPR2) gene that encodes a type 2 receptor for bone morphogenetic proteins (BMPs) are accepted as the most important predisposing factors for hereditary PAH (2,10,11). BMPs have a modulatory role in cell homeostasis, including apoptosis, proliferation, and differentiation, and also regulate a wide range of developmental functions (13). BMP ligands bind to type 1 and 2 serine/threonine kinase receptors (BMPR1 and BMPR2) when they have been processed into mature forms and secreted. Binding of the ligand results in a signal transduction cascade that induces the phosphorylation of a family of signaling proteins (Smads); the signal is then moved to the nucleus where it regulates the transcription of target genes (12-14). The expression of BMPR2 is reduced in the pulmonary arteries of patients with idiopathic PAH (18).
It has been shown that more than 50% of the cases of hereditary PAH are associated with BMPR2 (3, 4) mutations, leading to an increase in the proliferation of vascular smooth muscle cells and a reduction in apoptosis. Its genetic transition is autosomal dominant, and BMPR2 shows incomplete penetrance and variable expression (3, 4). The BMPR2 gene located on chromosome 2 at 2q33 has 13 exons (18-20). Of these exons, the exons 1–3 encode an extracellular ligand binding domain, the exon 4 encodes the transmembrane domain, the exons 5–11 encode a serine/threonine kinase domain, and the exons 12–13 encode an intracellular C-terminal region (cytoplasmic domain) (13,14).
More than 300 different BMPR2 mutations have been identified in patients with PAH associated with family history, sporadic disease, and other diseases (19). The frequency of this mutation is well addressed in European and American PAH registries from PAH referral centers (5, 6). However, there is no data from the Turkish patients with PAH. The aim of this study was to determine the presence of mutations in the serine-threonine kinase domain of the BMPR2 gene in a group of patients from a single PAH referral center in Turkey.
Methods
Patients
This cross-sectional study included 50 consecutive Turkish patients (26 women, 24 men, mean age: 36±13 years) with PAH who were monitored at the Ege University Medical School PAH-specialized referral center between 2011 and 2012. PAH was diagnosed according to the algorithm used in our center (7, 8). Patients were included if they had a hemodynamically measured mean pulmonary arterial pressure of >25 mm Hg with a mean pulmonary capillary wedge pressure of ≤15 mm Hg. Patients with severe left heart disease and/or pulmonary disease that could cause pulmonary hypertension were excluded. Of the enrolled 50 patients, 7 patients were excluded because of the presence of associated PAH [1 patient with connective tissue disease associated with PAH, 4 patients with chronic thromboembolic pulmonary hypertension, and 2 patients with group 5 pulmonary hypertension (sarcoidosis)]. The final study population consisted of 43 patients with Group I pulmonary hypertension [8 patients with idiopathic PAH and 35 patients with PAH associated with congenital heart disease (CHD)]. The study protocol was approved by the Clinical Research Ethics Committee; each participant gave written informed consent after appropriate genetic counseling in accordance with the ESC 2009 guidelines for the diagnosis and treatment of pulmonary hypertension (8, 9).
Molecular workup
DNA isolation: DNA was obtained from the peripheral blood of both patients using a Magna Pure LC Kit (Roche, Germany, Mannheim).
Polymerase chain reaction (PCR): To yield specific DNA material for further genetic analyses, we used the PCR technique. The genomic regions of the serine/threonine kinase domain (exons 5–11) in the BMPR2 gene from the DNA samples of patients were amplified using the primers listed in Table 1. Each PCR reaction was performed in a 25 mL volume containing 10xPCR buffer (50 mM KCl, 10 mM Tris-HCl, 1.5 mM MgCl2), 2 mM MgCl2, 0.8 µM each of primer, 200 µM dNTP mix, 1% DMSO, 0.5 U Taq DNA polymerase (Roche, Germany, Mannheim), and 50 ng genomic DNA. PCR amplification was performed in a DNA Thermal Cycler 9600 (Perkin Elmer, Oak Brook, IL, USA).
Table 1.
Primers of the serine/threonine kinase domain of BMPR2 for the PCR technique and DNA sequencing methods and the expected fragment size of the amplified regions. The primers were prepared using reference sequences (21, 30).
| Primer | Primer sequence | Expected amplification fragment size (bp) |
|---|---|---|
| Primers for Exon 5 of BMRP2 Gene | ||
| Forward | CTTGCTGCTAATCTTTCTGC | 300 |
| Rewerse | AAATGAATGAATGTCTTAATGAT | |
| Primers for Exon 6 of BMRP2 Gene | ||
| Forward | TGATAATGGAATAAACTGTAAG | 407 |
| Rewerse | GCATAAGCCACCACACCTG | |
| Primers for Exon 7 of BMRP2 Gene | ||
| Forward | TGCTAATTTACTCTTCATGTT | 316 |
| Rewerse | CAAACAACTGACTAATAATAAA | |
| Primers for Exon 8 of BMRP2 Gene | ||
| Forward | GTATGTTCATTTCATGTTCAATAGTCC | 276 |
| Rewerse | AATTATCATTTCAAAGTACATCAGTGTG | |
| Primers for Exon 9 of BMRP2 Gene | ||
| Forward | GGTCTAATGTCTGTTCTTCA | 300 |
| Rewerse | AAAGTTGAGTTAGGTACTATA | |
| Primers for Exon 10 of BMRP2 Gene | ||
| Forward | TATCAGAAATACCCCTGTTA | 303 |
| Rewerse | CGTTATTAACAGTCTATTTTTG | |
| Primers for Exon 11 of BMRP2 Gene | ||
| Forward | GAGCATGTTCCGTAATCC | 393 |
| Rewerse | TTGTTGGGTCTCAGTTTC | |
Amplification conditions: Initial denaturation at 95°C for 5 min followed by 30 cycles of denaturation at 95°C for 60 sec, annealing at 64°C for 60 sec, and extension at 72°C for 60 sec with a final extension at 72°C for 10 min for the serine/threonine kinase domain (exons 5–11) of the BMPR2 gene.
DNA sequencing: EDTA-blood samples were collected for genetic analysis in all patients. Human genomic DNA was obtained from peripheral blood lymphocytes. The BMPR2 gene consists of 13 exons, and the serine/threonine kinase domain of the gene covers the exons 5–11. Exons 5–11 of the BMPR2 gene were amplified by specific primers, and sequencing was performed with same primers using version 1.1 of the Big Dye terminator cycle sequencing kit on an ABI 3100 Genetic Analyzer (ABI 3100, Applied Biosystems, Foster City, CA). The sequencing results were analyzed with Chromas and Sequencing Analysis software. Mutations were identified by comparison with the NCBI human BMPR2 nucleotide reference sequence (NCBI: NM_001204).
Statistical analysis
All data were presented as mean ± standard deviation for continuous variables as appropriate, and numbers (percentage) for categorical variables. Associations between the categorical variables were determined using the chi-square test. Differences between the two subgroups were compared with the unpaired Student’s t-test for continuous variables. Statistical significance for all tests was accepted at the p<0.05 level.
Results
The clinical characteristics of the patients with PAH are presented in Table 2. The study population consisted of relatively young patients, particularly the CHD-PAH group. The mean age was 26±12 years at the time of diagnosis. Most of the patients were in WHO Functional Class II at the time of diagnosis. A family history of PAH was present only in one patient in the idiopathic PAH group.
Table 2.
Clinical characteristics of the study population
| All patients (n=43) | I-PAH (n=8) | CHD-PAH (n=35) | |
|---|---|---|---|
| Age, years (mean±SD) | 37±12 | 42±9 | 36±12 |
| Age at diagnosis, years (mean±SD) | 26±12 | 35±10 | 24±11 |
| Gender [n (%)] | |||
| Male | 21 (49) | 2 (25) | 19 (54) |
| Female | 22 (49) | 6 (75) | 16 (46) |
| WHO functional class [n (%)] | |||
| I | - | - | - |
| II | 18 (42) | 2 (25) | 16 (44) |
| III | 23 (53) | 4 (50) | 19 (56) |
| IV | 2 (5) | 2 (25) | - |
| Family history of PAH [n (%)] | 1 (2.3) | 1 (12.5) | 0 |
| Echocardiographic findings (mean±SD) | |||
| LV end-diastolic diameter, mm | 41±8 | 36±7 | 42±7 |
| LV end-systolic diameter, mm | 26±7 | 22±5 | 27±8 |
| RV end-diastolic diameter, mm | 36±10 | 36±11 | 35±10 |
| PA diameter, mm | 33±7 | 32±9 | 34±7 |
| Tricuspid regurgitation velocity | 4.1±0.7 | 3.7±1 | 4.2±0.5 |
| Systolic PA pressure, mm Hg | 78±21 | 74±24 | 79±20 |
| Mean PA pressure*, mm Hg | 70±20 | 59±17 | 72±20 |
measured by right heart catheterization
CHD-PAH - PAH associated with CHD; I-PAH - idiopathic PAH; LV - left ventricular; RV - right ventricular; PA - pulmonary artery; PAH - pulmonary arterial hypertension
BMPR2 gene
We did not identify any mutations in exons 5, 6, 7, 9, 10, and 11 of the serine/threonine kinase domain of the BMPR2 gene. We detected a missense mutation, p.C347Y (c.1040G>A), in exon 8 of the BMPR2 gene in one patient with idiopathic PAH. This patient was heterozygous for this mutation (Fig. 1). The rate of p.C347Y mutation in eight idiopathic PAH patients was 12.5%. No mutations were detected in the exons 5–11 in the PAH group associated with CHD (Table 3).
Figure 1.
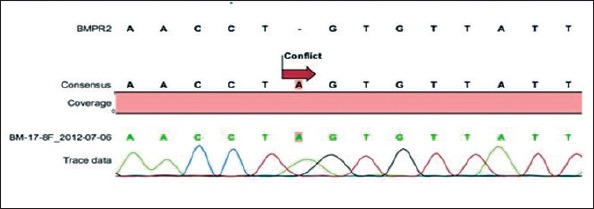
Sequence analysis of the BMPR2 serine/threonine region from one sample. A G allele was altered to an A allele. The alleles are heterozygotes.
Table 3.
Rate of p.C347Y missense mutation causing the codon conversion of nucleotides TGT>TAT (c.1040G>A) sequences (p.C347Y) and change of serine to tyrosine in exon 8 in one patient with idiopathic PAH among 43 patients with PAH.
| Mutation | Rate of mutation in | ||
|---|---|---|---|
| PAH (n=43) | Idiopathic PAH (n=8) | PAH associated CHD (n=35) | |
| p.C347Y | 1 (2.3%) | 1 (12.5%) | 0 (0%) |
CHD - congenital heart disease; PAH - pulmonary arterial hypertension
Values are expressed as the number (percentage) of patients [n (%)]
Clinical parameters of the patient with idiopathic PAH having a heterozygous p.C347Y mutation
A mutation was detected in a 27-year-old female with idiopathic PAH. Her dyspnea symptoms began 5 years previously after an abortion at 12 weeks of gestation. Her family history was remarkable for PAH. Her 18-year-old sister died because of an unknown disease with severe progressive shortness of breath, denoting probable PAH. However, we could not consider her as a familial case, as her sister died without a detailed examination to confirm the diagnosis of PAH. The marriage of the patient’s parents was consanguineous. The patient was referred to our PAH center with functional class III symptoms. Her six-minute walking distance was 238 meters. An ECG revealed right ventricular hypertrophy and an incomplete right bundle branch block. A chest X-ray of the patient showed a prominent pulmonary artery and enlargement of the right atrium and right ventricle (Fig. 2a). Echocardiographic findings were compatible with severe pulmonary hypertension. Her right ventricular fractional area contraction was 35%. She had severe pulmonary regurgitation and moderate tricuspid regurgitation with severely dilated right heart chambers. Her pulmonary artery was dilated (4 cm) (Fig. 2b). Her systolic pulmonary artery pressure was 76 mm Hg. Right heart catheterization revealed PAH with high mean pulmonary artery pressure (47 mm Hg) and normal pulmonary capillary wedge pressure. All other diagnostic workup was negative for other causes of pulmonary hypertension. She was put on PAH-specific treatment (bosentan, an endothelin receptor antagonist). Her response to treatment was very satisfactory, and she has been doing well with monotherapy for five years in functional class II. The patient and her relatives received detailed genetic counseling. Figure 3 shows the pedigree of the family.
Figure 2.
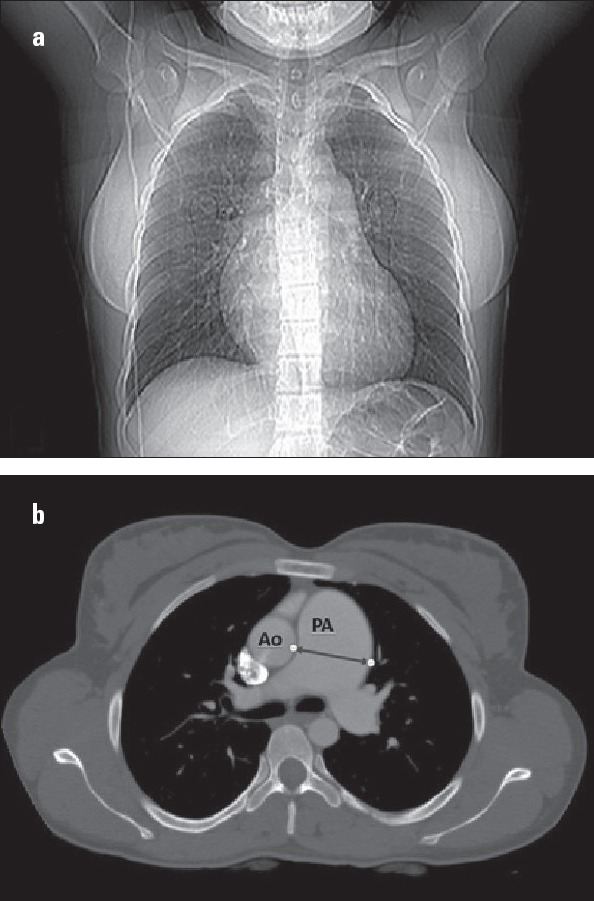
a, b. (a) Chest X-ray of the patient with PAH showing a prominent pulmonary artery and enlarged right atrium and right ventricle. (b) Thorax computed tomography showing an enlarged pulmonary artery with a diameter of 4 cm.
Ao - aorta; PA - pulmonary artery
Figure 3.
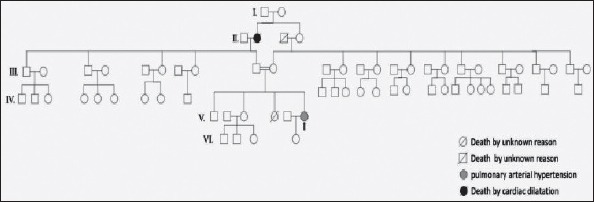
Pedigree of the family
Discussion
We performed the sequence analysis of exons 5–11 of the BMPR2 gene and detected a p.C347Y missense mutation located in the serine/threonine kinase domain, which is on the cytoplasmic side of the BMPR2 receptor gene (15). Mutations occur in various regions of BMPR2 at the ligand-binding domain, the kinase domain, and the long cytoplasmic tail. Point mutations leading to amino acid substitutions are generally located in either highly conserved or critically functional domains of the receptor; thus, they predict alterations in receptor function. BMPR2 gene mutations reflect a reduction in BMPR2 signaling. In addition, BMPR2 expression is also reduced in patients with PAH (18, 21). It has been suggested that p.C347Y mutation reverses the kinase activation of BMPR2 and inhibits BMPR1 receptor phosphorylation (15).
Mutations in the BMPR2 gene have been reported in 10%–20% of patients with idiopathic PAH and in up to 80% of the patients with familial PAH (10, 15). In line with these reports, we detected a missense BMPR2 mutation in one of the eight patients (12.5%) with idiopathic PAH. This was the only familial case within the study population. To the best of our knowledge, this is the first genetic analysis study of a group of Turkish patients with PAH. Although our PAH center has one of the leading PAH databases in Turkey, this ratio cannot be generalized to the broader community based on our study alone.
Molecular diagnosis of BMPR2 mutations is important for several reasons. Patients carrying BMPR2 mutations present approximately 10 years earlier than non-carriers. They are more hemodynamically compromised at diagnosis. They are also less likely to respond to acute vasodilator testing and unlikely to benefit from treatment with calcium channel blockers (4, 12, 21). However, our patient with a BMPR2 mutation had a good response to PAH-specific agents, probably due to an early initiation of treatment. Therefore, molecular screening of families with affected cases might have great value in identifying the disease at an early stage, leading to early PAH-specific treatment.
The type of mutation might also be important in the clinical courses of patients with PAH. Recent studies have suggested that compared with other BMPR2 mutation carriers, mutations affecting the cytoplasmic tail of BMPR2 were characterized by an older age of the patients at diagnosis, less severe hemodynamic characteristics, and a greater chance of long-term response to calcium channel blockers (22-24). An in vitro assay showed that mutations in the cytoplasmic tail domain do not interfere with the normal activation of the Smad pathway, whereas activation was abolished in the presence of mutations located in the kinase domain (13). BMPR2 signaling is associated with increased Smad1/5 phosphorylation in the BMP pathway, suggesting that cell surface trafficking of mutant BMPR2 may have therapeutic potential in PAH (22, 25).
Our study population also included 35 patients with PAH associated with CHD. None of the patients with CHD had BMPR2 mutations. BMPR2 mutation is occasionally reported in PAH with CHD (26, 27). Indeed, Limsuvan et al. (28) reported a lack of BMPR2 mutations in a cohort of 30 pediatric patients with PAH associated with CHD. However, as we have analyzed only the exons 5–11 of the 13 exons, we might have missed mutations in other exons. We preferred to study the exons 5–11 because they cover the active serine/kinase domain which is responsible for enzymatic kinase activation (3). Moreover, Treacy et al. (12) have decribed this domain as an essential domain of the BMPR2 gene, as any mutation in the serine/threonine kinase domain could affect the functional enzymatic activity of BMPR2 and could lead to the development of PAH.
Although genetic screening of the families with affected cases might have great value in identifying the disease at an early stage (29, 30), we could not perform genetic screening of the family of the proband. Moreover, current guidelines recommend genetic testing of the relatives of patients with a family history of PAH and also advising patients with idiopathic PAH about the availability of genetic testing (8, 30). However, the penetrance of BMPR2 mutation is incomplete, and PAH develops in only 27% of the mutation carriers, with a higher penetrance in women (42%) than men (14%) (8). Echocardiographic screening of first-degree family members should also be advised, particularly when the molecular analysis is not available or in the case of lack of consent.
Study limitations
The lack of whole gene sequencing and lack of assessment of receptor-like kinase 1 activity might be accepted as the major limitations of the study. Another important limitation is the lack of genetic screening of the family of the proband.
Conclusion
We detected a p.C347Y missense mutation located in the serine/threonine kinase domain in one of the eight patients with idiopathic PAH by sequencing the exons 5–11 of the BMPR2 gene. The mutation rate of 12.5% in this cohort of the Turkish patients with idiopathic PAH was similar to that observed in European registries. The index patient was a young female with a family history remarkable for PAH who had a good long-term response to PAH-specific treatment, probably due to early treatment. Genetic testing might have great value in identifying the disease at an early stage and should be advised for the relatives of patients with a family history of PAH.
Acknowledgements:
This study was supported by Ege University, APAK (Grant Number: 2.100.2012.0038).
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Author contributions: Concept - M.K., Z.E.; Design - M.K., Z.E., Z.M.; Supervision - M.K., Z.E., H.K., N.M.; Funding - M.K., Z.E., H.Kemal., S.N., B.E.; Materials - O.V., H.Kemal., S.N., N.M., B.E.; Data collection &/or processing - Z.M., O.V., H.Kemal., B.E., H.O.; Analysis and/or interpretation - Z.M., M.K., H.Kemal., S.N., H.O.; Literature search - Z.M., O.V., H.Kemal., S.N.; Writing - Z.M., M.K., Z.E., H.K.; Critical review - M.K., H.K., N.M., H.O.
References
- 1.Rubin LJ. Pathology and pathophysiology of primary pulmonary hypertension. Am J Cardiol. 1995;75:51A–4A. doi: 10.1016/s0002-9149(99)80383-x. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13s–24s. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alan B, Nalbantgil S. Genetic, cellular and molecular mechanisms of pulmonary arterial hypertension. Anatol J Cardiol. 2010;10:9–13. doi: 10.5152/akd.2010.114. [DOI] [PubMed] [Google Scholar]
- 5.Sztrymf B, Francoual J, Sitbon O, Labrune P, Jambou M, Pous C, et al. Clinical, haemodynamic and genetic features of familial pulmonary hypertension. Rev Mal Respir. 2004;21:909–15. doi: 10.1016/s0761-8425(04)71472-2. [DOI] [PubMed] [Google Scholar]
- 6.Rubin LJ. BMPR2 mutation and outcome in pulmonary arterial hypertension - Clinical relevance to physicians and patients. Am J Resp Crit Care. 2008;177:1300–1. doi: 10.1164/rccm.200804-495ED. [DOI] [PubMed] [Google Scholar]
- 7.Kayıkçıoğlu M, Kültürsay H. Current approach to the treatment of pulmonary arterial hypertension and our experience in the Cardiology Department Of Medicine Faculty of Ege University. Turk Kardiyol Dern Ars. 2009;37:580–90. [PubMed] [Google Scholar]
- 8.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2009;30:2493–537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 9.Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M, et al. Genetics and Genomics of Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2013;62:D13–21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 10.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 11.Humbert M, Nunes H, Sitbon O, Parent F, Herve P, Simonneau G. Risk factors for pulmonary arterial hypertension. Clin Chest Med. 2001;22:459. doi: 10.1016/s0272-5231(05)70284-7. [DOI] [PubMed] [Google Scholar]
- 12.Treacy CM, Buasso DT, Doughty N, Morrell NW, Pepke-Zaba J. Survival of idiopathic pulmonary arterial hypertension Bmpr2 mutation carriers Vs Bmpr2 Non Carriers. Thorax. 2012;67:A122. [Google Scholar]
- 13.Liu D, Morrell NW. Genetics and the molecular pathogenesis of pulmonary arterial hypertension. Curr Hypertens Rep. 2013;15:632–7. doi: 10.1007/s11906-013-0393-9. [DOI] [PubMed] [Google Scholar]
- 14.Yang J, Li X, Wu C, Morrell N. The Crosstalk of Pde inhibitor with bmp signalling pathway in human pulmonary arterial smooth muscle cells. Thorax. 2011;66:A48. [Google Scholar]
- 15.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 16.Chida A, Shintani M, Yagi H, Fujiwara M, Kojima Y, Sato H, et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. 2012;110:586–93. doi: 10.1016/j.amjcard.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Raimondi A, Blanco I, Pomares X, Barbera JA. Pulmonary arterial hypertension in a patient with hereditary hemorrhagic telangiectasia. Arch Bronconeumol. 2013;49:119–21. doi: 10.1016/j.arbres.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 19.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–32. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 20.Pousada G, Baloira A, Vilarino C, Cifrian JM, Valverde D. Novel mutations in BMPR2, ACVRL1 and KCNA5 genes and hemodynamic parameters in patients with pulmonary arterial hypertension. Plos One. 2014;9:e100261. doi: 10.1371/journal.pone.0100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai J, Pardali E, Sanchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. Febs Lett. 2012;586:1993–2002. doi: 10.1016/j.febslet.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin VV, Genthner DE, Panella MM, Rich S. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. New Engl J Med. 1998;338:273–7. doi: 10.1056/NEJM199801293380501. [DOI] [PubMed] [Google Scholar]
- 23.Galie N, Ussia G, Passarelli P, Parlangeli R, Branzi A, Magnani B. Role of pharmacological tests in the treatment of primary pulmonary hypertension. Am J Cardiol. 1995;75:55A–62A. doi: 10.1016/s0002-9149(99)80384-1. [DOI] [PubMed] [Google Scholar]
- 24.Girerd B, Coulet F, Jaïs X, Eyries M, Van Der Bruggen C, De Man F, et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of BMPR2. Chest. 2015;147:1385–94. doi: 10.1378/chest.14-0880. [DOI] [PubMed] [Google Scholar]
- 25.Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, et al. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: potential for rescue. Hum Mol Genet. 2008;17:3180–90. doi: 10.1093/hmg/ddn214. [DOI] [PubMed] [Google Scholar]
- 26.Roberts KE, McElroy JJ, Wong WPK, Yen E, Widlitz A, Barst RJ, et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24:371–4. doi: 10.1183/09031936.04.00018604. [DOI] [PubMed] [Google Scholar]
- 27.Pfarr N, Fischer C, Ehlken N, Becker-Grunig T, Lopez-Gonzalez V, Gorenflo M, et al. Hemodynamic and genetic analysis in children with idiopathic, heritable, and congenital heart disease associated pulmonary arterial hypertension. Resp Res. 2013;14:3. doi: 10.1186/1465-9921-14-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Limsuwan A, Choubtum L, Wattanasirichaigoon D. 5 ’ UTR repeat polymorphisms of the BMPR2 gene in children with pulmonary hypertension associated with congenital heart disease. Heart Lung Circ. 2013;22:204–10. doi: 10.1016/j.hlc.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Best DH, Austin ED, Chung WK, Elliott CG. Genetics of pulmonary hypertension. Curr Opin Cardiol. 2014;29:520–7. doi: 10.1097/HCO.0000000000000105. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Li W, Zhang WL, Sun K, Song XD, Gao S, et al. Novel promoter and exon mutations of the BMPR2 gene in Chinese patients with pulmonary arterial hypertension. Eur J Hum Genet. 2009;17:1063–9. doi: 10.1038/ejhg.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]