

Abstract
Background
Inappropriately sustained inflammation is a hallmark of chronic ischemic heart failure (HF); however, the pathophysiological role of T-lymphocytes is unclear.
Methods and Results
Permanent coronary ligation was performed in adult C57BL/6 mice. As compared with sham-operated mice, mice with HF (8 w after ligation) exhibited the following features: 1) significant (p<0.05) expansion of circulating CD3+CD8+ cytotoxic and CD3+CD4+ helper (Th) T-lymphocytes, together with increased Th1, Th2, Th17, and regulatory T-cell (Treg) CD4+ subsets; 2) significant expansion of CD8+ and CD4+ T-cells in failing myocardium, with increased Th1, Th2, Th17, and Treg CD4+ subsets, marked reduction of the Th1/Th2 ratio, augmentation of the Th17/Treg ratio, and upregulation of Th2 cytokines; and 3) significantly increased Th1, Th2, Th17 cells, and Tregs, in the spleen and mediastinal lymph nodes, with increased expansion of splenic antigen-experienced effector and memory CD4+ T cells. Antibody-mediated CD4+ T-cell depletion in HF mice (starting 4 w after ligation) reduced cardiac infiltration of CD4+ T-cells and prevented progressive LV dilatation and hypertrophy whereas adoptive transfer of splenic CD4+ T-cells (and, to a lesser extent, cardiac CD3+ T-cells) from donor mice with HF induced long-term LV dysfunction, fibrosis, and hypertrophy in naïve recipient mice.
Conclusions
CD4+ T-lymphocytes are globally expanded and activated in chronic ischemic HF, with Th2 (vs Th1) and Th17 (vs Treg) predominance in failing hearts, and with expansion of memory T-cells in the spleen. Cardiac and splenic T-cells in HF are primed to induce cardiac injury and remodeling, and retain this memory upon adoptive transfer.
Keywords: Heart failure, cardiac remodeling, T-lymphocytes, adaptive immunity, inflammation
Journal Subject Codes: Heart Failure, Inflammation, Remodeling, Myocardial Infarction
Heart failure (HF) is a state of chronic inflammation, characterized by heightened levels of circulating and myocardial pro-inflammatory cytokines that promote pathological left ventricular (LV) remodeling.1, 2 While pre-clinical and early human studies suggested a therapeutic role for cytokine antagonism in HF,1 large-scale clinical trials of antibody-based cytokine neutralization in HF failed to show benefit.3–5 The failure of immunomodulation trials suggests an incomplete understanding of the nature of inflammation in HF. Specifically, as plasma cytokine levels are in large part determined by the combined influences of innate and adaptive immune system activation, they may be less sensitive indexes of underlying disease pathogenesis than the direct effects (both locally and systemically) of specific immune cell populations on the heart. In this regard, we have recently demonstrated robust expansion of monocytes/macrophages and dendritic cells (DCs) in chronic ischemic HF in the context of a pro-inflammatory cardiosplenic axis, with activated splenocytes homing to failing heart to induce tissue injury.6 These observations further suggested that directly targeting specific leukocyte populations might represent a more fruitful approach to immunomodulation in HF.
The primary function of DCs is to process and present antigens to T-cells, thereby inducing T-cell activation.7 Therefore, the presence of global DC expansion in HF6 implies concomitant global T-cell activation. Several T-cell populations, including CD4+ T-cells,8 Foxp3+ regulatory T-cells (Tregs),9 and invariant natural killer T (iNKT) cells10 have been shown to favorably regulate inflammation, immune cell trafficking, angiogenesis, and cardiac remodeling early after myocardial infarction (MI). CD4+ T-cells also promote macrophage recruitment and arteriogenesis in acute hindlimb ischemia models.11 However, whether and how T-cells impact the pathophysiology of chronic ischemic cardiomyopathy and HF, which exhibit substantially different profiles of inflammatory activation and tissue injury than acute MI, are unknown. Notably, in murine models of non-ischemic, pressure-overload HF, activated CD4+ T-cells have been shown to infiltrate the heart and play a crucial role in promoting cardiac fibrosis, hypertrophy, and remodeling.12, 13 In human HF secondary to either ischemic and non-ischemic etiologies, a low circulating total lymphocyte count is predictive of poor outcomes and prognosis;14 however, the impact of specific lymphocyte subpopulations, and their relationship to HF disease stage, are poorly understood.
CD4+ T cells are comprised of a heterogeneous family of adaptive immune cells with specialized functions.15, 16 These include pro-inflammatory T helper (Th)1 cells that produce interferon(IFN)-γ and interleukin(IL)-2,17 anti-inflammatory Th2 cells that produce IL-4, IL-5 and IL-13,18 pro-inflammatory Th17 cells that secrete IL-17,19 and immunomodulatory regulatory T-cells (Tregs) that globally suppress activation of immune responses.20 Given that ischemic HF is a chronic inflammatory state, it follows that alterations in the activation and spatiotemporal distribution of T-cell subsets during the development of HF may have considerable impact on the progression of pathological LV remodeling. Accordingly, we characterized temporal changes in circulating T-lymphocyte profiles during the development of ischemic HF, the status of various CD4+ T-cell subsets in the heart, mediastinal lymph nodes (LNs), and spleen in chronic HF 8 w after MI, and, using a combination of antibody-mediated depletion and cardiac and splenic T-cell adoptive transfer, delineated the pathophysiological role of T-cells in HF progression. Our results indicate that T-cells are globally expanded and activated in chronic ischemic HF, and are indispensable for the progression of pathological LV remodeling.
Methods
All animal studies were approved by University of Alabama at Birmingham Institutional Animal Care and Use Committee and were compliant with the NIH Guide for the care and Use of Laboratory Animals (DHHS publication No. 85-23, revised 1996). A total of 138 mice were used for all the studies.
Mouse model and surgical protocol
Male, 8–10 week-old C57BL/6 mice (Jackson Laboratories, stock #000664) underwent left thoracotomy and either sham surgery (n=28) or left coronary artery ligation (n=80) to induce MI and ischemic HF, as previously described.6, 21–24 Peripheral blood was collected serially (1, 2, 4, 6 and 8 w post-MI) from the facial vein, and all the mice were euthanized at 8 w post MI. Heart, spleen and heart-draining mediastinal LNs were collected at the time of euthanasia and mononuclear cells were isolated either for T-cell analysis by flow cytometry or for adoptive transfer studies (see below).
Echocardiography
Murine echocardiography was performed under 1–2% isoflurane anesthesia using a VisualSonics Vevo 770 High-Resolution System, a heated, bench-mounted adjustable rail system (Vevo Imaging Station), and RMV707B scanhead as previously described.6
Immune cell isolation
Immune cell isolation from peripheral blood, myocardium, spleen, and LNs was performed as described previously.6 Blood (~100 μL) was collected into EDTA containing tubes and lysed with RBC buffer (600 μL) for 10 min (eBioscience). After lysis was complete, excess buffer was neutralized by adding 10 mL of cold PBS and centrifuged at 380g. The cell pellet was reconstituted in 100 μL of staining buffer and labeled for flow cytometry as described below. The heart underwent collagenase digestion followed by filtration through 40 μm filters to remove extracellular connective tissue debris prior to centrifugation and reconstitution. Inflammatory cells from lymphoid tissue (spleen and LNs) were extracted as detailed previously.6
Flow cytometry
Cell pellets were resuspended in staining buffer and incubated with a cocktail of anti-mouse CD3-FITC/PE (BD Biosciences), CD4-Qdot650NC/PECY7/eVolve 605/PerCP-Cy5.5 (eBioscience), CD8-Qdot605NC/eVolve 655 (eBioscience) and NK1.1-PE/AF700 (eBioscience) (and in separate experiments CD44-PE from BD Biosciences and, CD62L eVolve 605 and CD69-AF700 from eBioscience) fluorochrome-conjugated antibodies for 1 h. After washing with PBS, the cells were fixed using 1% paraformaldehyde, permeabilized for 30 min with 0.5% v/v tween-20, and stained with fluorochrome-conjugated antibodies (eBioscience) targeting the intracellular cytokines IFN-γ, IL-4, and IL-17, and the transcription factor Foxp3. In some experiments, cells were fixed prior to staining with antibodies for cell surface markers. After 1 h, the cells were washed with PBS and data was acquired by BD FACSdiva LSR-II Flow cytometer (BD Biosciences). FlowJo software version 10.0.6 was used for data analysis. From a separate group of mice, 100 μL blood, 15–20 mg spleen, and one mediastinal LN were analyzed for cell counts, whereas whole tissue was digested for absolute counts of cardiac T-cells. AccuCount beads were used to adjust for differences in events recorded by the flow cytometer. The total live cell SSC/FSC gate was used to normalize cell populations in the spleen, mediastinal LNs, and heart whereas the total lymphocyte-monocyte gate was used in peripheral blood. The pan T-cell marker CD3 was used to identify all lymphocytes. Helper and cytotoxic T-cells were defined as CD3+CD4+ and CD3+CD4−CD8+, respectively. Among CD4+ T-cells, the presence of intracellular IFN-γ, IL-4, IL-17, and Foxp3 was used to identify Th1, Th2, Th17, and Treg subsets, respectively. Also, CD3+CD4+CD44hi T-cells were considered antigen-experienced.25 CD44hiCD69+ were further defined as activated effector T-cells, CD44hiCD69− as total memory T-cells, and CD44hiCD62Lhi as central memory T-cells.26, 27
Measurement of circulating cytokines
Blood was collected from the facial vein, clotted and centrifuged at 4000g (10 mins at 4°C), and serum was separated and stored at −80 °C for analysis. Simultaneous analysis of circulating T-lymphocyte-related cytokines (IFN-γ, TNF, IL-17, IL-6, IL-10 and IL-4) was performed using the mouse Th1/Th2/Th17 Cytometric Bead Array (CBA) kit (BD Biosciences) following the manufacturer’s protocol. Briefly, 50 μL of serum or standard was incubated with 50 μL of bead mixture and PE conjugated detection antibody at RT. After 2 h, excess PE-conjugated reagent was removed by washing with 1 mL of wash buffer, and samples were subsequently analyzed on a BD LSRII flow cytometer. FCAP Array software was used to measure mean fluorescence intensity (MFI) of each cytokine and serum concentrations were calculated from standard curves prepared simultaneously.
Cardiac and splenic gene expression by quantitative real time PCR
RNA extraction from LV tissue (encompassing remote and border zone myocardium), cDNA synthesis, and quantitative real-time PCR were performed as previously described.21–23 Methodology for determination of splenocyte gene expression has been previously detailed.6 Briefly, splenic mononuclear cells were isolated by layering on a Ficoll-Paque gradient and incubated in serum-free DMEM media overnight. Adherent cells were collected and stored at −80°C in TRIzol reagent (Invitrogen) for subsequent RNA extraction and measurement of mRNA transcript levels. Gene expression was determined for IL-2, IL-4, IL-5, IL-10, IL-13, IL-17, IL-18, IL-33, IL-12Rβ2, IFN-γ, transforming growth factor(TGF)-β, and C-C chemokine receptor type 5 (CCR5). The forward and reverse primer pairs used to determine gene transcript levels are provided in Supplemental Table 1. Gene expression was normalized to β-actin for LV tissue or 18s rRNA expression for splenocytes using the ΔΔCT comparative method, and expressed as fold-changes. Th1/pro-inflammatory mediators considered were IFN-γ (Th1 T-cell specific cytokine),17 IL-12Rβ2 and CCR5 (receptors induced during Th1 polarization),28 IL-18 (promotes Th1 polarization),29 IL-17 (produced by Th17 cells 19), and IL-2 (released by T-cells30). For anti-inflammatory markers, we measured IL-4, IL-5 and IL-13 (cytokines released by Th-2 cells18), TGFβ and IL-10 (anti-inflammatory cytokines released by T-regs and Th-2 T-cells31) and IL-33 (inducer of Th-2 related cytokines32).
Histological analysis
Formalin-fixed, paraffin-embedded hearts from sham and HF mice were sectioned at 5 μm thickness, deparaffinized, and rehydrated. Histological staining was performed as previously described.6, 21, 23 Masson’s trichrome was used to evaluate tissue fibrosis and Alexa Fluor 488–conjugated wheat germ agglutinin (Invitrogen) to assess myocyte area, as quantified from 5 to 6 high-power fields per section in non-infarcted myocardium using Metamorph software version 6.3r5 (Molecular Devices).
To evaluate for tissue abundance of CD4+ and CD8+ T-cells, heart sections were embedded in OCT compound (Tissue-Tek OCT), and then kept at −80°C until sectioning. Sections (7 μm thickness) were fixed with 4% paraformaldehyde in PBS, and labeled with either rat anti-mouse CD4 (Clone GK1.5; eBiosciences) or CD8 antibody (Clone 4SM15; eBiosciences). Goat anti-rat antibody conjugated with Alex Fluor 555 and 488 (Life Technologies) were used as secondary antibodies for CD4 and CD8, respectively, and fluoroshield-containing DAPI (Sigma-Aldrich) was used as the mounting medium. CD4+ or CD8+ cells were quantified using confocal microscopy from 5–6 sections and 3–4 mice from each group. All measurements were conducted in a blinded manner. Confocal microscopy was performed on an LSM710 microscope (Zeiss).
Antibody-mediated in vivo CD4+ T-cell ablation
Four weeks after coronary ligation, C57BL/6 mice with comparable degrees of post-MI LV dilatation and dysfunction (as assessed by echocardiography) were randomized to receive either: 1) purified anti-CD4 antibody (100 μg/mouse i.p., GK 1.5 clone, eBioscience) for a total of two doses administered 10 d apart (n=10), or 2) IgG isotype control antibody at the same doses (n=6). Echocardiography and blood collection were performed prior to and 4 w after initiation of treatment (i.e., 4 w and 8 w post-MI) for serial evaluation of LV structure/function and circulating T-cells. The mice were then euthanized 8 w post-MI, with subsequent gravimetric and tissue analyses as described above.
Adoptive transfer studies
CD3+ T-cells from the heart and CD4+ T-cells from the spleen were harvested from naïve control (n=6) and HF mice (n=6; 8 w post-MI) and used for adoptive transfer into naïve recipient mice as described below.
Splenic CD4+ T-cells
Mononuclear splenocytes from naïve control and HF mice were isolated as described previously.6 Splenic CD4+ T-cells were separated and collected using the mouse CD4+ T-cell isolation kit (Miltenyi) following the manufacturer’s protocol. Cell recovery was 12 X 106 cells per spleen, with 85–90% purity of the CD4+ T-cell population by flow cytometry and 90–95% cell viability by trypan blue exclusion.
Cardiac CD3+ T-cells
Naïve control and HF hearts were minced and digested with collagenase, and cardiac mononuclear cells were isolated as described previously.6 Cells were labeled with CD3-FITC (BD Biosciences) for 30 min at 4°C. Viable CD3+ T-cells were identified by exclusion of the fluorochrome 7-amino actinomycin D (7-AAD) and then sorted by flow cytometry.
Due to the labor-intensive experimental setup, tissues from 3 animals were processed per day. Cells isolated from either 3 HF or control mice each day were pooled, divided into equal aliquots, and adoptively transferred i.v. to syngeneic naïve C57BL/6 recipient mice. The inter-day variability in the number of transferred splenic CD4+ T-cells was negligible, ranging from 11 to 13 X 106 cells. However, the number of cardiac CD3+ T-cells adoptively transferred to recipients varied on different days due to inconstant yield from control and failing hearts. The first group (n=3) of naïve recipients administered failing cardiac T-cells were given ~7,300 CD3+ cells i.v., whereas the second group (n=3) received ~48,100. Naïve recipients administered control cardiac T-cells were given between 15,400 and 18,200 CD3+cells (n=6 total). Recipient mice were followed by serial echocardiography performed at baseline (day 0) and 4 and 8 w after cell transfer. After 8 w, mice were euthanized and tissue harvested for gravimetric and histological analysis.
Statistical analysis
All quantitative analyses are mean ± SD. For statistical analyses, the variance of data sets was first assessed using the F-statistic, and the D’Agostino-Pearson test was used to assess normality. Statistical comparisons between HF and sham/control groups were then performed using the unpaired Student’s t-test with equal or unequal variance, for normally distributed data. For non-normally distributed data (observed with quantitative data of absolute T-cell number in the myocardium), logarithmic transformation was first performed to satisfy the assumption of normality, followed by analysis using the unpaired Student’s t-test with equal or unequal variance. Repeated measures ANOVA and Bonferroni post-test were used to analyze the adoptive transfer studies. GraphPad Prism version 7.0a was used for all analyses. A p value of < 0.05 was considered significant.
Results
Profound structural LV remodeling in chronic ischemic HF
Cardiac structural and functional parameters for control and HF mice 8 w after sham surgery or coronary ligation are shown in Figure S1. Representative gross heart specimens and M-mode echocardiograms of sham and failing hearts demonstrated cardiac enlargement, LV dilatation, and LV systolic dysfunction in HF mice. As seen in the group data (Figure S1A), HF mice exhibited markedly increased LV end-diastolic and end-systolic volume (EDV and ESV), severely reduced LV ejection fraction (EF), and significantly increased heart and wet lung weights consistent with cardiac hypertrophy and pulmonary congestion (Figure S1B). Increased spleen size and weight, along with increased total counts of splenic CD45+ leukocytes and CD3+ T-cells, were also evident in HF mice (Figure S1C), consistent with splenic remodeling occurring in HF, as demonstrated in our prior study.6 Hence, there was substantial pathological LV remodeling 8 w post-MI and evidence of chronic HF.
CD4+ T-lymphocytes are globally and progressively expanded in HF
CD3+CD4+ (helper) and CD3+CD4−CD8+ (cytotoxic) T-lymphocytes were serially measured in peripheral blood after ligation or sham surgery. As evident from Figures 1A–1B and S2A, both sham and HF mice exhibited time-dependent reduction in the frequency of circulating CD4+ and CD8+ T-cells after operation, approximating levels in non-surgical control mice by 2 w, a response likely associated with the surgical procedure. However, in ligated mice, blood CD4+ T-cell frequency was significantly higher than sham at nearly all time points after MI (only the 2 w post-MI interval did not reach significance, p = 0.064), and measured absolute circulating CD4+ cell count was elevated at both 1 w and 8 w after MI (Figure 1B). Similarly, the frequency of CD8+ T-cells was significantly increased over sham mice both early (1 w) post-MI and in established HF (≥ 4 w after MI), as was the absolute circulating CD8+ T-cell count 8 w after MI (Figure S2A).
Figure 1.


(A) Example flow cytometry lymphocyte-monocyte (lymph-mono) SSC/FSC live cell gate and scatter plots for peripheral blood CD3+CD4+ helper and CD3+CD4+Foxp3+ regulatory T-lymphocytes (Tregs) in control and heart failure (HF) mice 8 w after sham-operation or coronary ligation and myocardial infarction (MI). (B) Corresponding group data as %lymphocyte-monocyte gate at serial time points, as well as absolute cell counts (per μL blood) at 1 and 8 w, after operation. (C, D) Representative flow cytometry dot plots and group quantitation of cell frequency (C) and total cell counts (D) for CD4+ Th subsets - Th1 (IFN-γ+), Th2 (IL-4+), and Th17 (IL-17+) - in the same experimental groups. (E) Serum cytokines in mice 8 w after MI (HF) or sham surgery. N.S. Ctrl, non-surgical control.
Circulating CD4+ T-cell subsets were characterized as Th1 (CD3+CD4+NK1.1−IFN-γ+), Th2 (CD3+CD4+NK1.1−IL-4+), Th17 (CD3+CD4+NK1.1−IL-17+), and Tregs (CD3+CD4+Foxp3+). Since iNKT cells also produce both IFN-γ and IL-4,33 only NK1.1− cells were considered for Th1, Th2 and Th17 cell characterization. Similar to total CD4+ T-cells, the frequency of circulating Th1, Th2, and Th17 cells was increased in HF versus sham mice at all time points after MI (Figure 1C). Furthermore, absolute Th1, Th2, and Th17 cell counts were all significantly increased in HF mice as compared with sham at 8 w post-MI (Figure 1D). Th1 and Th2 cell counts were also elevated early (1w) post-MI. In contrast, the profile of circulating Tregs, an immunomodulatory CD4+ T-cell subset, was biphasic in HF mice, with no change in either frequency or absolute counts early (1 w) post-MI, and then augmented levels onward from 2 w post-MI, including increased total counts at 8 w as compared with sham mice. Consistent with the cell data, mice with HF (8 w post-MI) exhibited elevated serum levels of cytokines that induce either Th1 cells (IFN-γ, TNF), Th2 cells (IL-6, IL-10) or Tregs (IL-10)34, 35 as compared with sham, but without observed differences in serum IL-17 and IL-4 (Figure 1E).
CD4+ T-lymphocytes are augmented in the failing heart
Cardiac CD4 and CD8 immunostaining was performed in hearts harvested 8 w after coronary ligation or sham operation (Figures 2 and S2B). Both CD4+ and CD8+ T-cells were readily observed in failing hearts, at nearly 3–5 fold higher levels than in sham hearts (p = 0.018 and 0.022, respectively). Cardiac T-cell subpopulations were quantified by flow cytometry. As depicted in Figure 3A, >95% of cardiac mononuclear cells were viable and excluded the membrane impermeant fluorochrome 7-AAD. Scatter plots for CD4, CD8, and Foxp3 stained CD3+ T-cells indicated significantly augmented populations of CD4+ helper, CD8+ cytotoxic, and Foxp3+ regulatory T-cells in the failing heart (Figures 3B and S2C). Of the Th subsets, cardiac Th1, Th2, and Th17 populations were all robustly increased in HF as compared with sham (Figure 3C). The cardiac Th1/Th2 ratio was decreased in HF, indicating a Th2 predominant milieu in failing myocardium, and was accompanied by an augmented Th17/Treg ratio (Figure 3D). This cell profile was accompanied by diffuse mRNA upregulation of multiple Th2 (and Treg) polarizing inflammatory mediators in border and remote zone failing myocardium (IL-4, IL-13, IL-5, IL-33, TGF-β, and IL-10) but only select overexpression of Th1 and Th17 polarizing mediators as compared with sham (IL-18 and IL-17; no difference in IFN-γ, IL12Rβ2, CCR5, and IL-2) (Figure 3E).
Figure 2.
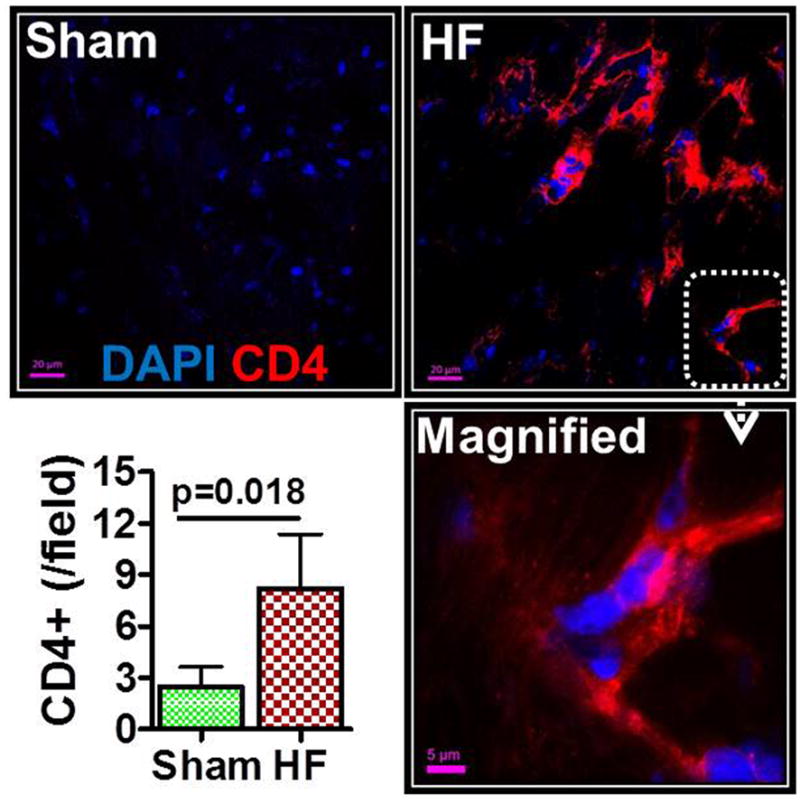
Representative confocal images of CD4 immunostained hearts from sham-operated and HF mice (8 w post-MI). CD4 positivity is indicated by red fluorescence; DAPI-labeled nuclei are blue. A magnified image is shown in the bottom right, and group quantitation of cardiac CD4+ cell abundance in the lower left.
Figure 3.

(A) Representative SSC/FSC live cell gate for cardiac mononuclear cells from a failing mouse heart and flow cytometry scatter plots after 7-AAD viability staining (live cells exclude 7-AAD). (B) Example scatter plots, and corresponding group data, for CD4+ helper and CD4+Foxp3+ regulatory T-cell counts in sham and HF hearts. (C) Representative density plots and quantitative group data for Th1, Th17, and Th2 CD4+ T-cell subsets, and (D) Th1/Th2 and Th17/Treg ratios in sham and failing hearts (n = 6–10 per group). (E) Th1- and Th2-related gene expression in border/remote zone myocardium from sham and HF mice (n = 5–6 per group). Fold changes are depicted using β-Actin as the endogenous control.
CD4+ T-lymphocytes are broadly activated in the HF spleen and mediastinal LNs
In HF mice, the hypertrophied spleen exhibited expansion of both CD4+ and CD8+ T-cells as compared with sham (Figures 4A and Figure S2D), and significant elevation of all CD4+ T-cell subsets examined - Th1, Th2, Th17, and Tregs (Figure 4A–4B). Splenocyte gene expression in established HF (8 w post-MI) exhibited upregulation of both Th1 polarizing mediators (IFN-γ, IL12Rβ2, CCR5, IL-18, IL-17, IL-2) and Th2 (and Treg) polarizing mediators (IL-4, IL-5, IL-10; no change in IL-13, IL-33, TGF-β) (Figure 4E). Splenic antigen-experienced CD4+ T-cells, defined by high CD44 expression25, were also expanded significantly in HF mice (Figure 4D). This expansion was comprised of increased levels of both CD44hiCD69+ activated effector T-cells and CD44hiCD69− total memory CD4+ T-cells27 (Figure 4E). Splenic central memory T-cells27 as identified by CD44hiCD62Lhi expression were also significantly elevated in HF mice as compared with sham (Figure 4F). Like the spleen, mediastinal LNs in HF mice were enlarged as compared with sham mice, with similar increases in cell frequencies and counts of CD4+ T-cells and the Th1, Th2, Th17, and Treg subsets, consistent with immune activation (Figure S3). The total cell number of CD8+ T-cells was also increased significantly in HF mediastinal LNs (Figure S2E), although the cell frequency (% SSC/FSC) was comparable to LNs from sham mice.
Figure 4.


(A) Example SSC/FSC live cell gate and scatter plots for CD3+CD4+ and CD3+CD4+Foxp3+ Tregs in mononuclear splenocytes isolated from sham and HF mice, and quantitative group data for the same, cell frequency previously published in Ismahil et al.6 (B) Flow cytometry scatter plots and frequency and cell count group data for CD4+ T-cell subsets in the spleen. (C) Th1 and Th2 related gene expression in splenocytes harvested from sham and HF mice (n=5 per group). Fold changes are shown using 18S rRNA as the endogenous control. (D) Representative flow histograms for splenic CD3+CD4+CD44+ T-cells (Left), scatter plots for identification of antigen-experienced CD44hi cells in sham-operated and HF mice (Middle), and corresponding quantitative group data (Right). (E) Flow cytometry scatter plots and group data for CD4+CD44hiCD69+ activated (effector) T-cells and CD4+CD44hiCD69− total memory T-cells, and (F) CD4+CD44hiCD62Lhi central memory T-cells in splenocytes from sham-operated and HF mice (n = 5–6/group).
CD4+ T-cells are obligatory for pathological LV remodeling in chronic HF
At 4 w post-MI, HF mice with equivalent degrees of LV dilatation and dysfunction (Figure S4) were randomly assigned to receive either anti-CD4 antibody or isotype IgG control antibody, and LV remodeling and T-cell subsets were assessed at 8 w (Figure 5A). Anti-CD4 treatment selectively depleted CD4+ T-cells in the blood, spleen, mediastinal LNs, and heart, without impacting circulating CD8+ T-cells (Figure 5B). Moreover, CD4+ T-cell depletion reduced LV and splenic remodeling, as evidenced by lower normalized heart, LV, and spleen weight as compared with control antibody (Figure 5C). Additionally, echocardiography revealed progressive LV chamber dilatation in HF mice treated with control antibody that was prevented by the administration of anti-CD4 antibody (Figure 5D), without significant effects on LV ejection fraction. Hence, CD4+ T-cells are necessary for the progression of pathological structural remodeling.
Figure 5.

(A) Protocol used for antibody-mediated CD4+ T-cell depletion in HF mice; PBC, peripheral blood cells. (B) Quantitative group data for CD4+ T-cells in blood, spleen, mediastinal LNs and hearts, and blood CD8+ T-cells, in HF mice treated with either anti-CD4 antibody or isotype control. (C) normalized heart, LV and spleen weight in HF mice treated as in B. (D) Representative end-diastolic echocardiographic images from HF mice before (4 w post-MI) and after (8 w post-MI) treatment with either isotype or anti-CD4 antibody, and quantitative group data for changes in LV end-diastolic and end-systolic volume (EDV and ESV) and ejection fraction (EF) (n = 6–10/group).
CD4+ and CD3+ T-cells are sufficient to induce cardiac remodeling
Splenic CD4+ T-cells and cardiac CD3+ T-cells isolated from HF mice (8 w post-MI) or naïve control mice were adoptively transferred (separately) into naïve recipient mice (Figure 6A). Splenic CD4+ T-cells were selected based on our prior work6 that demonstrated significant cardiac dysfunction in recipient mice after adoptive transfer of HF-activated splenocytes. Cardiac CD3+ T-cells were also tested as a T-cell population presumably enriched for activation against cardiac antigens. Recipient mice were followed for 8 w, with serial echocardiographic assessment of LV function every 4 w. As illustrated in Figure 6B, transfer of HF-activated splenic CD4+ T-cells induced significant LV systolic dysfunction (increased ESV and reduced EF) and LV hypertrophy (increased LV wall thickness) over this time, but without chamber dilatation (no change in EDV), as compared with mice that received control splenic CD4+ T-cells. Gravimetric analysis 8 w after cell transfer revealed a significant increase in normalized heart weight (Figure 6C), and histological analysis indicated a mild but significant augmentation of interstitial fibrosis and robust myocyte hypertrophy (Figure 6D) in mice receiving HF-activated (versus naïve) splenic CD4+ T-cells. Similar effects on LV function and interstitial fibrosis, but with less pronounced effects on cardiac hypertrophy, were observed upon adoptive transfer of HF-activated cardiac CD3+ T-cells into naïve recipients (Figure S5). The milder responses engendered by cardiac CD3+ T-cell transfer may be either a consequence of fewer and greater variability of transferred T-cells in this group (see Methods) or heterogeneity in the transferred T-cell subset pool between cardiac and splenic cells (i.e., cardiac cells included CD8+ cells, and were more enriched for Th2 and Th17 T-cells as compared with splenic T-cells). Collectively, however, these results indicate that T-cells in ischemic HF are primed to induce myocardial injury, and contribute importantly to progressive LV remodeling.
Figure 6.

(A) Protocol for adoptive transfer of splenic CD4+ and cardiac CD3+ T-cells from naïve control and HF mice to normal recipients. (B) Changes in LV ESV, EDV, and EF in recipient mice 4 and 8 w after splenic CD4+ T-cell transfer, along with measured septal and posterior wall (PW) thickness at 8 w. (C) Gross images of hearts isolated from mice 8 w after receiving either control or HF-activated splenic CD4+ T-cells and normalized gravimetric data for the tissues indicated; RV, right ventricle. (D) Representative Masson’s trichrome stains and quantitation of fibrosis (Top), and wheat germ-agglutinin stains (cell membranes are green, nuclei are blue with DAPI counterstaining; scale bar 200 μm) and quantitative data for myocyte size (Bottom), from mouse hearts 8 w after transfer of either naïve control or HF-activated splenic CD4+ T-cells. Arrows indicate example myocytes used for determination of cross-sectional area.
Discussion
There are several key and novel findings in this study. First, in chronic ischemic cardiomyopathy, there is systemic expansion of CD4+ and CD8+ T-cells, and CD4+ Th1, Th2, Th17, and Treg subsets, which encompasses the failing heart, circulation, and lymphoid organs. Second, there is temporal divergence in the behavior of Th subsets during the progression of ischemic HF, as well as organ-localized differences in Th cell distribution in established disease, with the failing myocardium exhibiting Th2 (when compared with Th1) and Th17 (when compared with Tregs) predominance (see below). Third, we have established that activated CD4+ T-cells are necessary for disease progression in ischemic HF, as CD4+ T-cell ablation alleviated pathological LV remodeling. Fourth, we have demonstrated that antigen-experienced splenic effector and memory T-cells are expanded in HF. Lastly, via adoptive transfer, we have provided direct evidence that both splenic CD4+ and cardiac CD3+ T-cells in HF are biologically active and primed for myocardial injury, and sufficient for the production of pathological remodeling. Taken together, we conclude that systemic CD4+ T-cell activation, presumably directed toward cardiac antigens, plays an indispensable role in the progression of adverse cardiac remodeling in chronic ischemic HF.
Chronic ischemic HF is a systemic CD4+ T-cell activated state
Multiple immune cell types infiltrate the acutely infarcted heart in a phasic manner during early post-MI remodeling.36–40 Following an early wave of neutrophils, infiltrating pro-inflammatory (Ly-6Chi) monocytes promote tissue digestion early (1–4 d) after MI and subsequently differentiate into reparative (Ly-6Clow) macrophages that resolve inflammation and promote fibrosis over several days. In concert with DCs,41 CD4+ T-cells,8 including Foxp3+ Tregs,9 infiltrate the heart and promote wound healing after MI by favorably influencing monocyte/macrophage trafficking and differentiation, and by facilitating collagen matrix formation and angiogenesis. Indeed, effective myocardial repair and remodeling after ischemic injury are critically linked to tissue-level responses mediated by both innate and adaptive immune cells.39 Importantly, early post-MI remodeling is ultimately marked by inflammation resolution and quiescence; consequently, the aforementioned studies provide little insight as to how T-cells might impact ischemic cardiomyopathy and HF, a disease state with a substantially different inflammatory profile.
We recently demonstrated that ischemic cardiomyopathy (well after completion of early post-MI remodeling) is characterized by sustained expansion of pro-inflammatory macrophages and DCs in the heart, together with splenic remodeling consistent with heightened antigen processing.6 Moreover, activated mononuclear splenocytes trafficked to the failing heart to promote apoptosis, fibrosis, and dysfunction, suggesting that adverse LV remodeling was in part immune–mediated. While T-cells were not rigorously examined, given that a primary function of DCs is the presentation of antigens to T-cells7 robust DC expansion intimated that T-cell activation, and perhaps T-cell mediated tissue injury in the heart, are also central to pathological remodeling. In this regard, recent studies of murine non-ischemic (pressure overload-induced) HF have indicated that heart-infiltrating, activated CD3+CD4+ T-cells impart detrimental effects on LV remodeling.12, 13 However, the role of CD4+ T-cells, and the behavior of various CD4+ T-cell subsets, in chronic ischemic HF, in which the heart exhibits much more robust initial cell death, has heretofore been poorly understood.
Our data establish (to our knowledge for the first time) that in chronic ischemic HF, there is robust T-cell expansion of both CD4+ and CD8+ T-cells, and CD4+ Th1, Th2, Th17, and Tregs, both within the failing myocardium and globally in the lymphoid organs and circulation. Interestingly, while all CD4+ T-cell subsets were expanded in the failing heart, the Th1/Th2 ratio was significantly diminished (classically anti-inflammatory) whereas the Th17/Treg ratio was increased (classically pro-inflammatory). Moreover, HF was characterized by robust pro-inflammatory gene expression in spleen, and augmented levels of splenic CD4+CD44hiCD69+ antigen-exposed effector T-cells,25, 27 CD4+CD44hiCD69− total memory T-cells27 and CD4+CD44hi CD62Lhi central memory T-cells, consistent with T-cell activation presumably in response to as-of-yet poorly defined self-antigens during MI or post-MI remodeling, with potential for re-activation of heart-directed adaptive immunity upon repeated exposures. These findings indicate that HF is not easily categorized as a “Th1” or “Th2” inflammatory state, but rather that T-cell alterations are spatiotemporally complex in the context of disease. This notion is in line with the understanding that CD4+ T-cells are comprised of a heterogeneous family of pro- and anti-inflammatory T-cells with diversely varying properties.15 In the failing heart, the reduced Th1/Th2 cell ratio suggests that expanded cardiac Th2 cells may induce local damage by secreting Th2 cytokines, which are known fibrogenic factors.42 In this regard, studies in human HF have demonstrated that urinary levels of IL-4 correlate with structural LV remodeling.43 Also, analogous to the failing heart in our study, the aging rodent heart exhibits a Th2-type phenotype, with augmented IL-4 and IL-13 levels that enhance collagen synthesis and interstitial fibrosis.44 Hence, Th2 T-cells may represent a therapeutic target in chronic ischemic HF to reduce tissue fibrosis and inflammation.
Our assessment of the T-cell network during the development of ischemic HF highlights two additional caveats. Tregs, immunomodulatory T-cells that suppress effector T-cell responses,45 exhibited a biphasic profile in the circulation, initially decreased in frequency early (1 w) after MI (without change in absolute cell counts), with subsequently elevated levels thereafter as compared with sham mice. While we did not specifically examine the role of Tregs in HF, in light of their potent immunoregulatory features, Treg modulation may represent a viable approach to indirectly modify CD4+ T-cell behavior and chronic inflammation. Moreover, we observed time dependent alterations in circulating CD4+ T-cells and helper T-cell subsets in sham-operated mice, as compared with non-surgical control mice, which generally approximated normal levels after ~2–4 w. Presumably these changes represent a generalized response to thoracotomy and subsequent post-operative healing (and not MI per se), and should be considered in the design of future experimental studies.
HF-activated CD4+ T-cells are both necessary and sufficient for pathological LV remodeling
Our findings of global CD4+ T-cell activation in murine ischemic HF are consistent with the prior demonstration of increased circulating CD4+ T-cells and CD4/CD8 T-cell ratios in human HF.46, 47 To evaluate the pathophysiological role of T-cell expansion, we first evaluated whether CD4+ T-cells are required for LV remodeling. Anti-CD4 antibody administration in established HF halted progressive LV dilatation (without impacting systolic function), and improved chamber hypertrophy and lung edema, indicating that activated CD4+ T-cells are obligatory for the progression of structural disease. Notably, early after acute MI, Hoffman et al8 have shown that CD4+ T-cells are beneficial for infarct wound healing and tissue repair. In contrast, our results show that in chronic ischemic HF, CD4+ T-cells impart pro-inflammatory and detrimental effects on the heart that promote adverse remodeling. Interestingly, antibody-mediated CD3+ T-cell depletion13 and genetic CD4+ (but not CD8+) T-cell disruption12 in the setting of pressure-overload prevented the long-term development of systolic dysfunction and fibrosis but did not impact hypertrophy, underscoring important differences regarding T-cell-mediated remodeling responses in ischemic vis-à-vis non-ischemic HF.
Lastly, to evaluate whether HF-activated T-cells are sufficient to drive tissue remodeling, we defined the effects of adoptive transfer of splenic CD4+ T-cells, and cardiac CD3+ T-cells, isolated from HF mice. Interestingly, the transfer of HF-activated T-cells recapitulated cardiac injury in naïve recipients over an extended follow-up period, with LV systolic dysfunction, chamber and/or myocyte hypertrophy, and interstitial fibrosis. Moreover, splenocytes exhibited marked upregulation of a broad array of pro-inflammatory mediators. These results are consistent with our prior work demonstrating a robust tissue-injurious cardiosplenic axis in ischemic HF,6 and also suggest that heart-localized T-cells are responsible for detrimental tissue responses. Importantly, the cardiac phenotype induced in recipient mice varied depending on splenic CD4+ versus cardiac CD3+ T-cell transfer. As alluded to above, this heterogeneity in response may be related to differences in the overall number of T-cells transferred, as well as disparities in the composition of T-cell subsets within transferred cardiac CD3+ T-cells and splenic CD4+ T-cells. Nonetheless, despite these limitations, these studies provide important proof-of-concept for T-cells as disease mediators in HF.
The spleen and LNs play several important roles in antigen-specific T-cell immunity, including naïve T-lymphocyte recruitment, the screening of antigens and antigen loaded DCs in blood and lymph delivered from inflamed sites, the provision of a suitable microenvironment for inducing effector responses (or antigen-specific tolerance) and homing characteristics, and fostering a niche for memory T-cells.48, 49 The transferability of cardiac-injurious effects of splenic CD4+ T-cells from HF mice to naïve mice suggests that T-cells in HF are activated against cardiac antigens, and retain memory for the induction of effector responses upon transfer. As both donor and recipient mice carried the CD45.2 allele, our data do not establish whether this is a direct effect of the transferred cells, or an indirect effect of secondary activation of immune cells in recipient mice. Also, the nature of the specific antigens responsible for T-cell activation remain undefined and need to be explored in future investigations. Nonetheless, considered together with our prior work,6 we have established that robust activation of both innate and adaptive immunity, probably in response to as-of-yet poorly defined self-antigens or alarmins released during cardiac injury, are central features and drivers of chronic inflammation in ischemic HF. Moreover, from a translational standpoint, our data suggests that targeting specific immune cell subsets at defined stages of disease may represent a better approach to therapeutic immunomodulation to improve HF.
In summary, chronic ischemic HF in mice is characterized by local and systemic adaptive immune activation of CD4+ and CD8+ T-cells, and CD4+ T-cell subsets, including Th1, Th2, Th17, and Treg cells. Moreover, failing hearts exhibit Th2 and Th17 cell predominance when compared with Th1 cells and Tregs, respectively, and the spleen exhibits broad expansion of antigen-experienced T-cells, including effector and memory T-cells. Most importantly, activated CD4+ T-cells are obligatory for disease progression in ischemic HF, and sufficiently activated in HF to induce cardiac tissue injury upon adoptive transfer. Hence, immunomodulation of CD4+ T-cells may represent a novel therapeutic strategy for chronic ischemic HF.
Supplementary Material
Clinical Impact.
Although heightened inflammation is a hallmark of heart failure (HF), therapeutic immunomodulation remains an unrealized goal. In particular, clinical trials of pro-inflammatory cytokine neutralization failed to show benefit. Classically, cytokine levels are influenced by innate and adaptive immune cells; however, their pathophysiological roles in HF are poorly understood. In this study, we demonstrate a central role for T-cells in the progression of cardiac remodeling after myocardial infarction in mice. Specifically, we found that both CD4+ helper (Th) and CD8+ cytotoxic T-cells are expanded in the blood, spleen, and mediastinal lymph nodes, and also accumulate in the failing heart. Moreover, both pro- and anti-inflammatory CD4+ T-cell subsets, including Th1, Th2, Th17 and regulatory T-cells, were increased in the heart, together with expansion of splenic antigen-experienced effector and memory CD4+ T cells. Most importantly, antibody depletion and adoptive transfer studies revealed that CD4+ T-cells are obligatory for disease progression, and sufficiently activated in HF to induce cardiac tissue injury. Therefore, we propose that ischemic cardiomyopathy should be thought of in part as an immune-mediated disease, as CD4+ T-cells, presumably activated against as-of-yet unidentified cardiac antigens, and are indispensable for long-term adverse remodeling. On the basis of these results, we further propose that circumscribed inhibition of CD4+ T-cells may represent a more fruitful therapeutic strategy in chronic ischemic HF. Our findings should stimulate an analogous evaluation of T-cell profiles in different stages of human ischemic cardiomyopathy, and the conceptual consideration of CD4+ T-cell immunomodulation as a viable approach to alleviate pathological cardiac remodeling.
Acknowledgments
Funding Sources: This work was supported by an American Heart Association Post-Doctoral Fellowship Grant 14POST20490323 (to S.S.B.), a VA Merit Award I01BX002706 (to S.D.P.), and National Institutes of Health grants 1K99HL132123 (to S.S.B.) and 1R01HL125735 (to S.D.P.).
The authors gratefully acknowledge the excellent technical assistance provided by Mr. Hai Zhong for mouse surgery and Mr. Yixin Wu for mouse echocardiography.
Footnotes
Disclosures: None.
References
- 1.Mann DL. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ Res. 2002;91:988–998. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 2.Prabhu SD, Chandrasekar B, Murray DR, Freeman GL. Beta-adrenergic blockade in developing heart failure: Effects on myocardial inflammatory cytokines, nitric oxide, and remodeling. Circulation. 2000;101:2103–2109. doi: 10.1161/01.cir.101.17.2103. [DOI] [PubMed] [Google Scholar]
- 3.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: Results of the anti-TNF therapy against congestive heart failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 4.Mann DL. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ Res. 2015;116:1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (renewal) Circulation. 2004;109:1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 6.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kushwah R, Hu J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology. 2011;133:409–419. doi: 10.1111/j.1365-2567.2011.03457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 9.Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. doi: 10.1161/CIRCRESAHA.115.303895. [DOI] [PubMed] [Google Scholar]
- 10.Sobirin MA, Kinugawa S, Takahashi M, Fukushima A, Homma T, Ono T, Hirabayashi K, Suga T, Azalia P, Takada S, Taniguchi M, Nakayama T, Ishimori N, Iwabuchi K, Tsutsui H. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ Res. 2012;111:1037–1047. doi: 10.1161/CIRCRESAHA.112.270132. [DOI] [PubMed] [Google Scholar]
- 11.Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, la Sala A, Miller JM, Shou M, Epstein SE, Fuchs S. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation. 2003;108:205–210. doi: 10.1161/01.CIR.0000079225.50817.71. [DOI] [PubMed] [Google Scholar]
- 12.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124. doi: 10.1161/CIRCULATIONAHA.113.007101. [DOI] [PubMed] [Google Scholar]
- 13.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. 2015;8:776–787. doi: 10.1161/CIRCHEARTFAILURE.115.002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy WC, Mozaffarian D, Linker DT, Sutradhar SC, Anker SD, Cropp AB, Anand I, Maggioni A, Burton P, Sullivan MD, Pitt B, Poole-Wilson PA, Mann DL, Packer M. The seattle heart failure model: Prediction of survival in heart failure. Circulation. 2006;113:1424–1433. doi: 10.1161/CIRCULATIONAHA.105.584102. [DOI] [PubMed] [Google Scholar]
- 15.Dong C. Helper t-cell heterogeneity: A complex developmental issue in the immune system. Cell Mol Immunol. 2010;7:163. doi: 10.1038/cmi.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, De Simone M, Pagani M, Abrignani S. Plasticity of human CD4 T cell subsets. Front Immunol. 2014;5:630. doi: 10.3389/fimmu.2014.00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Q, Ghilardi N, Wang H, Baker T, Xie M-H, Gurney A, Grewal IS, de Sauvage FJ. Development of Th1-type immune responses requires the type I cytokine receptor TCCR. Nature. 2000;407:916–920. doi: 10.1038/35038103. [DOI] [PubMed] [Google Scholar]
- 18.Paul WE, Zhu J. How are T(h)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 20.Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: Markers, mechanisms, and manipulation. FASEB J. 2012;26:2253–2276. doi: 10.1096/fj.11-193672. [DOI] [PubMed] [Google Scholar]
- 21.Hamid T, Gu Y, Ortines RV, Bhattacharya C, Wang G, Xuan YT, Prabhu SD. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: Role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119:1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Hamid T, Keith RJ, Zhou G, Partridge CR, Xiang X, Kingery JR, Lewis RK, Li Q, Rokosh DG, Ford R, Spinale FG, Riggs DW, Srivastava S, Bhatnagar A, Bolli R, Prabhu SD. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation. 2010;121:1912–1925. doi: 10.1161/CIRCULATIONAHA.109.905471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wysoczynski M, Solanki M, Borkowska S, van Hoose P, Brittian KR, Prabhu SD, Ratajczak MZ, Rokosh G. Complement component 3 is necessary to preserve myocardium and myocardial function in chronic myocardial infarction. Stem Cells. 2014;32:2502–2515. doi: 10.1002/stem.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baaten BJ, Tinoco R, Chen AT, Bradley LM. Regulation of antigen-experienced T cells: Lessons from the quintessential memory marker CD44. Front Immunol. 2012;3:23. doi: 10.3389/fimmu.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev Immunol. 1998;16:201–223. doi: 10.1146/annurev.immunol.16.1.201. [DOI] [PubMed] [Google Scholar]
- 27.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 28.Raines EW. Antigen-independent targeting of long-lived CD4+ cytolytic T effector cells to lesions of atherosclerosis. Circ Res. 2006;98:434–436. doi: 10.1161/01.RES.0000214353.38360.a2. [DOI] [PubMed] [Google Scholar]
- 29.Smeltz RB, Chen J, Hu-Li J, Shevach EM. Regulation of interleukin (IL)-18 receptor alpha chain expression on CD4(+) T cells during T helper (Th)1/Th2 differentiation. Critical downregulatory role of IL-4. J Exp Med. 2001;194:143–153. doi: 10.1084/jem.194.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao W, Lin J-X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol. 2011;12:551–559. doi: 10.1038/ni.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: Limiting collateral damage. J Clin Invest. 2004;114:1372–1378. doi: 10.1172/JCI23215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond) 2011;8:22. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Au-Yeung BB, Fowell DJ. A key role for Itk in both IFN gamma and IL-4 production by NKTcells. J Immunol. 2007;179:111–119. doi: 10.4049/jimmunol.179.1.111. [DOI] [PubMed] [Google Scholar]
- 34.Fuse K, Kodama M, Ito M, Okura Y, Kato K, Hanawa H, Aoki S, Aizawa Y. Polarity of helper T cell subsets represents disease nature and clinical course of experimental autoimmune myocarditis in rats. Clin Exp Immunol. 2003;134:403–408. doi: 10.1111/j.1365-2249.2003.02312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–1622. doi: 10.1161/CIRCRESAHA.114.303204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ismahil MA, Prabhu SD. Cardiac immune cell remodeling after myocardial infarction. J Mol Cell Cardiol. 2013;62:142–143. doi: 10.1016/j.yjmcc.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 38.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 41.Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234–1245. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 42.Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. 1992;90:1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosello-Lleti E, Rivera M, Bertomeu V, Cortes R, Jordan A, Gonzalez-Molina A. Interleukin-4 and cardiac fibrosis in patients with heart failure. Rev Esp Cardiol. 2007;60:777–780. [PubMed] [Google Scholar]
- 44.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248–256. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Q, Bluestone JA. The foxp3+ regulatory T cell: A jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maisel AS, Knowlton KU, Fowler P, Rearden A, Ziegler MG, Motulsky HJ, Insel PA, Michel MC. Adrenergic control of circulating lymphocyte subpopulations. Effects of congestive heart failure, dynamic exercise, and terbutaline treatment. J Clin Invest. 1990;85:462–467. doi: 10.1172/JCI114460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satoh S, Oyama J, Suematsu N, Kadokami T, Shimoyama N, Okutsu M, Inoue T, Sugano M, Makino N. Increased productivity of tumor necrosis factor-alpha in helper T cells in patients with systolic heart failure. Int J Cardiol. 2006;111:405–412. doi: 10.1016/j.ijcard.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 48.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 49.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.